

ABSTRACT
Limitations of antiretroviral therapy (ART) include poor patient adherence, drug toxicities, viral resistance, and failure to penetrate viral reservoirs. Recent developments in nanoformulated ART (nanoART) could overcome such limitations. To this end, we now report a novel effect of nanoART that facilitates drug depots within intracellular compartments at or adjacent to the sites of the viral replication cycle. Poloxamer 407-coated nanocrystals containing the protease inhibitor atazanavir (ATV) were prepared by high-pressure homogenization. These drug particles readily accumulated in human monocyte-derived macrophages (MDM). NanoATV concentrations were ∼1,000 times higher in cells than those that could be achieved by the native drug. ATV particles in late and recycling endosome compartments were seen following pulldown by immunoaffinity chromatography with Rab-specific antibodies conjugated to magnetic beads. Confocal microscopy provided cross validation by immunofluorescent staining of the compartments. Mathematical modeling validated drug-endosomal interactions. Measures of reverse transcriptase activity and HIV-1 p24 levels in culture media and cells showed that such endosomal drug concentrations enhanced antiviral responses up to 1,000-fold. We conclude that late and recycling endosomes can serve as depots for nanoATV. The colocalization of nanoATV at endosomal sites of viral assembly and its slow release sped antiretroviral activities. Long-acting nanoART can serve as a drug carrier in both cells and subcellular compartments and, as such, can facilitate viral clearance.
IMPORTANCE The need for long-acting ART is significant and highlighted by limitations in drug access, toxicity, adherence, and reservoir penetrance. We propose that targeting nanoformulated drugs to infected tissues, cells, and subcellular sites of viral replication may improve clinical outcomes. Endosomes are sites for human immunodeficiency virus assembly, and increasing ART concentrations in such sites enhances viral clearance. The current work uncovers a new mechanism by which nanoART can enhance viral clearance over native drug formulations.
INTRODUCTION
Long-acting nanoformulated antiretroviral therapy (nanoART) can result in improved patient adherence, decreased systemic toxicities, and sustained viral suppression (1–3). This is seen in nanoART's abilities to maintain consistent plasma and tissue drug levels, as demonstrated in our previous studies (4). Nonetheless, to facilitate clearance of human immunodeficiency virus type 1 (HIV-1), antiretroviral drugs need to be effectively delivered to viral sanctuaries (5). This can target persistent or restricted infection (6–8). With this in mind, our laboratories pioneered the use of monocytes and monocyte-derived macrophages (MDM) as nanoART carriers and drug depots. Macrophages can increase drug stability by preventing drug metabolic degradation, and because of their highly mobile nature, they may also be used for delivery of ART to and from lymphocytes and other viral reservoirs (9–11). How drug nanoparticles remain sequestered in macrophages for extended periods is incompletely understood. What is known is that nanoART can be delivered to endosomal organelles through clathrin-endosome pathways and can remain inside the cell for extended times (9). However, the virologic consequences of such a cell delivery system have not yet been elucidated. Investigations of nanoparticle interactions at the subcellular level remain of vital importance to the fields of long-acting antiretroviral pharmacokinetics and pharmacodynamics.
We reasoned that such mechanisms could be elucidated through investigations of nanoformulated viral protease inhibitors (PI). PI are substrate analogs for the HIV aspartyl protease enzyme involved in processing viral proteins by cleaving precursor proteins into smaller fragments and enabling the release of mature viral particles from infected cells. Once bound to the active site, they block the viral protease and in turn inhibit viral maturation, which blocks the formation of replication-competent virions (12, 13). Atazanavir (ATV), a U.S. Food and Drug Administration-approved PI for the treatment of HIV-1 infection, can selectively inhibit virus-specific processing of gag-pol polyproteins. As a consequence, PIs block viral assembly at action sites (14).
It is well known that subcellular organelles are utilized for HIV-1 assembly in mononuclear phagocytes (MP) (monocytes and tissue macrophages) (15, 16). Indeed, large caches of infectious HIV-1 released from MDM are produced in late endosomes (17). We thus reasoned that if nanoART can improve drug delivery to tissues and cells and affect viral clearance, its actions could be amplified if the PI is delivered to the late endosomal sites operative for viral assembly. Here, we demonstrate that nanoART enhances drug antiretroviral efficacy by being delivered to subcellular sites of active viral replication. By tracking endosomal nanoART transport and sequential immunoaffinity separations of cellular compartments and by developing computer-assisted mathematical models, we were able to track the antiretroviral drug activities inside macrophages. Antiretroviral responses, as measured by reverse transcriptase (RT) activity and HIV-1 p24 antigen, provided evidence for the colocalization of nanoART and progeny virus. Mathematical modeling uncovered the pathways for nanoparticle trafficking through endosomal compartments (18). Previous combinations of experimental and computational studies were limited by the inherent complexity of integrating simulated models with experimental results (19). The nanoparticle–subcellular-compartment modeling supports the idea that nanoformulations can facilitate the use of macrophages as drug carriers for ART and thus facilitate the establishment of drug depots and increase the speed of viral clearance.
MATERIALS AND METHODS
Reagents and antibodies.
ATV sulfate, purchased from Gyma Laboratories of America Inc. (Westbury, NY, USA), was free based with triethylamine. Poloxamer 407 (P407) and CF568-succinimidyl ester (CF568) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Sephadex LH-20 was obtained from GE Healthcare (Piscataway, NJ, USA). Pooled human serum was obtained from Innovative Biologics (Herndon, VA, USA). Macrophage colony-stimulating factor (M-CSF) was prepared from 5/9m alpha3-18 cells (ATCC; CRL-10154) cultured in ATCC complete growth medium as described previously (20). Rabbit anti-human antibodies to Rab5, Rab7, Rab11, and Rab14 and Alexa Fluor 488 goat anti-rabbit IgG were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Protein A/G mix magnetic beads were purchased from Millipore (Billerica, MA, USA). TRIzol reagent was obtained from Invitrogen (Grand Island, NY, USA).
NanoATV manufacture and characterization.
ATV nanoparticles were formulated by high-pressure homogenization (Avestin EmulsiFlex-C3; Avestin Inc., Ottawa, ON, Canada), using P407 to encase the drug crystals. The suspension containing free-based ATV (1% [wt/vol]) and P407 (0.5% [wt/vol]) in 10 mM HEPES buffer was premixed overnight at room temperature and then homogenized at 20,000 lb/in2 for ∼30 passes until the desired particle size of <300 nm was achieved. The size, zeta potential, and polydispersity (PDI) were determined by dynamic light scattering (DLS), using a Malvern Zetasizer Nano Series Nano-ZS (Malvern Instruments, Westborough, MA, USA). A minimum of four iterations were taken, which varied by <2%. After reaching the desired size (<300 nm), the sample was purified by centrifugation at 500 × g for 5 min to remove aggregated particles and then at 10,000 × g for 30 min to collect a purified particle pellet. The resulting particles were resuspended in a 0.2% (wt/vol) P407 surfactant solution for cell studies.
