

ABSTRACT
Herpes simplex virus (HSV) entry requires multiple interactions at the cell surface and activation of a complex calcium signaling cascade. Previous studies demonstrated that integrins participate in this process, but their precise role has not been determined. These studies were designed to test the hypothesis that integrin αvβ3 signaling promotes the release of intracellular calcium (Ca2+) stores and contributes to viral entry and cell-to-cell spread. Transfection of cells with small interfering RNA (siRNA) targeting integrin αvβ3, but not other integrin subunits, or treatment with cilengitide, an Arg-Gly-Asp (RGD) mimetic, impaired HSV-induced Ca2+ release, viral entry, plaque formation, and cell-to-cell spread of HSV-1 and HSV-2 in human cervical and primary genital tract epithelial cells. Coimmunoprecipitation studies and proximity ligation assays indicated that integrin αvβ3 interacts with glycoprotein H (gH). An HSV-2 gH-null virus was engineered to further assess the role of gH in the virus-induced signaling cascade. The gH-2-null virus bound to cells and activated Akt to induce a small Ca2+ response at the plasma membrane, but it failed to trigger the release of cytoplasmic Ca2+ stores and was impaired for entry and cell-to-cell spread. Silencing of integrin αvβ3 and deletion of gH prevented phosphorylation of focal adhesion kinase (FAK) and the transport of viral capsids to the nuclear pore. Together, these findings demonstrate that integrin signaling is activated downstream of virus-induced Akt signaling and facilitates viral entry through interactions with gH by activating the release of intracellular Ca2+ and FAK phosphorylation. These findings suggest a new target for HSV treatment and suppression.
IMPORTANCE Herpes simplex viruses are the leading cause of genital disease worldwide, the most common infection associated with neonatal encephalitis, and a major cofactor for HIV acquisition and transmission. There is no effective vaccine. These epidemiological findings underscore the urgency to develop novel HSV treatment or prevention strategies. This study addresses this gap by further defining the signaling pathways the virus usurps to enter human genital tract epithelial cells. Specifically, the study defines the role played by integrins and by the viral envelope glycoprotein H in entry and cell-to-cell spread. This knowledge will facilitate the identification of new targets for the development of treatment and prevention.
INTRODUCTION
Herpes simplex viruses (HSVs) are the leading cause of genital ulcer disease and neonatal encephalitis and a major cofactor in the HIV epidemic (1). These epidemiological findings highlight the need to develop new strategies for treatment and prevention. Defining the pathway of viral entry and cell-to-cell spread will promote the identification of targets for new drug or vaccine development. Entry into target cells by either serotype (HSV-1 or HSV-2) is complex, presumably reflecting the ability of virus to infect multiple cell types by either direct fusion or one of several endocytic pathways (2). Entry is initiated by attachment of HSV-1 glycoprotein C (gC) or HSV-2 gB to heparan sulfate moieties on syndecan proteoglycans (3–6) followed by engagement of one of several gD coreceptors, most commonly nectin-1 on epithelial cells (7–9). Engagement of the gD coreceptor is followed by the translocation of Akt to microdomains on the outer leaflet of the plasma membrane, where interactions with gB lead to Akt phosphorylation and release of calcium (Ca2+) near the plasma membrane (10). This initiates a signaling cascade that promotes the release of inositol-triphosphate receptor (IP3R)-dependent endoplasmic reticulum (ER) Ca2+ stores, leading to entry of viral capsids and tegument proteins and their transport to the nuclear pore (5, 11). The role played by gH in this Akt-Ca2+ entry pathway has not yet been delineated.
Glycoprotein H (which forms heteroligomers with gL) is also essential for viral entry and cell-to-cell spread and has been implicated in regulating the fusogenic activity of gB (12, 13). Several studies suggest that gH-gL may interact with integrins at the plasma membrane, although the findings have been inconsistent (5, 14–20). Viruses can induce conformational changes and/or clustering of integrins to elicit cell signaling, cytoskeletal rearrangement, and viral internalization (21). Since the sequence of gH contains the integrin binding motif Arg-Gly-Asp (RGD), it was previously proposed that gH might be a ligand for integrins (14, 20). A soluble form of HSV-1 gH-gL bound to Vero cells (monkey kidney epithelial cell line), and mutation of RGD to RGE blocked viral binding (14). However, a viral variant mutated in this sequence retained full infectivity, suggesting either that interactions with integrins are not essential or that the RGD motif may not be the only integrin binding partner (20). Other studies using CHO cells engineered to express different gD coreceptors found that integrin αvβ3 expression influenced the pathway of viral entry (18). More recently, it was found that integrin αvβ6 and αvβ8 promote HSV-1 endocytosis through engagement of gH in several different cell lines, including 293T cells (15). However, most of these prior studies have focused on HSV-1 and on cell lines where entry by endocytosis may predominate.
To address this gap, we explored the role integrins play in HSV-2 and HSV-1 entry into genital tract epithelial cells, where fusion of the viral envelope with the plasma cell membrane is presumed to predominate (2, 5). Studies were conducted using a human cervical cell line (CaSki) as well as primary genital tract human cells isolated from cervical tissue or cervicovaginal lavage (CVL) samples. We used small interfering RNA (siRNA) targeting different integrins, and cilengitide, a cyclic RGD peptide that binds to and inhibits the activities of the αvβ3 and αvβ5 integrins, and engineered a HSV-2 gH-null virus to examine the role gH-integrin interactions play in viral entry and Akt-Ca2+ signaling.
MATERIALS AND METHODS
Cells and viruses.
CaSki (human cervical epithelial cell line ATCC CRL-1550) and Vero (monkey kidney epithelial ATCC CCL-81) cells were passaged in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. Primary genital tract epithelial cells were isolated from cervical tissue obtained from women undergoing hysterectomy for benign conditions or from CVL samples obtained from healthy women participating in studies of genital tract mucosal immunity. Genital tract epithelial cells from cervical tissue were processed and expanded as previously described (12). The CVL samples were subjected to low-speed centrifugation, and the cellular pellet was resuspended in serum-free medium, subjected to a second centrifugation step to remove mucus, and grown in keratinocyte serum-free medium supplemented with epidermal growth factor (EGF) human recombinant and bovine pituitary extract (Life Technologies).
The laboratory isolates HSV-2 strain G [here designated HSV-2(G)], HSV-1(KOS), HSV-1(KVP26GFP), which contains a green fluorescent protein (GFP)-VP26 fusion protein (22), and HSV-2(333)ZAG, which expresses GFP under the control of a cytomegalovirus promoter inserted in an intergenic region between UL3 and UL4 (gift from P. Spear, Northwestern University) were propagated on Vero cells, and viral stocks were stored at −80°C. Stocks of an HSV-2(G) gD-2 deletion virus (ΔgD) (10) were propagated on complementing HSV-1 gD-expressing VD60 cells (23), and stocks of a previously described gB-2 deletion virus (ΔgB) were propagated on VgB2 cells (6). Stocks of a new gH-2-null virus (described below) were propagated on complementing HSV-1 gH-expressing F6 cells (24). One passage of each of the deletion viruses through Vero cells yields glycoprotein-negative virions. The complemented and noncomplemented viruses were purified on sucrose gradients, and relative particle numbers were determined by Western blotting and probing with an antibody (Ab) to the capsid protein, VP5 (5).
Construction of an HSV-2 gH-null virus.
The HSV-2 ΔgH virus was constructed using the recently described HSV-2 bacterial artificial chromosome (BAC) clone, bHSV2-BAC38 (25) and a linearized allelic exchange substrate. The allelic exchange plasmid was constructed by PCR amplification of 1-kb regions flanking UL22 with the primers described in Table 1. Purified DNA fragments were digested with Van91I (left homology region) and DraIII (right homology region) enzymes. Fragments were ligated with Van91I fragments of pYUB1471 using T4 DNA ligase (New England BioLabs, Ipswich, MA) (26). The allelic exchange plasmid contains a sacB-hygromycin counterselection/selection cassette driven by a prokaryotic promoter, which is flanked by γδ-resolvase sites. The resulting plasmid (pYUB1471-ΔUL22) was transformed into NEB 5-alpha competent cells, sequence verified, purified, and linearized at a single PacI site (NEB). The linearized product was electrotransformed into Escherichia coli SW102-containing bHSV2-BAC38, which was induced to express phage λ recombineering genes (25). Resulting colonies on chloramphenicol and hygromycin were screened for correct gene replacement, and DNA was purified using a Midiprep kit (Qiagen). Two primer sets were used to confirm appropriate gene replacement within the genome: left, primers gH2-L-check and sacB-Out; right, primers gH2-R-check and Hyg-Out (Table 1). Bacteria containing the desired allelic exchange plasmid were transformed with plasmid pYUB870, which encodes the gamma-delta-resolvase gene (tnpR) from transposon Tn1000 and a kanamycin-sacB selection/counterselection cassette. Transformed bacteria were grown on kanamycin, picked, grown at exponential phase, and plated on LB agar plates containing 10% sucrose. Colonies were then picked in replicates on plates containing chloramphenicol plus hygromycin or chloramphenicol alone. Colonies that grew only on plates containing chloramphenicol were expanded and screened for the loss of the sacB-hygromycin cassette. One microgram of this HSV-2 DNA was transfected into F6 cells with Effectene (Qiagen). Plates were screened for green plaques, and supernatants from positive wells were collected and overlaid onto fresh F6 cells for 1 h. Cells were then washed and covered with 1% low-melting-point agarose prepared in Optimem (Invitrogen). Single green fluorescent plaques were picked and purified three times using this method. HSV-2 ΔgH was then expanded on F6 cells to produce HSV-1 gH phenotypically complemented virus (designated ΔgH−/+ for phenotypically complemented virus or ΔgH−/− for noncomplemented virus). Genotypic confirmation of the gH deletion was performed by PCR using primers LL-V91I-UL22 and RR-DraIII-UL22. Phenotypic confirmation of the gH deletion was assessed by preparing Western blots of infected cell lysates or sucrose gradient-purified virus.
