

ABSTRACT
Human endogenous retrovirus type K (HERV-K) proviruses are scattered throughout the human genome, but as no infectious HERV-K virus has been detected to date, the mechanism by which these viruses replicated and populated the genome remains unresolved. Here, we provide evidence that, in addition to the RNA genomes that canonical retroviruses package, modern HERV-K viruses can contain reverse-transcribed DNA (RT-DNA) genomes. Indeed, reverse transcription of genomic HERV-K RNA into the DNA form is able to occur in three distinct times and locations: (i) in the virus-producing cell prior to viral release, yielding a DNA-containing extracellular virus particle similar to the spumaviruses; (ii) within the extracellular virus particle itself, transitioning from an RNA-containing particle to a DNA-containing particle; and (iii) after entry of the RNA-containing virus into the target cell, similar to canonical retroviruses, such as murine leukemia virus and HIV. Moreover, using a resuscitated HERV-K virus construct, we show that both viruses with RNA genomes and viruses with DNA genomes are capable of infecting target cells. This high level of genomic flexibility historically could have permitted these viruses to replicate in various host cell environments, potentially assisting in their many integration events and resulting in their high prevalence in the human genome. Moreover, the ability of modern HERV-K viruses to proceed through reverse transcription and package RT-DNA genomes suggests a higher level of replication competency than was previously understood, and it may be relevant in HERV-K-associated human diseases.
IMPORTANCE Retroviral elements comprise at least 8% of the human genome. Of all the endogenous retroviruses, HERV-K viruses are the most intact and biologically active. While a modern infectious HERV-K has yet to be found, HERV-K activation has been associated with cancers, autoimmune diseases, and HIV-1 infection. Thus, determining how this virus family became such a prevalent member of our genome and what it is capable of in its current form are of the utmost importance. Here, we provide evidence that HERV-K viruses currently found in the human genome are able to proceed through reverse transcription and historically utilized a life cycle with a surprising degree of genomic flexibility in which both RNA- and DNA-containing viruses were capable of mediating infection.
INTRODUCTION
The family Retroviridae constitutes a wide range of avian- and mammalian-tropic viruses, including medically relevant viruses such as human immunodeficiency virus (HIV) and human T-lymphotrophic virus (HTLV). Moreover, retroviral elements constitute at least 8% of the human genome, representing more than 4,000 proviruses (1–3). These viral sequences entered the human genome via infection of germ line cells, followed by vertical transmission into the greater population, over the course of millions of years (2). Subsequent to entering the human genome, most endogenous retroviruses (HERVs) acquired numerous mutations, deletions, or truncations interrupting the open reading frames (ORFs) of vital genes and rendering the resulting viruses noninfectious. Despite this, some HERVs have been found to impact basic human biology, including one HERV gene product that is critical for the fusion events that form the syncytial layer of the placenta during human reproduction (4–6). Furthermore, HERVs also have been found to be activated in, and associated with, several prevalent and severe human diseases, including HIV-1 infection, autoimmune diseases, and multiple malignancies (7–18).
Human endogenous retrovirus type K (HML-2) (referred to here as HERV-K) is the most recent family of HERVs to gain residence in the human genome, which occurred via infection events as recently as the last 200,000 years (19–23). As such, many of the known integrated HERV-K proviruses remain relatively intact (20). Indeed, some of these viruses have maintained several complete open reading frames and have been shown to produce not only mRNA and viral proteins but also virus-like particles in patients infected with HIV-1 and in patients with various malignancies, including lymphoma (7, 8, 14, 24–30). While to date no replication-competent HERV-K has been identified, it has long been presumed that historically these viruses utilized a canonical retrovirus life cycle, packaging an RNA genome that, upon entry into a target cell, is reverse transcribed into DNA that then integrates into the host genome (31). The recent resuscitation of replication-competent versions of HERV-K derived from consensus sequences of selected proviruses by two groups (termed HERV-KCON and Phoenix) allows for an in-depth examination of the HERV-K viral replication cycle for the first time (32, 33).
Retroviruses were named for their retrograde flow of genetic information, using reverse transcription of a viral RNA genome (vRNA) to eventually progress to an integrated DNA provirus. The retroviral life cycle (e.g., that of HIV) begins with extracellular progeny virions with a lipid envelope bearing attachment proteins surrounding a protein capsid that encases the vRNA genome and vital enzymes, such as reverse transcriptase (RT) and integrase (34). Upon attachment and entry into a suitable target cell, the vRNA is reverse transcribed by its own RT into a double-stranded DNA copy (termed early reverse transcription) that then can be integrated into the host genome, in turn utilizing the host cell transcription and translation machinery to produce new progeny virions containing vRNA (34, 35). Until recently, this general life cycle was considered to be ubiquitous across all vertebrate retroviruses; however, further study has shown that retroviral replication does not always follow the canonical pattern. Indeed, it has been recognized that partial DNA transcripts can be found associated with HIV and simian immunodeficiency virus particles (36, 37). Further, spumaviruses, retroviruses which include human and simian foamy viruses, have been found to produce progeny virions that package both RNA and DNA genomes, most likely in distinct particles (38–42). In the case of the viral DNA (vDNA)-containing progeny viruses, reverse transcription of RNA was shown to occur in the virus-producing cell, with the newly reverse-transcribed DNA (RT-DNA) being packaged into viral particles prior to budding and release (termed late reverse transcription) (40, 41). Moreover, in vitro studies done by Yu et al. and others found the spumavirus particles containing vDNA rather than vRNA to be the source of infectivity (40, 43). This contrasts with the vRNA-dominated mode of infection employed by all other known retroviruses. Hepadnaviruses, such as hepatitis B virus (HBV), while not classified as retroviruses, also use late reverse transcription in the producer cells to generate vDNA-containing progeny virions (44, 45). However, a distinguishing characteristic of the HBV life cycle is the partially double-stranded circular DNA genome in the mature particles, which is thought to arise from reverse transcription following encapsidation of the pregenomic RNA within the viral core proteins while in the cytoplasm of the producer cells prior to budding (44, 45).
Here, we examine the viral life cycle of HERV-K, employing teratocarcinoma cell lines known to produce HERV-K viral particles, plasma from lymphoma patients with quantifiable HERV-K viral loads, and the resuscitated, infectious HERV-KCON. Our findings suggest a mode of replication distinct from that of canonical retroviruses, spumaviruses, or hepadnaviruses. HERV-K appears to be a genomically versatile virus containing either vRNA or vDNA genomes within infectious particles. This versatility may offer a replicative advantage and at least partially explain how these viruses became such prevalent members of the current human genome. Further, this unexpected mode of replication must now be considered when assessing the role of HERV-K in human health and disease.
MATERIALS AND METHODS
Ethics statement.
Plasma samples were acquired from non-HIV-infected lymphoma patients with written informed consent and approval by the Institutional Review Board of the University of Michigan Medical School (IRBMED).
Cell lines and culture reagents.
NCCIT cells were cultured in RPMI 1640 with 10% fetal bovine serum (FBS) and supplemented with penicillin-streptomycin. Tera-1 cells were cultured in McCoy's 5a modified medium with 10% FBS and 1× penicillin-streptomycin. 293FT and BHK-21 cells were cultured in Dulbecco's modified Eagle medium (DMEM) with 10% FBS and 1× penicillin-streptomycin.
Patient samples.
Non-HIV-infected lymphoma patient blood samples were collected in calcium EDTA tubes, spun at 2,000 × g for 15 min to remove cellular debris, aliquoted, and stored at −80°C prior to use.
Viral load quantitation.