Synthesis of dye-labeled nanoATV.
For preparation of CF568-labeled nanoATV, CF568–P407 and P407 were dissolved in methanol at a weight ratio of 1:4 (6). The solvent was evaporated, and the mixture was resuspended with 10 mM HEPES to yield a 0.5% surfactant solution. Free-based ATV was added at a 1% weight ratio. The suspension was premixed overnight in a light-protected environment at room temperature. The suspension was homogenized by high-pressure homogenization and purified by centrifugation (10).
Human monocyte isolation and cultivation.
Human monocytes were obtained by leukapheresis from HIV-1-, HIV-2-, and hepatitis B virus-seronegative donors and then purified by countercurrent centrifugal elutriation. The recovered monocytes, >98% pure by Wright-stained cytosmears, were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% heat-inactivated pooled human serum, 1% glutamine, 50 μg/ml gentamicin, 10 μg/ml ciprofloxacin, and 1,000 U/ml recombinant human M-CSF for 7 days, facilitating cell differentiation into macrophages (MDM) (21).
Native and nanoformulated ATV cell uptake and retention.
MDM were treated with native ATV or nanoATV at 30 and 70 μg/ml. Uptake of drug was assessed without medium change for 24 h. Cell collection occurred at serial time points. After 24-h drug exposure, drug retention in MDM was evaluated. Adherent MDM were collected by washing three times with 1 ml of phosphate-buffered saline (PBS), followed by scraping the cells into 1 ml PBS. Samples were centrifuged at 950 × g for 10 min, and the supernatant was removed. The cell pellets were resuspended in 200 μl methanol and sonicated with a probe sonicator. The methanol extracts were centrifuged at 20,000 × g for 10 min prior to high-performance liquid chromatography (HPLC) analysis, as previously described (22). Triplicate 20-μl samples of cells were assessed by HPLC using a YMC Pack Octyl C8 column (Waters Inc., Milford, MA) with a C8 guard cartridge. The mobile phase, consisting of 47% acetonitrile-53% 25 mM KH2PO4, pH 4.15, was pumped at 0.4 ml/min with UV-visible (Vis) detection at 212 nm. ATV levels in cells were determined by peak area comparisons to those of a standard curve generated with free drug (0.025 to 100 μg/ml).
Immune isolation of endocytic subcellular compartments.
Immune isolation of endocytic compartments was performed as previously described (9). Briefly, MDM were treated with native or nanoATV for 16 h. The cells were washed three times in PBS to remove extracellular drug and then scraped into homogenization buffer [10 mM HEPES-KOH, pH 7.2, 250 mM sucrose, 1 mM EDTA, and 1 mM Mg(OAc)2]. The cells were then disrupted by 15 strokes in a Dounce homogenizer. Nuclei and unbroken cells were removed by centrifugation at 400 × g for 10 min at 4°C. Twenty microliters of slurry protein A/G paramagnetic beads conjugated to Rab5, Rab7, Rab11, or Rab14 antibodies (binding in 10% bovine serum albumin [BSA] in PBS for 12 h at 4°C) was incubated with cell supernatants. Following 24 h of incubation at 4°C, Rab5-, Rab7-, Rab11-, and Rab14-positive endocytic compartments were washed with PBS and then collected on a magnetic separator. The drug content of each compartment was determined by HPLC (9).
Western blotting.
Purification of isolated endocytic compartments was confirmed by Western blotting. Protein samples were quantified using the Pierce 660-nm Protein Assay and Prediluted Protein Assay BSA Standards as a standard curve. From each protein sample, 10 to 15 μg was loaded and electrophoresed on a NuPAGE Novex 4 to 12% Bis-Tris gel (Life Technologies), and the proteins were transferred to a polyvinylidene fluoride membrane. The membrane was blocked with 5% powdered milk-5% BSA in phosphate-buffered saline–Tween 20 (PBST) and probed with primary antibody (rabbit anti-human antibodies to Rab5, Rab7, Rab11, and Rab14; Santa Cruz), followed by secondary antibody (horseradish peroxidase [HRP]-conjugated goat anti-rabbit IgG; Novex-Life Technologies, Grand Island, NY, USA). Proteins were detected using SuperSignal West Pico Chemiluminescent substrate (Thermo Scientific) (9).
Detection of HIV-1 integration by PCR.
To estimate HIV integration, a modified version of the method developed by Liszewski and colleagues was employed (23). After 7 days of culture, MDM were treated with 100 μM native or nanoATV for 16 h and then challenged with HIV-1ADA at a multiplicity of infection (MOI) of 0.1 infectious viral particle/cell. Following viral infection for 4 h, the cells were cultured for an additional 14 days with half-medium exchanges every other day before the cells were scraped for collection. TRIzol Reagent was used to isolate DNA and RNA samples. The number of proviruses/cell was determined by a kinetic PCR assay. The standard curve was derived by running the nested-PCR protocol on several dilutions of the integrated standard nucleic acid samples (24). The concentrations of human genomes in the isolated sample DNA (or RNA) were determined by the optical density at 260 or 280 nm (OD260/280).
Antiretroviral activities.
Antiretroviral efficacy was determined by HIV-1 RT activity (21, 25). Briefly, MDM were treated with 100 μM native or nanoATV for 16 h and then challenged with HIV-1ADA at an MOI of 0.1. Following viral infection, the cells were cultured for 14 days with half-medium exchanges every other day. Medium samples were collected on days 2, 4, 6, 8, 10, 12, and 14 for measurement of progeny virion production, as assayed by RT activity. Here, 10-μl medium samples were mixed with 10 μl of a solution containing 100 mM Tris-HCl (pH 7.9), 300 mM KCl, 100 mM dithiothreitol, 0.1% NP-40, and water in a 96-well plate. The reaction mixture was incubated at 37°C for 15 min, and 25 μl of a solution containing 50 mM Tris-HCl (pH 7.9), 150 mM KCl, 5 mM dithiothreitol, 15 mM MgCl2, 0.05% NP-40, 10 μl/ml poly(A), 0.25 U/ml oligo(dT), and 10 μCi/ml [3H]dTTP was added to each well, and the plates were incubated at 37°C for 18 h. Following incubation, 50 μl of cold 10% trichloroacetic acid was added to each well, the wells were harvested onto glass fiber filters, and the filters were assessed for [3H]dTTP incorporation by beta-scintillation spectroscopy using a TopCount NXT (PerkinElmer Inc., Waltham, MA, USA).
Immunocytochemistry and confocal microscopy.