TABLE 1.
Primers used for construction of the gH-2 deletion virus
| Region amplified | Primer name | Sequence (5′ to 3′)a |
|---|---|---|
| Region homologous to left of UL22 | LL-Van91I-UL22 | TTTTTTTT CCAT AAA TTGG GTT CGT GCA AGT GAA GCA CAT CG |
| LR-Van91I-UL22 | TTTTTTTT CCAT TTC TTGG CGC GAA TAA ACG GGT GTG | |
| Region homologous to right of UL22 | RL-DraIII-UL22 | TTTTTTTT CAC AGA GTG GTC GTC CCG GCT GCC AGT C |
| RR-DraIII-UL22 | TTTTTTTT CAC CTT GTG CGT TCG TGG CCC TCA TGC C | |
| Left to UL22 | gH2-L-check | AAA TCA TGG GTG GAT GTG GTT CGC GAG C |
| gH2-WT-Left | ATC ACC CAC AAC GCC AGC TAC G | |
| sacB-Out | AGGATACAGGACCTGCCAAT | |
| Right to UL22 | gH2-R-check | GGT GAG GAT GCT TGG CCA GAA GCG G |
| gH2-WT-R-Out | GGC TGT CCC GCA TCG ATA TCA CG | |
| Hyg-Out | CTTCACCGATCCGGAGGAAC |
Underlining indicates Van91I and DraIII sites.
Antibodies and chemical reagents.
Primary antibodies and dilutions for Western blots, confocal microscopy, and coimmunoprecipitation were as follows: mouse monoclonal antibody (MAb) anti-VP16, 1:500 (catalog no. sc-7545; Santa Cruz Biotechnology [sc], Santa Cruz, CA); anti-VP5 MAb, 1:200 (sc-56989); anti-gB MAb, 1:500 (sc-69799), goat anti-gB, 1:200 (sc-22090); anti-gD MAb, 1:1,000 (sc-56988); anti-gC MAb, 1:200 (sc-69801); anti-gH-gL MAb, 1:200 (CH31, gift from R. Eisenberg and G. Cohen, University of Pennsylvania); anti-β-actin MAb, 1:5,000 (A-5441; Sigma-Aldrich, St. Louis, MO); anti-phospho-Akt (ser 473) MAb, 1:500 (4051; Cell Signaling Technology Inc., Danvers, MA); anti-histone H1 MAb, 1:250 (sc-8030), rabbit anti-total Akt123, 1:1,000 (sc-8312); rabbit anti-pY397 focal adhesion kinase (FAK), 1:1,000 (44-625G; Invitrogen), anti-total FAK MAb, 1:1,000 (610087; BD Bioscience), rabbit anti-goat immunoglobulin G (IgG), 1:500 (HAF017; R&D, Minneapolis, MN); goat polyclonal anti-integrin β8, 1:200 (sc-6638); goat polyclonal anti-integrin β6, 1:200 (sc-6633); rabbit polyclonal anti-integrin αv, 1:500 (sc6617-R); mouse anti-integrin αv, 1:200 (sc-9969); mouse anti-integrin αvβ3, 1:200 (sc7312); mouse anti-integrin β3, 1:50 (ab7167, Abcam, Cambridge, MA); rabbit anti-gH-gL, 1:500 (R137; gift from R Eisenberg and G. Cohen, University of Pennsylvania); rabbit polyclonal anti-PVRL1 (nectin-1), 1:100 (ab66985); and rabbit polyclonal anti-integrin β3, 1:500 (sc-14009). The secondary antibodies for Western blots were horseradish peroxidase-conjugated goat anti-mouse (170-5047, Bio-Rad, Hercules, CA), goat anti-rabbit (170-5046, Bio-Rad), or donkey anti-goat (sc-2020). The secondary antibodies for confocal microscopy were anti-mouse Alexa 488, Alexa 555, or Alexa 350 and anti-rabbit Alexa 488 or Alexa 350 (A11001, A211422, A11045, A11008, A21068; Invitrogen Molecular Probes). All secondary antibodies were diluted 1:1,000. Ionomycin was purchased from Invitrogen Molecular Probes (Bristol, United Kingdom) and cilengitide from MedKoo Biosciences (CedarLane, Burlington, NC).
Small interfering RNA and transfections.
Cells were transfected with a final concentration of 10 nM for each siRNA sequence in 12-well plates. For CaSki cells, the transfections were performed with the Lipofectamine RNAiMAX transfection reagent (Invitrogen, Carlsbad, CA) using the reverse transfection protocol provided by the manufacturer or HiPerFect transfection reagent (Qiagen). Primary cells were transfected using Effectene transfection reagent (Qiagen). The human integrin αv siRNA (identification no. s7568, s7569, s7570), integrin β3 siRNA (s7580, s7581, s7582), integrin α5 siRNA (s7547, s7548, s7549), integrin β5 siRNA (s7589, s7590, s7591), integrin β1 siRNA (s7574, s7575, s7576), control siRNA (Silencer negative-control siRNA no. 1, catalog no. AM4636) were purchased from Applied Biosystems (Applied Biosystems, Foster City, CA). Integrin β6 and integrin β8 siRNA (sc-43135 and sc-43137, respectively) were purchased from Santa Cruz Biotechnology. Cells were analyzed for protein expression by Western blots of cell lysates and for viral gene expression by quantitative reverse transcription-PCR (qRT-PCR) 72 h posttransfection. Total RNA was extracted using an Absolutely RNA Miniprep kit (Stratagene, La Jolla, CA). RNA was reverse transcribed with a High Capacity cDNA reverse transcription kit (Applied Biosystems). Real-time PCR amplification was performed using an ABI PRISM 7000 detection system and analyzed using sequence detector software. The reactions were performed using the TaqMan Universal PCR master mix (Applied Biosystems) and the commercially available probes for human integrin αv (Hs00233808_m1), integrin β3 (Hs00173978_m1), integrin α5 (Hs00233732_m1), integrin β5 (Hs00609896_m1), integrin β1 (Hs00559595_m1), Akt1 (Hs00178289_m1), Akt2 (Hs00609846_m1), Akt3 (Hs00178533_m1), interferon alpha (IFN-α; Hs00256882_s1), and the ribosomal large protein subunit (RPLPO) (4310879E) housekeeping gene obtained from Applied Biosystems. Quantification was normalized against the number of RPLPO transcripts in the same RNA extracts.
Western blots.
Cells were transfected with siRNA and were harvested and lysed in buffer containing 20 mM Tris (pH 7.5), 50 mM NaCl, 1% NP-40, and 0.05% sodium deoxycholate, supplemented with fresh protease and phosphatase inhibitors (Roche Diagnostics and Sigma-Aldrich, respectively). Proteins were separated by SDS-PAGE and transferred to membranes for immunoblotting with antibodies. Membranes were stripped between antibodies by incubating in Restore Western blot stripping buffer (Thermo Scientific) for 10 min at room temperature and washing 3 times in Tris-buffered saline (TBS)-Tween 20. Blots were scanned, and the band intensities were analyzed using a GelDoc2000 system. For Akt and FAK phosphorylation studies, the cells were transfected with siRNA as described above, and 48 h later, the medium was changed to serum-free medium and allowed to rest overnight. The cells were then exposed to virus (or medium as a mock-infection control), and at different times post-viral exposure, the cells were lysed and harvested for SDS-PAGE.
Viral binding, entry, and plaque assays.
For binding studies, cells were exposed to serial dilutions of purified virus for 5 h at 4°C. Unbound virus was removed by washing, and the cell-bound virus was analyzed by preparing Western blots of cell lysates and probing with anti-gD MAb (1103; Virusys, Sykesville, MD) (6). For plaque assays, cells were exposed to serial dilutions of virus for 1 h and then washed three times with phosphate-buffered saline (PBS), pH 7.4, and overlaid with 199 medium containing 1% pooled human IgG. Plaques were counted by immunoassay using an anti-human IgG antibody peroxidase conjugate (Calbiochem) (4). The ability of VP16, a viral tegument protein, to translocate to the nucleus following infection was assessed as a marker of viral entry. Cells were infected with the multiplicities of infection (MOI) of virus indicated in Fig. 2 at 37°C for 1 h and washed three times with PBS, and nuclear extracts were prepared and analyzed by Western blotting for VP16 and histone H1 (27).