NCCIT and Tera-1 supernatants, as well as patient plasma samples, were cleared of cellular debris by centrifugation at 2,000 rpm for 15 min at 4°C. The samples then were filtered using a Whatman 0.45-μm syringe filter. The clarified samples then were subjected to DNase treatment using the DNA-free kit (Ambion) and the included DNase inactivation reagent by following the manufacturer's protocol. Cleared and DNase-treated viral samples were lysed and the nucleic acids purified using the Purelink viral RNA/DNA minikit (Invitrogen) by following the manufacturer's protocol. The extracted viral nucleic acids then were either used immediately or stored at −80°C in small aliquots to prevent degradation. Where indicated (see Fig. S1B in the supplemental material), DNase-free RNase (Roche) was added at a 1:10 dilution to extracted nucleic acids and incubated for 30 min at 37°C to eliminate RNA. The HERV-K viral load was quantitated using the Brilliant III ultrafast SYBR green quantitative reverse transcription-PCR (qRT-PCR) kit (Agilent) with or without the included reverse transcriptase and using either HERV-K gag-specific primers (GagRTF, 5′-AGCAGGTCAGGTGCCTGTAACATT-3′; GagRTR, 5′-TGGTGCCGTAGGATTAAGTCTCCT-3′) or env-specific primers (env1RTF, 5′-GATGGTAACACCAGTCAC-3′; env1RTR, 5′-GGCAAGGTTTCCCTTTAG-3′; env2RTF, 5′-AGACACCGCAATCGAGCACCGTTGA-3′; env2RTR, 5′-ATCAAGGCTGCAAGCAGCATACTC-3′). The Applied Biosystems StepOnePlus real-time PCR system was used to quantify viral load by the standard curve method. Additionally, for viral load analyses involving HERV-KCON, murine leukemia virus (MLV), or HIV, a gfp-specific primer set was used (gfpRTF, 5′-TGACCCTGAAGTTCATCTGC-3′; gfpRTR, 5′-GAAGTCGTGCTGCTTCATGT-3′), and the analysis was done using a modified ΔΔCT method.
UNG treatment of viral samples.
Where indicated, uracil N-glycosylase (UNG) treatment was done after the extraction of viral nucleic acids but prior to qRT-PCR analysis. In these experiments, 1.5 μl of the UNG enzyme (New England BioLabs) or ultrapure water (mock) was combined with 1.5 μl of UNG buffer (New England BioLabs) and added to 15 μl of the viral nucleic acid sample. This solution was incubated for 10 min at 37°C to allow UNG-directed cleavage of uracil-containing DNA and then incubated for 10 min at 95°C to inactivate the enzyme. To further inactivate the enzyme, 1.5 μl of UNG inhibitor (New England BioLabs) was added to the reaction, and the solution was incubated for an additional 10 min at 37°C. The resulting samples then were analyzed by qRT-PCR as described above. Confirmation of the specificity of the UNG activity was made by examining the amplification of extracted cellular RNA templates and manually prepared DNA templates containing dTTP alone or a mixture of dTTP and dUTP.
HERV-K particle immunoprecipitation.
HERV-K viral supernatants from teratocarcinoma cell cultures and patient plasma samples were cleared of cellular debris and DNase treated as described above. Virus particles then were concentrated by ultracentrifugation at 25,000 rpm for 2 h at 4°C and resuspended in 500 μl of NH buffer (0.8% NaCl, 10 mM HEPES, pH 7.4). Two hundred μl of the resulting viral suspensions was incubated at 4°C with either mouse anti-HERV-K Env (Austral Biologicals) or control mouse IgG2a antibody for 2 h with rocking. Forty μl of protein A/G Plus agarose (Santa Cruz Biotechnology) was added to each tube and incubated for an additional 2 h at 4°C with rocking. The agarose beads were pelleted by centrifugation at 3,000 × g for 2 min and washed with 300 μl NH buffer three times. After washing, the beads were resuspended in a final volume of 200 μl NH buffer, which was split for nucleic acid extraction and qRT-PCR, completed as describe above, or Western blot analysis. Samples to be analyzed by Western blotting were pelleted once more and then resuspended in 50 μl of Laemmli sample buffer with dithiothreitol. The samples then were boiled for 5 min, subjected to SDS-PAGE, and transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane-bound proteins were probed with rabbit anti-HERV-K capsid (46) and a goat anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody. Visualization of the proteins was achieved using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) and autoradiography.
Differentiation of HERV-K vDNA species.
Viral nucleic acids were extracted from the viral particles within lymphoma patient plasma using the Invitrogen Purelink viral RNA/DNA minikit and immunoprecipitated as described above. These nucleic acids then were analyzed by long-range PCR using the Expand long-range dNTPack (Roche Applied Science) and the following primer sets: 1, envFor (5′-AAGTTCTACAATGAACCCATCGG-3′) and rRev (5′-GGGAACCAGCGTTCAGCATA-3′); 2, envFor (5′-AAGTTCTACAATGAACCCATCGG-3′) and u5Rev (5′-CACCTGTGGGTGTTTCTCGT-3′); and 3, u3For (5′-TGGGCAATGGAATGTCTCGG-3′) and envRev (5′-TCCGATGGGTTCATTGTAGAACT-3′). The same primer scheme also was used with the control full-length HERV-KCON plasmid as the template for amplification. Amplified products were analyzed by agarose gel electrophoresis and visualized using SybrSafe DNA gel stain (Invitrogen).
Recombinant virus production and infection.
The HERV-KCON construct encoding gfp in the place of its native env was kindly provided by Paul Bieniasz. Production of infectious HERV-KCON was done essentially as described previously (33). Briefly, plasmids encoding HERV-KCON-GFP, HERV-K Gag-Pol, HERV-K Rec, and vesicular stomatitis virus G protein (VSV-G) were transfected into 293FT cells (in the presence or absence of 50 μM AZT) using the XtremeGene HD reagent (no. 06366236001). After 24 h, the transfection media was removed, cells were washed at least five times with PBS in order to eliminate any residual plasmid, and the media were replaced with serum-free DMEM. Forty-eight h after initial transfection, viral supernatants were harvested, cleared, and filtered as previously described. To remove any residual reverse transcriptase inhibitor (RTI), viral supernatants then were centrifuged in an Ambion ultracentrifuge with a 10,000 nominal molecular weight limit (NMWL) filter, which allows the RTI to pass through the filter while the virus remains in the top fraction. The viral fractions then were resuspended in equal volumes of DMEM. These resuspended viral samples were incubated with fresh 293FT cells that had been plated in a 24-well dish 24 h earlier in 5 μg/ml of Polybrene in the presence or absence of 50 μM AZT. The plate then was spun at 800 × g for 1 h at room temperature, followed by incubation at 37°C. After 48 h, cells were trypsinized, pelleted at 4,000 rpm in a tabletop microcentrifuge, washed with PBS, and fixed with 2% paraformaldehyde. The fixed cells then were analyzed for green fluorescent protein (GFP) expression by flow cytometry. Similar strategies were used to produce recombinant MLV and HIV.
In-virion reverse transcriptase assay.
NCCIT supernatants were harvested after 48 h of growth. The supernatants then were cleared, filtered, and DNase treated as described above. The resulting cell-free sample then was incubated at 37°C, with the exception of 400 μl that was kept for immediate nucleic acid extraction as described above (time zero). After the indicated lengths of time (up to 168 h), 400 μl of the cell-free sample was removed and its viral nucleic acid extracted. Each extracted sample was immediately stored at −80°C, including the final extraction, prior to UNG treatment (where indicated) and viral load quantitation.
RESULTS
Teratocarcinoma cell lines produce HERV-K particles that contain RT-DNA.