For immunofluorescence staining, cells were washed three times with PBS and fixed with 4% paraformaldehyde (PFA) at room temperature for 30 min. The cells were treated with blocking/permeabilizing solution (0.1% Triton, 5% BSA in PBS) and quenched with 50 mM NH4Cl for 15 min. The cells were washed once with 0.1% Triton in PBS and sequentially incubated with primary and secondary antibodies at room temperature. Slides were covered in ProLong Gold AntiFade reagent with DAPI (4′,6-diamidino-2-phenylindole) and imaged using a 63× oil lens on an LSM 510 confocal microscope (Carl Zeiss Microimaging, Inc., Dublin, CA, USA) (26). The immunofluorescence was quantitated, and the percent overlap was determined using ImageJ software, the JACoP plug-in for percent overlap, and Zeiss LSM 510 Image browser AIM software version 4.2 for determining the number of pixels and the mean intensity of each channel, as previously described (6). The results are represented as means ± standard errors of the mean (SEM).
HIV-1 p24 staining.
Cells in different treatment groups were fixed with 4% phosphate-buffered PFA for 15 min at room temperature. The fixed cells were blocked with 10% BSA in PBS containing 1% Triton X-100 for 30 min at room temperature and incubated with mouse monoclonal antibodies to HIV-1 p24 (1:100; Dako, Carpinteria, CA, USA) for 3 h at room temperature. Binding of HIV-1 p24 antibody was detected using a Dako EnVision+ System, HRP-labeled polymer anti-mouse secondary antibody, and diaminobenzidine staining. Cell nuclei were counterstained with hematoxylin for 60 s. Images were taken using a Nikon TE300 microscope with a 40× objective. Quantitation of immunostaining was performed by densitometry using ImagePro Plus, v. 4.0 (27).
Statistics.
DNA, RNA, and HIV-1 p24 staining data were analyzed by one-way analysis of variance (ANOVA) and Tukey's multiple-comparison test using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA). HIV-1 RT activity in isolated Rab compartments following native or nanoATV treatment was analyzed by 2-way ANOVA and the Bonferroni multiple-comparisons test. Differences were considered significant at a P value of <0.05.
RESULTS
NanoART characterization.
The nanoATV particles were formulated as a nanosized drug crystals from free-base ATV and P407. Physical properties, including size, PDI, and zeta potential, were measured by DLS. The particle size was 371.7 nm, with a PDI of 0.194, which indicated that the majority of nanoATV particles were homogeneous. The zeta potential was −28.9 mV, and the negative charge was contributed by P407. Scanning electron microscopy revealed smooth rod-like morphologies for the nanoATV particles (data not shown) and confirmed the size measurements and distribution, which were consistent with our previous studies (9, 10).
Native and nanoATV cell uptake and retention.
MDM uptake and retention of drug were assessed by HPLC. Cells were exposed to native ATV or nanoATV for 24 h at 30 and 70 μg/ml for each treatment. Previous studies in our laboratory had shown that these concentrations of native and nanoATV are not toxic to MDM (5, 6). Substantial (about 3 orders of magnitude) differences were observed in the rate and extent of drug uptake between the native and nanoATV treatment groups (Fig. 1A). The uptake of nanoATV was found to increase with time, and the maximum uptake was observed at 16 h for the 70-μg/ml treatment group (53.5 μg ATV/106 cells) and 24 h for the 30-μg/ml group (23.7 μg ATV/106 cells). However, uptake of less than 0.1 μg ATV/106 cells was observed at 24 h for native ATV at either concentration, and this was not time dependent. Drug retention in MDM was determined 24 h after treatment. In MDM treated with nanoATV, drug levels of 28.5 μg/106 cells and 14.0 μg/106 cells in the 70- and 30-μg/ml treatment groups, respectively, were sustained over 24 h. Sustained release was expected for up to 15 days with nanoparticle treatment based on previous studies (6, 10). Much less ATV was detected 24 h following treatment with native ATV. Using these results, uptake and retention of native versus nanoART was mathematically modeled using difference equations represented by queues, where the input to the queue is described by one difference equation and the output by another difference equation (Fig. 1B). Connecting those queues into a network thus models a set of difference equations, where the lengths of the queues provide instantaneous solutions. Such a queuing network can be easily used to model scenarios where the arrival/service rates change dynamically depending on the lengths of some queues in the system or where changes happen randomly. This approach, successfully applied to modeling nonviral gene delivery and transfection processes (28), was implemented to model the flow of nanoparticles into and out of the cell.
FIG 1.

Comparison of native and nanoATV cellular drug uptake and antiretroviral activities. (A) Time course for MDM uptake of native or nanoATV. MDM were treated with native or nanoATV for 24 h. (B) Computational simulation of the time course of MDM drug uptake and retention. After 24 h treatment with native or nanoATV, cells were washed with PBS and treated with fresh medium for 24 h. (C and D) HIV-1 DNA (C) and RNA (D) levels were quantitated 14 days after viral infection (MOI = 0.1) of MDM loaded with various concentrations of native or nanoATV. The units for viral DNA and RNA are copies/103 cells and copies/cell, respectively. (E and F) HIV-1 RT activity was determined 14 days after HIV-1 infection in MDM loaded with various concentrations of native ATV (E) or nanoATV (F). The data are expressed as averages ± SEM for 3 replicate wells. The data presented are from one of three representative experiments. *, significantly different from non-drug-treated cells (P < 0.05); †, significantly different from the respective native-ATV treatment (P < 0.05).
Antiretroviral activities of native and nanoATV.
To estimate the levels of HIV-1 DNA and RNA, PCR amplification assays were employed. After 7 days of cultivation, MDM were treated with 100 μM native or nanoATV for 16 h and then challenged with HIV-1ADA at an MOI of 0.1. Following infection, cells were cultured for 14 days with half-medium exchanges every other day before cell collection. Isolated total cell DNA or RNA was quantitated by kinetic PCR, and nucleic acid levels were calculated using standard curves made by linear regression analyses. These data are shown in Fig. 1C and D. At day 14 after HIV-1 challenge, 1.15 × 104 viral DNA copies/103 MDM and 1.05 × 104 viral RNA copies/MDM were determined in infected samples. The number of viral copies paralleled the concentrations of native and nanoATV, where 1 μM native ATV failed to show significant antiretroviral activity. In contrast 1 μM nanoATV effectively suppressed viral DNA and RNA to 145.7 copies/103 MDM and 141.2 copies/MDM, respectively. Suppression of viral DNA and RNA was greatest with 100 μM nanoATV, where viral DNA and RNA copy numbers were 29.6/103 MDM and 36.1/MDM, respectively, indicating nearly complete suppression of viral replication.