FIG 2.

Silencing of integrin αvβ3 prevents HSV entry. (a) Primary genital tract cells generated from cervical tissue were transfected with siControl RNA or siRNA targeting integrin αvβ3 and were analyzed 72 h later for silencing by Western blotting, or (b) the silenced cells were inoculated with HSV-2(G) (MOI, 1 PFU/cell), and nuclear extracts were prepared 1 h p.i. and analyzed for the presence of the tegument protein VP16 and histone H1 (as a nuclear extract control). Blots shown are representative of results obtained in 2 independent experiments. (c) Transfected primary genital tract cells (siControl or siIntegrinαvβ3) were exposed to HSV-1(K26GFP) (MOI, 5 PFU/cell) and at 1 or 4 h p.i., cells were fixed and viewed by confocal microscopy; nuclei were stained with blue fluorescent Hoechst stain, and plasma membrane was stained with red fluorescent Alexa Fluor 594 wheat germ agglutinin stain. Top panels represent the XY image, bottom panels represent the XZ image. Bar =10 μm. (d) In parallel experiments, cells were infected in the presence of serum-free medium (control) or cilengitide (100 μM), and at 4 h p.i., the cells were fixed and viewed by confocal microscopy. (e) For both siRNA transfection and cilengitide treatment experiments, 100 cells from different fields were counted. Results are presented as the percentage of GFP-positive cells and are means (± SD) from 2 independent experiments; asterisks indicate significance (*, P < 0.05; **, P < 0.01) relative to the respective control.
Confocal microscopy.
Viral entry was also monitored by confocal microscopy. Cells were grown on glass coverslips in 12- or 24-well plates. The cells were transfected with siRNA and, at 48 h posttransfection, infected with KVP26GFP (MOI, 5 PFU/cell). To label plasma membranes and nuclei, the cells were stained for 10 min with red fluorescent Alexa Fluor 594 wheat germ agglutinin and blue fluorescent Hoechst 33342 stains as provided in an Image-It Live plasma membrane and nuclear labeling kit (Invitrogen). Cells were fixed with 4% paraformaldehyde solution (Electron Microscopy, Hatfield, PA) at the times postinfection (p.i.) indicated in the figure legends. Images were acquired by a ZeissLive/DuoScan laser confocal microscope equipped with a 100× oil immersion objective and a numerical aperture (NA) of 1.4. Images were captured in an optical slice of ∼0.5 μm with appropriate filters. Alexa Fluor 488 and GFP were excited using the 488-nm line of a krypton/argon laser and viewed with a 505- to 530-nm band-pass filter. Alexa Fluor 360 was excited with a 405-nm diode laser and collected with 420- to 475-nm filter, and Alexa Fluor 555 was excited using a 561-nm helium/neon laser and collected with a 575- to 655-nm filter. All images were captured using the multitrack mode of the microscope to decrease cross talk of fluorescent signals.
Calcium live-image microscopy.
Cells were grown in glass-bottom 35-mm culture dishes (P35G-1.5-10-C; MatTek Corporation, Ashland, MA), and cellular membranes were stained with lipophilic red fluorescent CellTrace Bodipy TR methyl ester stain and nuclei with Hoechst (blue) strain using an Image-It Live intracellular membrane and nuclear labeling kit (Invitrogen) or with Alexa Fluor 594 wheat germ agglutinin stain for plasma membranes and blue fluorescent Hoechst stain. The cells were then loaded with Calcium Green (2.5 μg/ml; Invitrogen) for 1 h at 37°C and infected with purified virus for 4 h at 4°C. Cells were washed three times with PBS, overlaid with 25 mM HEPES buffer, and placed into a temperature-regulated 37°C environmental chamber in a Zeiss Live DuoScan confocal microscope fitted with a 100×/1.4 NA oil objective. Images were acquired 3 min after the dishes were placed in the chamber. Z-sections were captured in an optical slice of 0.5 μm, and 15 to 20 cells were scanned per experiment. Image analysis was conducted using the laser scanning microscope (LSM) confocal software package (Carl Zeiss, Inc.), and intensity staining was quantified with image J software (National Institutes of Health, Bethesda, MD). Three-dimensional (3-D) images were generated using Volocity 5 confocal software (Improvision, Lexington, MA).
Calcium kinetic measurements.
CaSki cells (5 × 104) or primary vaginal cells (3 × 104) were seeded in 96-well black plates with clear bottoms (3340, CellBIND surface; Corning Inc., NY) and incubated with 25 μM fura-2 AM diluted in PBS (F1221; Invitrogen Molecular Probes) for 60 min at 37°C, rinsed thrice with PBS, placed on ice, and then exposed to cold purified HSV-2 (MOI, ∼5 PFU/cell) or control buffer (PBS). The cells were then transferred to a SpectraMaxMFe temperature-regulated chamber at 37°C (Molecular Devices) without washing; photometric data for [Ca2+] were generated by exciting cells at 340 and 380 nm and measuring emission at 510 nm every minute for 1 h. An intracellular calibration was performed with each experiment by determining the fluorescence ratio (340:380) in the presence of Ca2+-free 10 mM K2-EGTA buffer (Rmin) and 10 mM Ca-EGTA buffer containing 10 μM ionomycin (Rmax) (C-3008, calcium calibration buffer kit no. 1; Invitrogen Molecular Probes). The mean [Ca2+] was determined from four wells according to the manufacturer's recommendations using the following equation: [Ca2+] = Kd Q (R − Rmin)/(Rmax − R), where R represents the fluorescence intensity ratio Fλ1/Fλ2; λ1 (340 nm) and λ2 (380 nm) are the fluorescence detection wavelengths for ion-bound and ion-free indicators; Kd is the Ca2+ dissociation constant and equals 0.14 μM (fura and indo ratiometric calcium indicators, Invitrogen Molecular Probes); and Q is the ratio of Fmin to Fmax at λ2 (380 nm).
Immunoprecipitation assay.
Cells were serum starved for 24 h prior to being synchronously infected with purified virus (5 PFU/cell) and 2 and 15 min after being transferred to 37°C, the cells were placed back on ice, lysed by sonication in radioimmunoprecipitation assay (RIPA) buffer (Thermo Scientific) supplemented with complete protease inhibitors (Roche Diagnostics). The lysates were incubated with an equal amount (based on protein concentration provided by the manufacturer) of monoclonal anti-integrin αvβ3 or control mouse IgG (sc-7312 and sc-2343; Santa Cruz) overnight at 4°C and then immune complexes were isolated following 4 h of incubation with protein A agarose beads (Thermo Scientific). The precipitated complexes (pellet), supernatants, or an aliquot of the nonimmunoprecipitated cell lysate were analyzed by Western blotting for gH (rabbit serum R137) or gB (goat anti-gB, sc-22090). Reciprocal immunoprecipitation studies were performed by immunoprecipitating with anti-gH-gL MAb (CH31) and performing Western blots probing for integrin αv (rabbit sc-6617).
PLA.
CaSki cells (∼105 cell/well) were seeded on microscope glass cover slides in a 24-well plate (Fisher Scientific) and incubated with purified viruses (MOI, 5 to 10 PFU/cell) or mock infected for 4 h at 4°C, and the unbound virus was removed by washing thrice in cold PBS. The cells were then transferred to a 37°C water bath for 15 min, placed back at 4°C, and fixed with cold 4% paraformaldehyde. Blocking solution from Duolink in situ was used to prevent nonspecific interactions. Cells were incubated with primary Abs (1:100, 3% bovine serum albumin [BSA] in PBS) for 1 h at room temperature. Proximity ligation assay (PLA) probes used in this study were anti-mouse Minus (Duolink in situ, DUO092004; Sigma-Aldrich) and anti-rabbit Plus (Duolink in situ; DUO092002, Sigma-Aldrich). Hybridization, ligation, amplification, and detection steps were performed following the manufacturer's protocol (Detection Reagents Orange, Art. no. DUO092007, mounting medium with 4′,6′-diamidino-2-phenylindole [DAPI], Duolink, 82040-0005; Sigma-Aldrich). Fluorescence signals were detected using a Zeiss Live DuoScan confocal microscope fitted with a 64×/1.4 NA oil objective. Z-sections were captured in an optical slice of 0.44 μm. Extended-focus images were generated using Volocity 5 confocal software (Improvision, Lexington, MA).
Infectious center assays.