In order to determine the genomic content of HERV-K viral particles, we collected the supernatant from two teratocarcinoma cell lines (NCCIT and Tera-1) that have previously been shown to produce HERV-K virus particles (26–28). To prevent contamination by extraviral DNA (cellular genomic or extraneous), the supernatants were cleared by centrifugation and filtration to remove cellular debris and then treated with DNase using a DNA-free kit with a bead-based inactivation reagent that removes residual DNase prior to viral lysis. The effectiveness of this DNase treatment to remove extraviral DNA without affecting the viral genome protected within the viral particle was confirmed by the addition of plasmid DNA to cellular supernatant prior to DNase treatment (see Fig. S1A in the supplemental material). The viral particles then were lysed, and their nucleic acids (both RNA and DNA) were extracted from the cleared, DNase-treated cell supernatants and subjected to quantitative PCR (qPCR; only amplifies products from DNA templates) or qRT-PCR (amplifies products from both RNA and DNA templates) for HERV-K gag to compare the quantity of DNA and RNA HERV-K genomes. Figure 1A shows representative experiments for NCCIT and Tera-1 viral supernatants (32% and 46% DNA, respectively). Of note, while results similar to those shown in Fig. 1A were obtained in multiple experiments, the percentage of DNA genomes varied considerably between viral supernatants (from below 1% up to 80% of all nucleic acids), likely due to cell growth variations such as cell cycle, cell density, and passage number. RNase treatment of the extracted viral nucleic acids from NCCIT and Tera-1 supernatants prior to quantitative analysis further verified the presence of HERV-K DNA genomes (see Fig. S1B in the supplemental material).
FIG 1.

HERV-K particles containing RT-DNA are produced by teratocarcinoma cells in tissue culture. (A) Supernatants from NCCIT and Tera-1 cells were cleared of debris and contaminating DNA, concentrated, and subjected to qPCR or qRT-PCR for HERV-K gag. (i) HERV-K viral load in the supernatant from one representative experiment for each cell type on a log scale. (ii) Percentages of HERV-K vRNA and vDNA assuming 100% RT efficiency from the same experiments. (B) Supernatants from NCCIT or Tera-1 cells were prepared as described for panel A, either mock treated or UNG treated, and analyzed by qRT-PCR for HERV-K gag. The bars shown represent the percentages of HERV-K gag copies normalized to values for mock treatment of the respective cells from one representative experiment with Tera-1 cells and the averages from seven experiments with NCCIT cells. Statistical significance was determined by the Student t test comparing mock- and UNG-treated samples (*, P < 0.01). (C) NCCIT cells were cultured in the presence or absence of a reverse transcriptase inhibitor (3TC at 50 μM). The supernatant (Sup) was collected, cleared, concentrated, either mock or UNG treated, and then analyzed for HERV-K viral load as described above. The data shown display the effect of UNG relative to the level of HERV-K gag copies in each specific treatment. (D, i) Concentrated (Conc.) NCCIT cell supernatants were maintained or immunoprecipitated (IP) with a control IgG2A- or HERK-Env-specific capture antibody. The relative fold enrichment of HERV-K genomes as measured by qRT-PCR for HERV-K gag with respect to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was calculated using the ΔΔCT method. (ii) HERV-K particles from concentrated NCCIT supernatants were immunoprecipitated as described above and then probed by Western blotting (WB) for HERV-K capsid protein. Equivalent volumes were loaded into the lanes for the immunoprecipitated samples. (iii) NCCIT cells were cultured in the absence or presence of the reverse transcription inhibitor (RTI; 3TC at 50 μM), and HERV-K viral particles were immunoprecipitated as described above. Viral load was analyzed by qRT-PCR for HERV-K gag, and the effect of UNG was normalized to values for mock treatment for each sample independently.
The most straightforward means by which HERV-K viruses could contain DNA genomes is through reverse transcription from HERV-K RNA while still in the virus-producing cells, as is seen during spumavirus and hepadnavirus replication. To further analyze the HERV-K viruses produced by these teratocarcinoma cell lines, we took advantage of the fact that reverse transcriptase occasionally incorporates uracil in the place of thymine within the newly synthesized DNA (RT-DNA). The enzyme UNG specifically hydrolyzes uracil-glycosidic bonds in DNA but not RNA; thus, it can be used to differentiate newly reverse transcribed DNA from all other nucleic acids (47, 48). This strategy has been used previously in one other report to suggest the possibility of DNA genomes in a specific HERV-K (HERV-K102) (49), and the enzyme's specificity was further confirmed in our hands (see Fig. S2 in the supplemental material). UNG treatment of extracted viral nucleic acids from both NCCIT and Tera-1 supernatants resulted in a decreased viral load compared to that of mock-treated samples analyzed by qRT-PCR for HERV-K gag (Fig. 1B) and env (see Fig. S3 in the supplemental material), primer sets that are specific for disparate ends of the HERV-K coding region. To confirm that the UNG treatment eliminated the amplification of new reverse transcripts, we analyzed the viral supernatants from NCCIT cultured for 48 h in the presence or absence of an RTI (50 μM 3TC). As expected, the treatment of these cells with an RTI eliminated the population of HERV-K genomes that was sensitive to UNG (Fig. 1C).
Finally, to further verify that the HERV-K genomes analyzed in these experiments were in fact inside HERV-K particles, we developed a method to immunoprecipitate HERV-K particles using a capture antibody specific to the mature HERV-K envelope protein (Env) that studs the viral particle. Immunoprecipitation with HERV-K Env antibody resulted in a 27.6-fold enrichment of HERV-K genomes over a control antibody, as analyzed by qRT-PCR (Fig. 1Di). Furthermore, we verified the enrichment of HERV-K particles by Western blotting using an HERV-K capsid-specific antibody (Fig. 1Dii). UNG treatment of the viral nucleic acids from HERV-K Env-immunoprecipitated samples recapitulated our previous data, resulting in a significant decrease in viral load compared to that of mock-treated samples (Fig. 1Diii). This effect was reduced when the HERV-K-producing cells were cultured in the presence of RTI. Together, these data suggest that a significant portion of the HERV-K particles produced by teratocarcinoma cells contain newly reverse transcribed DNA genomes.
Reverse transcription within extracellular HERV-K virions.
Although poorly understood, it has been previously reported that low levels of reverse transcription may be able to occur within extracellular retroviral particles (50–53). To test if this was true for HERV-K, we once again utilized the HERV-K-producing teratocarcinoma NCCIT cell line. NCCIT cells were allowed to grow and produce HERV-K particles for a minimum of 48 h, at which point the viral supernatants were harvested, cleared of cells and cellular debris by centrifugation and filtration, and DNase treated as described above (at the 0-h time point). The resulting cell-free viral supernatants then were incubated in the culture medium at 37°C for various lengths of time without the addition of any additional deoxynucleoside triphosphates (dNTPs), polyamines, or detergents that apparently are required for the in-virion reverse transcription of other retroviruses (53, 54). Viral nucleic acids were extracted and subjected to mock or UNG treatment and the viral loads analyzed by qRT-PCR. Such experiments were conducted at least five times, although with different time points being analyzed. Figure 2A shows the results of one such experiment. In this experiment, there was an initial decrease in both total HERV-K viral load and RT-DNA levels at the 24-h time point, most likely due to viral instability and degradation. A similar decrease from initial viral load was seen in all experiments, although the time at which this was seen varied (6 to 48 h). However, as seen in Fig. 2A, the total HERV-K viral load then increased over time, due almost exclusively to an increase in HERV-K RT-DNA levels. The primer set used for these experiments is specific for gag in the 5′ end of the HERV-K coding region, which is actually a downstream sequence in the reverse transcription process, and suggests that, surprisingly, the reverse transcription occurring within these virions yields a nearly full-length product. A second experiment, shown in Fig. 2B, displays RT-DNA levels using additional primers specific to the env gene in the 3′ end of the HERV-K coding region (one set specific for type 1 HERV-K and one for type 2, the former of which has a 292-bp deletion in the env region); thus, it is an earlier marker of reverse transcription. These data recapitulate those shown in Fig. 2A, further suggesting that some reverse transcription is taking place in the HERV-K virion itself as well as in the virus-producing cell.