To determine the levels of progeny virus produced in native-ATV- and nanoATV-treated cultures, we treated MDM with native or nanoATV in increasing drug concentrations of 0.01, 0.1, 1, 10, and 100 μM. Treatment was for 16 h, with subsequent HIV-1ADA challenge at an MOI of 0.1. The same formulation used for cell uptake and retention was tested for antiretroviral activities to ensure that the results were comparable. Infected cells were cultured for 14 days with a half-medium change every other day. Culture supernatants were collected at days 2, 4, 6, 8, 10, 12, and 14 for determination of progeny virion production assayed by RT activity. A dose-dependent effect on RT activity for native and nanoATV at all time points was observed (Fig. 1E and F, respectively). Clear differences between native and nanoATV were seen. For native-ATV-treated cells, suppression of RT activity was maintained over time only at 100 μM; most of the other native-drug concentrations failed to provide substantial viral suppression compared to untreated HIV-1-infected controls beyond day 8. In contrast, nanoATV reduced RT activity effectively over time, starting at 1 μM. A 2-log-unit increase in potency for viral suppression was seen with nanoATV compared to native ATV.
NanoATV effects on HIV-1 p24 antigen.
Cross validation of the results described above was done through evaluation of viral-antigen expression following native and nanoATV treatments in infected cell cultures. The expression of HIV-1 p24 antigen was used to determine antiretroviral activity in MDM that were treated with native or nanoATV and subsequently challenged with HIV-1ADA at an MOI of 0.1. Evaluation of p24 expression in infected MDM treated with native or nanoATV showed a concentration-dependent effect on HIV-1 p24 expression at day 14 following viral exposure. Higher treatment concentrations resulted in lower p24 expression for both treatment groups (Fig. 2A to H). Formation of multinucleated giant cells was also observed. Significant differences between native and nanoATV were seen at all concentrations (P < 0.05). The expression of HIV-1 p24 antigen decreased slightly in cells treated with 1 or 10 μM native ATV, but an 89.4% decrease was found in cells treated with 100 μM native ATV (Fig. 2I). HIV-1 p24 expression in cells was reduced at all concentrations of nanoATV. Viral suppression to nearly asymptomatic levels was achieved with nanoATV treatment, i.e., 90.9, 94.2 and 95.7% for 1, 10, and 100 μM nanoATV treatment, respectively (Fig. 2I).
FIG 2.
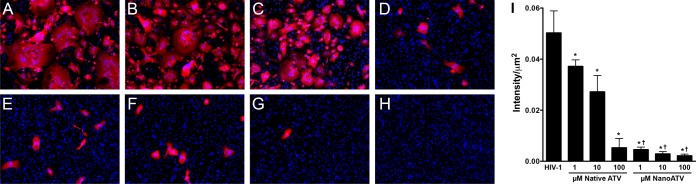
HIV-1 p24 staining of virus-infected MDM pretreated with native or nanoATV. MDM were treated with native or nanoATV for 16 h and then challenged with HIV-1 (MOI = 0.1). Fourteen days later, the cells were fixed and stained for HIV-1 p24 antigen. The treatment groups were HIV-1-infected controls (A); 1, 10, and 100 μM native ATV (B to D, respectively); 1, 10, and 100 μM nanoATV (E to G, respectively); and uninfected MDM (H). Red, HIV-1 p24 staining; blue, cell nuclei stained with DAPI. (I) Quantitation of immunostaining by densitometry using ImagePro Plus v. 4.0. The data are expressed as averages and SEM. *, significant differences from HIV-1-infected controls (P < 0.05); †, significant differences from native-ATV treatment (P < 0.05).
NanoATV subcellular distributions.
Confocal microscopy enabled both visualization and quantitation of the subcellular nanoATV distribution in early (Rab5), late (Rab7), and recycling (Rab11 and Rab14) endocytic compartments (29). In these experiments, MDM were treated with 100 μM nanoATV fluorescently labeled with CF568. Immunostaining was performed 16 h after particle incubation for visualization of endocytic compartments and nanoATV colocalization. These experiments showed nanoATV distributed in a punctate pattern throughout the cytoplasm and perinuclear cell regions. NanoATV was found predominantly in late and recycling endosomes of uninfected macrophages (Fig. 3A). Quantitation of the fluorophore-labeled Rab species in endosomes with CF568-labeled nanoATV was done by adapting Pearson's correlation coefficient to measure only positive colocalization coefficients (i.e., M1 and M2) of the two fluorophores at different emission wavelengths. Based on this, we showed significant accumulation (P < 0.001) of nanoATV within Rab5-positive (35.4% ± 5%), Rab7-positive (65.1% ± 7%), Rab11-positive (70.3% ± 11%), and Rab14-positive (56.9% ± 12%) compartments (Fig. 3C). To assess subcellular colocalization of nanoATV in HIV-1-infected cells, MDM were treated with 100 μM CF568-labeled nanoATV for 16 h following 14 days of HIV-1ADA infection. Identical Rab-specific antibodies were employed to immunostain endocytic compartments in multinucleated giant cells. Quantitation of fluorophore in Rab compartments showed clear colocalization between virus and the nanoparticles (Fig. 3B and D). The greatest amount of colocalization was found in Rab7-immunopositive late endosomes. These data indicated that after HIV-1 infection, nanoATV persists in late endosomal compartments, the site of active viral assembly, but at half the level present in recycling endosomes of uninfected macrophages (Fig. 3C versus D).
FIG 3.

NanoATV subcellular distribution. (A and B) Uninfected MDM (A) and HIV-1-infected MDM (B) were treated with 100 μM CF568-labeled nanoATV (red) (28) for 16 h and then immunostained with Rab5, Rab7, Rab11, or Rab14 antibodies and Alexa Fluor 488-labeled secondary antibody (green) to visualize nanoparticle and organelle coregistration. The arrowheads indicate overlap (yellow) of nanoATV and Rab compartments. DAPI (blue) stain indicates cell nuclei. (C and D) Quantitation of the percent overlap of nanoATV and Rab compartments in uninfected (C) and HIV-1-infected (D) MDM. The data are expressed as averages and SEM of 10 replicates.
NanoATV trafficking in endosomal subcellular comparments.
To assess the location of nanoATV at the subcellular level, we analyzed individual endosomal compartments within MDM by immunoaffinity techniques. MDM exposed to nanoATV were mechanically disrupted at specific time points, and subcellular compartments, including early, late, and recycling endosomes, were immunoisolated using protein A/G paramagnetic beads conjugated to Rab5, Rab7, Rab11, or Rab14 antibodies. Endocytic compartments bound to beads were collected by magnetic separation, digitally imaged, and then analyzed by HPLC for drug content. The purity of the isolated endocytic compartments was assessed by Western blotting, as shown in Fig. 4A. Specific endocytic compartments were isolated by Rab5, Rab7, Rab11, or Rab14 antibodies. Western blots demonstrated little contamination of Rab5, Rab11, and Rab14 compartments with Rab7 protein and Rab5, Rab7, and Rab11 compartments with Rab14 protein, indicating the high purity of the isolated endosomal compartments. Time-dependent endocytic uptake was observed in all labeled endosomes, and the data were used to generate 48-h subcellular uptake curves (Fig. 4B). At 48 h, the maximum nanoATV uptake was 10.6 ± 1.4 μg/106 cells in Rab14 (early recycling endosome) compartments, followed by 8.3 ± 0.7 μg/million cells in Rab11 (late recycling endosome) compartments. The lowest nanoATV concentration was found in Rab5 compartments, representing early endosomes, regardless of HIV-1 infection, which was consistent with the colocalization confocal microscopy tests.