CaSki cells (donor cells) were labeled with 25 nM MitoTracker Orange CMTMRos (M7510; Invitrogen) for 15 min, washed, and infected with HSV-2(333)ZAG at a MOI of 5 to 10 PFU/cell. The cells were washed with citrate buffer to inactivate residual extracellular virus at 2 h p.i., detached with trypsin-EDTA at 4 to 5 h p.i., plated at a ratio of 1:5 with target cells that had been transfected with 10 nM siRNAs 72 h earlier, and then replated on MatTek 35-mm glass-bottom dishes (MatTek, Ahland, MA) 1 day prior to coculture. The cocultures were performed in medium containing pooled human IgG (Sigma), which neutralizes infection by virus released into the medium. The cocultures were incubated for 12 h, washed, fixed with 4% paraformaldehyde solution (Electron Microscopy, Hatfield, PA), and mounted in anti-fade reagent with DAPI (Invitrogen). Images were examined using a Leica SP2 AOBS confocal microscope (Leica Microsystems GmbH, Germany), and at least 10 randomly selected fields were counted.
Statistical analyses.
Analysis of variance or Student's t tests were performed using GraphPad Prism 6 (GraphPad Software, San Diego, California, USA) with adjustments for multiple comparisons, and P values of <0.05 were considered significant.
RESULTS
Silencing of integrin αvβ3, but not other integrins, reduces HSV plaque formation.
CaSki cells were transfected with siRNA sequences against integrin αv, α5, β1, β3, β5, β6, or β8 alone or a combination of αvβ3 and were infected 72 h later with HSV-2(G). Silencing of integrin αv and β3 or the combination, but that not the other integrins, significantly inhibited HSV-2 plaque formation (Fig. 1a). RT-PCR and Western blots were performed to assess the efficacy and specificity of silencing of integrin protein and gene expression (Fig. 1b and c, respectively). Each targeted integrin subunit was silenced specifically, although a reduction in αv expression was observed following transfection with siRNA targeting β3. The siRNAs were not cytotoxic, as measured by a cell proliferation assay (CellTiter96; Promega, CA, USA) and did not induce an IFN-α response (not shown). Similarly, transfection of primary genital tract epithelial cells with siRNA targeting integrin αvβ3 significantly reduced the number of infected cells, which were monitored by microscopy for GFP expression after exposing cells (3 × 104 cells per well) to HSV-2(333)ZAG (MOI, 0.1 PFU/cell) (Fig. 1d). No integrin β6 or β8 expression was detected in primary cells by Western blotting (not shown).
FIG 1.

Silencing of integrin αvβ3, but not other integrins, reduces HSV plaque formation. (a) CaSki cells were transfected with siRNA targeting αv, α5, β1, β3, β6, or β8 alone or a combination of αvβ3 and were infected 72 h later with serial dilutions of HSV-2(G). Viral plaques were counted at 48 h p.i. Results are presented as PFU on siRNA-transfected cells as a percentage of PFU on nontransfected cells and are means (± standard deviations [SD]) from at least 3 independent experiments conducted in duplicate. Only wells in which the number of plaques ranged between 25 and 200 plaques were used to calculate the viral titer. (b) CaSki cells were transfected with siRNA targeting the indicated integrins, and gene expression was determined by RT-PCR at 72 h posttransfection. Results are presented as percent expression relative to that of nontransfected cells and are means (± SD) from at least 3 independent experiments. (c) CaSki cells were transfected with siControl (siCtrl) or siRNA targeting integrin αvβ3 (here termed siIntegrinαvβ3 or siInαvβ3), siIntegrin β3 (siIntβ3), siIntegrin β6 (siIntβ6), or siIntegrin β8 RNA (siIntβ8), and protein expression was evaluated by Western blotting at 72 h posttransfection; the blots are representative of at least 2 independent experiments. (d) Primary vaginal cells cultivated from CVL pellets were transfected with the indicated siRNA and were infected 72 h later with HSV-2(333)ZAG (MOI, 0.1 PFU/cell) and monitored for GFP expression; images are representative of 2 independent experiments. Asterisks indicate significant difference relative to the control (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Silencing of integrin αvβ3 prevents HSV entry.
Several independent approaches were applied to determine if integrin αvβ3 contributes to viral entry. First, primary genital tract cells were transfected with siRNA (αvβ3 or control), and silencing was confirmed 72 h later by Western blotting (Fig. 2a). The cells were then infected with HSV-2(G), and after 1 h of incubation, nuclear extracts were prepared and evaluated for VP16 by Western blotting; histone H1 was included as a control for the nuclear extracts. VP16 is a viral tegument protein, which is delivered to the nucleus following HSV entry; thus, its nuclear transport provides a surrogate of entry. Silencing of integrin αvβ3 reduced the nuclear transport of VP16, which is consistent with a block to viral entry (Fig. 2b).
To further assess the impact of αvβ3 silencing on viral entry, confocal studies were conducted with purified K26GFP, an HSV-1 viral variant in which GFP has been fused in frame with the gene for the viral capsid protein VP26 (22). Cells were transfected with siRNA (control [here termed siControl] or αvβ3 [siαvβ3]) and 72 h later, the cells were infected (MOI, 5 PFU/cell). Cells were fixed and stained at 1 and 4 h p.i. and viewed by confocal microscopy. Capsids (GFP, green) were readily detected in the nuclei (DAPI, blue) of siControl, but not siαvβ3, cells (Fig. 2c). Specifically, colocalization of GFP and DAPI staining was observed for 85% of siControl compared to 10% of siαvβ3 cells (n = 100 cells counted) at 4 h p.i. (Fig. 2e). Similarly, treatment of cells with cilengitide (100 μM), a specific inhibitor of αvβ3 signaling, also decreased the number of internalized viral capsids (Fig. 2d and e). Cilengitide was not cytotoxic as measured by a cell proliferation assay of CaSki and primary genital tract cells (not shown).
Integrin αvβ3 contributes to the HSV-mediated cytoplasmic Ca2+ responses post-Akt phosphorylation.
Recent studies with human epithelial cells indicate that HSV induces Akt phosphorylation and the release of Ca2+ near the plasma membrane (10). The Akt activation was retained in CaSki cells transfected with siRNA targeting integrin αvβ3 (Fig. 3a). However, only a small amount of Ca2+ was detected in response to HSV-2 in siαvβ3-transfected cells by confocal microscopy, which was localized primarily near the plasma membrane (Fig. 3b). In contrast, HSV-2 triggered an increase in both plasma membrane and cytosolic Ca2+ in cells transfected with a control siRNA (siα5). The reduced Ca2+ response to virus in the siαvβ3-transfected cells did not reflect a loss of intracellular stores, as ionomycin triggered comparable responses in siαvβ3- and siα5-transfected cells.
FIG 3.

Integrin αvβ3 contributes to the virus-induced cytoplasmic Ca2+ response post-Akt phosphorylation. (a) CaSki cells transfected with siIntegrinαvβ3 RNA were mock infected (serum free medium) or infected with purified HSV-2(G) (10 PFU/cell), and cell lysates were prepared for Western blot analysis at the indicated times p.i. Blots were incubated with anti pS473-Akt123 and then stripped and probed with anti-total Akt123. A representative blot from 3 independent experiments is shown. (b) CaSki cells were loaded with Calcium Green 72 h posttransfection with the indicated siRNA and synchronously mock infected or infected with purified HSV-2(G) (5 PFU/cell). Live images were acquired 3 min after a temperature shift to 37°C. Cellular membranes were stained with CellTrace (red), nuclei were stained with Hoechst (blue), and Ca2+ is green. Representative XYZ images from 3 independent experiments are shown. Bars = 9.2 μm. (c) Transfected CaSki cells were loaded with fura-2, infected with purified HSV-2(G) (2 PFU/cell), or mock infected. To assess whether siRNA transfections impacted the intracellular Ca2+ stores, uninfected cells were treated with 1 μM ionomycin. The mean intracellular Ca2+ concentration (nM) over 1 h was calculated from 4 wells, each containing 5 × 104 cells; the asterisk indicates a significant increase in Ca2+ concentration relative to the mock-infected control (P < 0.05). (d) Parallel studies were conducted with cells treated with calcium-free buffer (control) or buffer supplemented with cilengitide (100 μM), and the Ca2+ response over 1 h p.i. was compared to that of mock-infected cells.
While confocal microscopy allows one to detect responses in individual cells and identify the subcellular localization of Ca2+ responses, it is not quantitative. Therefore, quantitative kinetic fluorometric studies were conducted in parallel. Transfected CaSki cells (siControl or siαvβ3) were loaded with fura-2 and infected with purified HSV-2(G) (MOI, 2 PFU/cell), and the mean intracellular Ca2+ concentration ([Ca2+]) over 1 h was calculated from 4 wells, each containing 5 × 104 cells. HSV-2 induced a significant increase (∼5.4-fold) in [Ca2+] compared to that of mock-infected cells transfected with a control siRNA. In contrast, HSV-2 triggered only a small (∼1.7-fold) increase in [Ca2+] in cells transfected with siαvβ3, which did not achieve statistical significance (Fig. 3c). This small response presumably reflects the Akt-associated response at the plasma membrane. Cilengitide also inhibited the Ca2+ response to HSV-2 in primary genital tract epithelial cells (Fig. 3d). Together, these findings suggest that integrin is activated downstream of virus-induced Akt phosphorylation and contributes to the cytosolic Ca2+ signaling pathway required for HSV entry.