FIG 2.
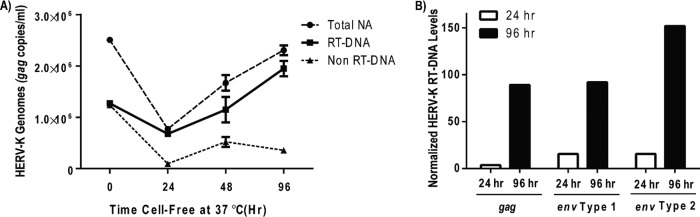
Reverse transcription occurs within extracellular HERV-K virions. (A) Viral supernatants from NCCIT cells were collected, cleared, and DNase treated as described for Fig. 1. The resulting cell-free viral suspensions then were incubated at 37°C for various times prior to extracting the viral nucleic acid, mock or UNG treatment, and viral load analysis by qRT-PCR for HERV-K gag. The three lines represent the mock-treated sample (Total NA), the UNG-treated sample (Non RT-DNA), and the difference between the two (RT-DNA). (B) In a similar but separate experiment, the HERV-K RT-DNA levels were examined over time with three distinct primer sets (the gag product is from the 5′ end of the HERV-K coding region, and the two env primer sets amplify products in the 3′ end of the HERV-K coding region). For each primer set, time zero is set at 100. Of note, at later time points (>168 h) the overall viral load was significantly diminished, as would be expected with long-term exposure at 37°C due to general virus instability. A single representative experiment (of eight in total) is shown for both panels A and B.
HERV-K particles containing DNA genomes are present in the plasma of lymphoma patients.
To determine the physiological relevance of our previous findings, the HERV-K genomic profile of plasma samples from lymphoma patients was examined, as previous work from our laboratory has demonstrated quantifiable HERV-K viral loads in the plasma of these individuals (7). In Fig. 3A, the plasma from a representative lymphoma patient was analyzed for HERV-K RNA and DNA genomes as described above. In this patient, approximately 38% of the total HERV-K genomes were DNA genomes. Indeed, examining plasma samples from multiple lymphoma patients presenting with significant viral loads of HERV-K (greater than 104 particles/ml based on qRT-PCR) with and without RT yielded a range of DNA genomes from 12% to 52% of the total HERV-K viral load (Fig. 3B). Further, an additional DNase treatment of viral nucleic acids after lysis of the viral particles decreased the viral load proportionately (Fig. 3C), strongly suggesting that the vDNA is protected within an intact viral particle and corroborating the data shown in Fig. S1 in the supplemental material.
FIG 3.
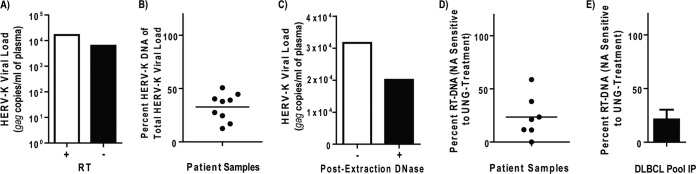
HERV-K vDNA is present in the plasma of patients with lymphoma. (A) Plasma from a single representative patient was treated with DNase and then assayed for HERV-K genomes with gag-specific primers with or without reverse transcriptase (RT) as described for Fig. 2. (B) Multiple patients were assayed as described for panel A, and the percentage of HERV-K vDNA genomes in the population was calculated by dividing the no-RT viral load by the RT viral load. (C) Lymphoma patient plasma (distinct from those shown in panels A and B) was treated with DNase prior to the extraction of viral nucleic acids and then treated with DNase again after viral lysis prior to assay by qRT-PCR. The data from one representative experiment are shown. (D) A subset of plasma from patients with lymphoma was analyzed for their sensitivity to UNG treatment as described in the legend to Fig. 1. The percentage of HERV-K nucleic acids susceptible to UNG (RT-DNA genomes) is shown for each patient. (E) Plasma from four patients with diffuse large B cell lymphoma (DLBCL) were pooled, and then HERV-K particles were immunoprecipitated as described for Fig. 1. HERV-K nucleic acids were extracted and subjected to mock or UNG treatment prior to qRT-PCR for HERV-K gag. The bar shown represents the percentage of HERV-K genomes susceptible to UNG (RT-DNA genomes) of total HERV-K genomes from a single representative experiment.
When a subset of the samples from the lymphoma patients used for Fig. 3B were mock or UNG treated, as shown in Fig. 1B, between 0% and 59% (mean value of 27%) of HERV-K genomes were found to be UNG sensitive; thus, they were considered RT-DNA (Fig. 3D). Moreover, a similar percentage of RT-DNA genomes was found when the plasma from four patients with diffuse large B cell lymphoma (DLBCL) were pooled and immunoprecipitated with the HERV-K Env capture antibody, as described for Fig. 1D (Fig. 3E). These data substantiate those found for the teratocarcinoma cell lines and suggest that HERV-K replication is able to minimally progress through first-strand synthesis by reverse transcriptase prior to entry into a target cell, as is seen in the spumavirus family.
dsDNA HERV-K genomes are present in the plasma of lymphoma patients.
In order to confirm and extend our findings that lymphoma patients produce HERV-K virions that contain RT-DNA genomes, we designed a PCR primer scheme that would allow us to take advantage of the stepwise process of reverse transcription that progresses from vRNA through first-strand cDNA to a complete double-stranded DNA (dsDNA) viral genome (linear, as is the case for most retroviruses, or circular, as is the case for the hepadnaviruses) (31, 35). The three primer sets utilized, their regions of specificity on the various potential species of HERV-K genomes, and the expected sizes of their amplification products are depicted in Fig. 4A. Figure 4B shows a DNA gel of the PCR products using these primer sets with either HERV-K particles immunoprecipitated from lymphoma patient plasma or control HERV-KCON plasmid DNA as the template. Appropriately sized bands for primer sets 1 to 3 were seen using the control plasmid, with the expected absence of bands that would indicate a closed circular dsDNA genome (2 long terminal repeat [2LTR] circles) not found in this plasmid. Similar results were seen when the PCR template was HERV-K particles immunoprecipitated from lymphoma patient plasma, suggesting the presence of linear dsDNA genomes but not 2LTR circles. Sequence verification confirmed these bands to be HERV-K. Notably, although not quantifiable in the gel analysis used, the strength of the dsDNA band from the patient plasma was markedly less than that seen with the HERV-KCON template or with the primer set that would amplify all DNA (cDNA as well as dsDNA), suggesting that only a portion of the DNA genomes are fully dsDNA. Additionally, it is important to note that while 2LTR circular HERV-K genomes have been detected in cells infected with the HERV-KCON virus (and several other infectious retroviruses) (55–58), we see no evidence of those genomes being present in extracellular virions. However, the possibility of circular HERV-K genomes being packaged cannot be fully dismissed from this experiment alone due to the innate bias of PCR amplification for shorter amplicons, although cycling conditions were optimized for the longer product expected if 2LTR circles were present. Further, it is formally possible that recombination could occur that leads to circular HERV-K genomes with a single LTR, which would not be differentiated from linear dsDNA by this experiment. However, single LTR circles most likely would be incapable of integration within downstream target cells, which is expected to be required for productive retroviral infection (59).
FIG 4.