FIG 4.

Kinetics of nanoparticle trafficking in subcellular compartments. (A) The purity of Rab5, Rab7, Rab11, and Rab14 isolated compartments was confirmed by Western blotting using antibodies to Rab7 and Rab14 proteins. (B) ATV levels in different endosomal compartments in MDM exposed to 100 μM nanoATV for 48 h. (C) Simulated subcellular nanoATV uptake in different endosomal compartments over the same time frame. (D) Simulated subcellular nanoATV uptake pathways. The data are expressed as averages ± SEM of 3 replicates.
Simulation of nanoATV at the subcellular level.
Mathematical means to simulate drug particle uptake and retention at both the cellular and subcellular levels were developed based upon our results. These models demonstrated that trafficking of nanoformulated particles in endosomal cell compartments provided an accurate and reliable view of their behavior in real time. The drug levels in specific compartments at different time points, together with the confirmation that more nanoATV than native ATV could accumulate in late and recycling endosomes, proved these to be the major subcellular depots for nanoATV (Fig. 4C). The modeled flow of nanoparticles is presented in Fig. 4D, where outside the cell and the specific subcellular compartments involved in trafficking of the majority of nanoATV are represented as the graph nodes: “Out” is the outside of the cell, and Rab5, Rab7, Rab11, and Rab14 denote the compartments identified by the respective Rab proteins. The instantaneous concentration of nanoATV outside the cell, Q1, is associated with the node Out, and the instantaneous concentrations of the respective Rab proteins, Q2 to Q5, are associated with the nodes Rab5, Rab7, Rab11, and Rab14. It has been noted that the total number of observed nanoATV/fluorescence-labeled particles in the experiment decreases over time. This has been taken into account in the model as an open path marked δ in the graph in Fig. 4D.
Based on the graph topology, the time changes of the concentrations in the model are controlled by the set of 5 difference equations: Q1i = Q1i − 1 − V1i − 1ΔT + V6i − 1ΔT + V7i − 1ΔT; Q2i = Q2i − 1 + V1i − 1ΔT − V2i − 1ΔT − V3i − 1ΔT − V4i − 1ΔT; Q3i = Q3i − 1 + V3i − 1ΔT − V6i − 1ΔT; Q4i = Q4i − 1 + V2i − 1ΔT − V5i − 1ΔT; and Q5i = Q5i − 1 + V4i − 1ΔT + V5i − 1ΔT − V7i − 1ΔT − V8i − 1ΔT, where ΔT denotes the time increment in simulations and (·)j denotes the value of (·) at the jth step of simulation.
Based on the experimental results, using multiple linear regression and curve-fitting methods implemented as standard MATLAB functions, the following relationships between the concentrations Q1 to Q5 and the rates of changes, V1 to V8, were established: V1 = 0.04269Q1 + 0.7778; V2 = 0.1000Q2 − 0.085Q3 + 0.0814; V3 = 0.0367Q2 − 0.2714Q4 + 2.238; V4 = 0.003Q1 + 0.0367Q2 − 0.155Q3 − 0.1263Q5 + 0.7366; V5 = −0.003Q1 + 0.155Q3 − 0.33; V6 = −0.0031Q1 + 0.2344Q4 − 0.22; V7 = −0.0031Q1 + 0.01785Q5 − 0.112; and V8 = 0.01exp (0.01t)Q5, where t is the time from the experiment start. The units for all the rates are μg/106 cells/h and μg/106 cells for concentrations. The simulation model based on the set of difference equations was implemented in MATLAB using a technique similar to those applied previously (28).
Subcellular antiretroviral activity.
To determine in which compartments the virus persists and the antiretroviral efficacy of our nanoATV at a subcellular level, MDM were challenged with HIV-1ADA for 4 h at an MOI of 0.1 and then treated with 100 μM native ATV or nanoATV for 16 h. MDM were mechanically disrupted at days 7 and 14, and subcellular compartments were immunoisolated using protein A/G paramagnetic beads conjugated to Rab5, Rab7, Rab11, or Rab14 antibodies. Endocytic compartments bound to beads were collected by magnetic separation for the RT assay. A schematic for nanoATV and HIV-1 subcellular trafficking is illustrated in Fig. 5A. The highest RT activity was found in Rab11 compartments, representing recycling endosomes, and Rab7 compartments, representing late endosomes, in nontreated cells after 7 and 14 days of HIV-1 challenge (Fig. 5B), respectively, which suggested that the virus was located mainly in these endosomal compartments. NanoATV and native ATV at 100 μM both exhibited antiretroviral efficacy; however, the suppression of RT activity in all endosome compartments was greater with nanoATV than native ATV, with a 2-fold difference in RT suppression in Rab7 and Rab11 fractions at 14 days. Based on the confocal results, greater overlap of nanoATV and cellular compartments was found in late and recycling endosomes, which could explain why nanoATV was more effective in suppressing HIV-1 in these compartments.
FIG 5.

Intracellular pathways for HIV-1 progeny virion production and nanoATV trafficking. (A) Schematic diagram of HIV-1 and nanoATV trafficking. (B) HIV RT activity in subcellular endosomal compartments. MDM were infected with HIV-1 for 4 h and then treated with 100 μM native or nanoATV for 16 h. Endosomal compartments were isolated using specific Rab antibody-coated magnetic beads on day 7 and day 14 after infection, and RT activity was measured in the endosomal compartments. R5, R7, R11, and R14 represent Rab5, Rab7, Rab11, and Rab14, respectively. The data are expressed as the averages of 5 experimental replicates.
DISCUSSION
Nanocrystals are broadly used for drug delivery due to their high drug-loading capacity, increased dissolution in solution, and enhanced bioavailability (30–33). Our previous studies described the manufacture and uptake of P407-coated nanoATV by homogenization and demonstrated that its physical properties, such as size, surface charge, and shape, can facilitate macrophage uptake (6). Indeed, we demonstrate in the present study that nanoATV is readily internalized by macrophages and produces sustained drug release (6). This serves to improve drug efficacy by increasing subcellular bioavailability. Nanosize drug crystals coated with surfactant exhibit enhanced cellular uptake and facilitate the cellular maintenance of drug particles for prolonged times. Particle coating serves to increase intracellular drug stability. Notably, nanoATV leads to effective inhibition of HIV-1 replication for prolonged times. In contrast to native ATV, nanoATV shows weeks of viral suppression linked to its abilities to increase drug delivery to subcellular macrophage compartments. This is facilitated by sustained antiretroviral drug release rates.