Integrin αvβ3 interacts with glycoprotein H.
It was previously suggested that gH may be a ligand for integrins based, for example, on surface plasmon resonance studies with soluble proteins (15), but direct evidence with virus is lacking. To address this, coimmunoprecipitation studies were performed. CaSki cells were synchronously infected with purified HSV-2(G) (MOI, 5 PFU/cell), and cell lysates were prepared 2 and 15 min after the shift in temperature and were incubated with a MAb to integrin αvβ3 or an isotype control MAb. The immune complexes were precipitated with protein A-agarose and analyzed by Western blotting with rabbit polyclonal antibody to gH-gL or, as a control, with a goat polyclonal antibody to gB. Glycoprotein H, but not gB, was detected in the pellet following precipitation with anti-integrin αvβ3 antibodies, but gH was retained in the supernatant when proteins were precipitated with an isotype control antibody (Fig. 4a, left and middle panels). Conversely, integrin αv was detected in the pellet (and supernatant) following precipitation with an anti-gH-gL MAb (CH31) (Fig. 4a, right). As a complementary approach, proximity ligation assays were performed. CaSki cells were synchronously infected (no siRNA) or first transfected with integrin αvβ3 or control siRNA and then synchronously infected 72 h later by exposing the cells to HSV-2(G) (MOI, 5 PFU/cell) at 4°C to allow viral binding, washing the cells, and then transferring them to a water bath at 37°C for 15 min to allow viral entry. The cells were then placed back on ice, fixed, and stained with murine monoclonal antibodies (MAbs) directed against integrin αvβ3 and rabbit polyclonal serum (rabbit immunoglobulin [RIg]) against gH-gL and probed with species-specific proximity ligation secondary antibodies. As a positive control for the assay, nontransfected HSV-2 synchronously infected cells were stained with a MAb to gD and RIg to nectin-1. A high concentration of fluorescence (red signal) was detected in the majority of nontransfected and siControl-infected cells, but not siαvβ3-transfected cells, supporting an interaction between gH and integrin αvβ3 (Fig. 4b). A similar signal was observed with the gD-nectin-1 Ab-stained cells. The polyclonal rabbit anti-gH-gL Ab did not interact with MAbs directed against syndecan-2 or Akt, and conversely, rabbit anti-integrin αvβ3 did not interact with gB or gD (Fig. 4c).
FIG 4.

Integrin αvβ3 interacts with glycoprotein H. (a) CaSki cells were synchronously infected with purified HSV-2 (5 PFU/cell), and cell lysates were harvested 2 and 15 min post-temperature shift and incubated with monoclonal mouse anti-integrin αvβ3 (left panels) or an isotype control MAb (middle panel). Immune complexes were precipitated, and equivalent volumes of the whole-cell lysate (starting material), supernatant, and pellet were subjected to Western blotting with rabbit anti-gH-gL (upper left) or goat anti-gB (lower left) antibodies. In reciprocal experiments, lysates were precipitated with monoclonal antibodies to gH-gL and analyzed by Western blotting with rabbit polyclonal antibodies to integrin αv (right panel). Controls included uninfected cell lysates; blots shown are representative of 5 independent experiments. (b) CaSki cells were synchronously infected with purified HSV-2(G) (5 PFU/cell) (no siRNA) 72 h after being transfected with the indicated siRNA. The cells were subsequently fixed and probed with monoclonal mouse antibodies (MAb) to integrin αvβ3 or gD and rabbit sera (RIg) to gH-gL or nectin-1 and assessed in a proximity ligation assay. (c) Additional proximity ligation studies were conducted with nontransfected CaSki cells that were synchronously infected with HSV-2(G) as in panel b and fixed and probed with the indicated antibodies. Proximity ligation results are representative of 2 independent experiments. Bar = 10 μM.
An HSV-2 gH-null virus is impaired in viral entry.
To further address the role of gH-integrin interactions in promoting the signaling cascade associated with HSV entry, a virus with a deletion of gH-2 was constructed using a recently described HSV-2 BAC clone, bHSV2-BAC38 (25). The virus was expanded on gH-1-expressing F6 cells to produce phenotypically complemented virus (designated ΔgH−/+) and passaged once on Vero cells to produce phenotypically gH-null virus (ΔgH−/−). Genotypic confirmation of the gH deletion was performed by PCR (Fig. 5a). The phenotype was assessed by performing Western blots of purified virus after passage of the virus on F6 (ΔgH−/+) or Vero (ΔgH−/−) cells and by examining plaque formation with fluorescence microscopy of the complementing and noncomplementing cells. The gH protein was detected only in complemented virus, whereas gD, gB, gC, and VP5 were detected in virus purified from both F6 and Vero cells (Fig. 5b). Plaques were readily detected when F6 cells were infected with the complemented ΔgH−/+ virus, but only rare singly infected cells were observed for the Vero cells (Fig. 5c). The ΔgH−/+ virus was competent for viral binding, which is mediated by gB-2 (Fig. 5d), but it is impaired in entry as evidenced by decreased detection of nuclear VP16 following exposure of CaSki cells to ΔgH−/− (Fig. 5e). Moreover, gH-integrin αvβ3 interactions were observed by proximity ligation assay when CaSki cells were exposed to ΔgH−/+, but not ΔgH−/− (Fig. 5f). Virus purified on F6 cells had a 10,000 higher titer for F6 cells than that for Vero cells (Table 2).
FIG 5.

Construction and characterization of an HSV-2 gH-null virus. (a) Genotypic characterization of ΔgH-2 was performed by PCR using two primer sets to confirm appropriate replacement of gH. The left flank of UL22 was tested with primers gH2-L-check and sacB-Out, while the right flank of this gene was assessed with primers gH2-R-check and Hyg-Out (Table 1). (b) The gH-null virus was propagated on gH-1-expressing F6 cells to yield complemented virus (ΔgH−/+) or on Vero cells to yield noncomplemented virus (ΔgH−/−). The viruses were purified on sucrose gradients, and equivalent numbers of viral particles (estimated by comparing VP5 expression on Western blots) were analyzed for expression of viral proteins by immunoblotting with rabbit polyclonal Ab to gH-gL or murine MAbs to gB, gD, gC, and VP5. (c) Representative fluorescence microscopy image obtained 36 h p.i. of F6 or Vero cells with ΔgH−/+. (d) To evaluate whether deletion of gH impacted binding, CaSki cells were exposed to serial 2-fold dilutions of relatively equal numbers of purified complemented or noncomplemented gH-null viruses (starting with viral particle numbers equivalent to an MOI of 5 PFU/cell) at 4°C for 4 h. Binding was assessed by performing Western blots of cellular lysates with gD and β-actin, and results shown are representative of 3 independent experiments. (e) CaSki cells were inoculated with purified virus (relative particle numbers equivalent to an MOI of 1 PFU/cell on F6 cells), and nuclear extracts were prepared 1 h p.i. and probed for the tegument protein VP16 and histone 1 (H1). A blot representative of results from 3 independent experiments is shown. (f) CaSki cells were synchronously infected with purified ΔgH−/+ or ΔgH−/− (equivalent to 5 PFU/cell) and fixed and probed with monoclonal mouse antibodies to integrin αvβ3 and rabbit sera to gH-gL and assessed in a proximity ligation assay. Results are representative of 3 independent experiments.
TABLE 2.
Titer of the HSV-2 gH null virus purified on complementing gH-1-expressing F6 cells or noncomplementing Vero cells
| Cell line virus purified on: | Titer (PFU/ml) on: |
|
|---|---|---|
| Vero cells | F6 cells | |
| F6 cells (ΔgH−/+) | 5 × 103 | 3 × 107 |
| Vero cells (ΔgH−/−) | 4 × 103 | 6 × 103 |
Integrin αvβ3-gH interactions are required for cytosolic Ca2+ responses and FAK phosphorylation.
Exposure of cells to either ΔgH−/+ or ΔgH−/− did not prevent the release of a small amount of Ca2+ at the plasma membrane as seen by confocal microscopy (Fig. 6a). However, there was no significant increase in cytosolic [Ca2+] following exposure of CaSki cells (Fig. 6b, upper panel) or primary genital tract epithelial cells (Fig. 6b, lower panel) to ΔgH−/− virus, whereas the complemented virus (ΔgH−/+) triggered a significant increase in [Ca2+] in both cell types (3.8- and 3.0-fold increase, respectively). Furthermore, using a proximity ligation assay with a mouse MAb to gB and rabbit polyclonal antibody to Akt, we observed that inoculation of cells with either ΔgH−/+ or ΔgH−/− did not block the interaction between Akt and gB (Fig. 6c). These findings contrast with previously described results for ΔgB−/− and ΔgD−/− mutants (10) and support the findings that Akt phosphorylation is preserved in integrin αvβ3-silenced cells (Fig. 3a).
FIG 6.