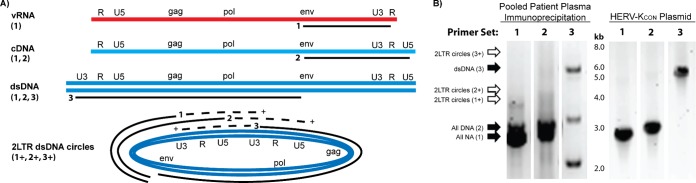
HERV-K particles from pooled lymphoma patient plasma contain dsDNA genomes. (A) Three different primer sets were employed to take advantage of differences in the viral genomes before, during, and after reverse transcription. Primer set 1 (env forward and R reverse) will amplify a 2.9-kb product in all forms of HERV-K genomes and an additional 3.9-kb product in 2LTR dsDNA circles (1+). Primer set 2 (env forward and U5 reverse) will amplify a 3-kb product in all species except vRNA and an additional 4-kb product in 2LTR circles (2+). Primer set 3 (U3 forward and env reverse) will amplify a 5.9-kb product in dsDNA and 2LTR circles and an additional 6.9-kb product in 2LTR circles (3+; depicted on the 2LTR circular DNA by the dashed line). (B) RT-PCR (primer set 1) or PCR (primer sets 2 and 3) was done with both pooled immunoprecipitated patient plasma (as described for Fig. 1D) and a control HERV-KCON plasmid (which should yield all amplification products except for the 2LTR circular DNA form). The lanes shown are labeled with the primer set used, are from the same experiment, and were run on the same gel. Solid arrows indicate a DNA band at the expected size in the indicated lane, while empty arrows indicate no band of the predicted size, suggesting the absence of 2LTR circles in this experiment. Bands of interest were confirmed as HERV-K by sequence verification. Note that the doublets seen in the patient serum samples with primer sets 1 and 2 represent HERV-K type 1 (smaller) and 2 (larger) genomes.
HERV-K particles with RNA or RT-DNA genomes are capable of productive infection.
To investigate the importance of our finding of RT-DNA genomes in the historical replication cycle of HERV-K, we utilized the resuscitated HERV-KCON construct. The HERV-KCON sequence was generated as a consensus sequence from several of the most intact HERV-K proviruses scattered throughout the human genome, and its ability to infect human tissue culture cells was reported previously (33). Following the previously published methodology, we produced HERV-KCON particles bearing VSV-G and encoding a GFP reporter in 293FT cells (see Fig. S4 in the supplemental material) in the absence or presence of RTI (50 μM AZT), which would prevent the late reverse transcription in the virus-producing cells as seen in the spumavirus family. Viral supernatants then were cleared and filtered, and residual RTI was removed by centrifugation in an Amicon Ultra 10,000 NMWL filter unit. Similarly, we produced control retroviral MLV and HIV, also bearing VSV-G and encoding a GFP reporter (see Fig. S4). HERV-KCON, MLV, and HIV (produced in mock- and RTI-treated cells) then were allowed to infect 293FT cells in the absence or presence of RTI, which would prevent the early reverse transcription in target cells used by canonical retroviruses. Figure 5A details the results of the experiments analyzing HERV-KCON and MLV. While MLV infection decreased (by 99.7%) only when the RTI was present in the target cells (inhibiting early reverse transcription), HERV-KCON infection was reduced by approximately 51% by RTI treatment of the virus-producing cells (inhibiting late reverse transcription). HERV-KCON infection was decreased by treatment of the target cells alone with RTI as well, but not to the same extent as was MLV. Interestingly, even with RTI present in both the virus-producing cells and the target cells, a low level of HERV-KCON infectivity still remained. While this could be due to incomplete inhibition of the HERV-K reverse transcriptase, the fact that the MLV infection was so completely reduced by the same treatment suggests that extracellular reverse transcription within the virions (as seen in Fig. 2) is responsible for this residual infectivity. Taken together, these data suggest that while infection by canonical retroviruses, as seen here with MLV and with a recombinant HIV construct (see Fig. S5 in the supplemental material), is dependent solely on virions containing vRNA genomes and early reverse transcription within the target cell, HERV-K can utilize either vRNA genomes (by early reverse transcription in the target cell) or vDNA genomes (made by late reverse transcription in the virus-producing cell and potentially extracellular, in-virion reverse transcription) to infect cells.
FIG 5.
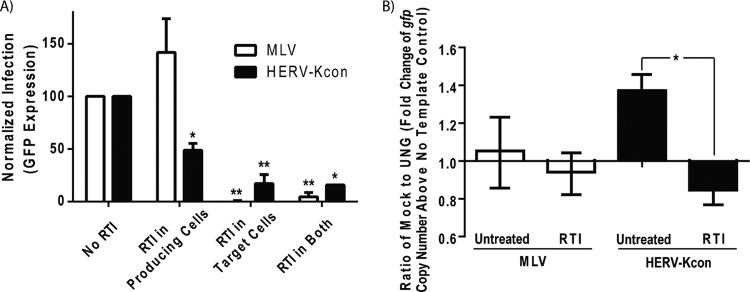
RT-DNA- and RNA-containing HERV-KCON particles both yield productive infection. HERV-KCON particles bearing VSV-G and encoding a GFP reporter were produced in 293FT cells as described in reference 33. Recombinant MLV particles bearing VSV-G and encoding a GFP reporter were produced in parallel. (A) Analysis of the effects of RTI on infection levels of HERV-K and MLV. Viruses were produced in cells in the presence or absence of a reverse transcriptase inhibitor (AZT at 50 μM) for 48 h, and the supernatant was collected, cleared for debris by centrifugation, and filtered through a 0.45-μm Whatman filter. 293FT cells in the presence or absence of reverse transcriptase inhibitor (AZT at 50 μM) were spinfected with the resulting cleared viral supernatants for 1 h at 800 × g and incubated at 37°C for an additional 36 h to allow infection to occur. The cells then were prepared for flow cytometry, and infection was assessed by GFP reporter expression. Overall infection levels for HERV-KCON were low (100 to 1,000 IU/ml) but reproducible. The bars shown are the normalized averages from three experiments. Statistical significance was determined by a Student t test comparing the various RTI treatments to no RTI (*, P < 0.01; **, P < 0.001). (B) Analysis of the genomic constituents of infectious virus particles. Viral preparations used for panel A were treated with DNase prior to extraction of the viral nucleic acids and subjected to mock or UNG treatment prior to qRT-PCR with primers specific to the gfp reporter gene found in both recombinant viruses. The ΔΔCT method was used to determine the fold change of the gfp gene copy number above the no-template control for the mock- and UNG-treated samples (representing the relative levels of the viral genomes). The bars shown represent the fold change from mock-treated samples divided by that from UNG-treated samples. A value of 1 suggests that no RT-DNA-containing particles were present in the viral population (the quantity of viral genomes was the same in mock- and UNG-treated samples), and a value significantly above 1 suggests the presence of RT-DNA genomes in the population (UNG treatment reduced the number of viral genomes amplified). Significance was determined by the Student t test comparing the fold expression of the untreated and RTI-treated virus-producing cells within each virus type (*, P < 0.001).
To confirm that the mechanism by which the RTI treatment in virus-producing cells was reducing infectivity was by inhibiting the production of RT-DNA genomes, we analyzed the genomic contents of the viral supernatants used in the infection experiments. To do this, we employed qRT-PCR with a primer set specific for the gfp reporter sequence found in both HERV-KCON and MLV and compared mock- to UNG-treated samples to determine their fold expression of gfp above background levels. Figure 5B shows the results of this analysis, where a ratio of 1 represents viral genomes that are not sensitive to UNG treatment and contain no RT-DNA, whereas a ratio of greater than 1 suggests the presence of RT-DNA. While MLV produced in the presence or absence of RTI did not have a ratio significantly different from 1 (suggesting little to no RT-DNA), the untreated HERV-KCON viral supernatant's ratio was significantly higher than 1 (P < 0.001), confirming the presence of RT-DNA genomes. As expected, the ratio of the HERV-KCON genomes produced in RTI-treated cells was ≤1 and was significantly less than that of the untreated HERV-KCON population (P < 0.001), suggesting the effective inhibition of reverse transcriptase and a loss of RT-DNA-containing particles.