Interestingly, HIV-1 infection affects the size and or number of endosomes as a reflection of Rab protein expression (G. Zhang, unpublished observation). We posit that such virus-induced alterations in endosomes are biologically relevant to sustain viral growth. Indeed, endosomes are sites of active viral assembly and are the cellular substructures where large numbers of virions accumulate during productive infection (17). Such a function has added strategic advantages for the nanoformulated antiretroviral drug, as it enables the drug particle to reside at the specific site where viral maturation occurs in the cell. Taken together, such events certainly facilitate antiretroviral drug activities. This may also be applicable to a broad number of endosomal compartments that affect “transport” of antimicrobial drugs and is likely a common pathway for macrophage scavenging functions. Nevertheless, we also accept that the pathways for endosomal trafficking and HIV are not perfectly congruent. Further investigations will determine the mechanisms behind each trafficking scheme. Such observations, nonetheless, provide the means to improve antimicrobial therapies beyond what is reported in the present study.
Over the past decade, our laboratory has pioneered the development of long-acting nanoART. NanoATV serves as a model for macrophage-based nanoparticle drug delivery, as descriptions of cell uptake and intracellular localization of ART content parallel one another. To better understand formulation trafficking at the cellular and subcellular levels, we developed a mathematical means to simulate endosomal trafficking. The model is simple to implement and can provide assessment of putative drug activities based on changes in the concentration of nanoATV and/or Rab proteins in real time. Based on the model, divergent behaviors between native and nanoATV were readily observed. NanoATV was taken up efficiently into macrophages and effected sustained drug release, while native ATV was minimally internalized. This helps to explain why nanoATV possesses distinct long-acting antiretroviral efficacy. Through our subcellular uptake simulation model, nanoATV subcellular distributions were visualized, and late and recycling endosomes were considered the nanoATV depots. Indeed, large amounts of nanoATV were deposited in these endocytic compartments. However, it is also noted that differences exist between HIV-1 and nanoparticle drug subcellular accumulation, including recycling endosomes. Whether this is a clever means for the virus to alter the transport properties of nanoparticles or a compensatory host mechanism in an attempt to contain the virus is not yet known. Further studies are certainly needed to assess the unique transport properties of the virus and the drug-laden particle.
Harnessing macrophage transport properties for drug delivery can improve clinical drug responses. Indeed, cell-based nanocarriers have been developed for not only cancer chemotherapy, but also a wide range of microbial infections (34–36). Macrophage-based nanomedicine delivery schemes may have several advantages over more conventional drug delivery, including enhanced cure rates, reduced side effects, increased drug stability, and effective subcellular targeting (26, 27, 37, 38). Using macrophages as a nanocarriage vehicle can permit investigation of nanoATV entry, intracellular trafficking, and drug release kinetics.
NanoATV contained within endocytic compartments provides a protected environment to facilitate drug release with unaltered antiretroviral activities. Both late and recycling endosomes were able to retain a large number of nanoparticles. Macrophages likely play a vital role in HIV pathogenesis, as they are among the first cells infected (39, 40). Within an infected macrophage, virions are formed in a temporally and spatially coordinated manner where the components that make up the virus assemble in association with a specific cellular membrane from which the viral envelope is derived (17). HIV-1 virions bud directly into late endosomes and thereby acquire late endosomal membrane proteins, such as Rab7, LAMP-1, and CD63 (16). Macrophages secrete virions from virus-containing intracellular vacuoles (41). Late endosomes are the principal locations for HIV assembly (42–46). Importantly, nanoATV retains full antiretroviral activity in late endosomes, as RT activity was substantially decreased in Rab7 and Rab11 vesicles.
In summary our results demonstrate that nanoATV and HIV target overlapping subcellular compartments. Entry of virus and particles inside macrophages is facilitated through clathrin-mediated pathways (47–50). NanoATV particles are stored within late endosomes or recycling compartments, which serves to minimize intracellular degradation. For release, nanoATV is slowly recycled to the plasma membrane (51–54). This provides a means of escape from phagolysosomal degradation and effective delivery of drug to action sites, which in the end has a net effect of improved therapeutic efficacy.
Conclusions.
NanoATV has now been demonstrated to effect increases in cellular drug uptake and retention in macrophages. The long-acting antiretroviral efficacy of the crystalline nanoformulated drug is significantly enhanced over its native-drug counterpart. Macrophages act as carriers of nanoATV to improve drug bioavailability. Computer-assisted mathematical modeling can simulate subcellular trafficking of nanoATV to late and recycling endosomes that serve as drug particle depots. Overall, nanoATV and HIV utilize similar subcellular pathways. Delivery of drug through subcellular compartments increases its antiretroviral responses.
ACKNOWLEDGMENTS
We thank Janice Taylor and James R. Talaska of the Confocal Laser Scanning Core facility at UNMC for assistance with confocal microscopy.
This work was supported by the University of Nebraska Foundation, which includes individual donations from Carol Swarts and Frances and Louie Blumkin; the Vice Chancellor's office of the University of Nebraska Medical Center; ViiV Healthcare; and National Institutes of Health grants P01 DA028555, R01 NS36126, P01 NS31492, 2R01 NS034239, P01 MH64570, P01 NS43985, P30 MH062261, and R01 AG043540.