Integrin αvβ3-gH interactions trigger release of cytosolic Ca2+. (a) CaSki cells were loaded with Calcium Green and synchronously infected with ΔgH2−/+ or ΔgH2−/− viruses (equivalent to 5 PFU/cell) or mock infected (medium). Live images were acquired at the indicated time post-temperature shift. Nuclei were stained with Hoechst (blue), and plasma membrane was stained with red fluorescent Alexa Fluor 594 wheat germ agglutinin. Representative extended-focus images from 3 independent experiments are shown. (b) CaSki or primary genital tract cells were loaded with fura-2 and exposed to ΔgH2−/+ or ΔgH2−/− viruses (equivalent to 5 PFU/cell), and the mean intracellular Ca2+ concentration (nM) over 1 h was calculated from 4 wells, each containing 5 ×104 CaSki (upper panel) or 3 × 104 primary (lower panel) cells; the asterisk indicates significant increase in Ca2+ concentration relative to that seen with mock infection (P < 0.01). (c) CaSki cells were exposed to ΔgH2−/+ or ΔgH2−/− viruses (equivalent to 2 PFU/cell) for 4 h at 4°C, unbound virus was removed by washing, and cells were shifted to 37°C for 15 min (synchronous infection). The cells were then fixed and stained for a proximity ligation assay with mouse MAb to gB and rabbit polyclonal antibodies to Akt. Images are representative of results of 2 independent experiments.
Consistent with these findings, Akt was detected on the surface of cells following exposure to phenotypically complemented viruses and following exposure to both ΔgH−/+ or ΔgH−/−, but not in mock-infected cells or cells exposed to noncomplemented ΔgB−/− and ΔgD−/− mutants (Fig. 7a and 7b, green staining). Akt was assessed and detected under all conditions in permeabilized cells (only shown for mock-infected cells). Notably, there was a parallel increase in the detection of surface integrin αvβ3 in cells in response to the same viruses (Fig. 7a and c, red staining). These findings confirm previous studies indicating that binding of gD and gB are sufficient to trigger a redistribution of Akt (10) and indicate that this is associated with increased integrin αvβ3 detection near the cell surface, which may facilitate subsequent interactions with gH.
FIG 7.

Glycoprotein H is not required for Akt or integrin αvβ3 relocalization. CaSki cells were synchronously infected with ΔgH2−/+ or ΔgH2−/− viruses (equivalent to 5 PFU/cell), and at 15 min post-temperature shift, the cells were fixed and stained with rabbit polyclonal Ab to Akt123 (green) and mouse MAb to integrin αvβ3 (red). Nuclei were stained with DAPI (blue). For comparison, mock-infected cells were also permeabilized (perm) with 1% Triton X-100. Representative XYZ images from 2 independent experiments are shown in panel a, and the mean (± standard error of the mean [SEM]) fluorescence intensity (MFI) per cell calculated from 100 cells for Akt (green) is shown in panel b and that for integrin (red) in panel c. The asterisks indicate significant increases in MFI relative to the nonpermeabilized mock-infected cells (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
FAK phosphorylation, which has been previously shown to promote transport of viral capsids to the nuclear pore (11), was reduced when cells were transfected with siRNA targeting integrin αv and/or β3 (Fig. 8a). A modest decrease in FAK phosphorylation was also observed in cells transfected with siRNA targeting β1, although this did not translate to other phenotypic changes. Similarly, the gH-null virus did not induce FAK phosphorylation; phosphorylation (which was observed as early as 5 min p.i.) was restored when cells were exposed to complemented virus (Fig. 8b). Thus, while gB and gD are required to initiate the cellular signaling responses, gH must engage integrin αvβ3 to complete the internalization process, including the release of intracellular Ca2+ stores and activation of FAK to facilitate viral entry and capsid transport.
FIG 8.
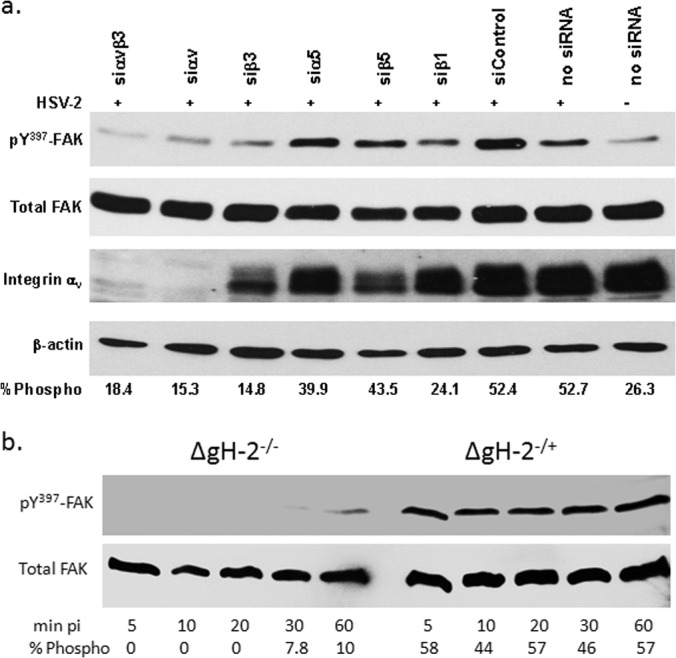
Integrin αvβ3-gH interactions are required for FAK phosphorylation. (a) CaSki cells transfected with the indicated siRNA and were exposed 72 h later to serum-free medium (mock-infected) or infected with HSV-2(G) (MOI, 10 PFU/cell) and cellular lysates prepared 5 min p.i. Western blots were performed and probed for phosphorylated FAK (pY397FAK), total FAK, integrin αv, and β-actin. A blot representative of 3 independent experiments is shown. The blots were scanned, and the mean percentage of phosphorylated FAK relative to total FAK from the 3 independent experiments is indicated. (b) To assess the role of gH, CaSki cells were infected with ΔgH2−/+ or ΔgH2−/− viruses (equivalent to 5 PFU/cell), and at the indicated times p.i., analyzed for phosphorylated and total FAK. Results are representative of 2 independent experiments.
Integrin αvβ3 is important for HSV cell-to-cell spread.
Silencing of integrin αvβ3, but not other integrin subunits, reduced the number and size of plaques (Fig. 1a and Fig. 9a). Based on this observation, we investigated whether integrin αvβ3 plays a role in HSV cell-to-cell spread. A modified infectious center was performed whereby CaSki cells were labeled with Mitotracker Orange, infected with HSV-2(333)ZAG (MOI, 5) for 4 h (donor cells) and then cocultured (ratio 1:5) with uninfected unlabeled target cells, that had been transfected with integrin-specific or control siRNAs 72 h earlier. Cells were cocultured for 12 h in the presence of pooled human immunoglobulin, and the number of unlabeled targets expressing GFP (indicative of cell-to-cell spread) was quantified. Silencing of integrin αvβ3 reduced the number of GFP-positive target cells per infected donor cell 19-fold, from 2.97 ± 0.24 in siα5-transfected targets to 0.19 ± 0.06 in siαvβ3-transfected targets (10 fields of cells were counted from a minimum of 2 experiments). Representative images are shown in Fig. 9b. Similarly, deletion of gH also prevented cell-to-cell spread as evidenced by the size of plaques on noncomplementing cells (Fig. 5). Together, these findings indicate that gH-integrin interactions are required for both viral entry and cell-to-cell spread.
FIG 9.
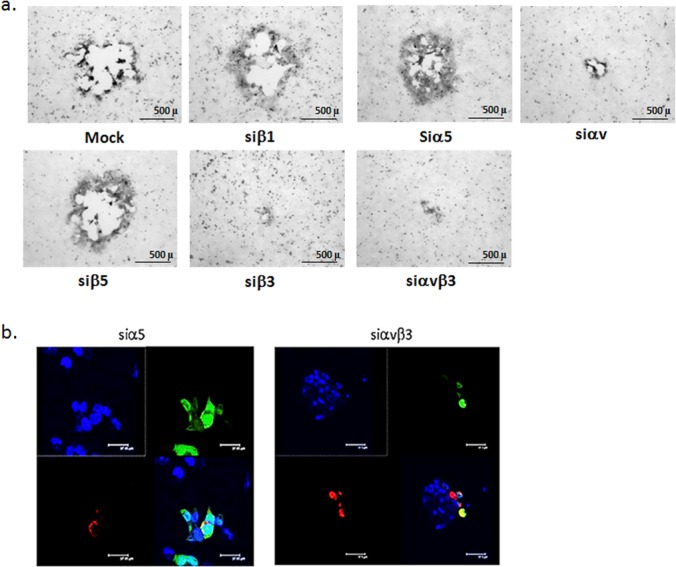
Integrin αvβ3 is important for cell-to-cell spread. (a) CaSki cells were transfected with the indicated siRNA and 72 h later were infected with HSV-2(G). Plaques were visualized by immunostaining and representative images are shown. (b) To further assess the impact of integrin αvβ3 on cell-to-cell spread, a modified infectious center assay was performed in which “donor” cells were labeled with Mitotracker Orange, infected with HSV-2(333)ZAG, treated with a low-pH citrate buffer at 1 h p.i. to inactivate any extracellular virus. At 4 h p.i., the donor cells were trypsinized and plated at a ratio of 1:5 on “receiver” cells that had been transfected 72 h earlier with the indicated siRNA and then cocultured in medium containing pooled human immunoglobulin. The cocultures were incubated for 12 h, washed, fixed, and mounted in anti-fade reagent with DAPI to stain nuclei (blue). Representative images from 10 different fields and a minimum of 2 independent experiments are shown. Upper left, DAPI channel; upper right, GFP channel; lower left, Mitotracker Orange; and lower right, merge.