DISCUSSION
Traditionally, members of the family Retroviridae were thought to replicate via a common pathway in which a virus particle containing two single-stranded RNA genomes and the reverse transcriptase enzyme (in addition to other viral components) would enter its target cell, its genome would undergo reverse transcription into a dsDNA form that could integrate into the host genome, and then it would utilize the host machinery to produce its viral components to be assembled and released as extracellular viral progeny, again containing RNA genomes (Fig. 6) (34, 35). However, a few studies suggested that in certain specific situations (e.g., quiescent target cells), a low level of infection can be derived from retroviral particles with genomes reverse transcribed prior to entry into the target cell (54, 60). Further, recent studies examining the replicative mechanisms of spumaviruses showed that these retroviruses actually are able to produce virions that contain DNA as well as RNA genomes. Whether these genomes are in the same or distinct particles still is unclear. Moreover, it was shown that the particles containing DNA genomes produced through a late reverse transcription event in the virus-producing cell are almost solely responsible for spumavirus infectivity (38–43). An additional variation on the theme of viral reverse transcription is seen in hepadnaviruses, not members of the family Retroviridae, which create circular dsDNA viral genomes that get packaged into progeny virions (44, 45). The data presented in this study support a fourth strategy of retroviral replication, which is utilized by HERV-K family members. The fundamental concept of this HERV-K replication strategy is genomic flexibility. Our data suggest that HERV-K viruses, in their current and ancestral forms, are able to undergo reverse transcription of their genome at three distinct times and locations: (i) upon entering a target cell, as in canonical retroviruses; (ii) prior to release from the virus-producing cell, likely before or during viral packaging, as in spumaviruses; and (iii) within the extracellular virus particles themselves (Fig. 6). Distinct from the canonical retroviruses, spumaviruses, or hepadnaviruses, HERV-K viruses, in their historical form, are able to infect actively growing target cells using either RNA- or DNA-containing virions. This represents a previously unobserved level of life cycle versatility in the field of virology.
FIG 6.
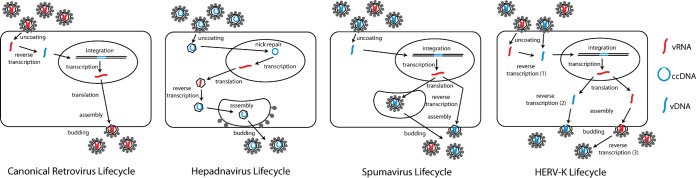
Model for the viral life cycle of HERV-K compared to those of the canonical Retroviridae, Hepadnaviridae, and Spumaviridae. The three distinct replication pathways used by retroviruses, hepadnaviruses, and spumaviruses are depicted. Reverse transcription of the vRNA (red) genome occurs early in the infection process for canonical retroviruses and late during infection in hepadnaviruses and spumaviruses, so that the latter two virus families produce progeny virions containing vDNA (blue) genomes. Of note, while the spumavirus particles with vDNA genomes are much more important for downstream infection, vRNA genomes also can be packaged into viral particles. Alternatively, putative infectious HERV-K viruses can undergo reverse transcription either early or late in the infection process (and potentially in extracellular virions).
Previous research in the field of human endogenous retroviruses has been hampered by the presence of multiple copies of highly similar proviruses integrated throughout the human genome. This makes tracking infection by sequence-based techniques very challenging and, importantly, represents an overwhelming source of potentially contaminating HERV DNA. We have overcome these issues in several ways. All quantitative PCR-based experiments were prepared within an AirClean 600 PCR workstation. Furthermore, all samples (unless otherwise indicated) were treated with DNase in a manner that differs in two important ways from the methods used in many previous studies (61). First, the DNase treatment was conducted directly on the viral supernatants or patient plasma samples prior to viral lysis. This timing is crucial for eliminating extraviral DNA contamination, while viral DNA genomes remain protected within the virus particles themselves. Second, the DNase treatment was performed using the DNA-free kit, which employs a bead-based inactivation reagent that binds and removes any residual DNase after treatment but prior to viral lysis. This prevents DNase carryover, which then would be able to digest any viral DNA once the virus particles were lysed during the nucleic acid extraction procedure. The efficacy of this manner of DNase treatment is depicted in Fig. 3 (also see Fig. S1 in the supplemental material). Moreover, the UNG- and RTI-based studies provide additional confirmation that what we are detecting in our PCR (and RT-PCR) studies is not contaminating genomic proviral DNA. We further confirmed that we were detecting HERV-K DNA within bona fide HERV-K viral particles through the development of an immunoprecipitation protocol that utilized a capture antibody specific for the HERV-K Env protein that studs viral particles. Formerly, detection of endogenous retrovirus DNA in cell-free fluids was widely thought to be exclusively the result of contaminating cellular DNA. Our studies, however, show that the need to differentiate contaminant DNA and viral RT-DNA may be vital in studying the replication of all endogenous retroviruses.
In this study, we found that HERV-K particles from teratocarcinoma cell lines, lymphoma patient plasma, and the reconstituted HERV-KCON all produced viral populations with both RNA and DNA genomes. The percentage of viral genomes that were DNA compared to RNA varied both within and among sample types, most likely attributable to the cellular environment (cell cycle, cellular age, growth conditions, etc.) in which the HERV-K virions were being produced. Further study is required to determine the exact mechanisms on which this variation is dependent. Moreover, our infection studies using HERV-KCON suggest that, historically, HERV-K viruses could use either type of genomic material to initiate replication in a target cell. The benefits of such genomic flexibility for viral replication are manifold. For one, the virus can undergo reverse transcription at any point when it encounters suitable conditions in which to do so. Thus, the virus may be able to avoid host cell restriction factors that are present in target cells but absent from the virus-producing cells or vice versa. Additionally, while not examined in this study, the increased stability of DNA compared to RNA may have allowed virus particles with DNA genomes to survive longer than their RNA counterparts, resulting in increased dissemination. Indeed, this genomic flexibility may be one of the features that allowed HERV-K viruses, and possibly other HERV families, to populate the genome at such a high multitude (at least 91 full-length HERV-K proviruses in the current human genome and thousands of HERV-K solo LTRs) (20). Further, our studies using samples from present-day lymphoma patients that show the presence of HERV-K viruses with RT-DNA genomes suggest that these viruses are more competent than previously expected, as they have the ability to at least undergo reverse transcription if not yield productive infection in downstream target cells. Additional studies will be needed to examine the importance of the genomic flexibility of HERV-K and its correlation with HERV-K-associated diseases, such as cancer, HIV infection, and autoimmune disease.
Supplementary Material
ACKNOWLEDGMENTS
We thank Paul Bieniasz (Rockefeller University) for providing the HERV-KCON constructs, Norbert Bannert (Robert Koch Institute) for providing the HERV-K capsid antibody, and Mark Kaminski and Judith Estes for their aid in acquiring the lymphoma patient plasma samples. We also thank Miriam Laderoute and Gilbert Omenn for insightful discussions.
The majority of this work was supported by RO1 CA144043 to D.M.M. from the National Institutes of Health. Further, D.D. was supported by a Molecular Mechanisms of Microbial Pathogenesis (MMMP) training grant (5T32AI007528-13) and an NIH Ruth L. Kirschstein NRSA Individual Postdoctoral Fellowship (1 F32 AI106189-01). R.C.-G. was supported by Research Supplement to Promote Diversity in Health-Related Research grant R01CA144043-03S1 from the NIH, M.J.G.-H. by an MMMP training grant and an NIH Ruth L. Kirschstein NRSA Individual Predoctoral Fellowship to Promote Diversity in Health-Related Research grant (1F31CA150523-01), and S.D.G. by the Veterans Education and Research Association of Michigan (VERAM) Award. M.H.K. was supported by a grant from Concerned Parents for AIDS Research.
Footnotes
Published ahead of print 11 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01147-14.