Footnotes
Published ahead of print 11 June 2014
REFERENCES
- 1.Wainberg MA. 2011. AIDS: drugs that prevent HIV infection. Nature 469:306–307. 10.1038/469306a [DOI] [PubMed] [Google Scholar]
- 2.Pretorius E, Klinker H, Rosenkranz B. 2011. The role of therapeutic drug monitoring in the management of patients with human immunodeficiency virus infection. Ther. Drug Monit. 33:265–274. 10.1097/FTD.0b013e31821b42d1 [DOI] [PubMed] [Google Scholar]
- 3.Beyrer C, Malinowska-Sempruch K, Kamarulzaman A, Kazatchkine M, Sidibe M, Strathdee SA. 2010. Time to act: a call for comprehensive responses to HIV in people who use drugs. Lancet 376:551–563. 10.1016/S0140-6736(10)60928-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dash P, Gendelman HE, Roy U, Balkundi S, Alnouti Y, Mosley RLM, Gelbard H, McMillan J, Gorantla S, Poluektova L. 2012. Long-acting nanoformulated antiretroviral therapy elicits potent antiretroviral and neuroprotective responses in HIV-1 infected humanized mice. AIDS 26:2135–2144. 10.1097/QAD.0b013e328357f5ad [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nowacek AS, McMillan J, Miller R, Anderson A, Rabinow B, Gendelman HE. 2010. Nanoformulated antiretroviral drug combinations extend drug release and antiretroviral responses in HIV-1-infected macrophages: implications for neuroAIDS therapeutics. J. Neuroimmune Pharmacol. 5:592–601. 10.1007/s11481-010-9198-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Puligujja P, McMillan J, Kendrick L, Li T, Balkundi S, Smith N, Veerubhotla RS, Edagwa BJ, Kabanov AV, Bronich T, Gendelman HE, Liu XM. 2013. Macrophage folate receptor-targeted antiretroviral therapy facilitates drug entry, retention, antiretroviral activities and biodistribution for reduction of human immunodeficiency virus infections. Nanomedicine 9:1263–1273. 10.1016/j.nano.2013.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X, Li J, Wang Y, Cho KJ, Kim G, Gjyrezi A, Koenig L, Giannakakou P, Shin HJ, Tighiouart M, Nie S, Chen ZG, Shin DM. 2009. HFT-T, a targeting nanoparticle, enhances specific delivery of paclitaxel to folate receptor-positive tumors. ACS Nano. 3:3165–3174. 10.1021/nn900649v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kabanov AV, Gendelman HE. 2007. Nanomedicine in the diagnosis and therapy of neurodegenerative disorders. Prog. Polym. Sci. 32:1054–1082. 10.1016/j.progpolymsci.2007.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kadiu I, Nowacek A, McMillan J, Gendelman HE. 2011. Macrophage endocytic trafficking of antiretroviral nanoparticles. Nanomedicine 6:975–994. 10.2217/nnm.11.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nowacek AS, Miller RL, McMillan J, Kanmogne G, Kanmogne M, Mosley RL, Ma Z, Graham S, Chaubal M, Werling J, Rabinow B, Dou H, Gendelman HE. 2009. NanoART synthesis, characterization, uptake, release and toxicology for human monocyte-macrophage drug delivery. Nanomedicine 4:903–917. 10.2217/nnm.09.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kingsley JD, Dou H, Morehead J, Rabinow B, Gendelman HE, Destache CJ. 2006. Nanotechnology: a focus on nanoparticles as a drug delivery system. J. Neuroimmune Pharmacol. 1:340–350. 10.1007/s11481-006-9032-4 [DOI] [PubMed] [Google Scholar]
- 12.Wood R. 2008. Atazanavir: its role in HIV treatment. Expert Rev. Anti Infect. Ther. 6:785–796. 10.1586/14787210.6.6.785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwarcz SK, Hsu LC, Vittinghoff E, Katz MH. 2000. Impact of protease inhibitors and other antiretroviral treatments on acquired immunodeficiency syndrome survival in San Francisco, California, 1987–1996. Am. J. Epidemiol. 152:178–185. 10.1093/aje/152.2.178 [DOI] [PubMed] [Google Scholar]
- 14.Arribas JR, Eron J. 2013. Advances in antiretroviral therapy. Curr. Opin. HIV AIDS 8:341–349. 10.1097/COH.0b013e328361fabd [DOI] [PubMed] [Google Scholar]
- 15.Rowell JF, Stanhope PE, Siliciano RF. 1995. Endocytosis of endogenously synthesized HIV-1 envelope protein. Mechanism and role in processing for association with class II MHC. J. Immunol. 155:473–488 [PubMed] [Google Scholar]
- 16.Raposo G, Moore M, Innes D, Leijendekker R, Leigh-Brown A, Benaroch P, Geuze H. 2002. Human macrophages accumulate HIV-1 particles in MHC II compartments. Traffic 3:718–729. 10.1034/j.1600-0854.2002.31004.x [DOI] [PubMed] [Google Scholar]
- 17.Pelchen-Matthews A, Kramer B, Marsh M. 2003. Infectious HIV-1 assembles in late endosomes in primary macrophages. J. Cell Biol. 162:443–455. 10.1083/jcb.200304008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goncalves E, Bucher J, Ryll A, Niklas J, Mauch K, Klamt S, Rocha M, Saez-Rodriguez J. 2013. Bridging the layers: towards integration of signal transduction, regulation and metabolism into mathematical models. Mol. Biosyst. 9:1576–1583. 10.1039/c3mb25489e [DOI] [PubMed] [Google Scholar]
- 19.Karr JR, Sanghvi JC, Macklin DN, Gutschow MV, Jacobs JM, Bolival B, Jr, Assad-Garcia N, Glass JI, Covert MW. 2012. A whole-cell computational model predicts phenotype from genotype. Cell 150:389–401. 10.1016/j.cell.2012.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clark SC. 1989. Interleukin-6. Multiple activities in regulation of the hematopoietic and immune systems. Ann. N. Y. Acad. Sci. 557:438–443 [PubMed] [Google Scholar]
- 21.Gendelman HE, Orenstein JM, Martin MA, Ferrua C, Mitra R, Phipps T, Wahl LA, Lane HC, Fauci AS, Burke DS, Skillman D, Meltzer MS. 1988. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J. Exp. Med. 167:1428–1441. 10.1084/jem.167.4.1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nowacek AS, Balkundi S, McMillan J, Roy U, Martinez-Skinner A, Mosley RL, Kanmogne G, Kabanov AV, Bronich T, Gendelman HE. 2011. Analyses of nanoformulated antiretroviral drug charge, size, shape and content for uptake, drug release and antiviral activities in human monocyte-derived macrophages. J. Control Release 150:204–211. 10.1016/j.jconrel.2010.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liszewski MK, Yu JJ, O'Doherty U. 2009. Detecting HIV-1 integration by repetitive-sampling Alu-gag PCR. Methods 47:254–260. 10.1016/j.ymeth.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar R, Vandegraaff N, Mundy L, Burrell CJ, Li P. 2002. Evaluation of PCR-based methods for the quantitation of integrated HIV-1 DNA. J. Virol. Methods 105:233–246. 10.1016/S0166-0934(02)00105-2 [DOI] [PubMed] [Google Scholar]
- 25.Kalter DC, Nakamura M, Turpin JA, Baca LM, Hoover DL, Dieffenbach C, Ralph P, Gendelman HE, Meltzer MS. 1991. Enhanced HIV replication in macrophage colony-stimulating factor-treated monocytes. J. Immunol. 146:298–306 [PubMed] [Google Scholar]