DISCUSSION
HSV infects many different cell types and likely enters both by direct fusion and fusion following endocytosis or phagocytosis (28, 29). These processes involve interactions with multiple cellular receptors and activation of Ca2+ signaling pathways that facilitate viral entry and transport of viral capsids to the nuclear pore (5, 10, 11, 27). A role for integrins in HSV entry was suggested previously based on the observation that gH has an RGD sequence, but studies of cell lines that are not the primary target of HSV, such as Vero, CHO, and integrin-negative K562 (myeloid leukemia) cells and with an HSV-1 variant carrying a mutation in the RGD sequence have yielded conflicting results (14, 15, 18, 19). We therefore focused our efforts on human cells derived from the female genital tract (CaSki and primary cells) because the genital tract is an important site of infection and engineered a gH-2-null virus (propagated on a gH-1-expressing cell line) to test the hypothesis that gH engages integrins to promote Ca2+ signaling and viral entry and to identify the specific integrin subunits. The results obtained extend a model of HSV entry that involves activation of a complex Ca2+ signaling cascade. Specifically, the data indicate that gH from either serotype interacts with integrin αvβ3 downstream of virus-induced Akt phosphorylation to trigger the release of intracellular Ca2+ stores and FAK phosphorylation promoting viral entry and capsid transport. Silencing of other integrins had little effect on viral infection, although it should be noted that CaSki (Fig. 1) and primary genital tract cells (not shown) expressed very low levels of integrin β6 or β8.
Integrins are multifunctional molecules expressed in many cell types that respond to signals from outside (“outside-in”) and inside (“inside-out”) the cell. Integrin clustering and engagement by ligands activate several intracellular signaling pathways involving molecules such as Akt and FAK (30). HSV appears to usurp this signaling network to promote viral entry, but in an unusual manner as illustrated by the activation of Akt upstream of integrin αvβ3 signaling. This differs from the more commonly described scenario in which virus-induced integrin signaling activates Akt downstream, as observed for vaccinia virus and human cytomegalovirus (HCMV) (31, 32). Moreover, exposure of cells to HSV triggers a relocalization of Akt to the outer plasma membrane as evidenced here by microscopy of nonpermeabilized cells (Fig. 7) and previously by biotinylation of cell surface proteins (10). The relocalization and activation of Akt was associated with increased detection of integrin αvβ3 at the plasma membrane, which may facilitate interactions with gH. Translocation of integrin αvβ3 to lipid rafts has been described during HCMV entry (32) and increased migration and expression of integrin αvβ3 at the cell surface was also described in response to transforming growth factor β (TGF-β) on human chondrosarcoma cells (33). The cellular responses to HSV facilitate infection, since perturbations in Akt or integrin signaling with siRNA or by the addition of pharmacological inhibitors impede HSV entry (10).
While gH-integrin αvβ3 interactions are not required for the HSV-induced activation of Akt and the associated small increase in Ca2+ near the plasma membrane, this pairing is required for the significant increase in intracellular Ca2+ concentration, which is observed by confocal microscopy and Ca2+ fluorometry. Furthermore, the entry process is aborted when integrin αvβ3 (but not other integrins) are silenced or following exposure to a gH-2-null virus. These findings are consistent with a known link between integrin and Ca2+ responses. Integrin ligands trigger the activation of phosphoinositide-specific phospholipase C enzymes, which in turn catalyze the hydrolysis of phosphatidylinositol 4,5-bisphosphate to inositol 1,4,5-trisphosphate (IP3) and diacylglycerol; the former binds to the IP3 receptor to trigger the release of endoplasmic reticulum (ER) Ca2+ stores (5, 34). Activation of this pathway often occurs in response to inside-out signals or through G-protein-coupled receptors (35). The response to HSV, which is triggered by an outside (gH) signal, also differs from the response described for Madin-Darby canine kidney epithelial cells, where RGD triggered a Ca2+ influx independent of IP3 as well as the release of IP3-mediated intracellular stores (34). Our prior work indicates that the predominant source of the increased intracellular Ca2+ is the release of IP3-dependent ER stores and not the Ca2+ influx. This is supported by the findings that the HSV-induced Ca2+ response is blocked when intracellular, but not extracellular, Ca2+ is chelated and by silencing of or pharmacological blockade of the IP3R (27).
The observation that integrin binding and aggregation lead to the activation of FAK has been previously described with other integrin ligands (36). For example, viruses that bind integrin receptors, such as human herpesvirus 8 (HHV-8), trigger FAK phosphorylation (37). We previously showed that HSV-1 and HSV-2 induce FAK and proline-rich kinase 2 (Pyk2) phosphorylation to promote the transport of viral capsids and tegument proteins to the nuclear pore (11). However, while the earlier studies showed that FAK phosphorylation occurred downstream of Ca2+ responses, the signal pathways that triggered FAK phosphorylation were not identified. The current studies fill this gap and link FAK phosphorylation to αvβ3-gH interactions. We used a newly engineered gH-2-null virus, rather than the previously described HSV-1 variant in which the RGD motif in gH was mutated to RGE or recombinant proteins, to test the role of gH and integrin signaling in viral infection. The different outcomes obtained with the gH-null virus compared to those with the RGE mutant virus suggest that the RGD motif is not essential, as the latter virus retained infectivity (14). Deletion of gH from the viral envelope prevented viral interactions with integrin αvβ3 and blocked viral entry; the phenotype was restored when the virus was complemented with HSV-1 gH-1 by growing the virus on F6 cells.
We also demonstrate for the first time by a combination of coimmunoprecipitation and proximity ligation assays that gH-2 [HSV-2(G)] and gH-1 (gH-2-null virus complemented with F6 cells) are ligands for integrin αvβ3. Importantly, these studies were performed with virus and human cells, rather than recombinant and/or tagged viral proteins, as the structure and function of individual components may be impacted by virus-cell interactions and other envelope glycoproteins. Indeed, no interaction between gH and αvβ3 was detected by proximity ligation assay in response to ΔgD or ΔgB virus (expressing gH), although soluble gH-gL can bind to immobilized integrin αvβ3 (16).
The current studies focused on the impact of gH-αvβ3 interactions on viral entry. However, other work also links the virus-induced Akt/integrin/Ca2+ cascade to innate immune responses. For example, using 293T cells, Campadelli-Fiume and colleagues have shown that integrin αvβ3 relocates HSV-1 to cholesterol-rich membrane microdomains where the virus activates innate responses through Toll-like receptor 2 (TLR2)-dependent and TLR2-independent pathways (16). Moreover, HSV virus-like particles induced the expression of interferon-stimulated genes, including CXCL10 in dendritic cells, but the response was dependent on Akt activation, as drugs that blocked Akt phosphorylation prevented the CXCL10 response (38). Possibly, the signaling pathway described here to promote HSV entry into genital tract epithelial cells also modulates innate immune responses, as genital tract epithelium expresses Toll-like receptors, chemokines, cytokines, and interferons. Thus, targeting the virus-cell signaling pathways may not only lead to new strategies to prevent HSV infection but also could modulate host immune responses.
ACKNOWLEDGMENTS
We thank Prashant Desai (Johns Hopkins University) for the KVP26GFP virus, Patricia Spear for the HSV-2(333)ZAG virus, and Jeff Cohen (NIH) for the BAC. We also thank Roselyn Eisenberg and Gary Cohen for antibodies.
This work was supported in part by NIH AI-061679, AI-065309, and the Center for AIDS Research at the Albert Einstein College of Medicine and Montefiore Medical Center (NIH AI-51519).