REFERENCES
- 1.de Parseval N, Heidmann T. 2005. Human endogenous retroviruses: from infectious elements to human genes. Cytogenet. Genome Res. 110:318–332. 10.1159/000084964 [DOI] [PubMed] [Google Scholar]
- 2.Bannert N, Kurth R. 2004. Retroelements and the human genome: new perspectives on an old relation. Proc. Natl. Acad. Sci. U. S. A. 101(Suppl 2):S14572–S14579. 10.1073/pnas.0404838101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lower R, Lower J, Kurth R. 1996. The viruses in all of us: characteristics and biological significance of human endogenous retrovirus sequences. Proc. Natl. Acad. Sci. U. S. A. 93:5177–5184. 10.1073/pnas.93.11.5177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kitamura M, Maruyama N, Shirasawa T, Nagasawa R, Watanabe K, Tateno M, Yoshiki T. 1994. Expression of an endogenous retroviral gene product in human placenta. Int. J. Cancer 58:836–840. 10.1002/ijc.2910580615 [DOI] [PubMed] [Google Scholar]
- 5.Mi S, Lee X, Li X, Veldman GM, Finnerty H, Racie L, LaVallie E, Tang XY, Edouard P, Howes S, Keith JC, Jr, McCoy JM. 2000. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403:785–789. 10.1038/35001608 [DOI] [PubMed] [Google Scholar]
- 6.Frendo JL, Olivier D, Cheynet V, Blond JL, Bouton O, Vidaud M, Rabreau M, Evain-Brion D, Mallet F. 2003. Direct involvement of HERV-W Env glycoprotein in human trophoblast cell fusion and differentiation. Mol. Cell. Biol. 23:3566–3574. 10.1128/MCB.23.10.3566-3574.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Contreras-Galindo R, Kaplan MH, Leissner P, Verjat T, Ferlenghi I, Bagnoli F, Giusti F, Dosik MH, Hayes DF, Gitlin SD, Markovitz DM. 2008. Human endogenous retrovirus K (HML-2) elements in the plasma of people with lymphoma and breast cancer. J. Virol. 82:9329–9336. 10.1128/JVI.00646-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Contreras-Galindo R, Kaplan MH, Markovitz DM, Lorenzo E, Yamamura Y. 2006. Detection of HERV-K(HML-2) viral RNA in plasma of HIV type 1-infected individuals. AIDS Res. Hum. Retrovir. 22:979–984. 10.1089/aid.2006.22.979 [DOI] [PubMed] [Google Scholar]
- 9.Abraham GN, Khan AS. 1990. Human endogenous retroviruses and immune disease. Clin. Immunol. Immunopathol. 56:1–8. 10.1016/0090-1229(90)90163-K [DOI] [PubMed] [Google Scholar]
- 10.Antony JM, Deslauriers AM, Bhat RK, Ellestad KK, Power C. 2011. Human endogenous retroviruses and multiple sclerosis: innocent bystanders or disease determinants? Biochim. Biophys. Acta 1812:162–176. 10.1016/j.bbadis.2010.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balada E, Ordi-Ros J, Vilardell-Tarres M. 2009. Molecular mechanisms mediated by human endogenous retroviruses (HERVs) in autoimmunity. Rev. Med. Virol. 19:273–286. 10.1002/rmv.622 [DOI] [PubMed] [Google Scholar]
- 12.Balada E, Vilardell-Tarres M, Ordi-Ros J. 2010. Implication of human endogenous retroviruses in the development of autoimmune diseases. Int. Rev. Immunol. 29:351–370. 10.3109/08830185.2010.485333 [DOI] [PubMed] [Google Scholar]
- 13.Brodsky I, Foley B, Gillespie D. 1993. Expression of human endogenous retrovirus (HERV-K) in chronic myeloid leukemia. Leuk. Lymphoma 11(Suppl 1):S119–S123 [DOI] [PubMed] [Google Scholar]
- 14.Buscher K, Trefzer U, Hofmann M, Sterry W, Kurth R, Denner J. 2005. Expression of human endogenous retrovirus K in melanomas and melanoma cell lines. Cancer Res. 65:4172–4180. 10.1158/0008-5472.CAN-04-2983 [DOI] [PubMed] [Google Scholar]
- 15.Christensen T, Moller-Larsen A. 2003. Human endogenous retroviruses and disease? Ugeskr. Laeger. 165:556–561 [PubMed] [Google Scholar]
- 16.Dolei A. 2006. Endogenous retroviruses and human disease. Expert Rev. Clin. Immunol. 2:149–167. 10.1586/1744666X.2.1.149 [DOI] [PubMed] [Google Scholar]
- 17.Kurth R, Bannert N. 2010. Beneficial and detrimental effects of human endogenous retroviruses. Int. J. Cancer 126:306–314. 10.1002/ijc.24902 [DOI] [PubMed] [Google Scholar]
- 18.Nelson PN, Carnegie PR, Martin J, Davari Ejtehadi H, Hooley P, Roden D, Rowland-Jones S, Warren P, Astley J, Murray PG. 2003. Demystified. Human endogenous retroviruses. Mol. Pathol. 56:11–18. 10.1136/mp.56.1.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Contreras-Galindo R, Kaplan MH, He S, Contreras-Galindo AC, Gonzalez-Hernandez MJ, Kappes F, Dube D, Chan SM, Robinson D, Meng F, Dai M, Gitlin SD, Chinnaiyan AM, Omenn GS, Markovitz DM. 2013. HIV infection reveals wide-spread expansion of novel centromeric human endogenous retroviruses. Genome Res. 23:1505–1513. 10.1101/gr.144303.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Subramanian RP, Wildschutte JH, Russo C, Coffin JM. 2011. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology 8:90. 10.1186/1742-4690-8-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Macfarlane C, Simmonds P. 2004. Allelic variation of HERV-K(HML-2) endogenous retroviral elements in human populations. J. Mol. Evol. 59:642–656. 10.1007/s00239-004-2656-1 [DOI] [PubMed] [Google Scholar]
- 22.Mager DL, Medstrand P. 2003. Retroviral repeat sequences, p 57–63 In Cooper D. (ed), Nature encyclopedia of the human genome. Macmillan Publishers Ltd., Hampshire, England [Google Scholar]
- 23.Seifarth W, Baust C, Murr A, Skladny H, Krieg-Schneider F, Blusch J, Werner T, Hehlmann R, Leib-Mosch C. 1998. Proviral structure, chromosomal location, and expression of HERV-K-T47D, a novel human endogenous retrovirus derived from T47D particles. J. Virol. 72:8384–8391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Contreras-Galindo R, Almodovar-Camacho S, Gonzalez-Ramirez S, Lorenzo E, Yamamura Y. 2007. Comparative longitudinal studies of HERV-K and HIV-1 RNA titers in HIV-1-infected patients receiving successful versus unsuccessful highly active antiretroviral therapy. AIDS Res. Hum. Retrovir. 23:1083–1086. 10.1089/aid.2007.0054 [DOI] [PubMed] [Google Scholar]
- 25.Contreras-Galindo R, Lopez P, Velez R, Yamamura Y. 2007. HIV-1 infection increases the expression of human endogenous retroviruses type K (HERV-K) in vitro. AIDS Res. Hum. Retrovir. 23:116–122. 10.1089/aid.2006.0117 [DOI] [PubMed] [Google Scholar]
- 26.Bieda K, Hoffmann A, Boller K. 2001. Phenotypic heterogeneity of human endogenous retrovirus particles produced by teratocarcinoma cell lines. J. Gen. Virol. 82:591–596 [DOI] [PubMed] [Google Scholar]
- 27.Boller K, Konig H, Sauter M, Mueller-Lantzsch N, Lower R, Lower J, Kurth R. 1993. Evidence that HERV-K is the endogenous retrovirus sequence that codes for the human teratocarcinoma-derived retrovirus HTDV. Virology 196:349–353. 10.1006/viro.1993.1487 [DOI] [PubMed] [Google Scholar]
- 28.Li MD, Bronson DL, Lemke TD, Faras AJ. 1995. Restricted expression of new HERV-K members in human teratocarcinoma cells. Virology 208:733–741. 10.1006/viro.1995.1205 [DOI] [PubMed] [Google Scholar]