- 26.Dou H, Destache CJ, Morehead JR, Mosley RL, Boska MD, Kingsley J, Gorantla S, Poluektova L, Nelson JA, Chaubal M, Werling J, Kipp J, Rabinow BE, Gendelman HE. 2006. Development of a macrophage-based nanoparticle platform for antiretroviral drug delivery. Blood 108:2827–2835. 10.1182/blood-2006-03-012534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dou H, Grotepas CB, McMillan JM, Destache CJ, Chaubal M, Werling J, Kipp J, Rabinow B, Gendelman HE. 2009. Macrophage delivery of nanoformulated antiretroviral drug to the brain in a murine model of neuroAIDS. J. Immunol. 183:661–669. 10.4049/jimmunol.0900274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin TM, Wysocki BJ, Beyersdorf JP, Wysocki TA, Pannier AK. Integrating mitosis, toxicity, and transgene expression in a telecommunications packet-switched network model of lipoplex-mediated gene delivery. Biotechnol. Bioeng., in press [DOI] [PubMed] [Google Scholar]
- 29.Hutagalung AH, Novick PJ. 2011. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 91:119–149. 10.1152/physrev.00059.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Junghanns JU, Muller RH. 2008. Nanocrystal technology, drug delivery and clinical applications. Int. J. Nanomedicine 3:295–309. 10.2147/IJN.S595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar L, Verma S, Prasad DN, Bhardwaj A, Vaidya B, Jain AK. 25 February 2014. Nanotechnology: a magic bullet for HIV AIDS treatment. Artif. Cells Nanomed. Biotechnol. 10.3109/21691401.2014.883400 [DOI] [PubMed] [Google Scholar]
- 32.Ramana LN, Sharma S, Sethuraman S, Ranga U, Krishnan UM. 2014. Evaluation of chitosan nanoformulations as potent anti-HIV therapeutic systems. Biochim. Biophys. Acta 1840:476–484. 10.1016/j.bbagen.2013.10.002 [DOI] [PubMed] [Google Scholar]
- 33.Rabinow BE. 2004. Nanosuspensions in drug delivery. Nat. Rev. Drug Discov. 3:785–796. 10.1038/nrd1494 [DOI] [PubMed] [Google Scholar]
- 34.Gollapudi K, Galet C, Grogan T, Zhang H, Said JW, Huang J, Elashoff D, Freedland SJ, Rettig M, Aronson WJ. 2013. Association between tumor-associated macrophage infiltration, high grade prostate cancer, and biochemical recurrence after radical prostatectomy. Am. J. Cancer Res. 3:523–529 [PMC free article] [PubMed] [Google Scholar]
- 35.Schulz R, Moll UM. 2014. Targeting the heat shock protein 90: a rational way to inhibit macrophage migration inhibitory factor function in cancer. Curr. Opin. Oncol. 26:108–113. 10.1097/CCO.0000000000000036 [DOI] [PubMed] [Google Scholar]
- 36.Goshima F, Esaki S, Luo C, Kamakura M, Kimura H, Nishiyama Y. 2014. Oncolytic viral therapy with a combination of HF10, a herpes simplex virus type 1 variant and granulocyte-macrophage colony-stimulating factor for murine ovarian cancer. Int. J. Cancer 134:2865–2877. 10.1002/ijc.28631 [DOI] [PubMed] [Google Scholar]
- 37.Gautam N, Roy U, Balkundi S, Puligujja P, Guo D, Smith N, Liu XM, Lamberty B, Morsey B, Fox HS, McMillan J, Gendelman HE, Alnouti Y. 2013. Preclinical pharmacokinetics and tissue distribution of long-acting nanoformulated antiretroviral therapy. Antimicrob. Agents Chemother. 57:3110–3120. 10.1128/AAC.00267-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beduneau A, Ma Z, Grotepas CB, Kabanov A, Rabinow BE, Gong N, Mosley RL, Dou H, Boska MD, Gendelman HE. 2009. Facilitated monocyte-macrophage uptake and tissue distribution of superparmagnetic iron-oxide nanoparticles. PLoS One 4:e4343. 10.1371/journal.pone.0004343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meltzer MS, Gendelman HE. 1992. Mononuclear phagocytes as targets, tissue reservoirs, and immunoregulatory cells in human immunodeficiency virus disease. Curr. Top. Microbiol. Immunol. 181:239–263 [DOI] [PubMed] [Google Scholar]
- 40.Meltzer MS, Nakamura M, Hansen BD, Turpin JA, Kalter DC, Gendelman HE. 1990. Macrophages as susceptible targets for HIV infection, persistent viral reservoirs in tissue, and key immunoregulatory cells that control levels of virus replication and extent of disease. AIDS Res. Hum. Retroviruses 6:967–971 [DOI] [PubMed] [Google Scholar]
- 41.Feng Y, Press B, Wandinger-Ness A. 1995. Rab 7: an important regulator of late endocytic membrane traffic. J. Cell Biol. 131:1435–1452. 10.1083/jcb.131.6.1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meng B, Lever AM. 2013. Wrapping up the bad news: HIV assembly and release. Retrovirology 10:5. 10.1186/1742-4690-10-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bell NM, Lever AM. 2013. HIV Gag polyprotein: processing and early viral particle assembly. Trends Microbiol. 21:136–144. 10.1016/j.tim.2012.11.006 [DOI] [PubMed] [Google Scholar]
- 44.Bell NM, L'Hernault A, Murat P, Richards JE, Lever AM, Balasubramanian S. 2013. Targeting RNA-protein interactions within the human immunodeficiency virus type 1 lifecycle. Biochemistry 52:9269–9274. 10.1021/bi401270d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benaroch P, Billard E, Gaudin R, Schindler M, Jouve M. 2010. HIV-1 assembly in macrophages. Retrovirology 7:29. 10.1186/1742-4690-7-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Varthakavi V, Smith RM, Martin KL, Derdowski A, Lapierre LA, Goldenring JR, Spearman P. 2006. The pericentriolar recycling endosome plays a key role in Vpu-mediated enhancement of HIV-1 particle release. Traffic 7:298–307. 10.1111/j.1600-0854.2005.00380.x [DOI] [PubMed] [Google Scholar]
- 47.Kumari S, Mg S, Mayor S. 2010. Endocytosis unplugged: multiple ways to enter the cell. Cell Res. 20:256–275. 10.1038/cr.2010.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McMahon HT, Boucrot E. 2011. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell. Biol. 12:517–533. 10.1038/nrm3151 [DOI] [PubMed] [Google Scholar]
- 49.Jordens I, Marsman M, Kuijl C, Neefjes J. 2005. Rab proteins, connecting transport and vesicle fusion. Traffic 6:1070–1077. 10.1111/j.1600-0854.2005.00336.x [DOI] [PubMed] [Google Scholar]
- 50.Sonnichsen B, De Renzis S, Nielsen E, Rietdorf J, Zerial M. 2000. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J. Cell Biol. 149:901–914. 10.1083/jcb.149.4.901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schonteich E, Wilson GM, Burden J, Hopkins CR, Anderson K, Goldenring JR, Prekeris R. 2008. The Rip11/Rab11-FIP5 and kinesin II complex regulates endocytic protein recycling. J. Cell Sci. 121:3824–3833. 10.1242/jcs.032441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parent A, Hamelin E, Germain P, Parent JL. 2009. Rab11 regulates the recycling of the beta2-adrenergic receptor through a direct interaction. Biochem. J. 418:163–172. 10.1042/BJ20080867 [DOI] [PubMed] [Google Scholar]
- 53.Yamamoto H, Koga H, Katoh Y, Takahashi S, Nakayama K, Shin HW. 2010. Functional cross-talk between Rab14 and Rab4 through a dual effector, RUFY1/Rabip4. Mol. Biol. Cell 21:2746–2755. 10.1091/mbc.E10-01-0074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zerial M, McBride H. 2001. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell. Biol. 2:107–117. 10.1038/35052055 [DOI] [PubMed] [Google Scholar]