Footnotes
Published ahead of print 18 June 2014
REFERENCES
- 1.Corey L, Wald A, Celum CL, Quinn TC. 2004. The effects of herpes simplex virus-2 on HIV-1 acquisition and transmission: a review of two overlapping epidemics. J. Acquir. Immune Defic. Syndr. 35:435–445. 10.1097/00126334-200404150-00001 [DOI] [PubMed] [Google Scholar]
- 2.Spear PG. 2004. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol. 6:401–410. 10.1111/j.1462-5822.2004.00389.x [DOI] [PubMed] [Google Scholar]
- 3.WuDunn D, Spear PG. 1989. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 63:52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herold BC, WuDunn D, Soltys N, Spear PG. 1991. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 65:1090–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheshenko N, Liu W, Satlin LM, Herold BC. 2007. Multiple receptor interactions trigger release of membrane and intracellular calcium stores critical for herpes simplex virus entry. Mol. Biol. Cell 18:3119–3130. 10.1091/mbc.E07-01-0062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheshenko N, Herold BC. 2002. Glycoprotein B plays a predominant role in mediating herpes simplex virus type 2 attachment and is required for entry and cell-to-cell spread. J. Gen. Virol. 83:2247–2255 [DOI] [PubMed] [Google Scholar]
- 7.Carfi A, Gong H, Lou H, Willis SH, Cohen GH, Eisenberg RJ, Wiley DC. 2002. Crystallization and preliminary diffraction studies of the ectodomain of the envelope glycoprotein D from herpes simplex virus 1 alone and in complex with the ectodomain of the human receptor HveA. Acta Crystallogr. D Biol. Crystallogr. 58:836–838. 10.1107/S0907444902001270 [DOI] [PubMed] [Google Scholar]
- 8.Connolly SA, Landsburg DJ, Carfi A, Whitbeck JC, Zuo Y, Wiley DC, Cohen GH, Eisenberg RJ. 2005. Potential nectin-1 binding site on herpes simplex virus glycoprotein d. J. Virol. 79:1282–1295. 10.1128/JVI.79.2.1282-1295.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warner MS, Geraghty RJ, Martinez WM, Montgomery RI, Whitbeck JC, Xu R, Eisenberg RJ, Cohen GH, Spear PG. 1998. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 246:179–189. 10.1006/viro.1998.9218 [DOI] [PubMed] [Google Scholar]
- 10.Cheshenko N, Trepanier JB, Stefanidou M, Buckley N, Gonzalez P, Jacobs W, Herold BC. 2013. HSV activates Akt to trigger calcium release and promote viral entry: novel candidate target for treatment and suppression. FASEB J. 27:2584–2599. 10.1096/fj.12-220285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheshenko N, Liu W, Satlin LM, Herold BC. 2005. Focal adhesion kinase plays a pivotal role in herpes simplex virus entry. J. Biol. Chem. 280:31116–31125. 10.1074/jbc.M503518200 [DOI] [PubMed] [Google Scholar]
- 12.Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. U. S. A. 104:18718–18723. 10.1073/pnas.0707452104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 84:12292–12299. 10.1128/JVI.01700-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parry C, Bell S, Minson T, Browne H. 2005. Herpes simplex virus type 1 glycoprotein H binds to αvβ3 integrins. J. Gen. Virol. 86:7–10. 10.1099/vir.0.80567-0 [DOI] [PubMed] [Google Scholar]
- 15.Gianni T, Salvioli S, Chesnokova LS, Hutt-Fletcher LM, Campadelli-Fiume G. 2013. αvβ6- and αvβ8-integrins serve as interchangeable receptors for HSV gH/gL to promote endocytosis and activation of membrane fusion. PLoS Pathog. 9:e1003806. 10.1371/journal.ppat.1003806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gianni T, Leoni V, Chesnokova LS, Hutt-Fletcher LM, Campadelli-Fiume G. 2012. αvβ3-integrin is a major sensor and activator of innate immunity to herpes simplex virus-1. Proc. Natl. Acad. Sci. U. S. A. 109:19792–19797. 10.1073/pnas.1212597109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gianni T, Leoni V, Campadelli-Fiume G. 2013. Type I interferon and NF-κB activation elicited by herpes simplex virus gH/gL via αvβ3 integrin in epithelial and neuronal cell lines. J. Virol. 87:13911–13916. 10.1128/JVI.01894-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gianni T, Gatta V, Campadelli-Fiume G. 2010. αVβ3-integrin routes herpes simplex virus to an entry pathway dependent on cholesterol-rich lipid rafts and dynamin2. Proc. Natl. Acad. Sci. U. S. A. 107:22260–22265. 10.1073/pnas.1014923108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gianni T, Cerretani A, Dubois R, Salvioli S, Blystone SS, Rey F, Campadelli-Fiume G. 2010. Herpes simplex virus glycoproteins H/L bind to cells independently of αVβ3 integrin and inhibit virus entry, and their constitutive expression restricts infection. J. Virol. 84:4013–4025. 10.1128/JVI.02502-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galdiero M, Whiteley A, Bruun B, Bell S, Minson T, Browne H. 1997. Site-directed and linker insertion mutagenesis of herpes simplex virus type 1 glycoprotein H. J. Virol. 71:2163–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stewart PL, Nemerow GR. 2007. Cell integrins: commonly used receptors for diverse viral pathogens. Trends Microbiol. 15:500–507. 10.1016/j.tim.2007.10.001 [DOI] [PubMed] [Google Scholar]
- 22.Desai P, Person S. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 72:7563–7568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ligas MW, Johnson DC. 1988. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by beta-galactosidase sequences binds to but is unable to penetrate into cells. J. Virol. 62:1486–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forrester A, Farrell H, Wilkinson G, Kaye J, Davis-Poynter N, Minson T. 1992. Construction and properties of a mutant of herpes simplex virus type 1 with glycoprotein H coding sequences deleted. J. Virol. 66:341–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang K, Kappel JD, Canders C, Davila WF, Sayre D, Chavez M, Pesnicak L, Cohen JI. 2012. A herpes simplex virus 2 glycoprotein D mutant generated by bacterial artificial chromosome mutagenesis is severely impaired for infecting neuronal cells and infects only Vero cells expressing exogenous HVEM. J. Virol. 86:12891–12902. 10.1128/JVI.01055-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jain P, Hsu T, Arai M, Biermann K, Thaler DS, Nguyen A, Gonzalez PA, Tufariello JM, Kriakov J, Chen B, Larsen MH, Jacobs WR., Jr 2014. Specialized transduction designed for precise high-throughput unmarked deletions in Mycobacterium tuberculosis. mBio 5:e01245–14. 10.1128/mBio.01245-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheshenko N, Del Rosario B, Woda C, Marcellino D, Satlin LM, Herold BC. 2003. Herpes simplex virus triggers activation of calcium-signaling pathways. J. Cell Biol. 163:283–293. 10.1083/jcb.200301084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heldwein EE, Krummenacher C. 2008. Entry of herpesviruses into mammalian cells. Cell. Mol. Life Sci. 65:1653–1668. 10.1007/s00018-008-7570-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clement C, Tiwari V, Scanlan PM, Valyi-Nagy T, Yue BY, Shukla D. 2006. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J. Cell Biol. 174:1009–1021. 10.1083/jcb.200509155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schlaepfer DD, Hauck CR, Sieg DJ. 1999. Signaling through focal adhesion kinase. Progress Biophys. Mol. Biol. 71:435–478. 10.1016/S0079-6107(98)00052-2 [DOI] [PubMed] [Google Scholar]
- 31.Izmailyan R, Hsao JC, Chung CS, Chen CH, Hsu PW, Liao CL, Chang W. 2012. Integrin β1 mediates vaccinia virus entry through activation of PI3K/Akt signaling. J. Virol. 86:6677–6687. 10.1128/JVI.06860-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X, Huang DY, Huong SM, Huang ES. 2005. Integrin αvβ3 is a coreceptor for human cytomegalovirus. Nat. Med. 11:515–521. 10.1038/nm1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeh YY, Chiao CC, Kuo WY, Hsiao YC, Chen YJ, Wei YY, Lai TH, Fong YC, Tang CH. 2008. TGF-β1 increases motility and αvβ3 integrin up-regulation via PI3K, Akt and NF-κB-dependent pathway in human chondrosarcoma cells. Biochem. Pharmacol. 75:1292–1301. 10.1016/j.bcp.2007.11.017 [DOI] [PubMed] [Google Scholar]
- 34.Sjaastad MD, Lewis RS, Nelson WJ. 1996. Mechanisms of integrin-mediated calcium signaling in MDCK cells: regulation of adhesion by IP3- and store-independent calcium influx. Mol. Biol. Cell 7:1025–1041. 10.1091/mbc.7.7.1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Calderwood DA. 2004. Integrin activation. J. Cell Sci. 117:657–666. 10.1242/jcs.01014 [DOI] [PubMed] [Google Scholar]
- 36.Kornberg L, Earp HS, Parsons JT, Schaller M, Juliano RL. 1992. Cell adhesion or integrin clustering increases phosphorylation of a focal adhesion-associated tyrosine kinase. J. Biol. Chem. 267:23439–23442 [PubMed] [Google Scholar]
- 37.Naranatt PP, Akula SM, Zien CA, Krishnan HH, Chandran B. 2003. Kaposi's sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-ζ-MEK-ERK signaling pathway in target cells early during infection: implications for infectivity. J. Virol. 77:1524–1539. 10.1128/JVI.77.2.1524-1539.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holm CK, Jensen SB, Jakobsen MR, Cheshenko N, Horan KA, Moeller HB, Gonzalez-Dosal R, Rasmussen SB, Christensen MH, Yarovinsky TO, Rixon FJ, Herold BC, Fitzgerald KA, Paludan SR. 2012. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat. Immunol. 13:737–743. 10.1038/ni.2350 [DOI] [PMC free article] [PubMed] [Google Scholar]