- 29.Lower R, Boller K, Hasenmaier B, Korbmacher C, Muller-Lantzsch N, Lower J, Kurth R. 1993. Identification of human endogenous retroviruses with complex mRNA expression and particle formation. Proc. Natl. Acad. Sci. U. S. A. 90:4480–4484. 10.1073/pnas.90.10.4480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muster T, Waltenberger A, Grassauer A, Hirschl S, Caucig P, Romirer I, Fodinger D, Seppele H, Schanab O, Magin-Lachmann C, Lower R, Jansen B, Pehamberger H, Wolff K. 2003. An endogenous retrovirus derived from human melanoma cells. Cancer Res. 63:8735–8741 [PubMed] [Google Scholar]
- 31.Berkhout B, Jebbink M, Zsiros J. 1999. Identification of an active reverse transcriptase enzyme encoded by a human endogenous HERV-K retrovirus. J. Virol. 73:2365–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dewannieux M, Harper F, Richaud A, Letzelter C, Ribet D, Pierron G, Heidmann T. 2006. Identification of an infectious progenitor for the multiple-copy HERV-K human endogenous retroelements. Genome Res. 16:1548–1556. 10.1101/gr.5565706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee YN, Bieniasz PD. 2007. Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog. 3:e10. 10.1371/journal.ppat.0030010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nisole S, Saib A. 2004. Early steps of retrovirus replicative cycle. Retrovirology 1:9. 10.1186/1742-4690-1-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coffin JM. 1997. Retroviruses. Cold Spring Harbor Laboratory Press, Plainview, NY: [PubMed] [Google Scholar]
- 36.Trono D. 1992. Partial reverse transcripts in virions from human immunodeficiency and murine leukemia viruses. J. Virol. 66:4893–4900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lori F, di Marzo Veronese F, de Vico AL, Lusso P, Reitz MS, Jr, Gallo RC. 1992. Viral DNA carried by human immunodeficiency virus type 1 virions. J. Virol. 66:5067–5074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Linial ML. 1999. Foamy viruses are unconventional retroviruses. J. Virol. 73:1747–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rethwilm A. 2003. The replication strategy of foamy viruses. Curr. Top. Microbiol. Immunol. 277:1–26. 10.1007/978-3-642-55701-9_1 [DOI] [PubMed] [Google Scholar]
- 40.Yu SF, Sullivan MD, Linial ML. 1999. Evidence that the human foamy virus genome is DNA. J. Virol. 73:1565–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu SF, Baldwin DN, Gwynn SR, Yendapalli S, Linial ML. 1996. Human foamy virus replication: a pathway distinct from that of retroviruses and hepadnaviruses. Science 271:1579–1582. 10.1126/science.271.5255.1579 [DOI] [PubMed] [Google Scholar]
- 42.Delelis O, Saib A, Sonigo P. 2003. Biphasic DNA synthesis in spumaviruses. J. Virol. 77:8141–8146. 10.1128/JVI.77.14.8141-8146.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moebes A, Enssle J, Bieniasz PD, Heinkelein M, Lindemann D, Bock M, McClure MO, Rethwilm A. 1997. Human foamy virus reverse transcription that occurs late in the viral replication cycle. J. Virol. 71:7305–7311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nassal M. 2008. Hepatitis B viruses: reverse transcription a different way. Virus Res. 134:235–249. 10.1016/j.virusres.2007.12.024 [DOI] [PubMed] [Google Scholar]
- 45.Locarnini S, Zoulim F. 2010. Molecular genetics of HBV infection. Antivir. Ther. 15(Suppl 3):S3–S14. 10.3851/IMP1619 [DOI] [PubMed] [Google Scholar]
- 46.George M, Schwecke T, Beimforde N, Hohn O, Chudak C, Zimmermann A, Kurth R, Naumann D, Bannert N. 2011. Identification of the protease cleavage sites in a reconstituted Gag polyprotein of an HERV-K(HML-2) element. Retrovirology 8:30. 10.1186/1742-4690-8-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Longo MC, Berninger MS, Hartley JL. 1990. Use of uracil DNA glycosylase to control carry-over contamination in polymerase chain reactions. Gene 93:125–128. 10.1016/0378-1119(90)90145-H [DOI] [PubMed] [Google Scholar]
- 48.Lindahl T, Ljungquist S, Siegert W, Nyberg B, Sperens B. 1977. DNA N-glycosidases: properties of uracil-DNA glycosidase from Escherichia coli. J. Biol. Chem. 252:3286–3294 [PubMed] [Google Scholar]
- 49.Laderoute MP, Giulivi A, Larocque L, Bellfoy D, Hou Y, Wu HX, Fowke K, Wu J, Diaz-Mitoma F. 2007. The replicative activity of human endogenous retrovirus K102 (HERV-K102) with HIV viremia. AIDS 21:2417–2424. 10.1097/QAD.0b013e3282f14d64 [DOI] [PubMed] [Google Scholar]
- 50.Yamada Y, Cancelas JA, Rothenberg ME. 2009. Murine model of hypereosinophilic syndromes/chronic eosinophilic leukemia. Int. Arch. Allergy Immunol. 149(Suppl 1):S102–S107. 10.1159/000211381 [DOI] [PubMed] [Google Scholar]
- 51.Rothenberg E, Smotkin D, Baltimore D, Weinberg RA. 1977. In vitro synthesis of infectious DNA of murine leukemia-virus. Nature 269:122–126. 10.1038/269122a0 [DOI] [PubMed] [Google Scholar]
- 52.Borroto-Esoda K, Boone LR. 1991. Equine infectious anemia virus and human immunodeficiency virus DNA synthesis in vitro: characterization of the endogenous reverse transcriptase reaction. J. Virol. 65:1952–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang H, Bagasra O, Niikura M, Poiesz BJ, Pomerantz RJ. 1994. Intravirion reverse transcripts in the peripheral blood plasma on human immunodeficiency virus type 1-infected individuals. J. Virol. 68:7591–7597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang H, Dornadula G, Pomerantz RJ. 1996. Endogenous reverse transcription of human immunodeficiency virus type 1 in physiological microenviroments: an important stage for viral infection of nondividing cells. J. Virol. 70:2809–2824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brady T, Lee YN, Ronen K, Malani N, Berry CC, Bieniasz PD, Bushman FD. 2009. Integration target site selection by a resurrected human endogenous retrovirus. Genes Dev. 23:633–642. 10.1101/gad.1762309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee YM, Coffin JM. 1990. Efficient autointegration of avian retrovirus DNA in vitro. J. Virol. 64:5958–5965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Farnet CM, Haseltine WA. 1991. Circularization of human immunodeficiency virus type 1 DNA in vitro. J. Virol. 65:6942–6952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schacker T. 2010. New tools to track HIV. Nat. Med. 16:373–374. 10.1038/nm0410-373 [DOI] [PubMed] [Google Scholar]
- 59.Sloan RD, Wainberg MA. 2011. The role of unintegrated DNA in HIV infection. Retrovirology 8:52. 10.1186/1742-4690-8-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dornadula G, Zhang H, Bagasra O, Pomerantz RJ. 1997. Natural endogenous reverse transcription of simian immunodeficiency virus. Virology 227:260–267. 10.1006/viro.1996.8317 [DOI] [PubMed] [Google Scholar]
- 61.Esqueda D, Xu F, Moore Y, Yang Z, Huang G, Lennon PA, Hu PC, Dong J. 2013. Lack of correlation between HERV-K expression and HIV-1 viral load in plasma specimens. Ann. Clin. Lab. Sci. 43:122–125 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.