

ABSTRACT
The herpes simplex virus 1 (HSV-1) virion DNA contains nicks and gaps, and in this study a novel assay for estimating the size and number of gaps in virion DNA was developed. Consistent with previous reports, we estimate that there are approximately 15 gaps per genome, and we calculate the average gap length to be approximately 30 bases. Virion DNA was isolated and treated with DNA-modifying enzymes in order to fill in the gaps and modify the ends. Interestingly, filling in gaps, blunting the ends, or adding random sequences to the 3′ ends of DNA, producing 3′ flaps, did not impair the infectivity of treated DNA following transfection of Vero cells. On the other hand, the formation of 5′ flaps in the DNA following treatment resulted in a dramatic reduction (95 to 100%) in infectivity. Virion DNA stimulated DNA-PKcs activity in transfected cells, and DNA with 5′ flaps stimulated a higher level of DNA-PKcs activity than that observed in cells transfected with untreated virion DNA. The infectivity of 5′-flapped DNA was restored in cells that do not express DNA-PKcs and in cells cotransfected with the immediate early protein ICP0, which degrades DNA-PKcs. These results are consistent with previous reports that DNA-dependent protein kinase (DNA-PK) and the nonhomologous end joining (NHEJ) repair pathway are intrinsically antiviral and that ICP0 can counteract this effect. We suggest that HSV-1 DNA with 5′ flaps may induce an antiviral state due to the induction of a DNA damage response, primarily mediated by NHEJ, that renders the HSV-1 genome less efficient for lytic infection.
IMPORTANCE For productive lytic infection to occur, HSV-1 must counteract a variety of cellular intrinsic antiviral mechanisms, including the DNA damage response (DDR). DDR pathways have been associated with silencing of gene expression, cell cycle arrest, and induction of apoptosis. In addition, the fate of viral genomes is likely to play a role in whether viral genomes adopt a configuration suitable for lytic DNA replication. This study demonstrates that virion DNA activates the cellular DDR kinase, DNA-PK, and that this response is inhibitory to viral infection. Furthermore, we show that HSV-1 ubiquitin ligase, ICP0, plays an important role in counteracting the negative effects of DNA-PK activation. These findings support the notion that DNA-PK is antiviral and suggest that the fate of incoming viral DNA has important consequences for the progression of lytic infection. This study underscores the complex evolutionary relationships between HSV and its host.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) has a double-stranded linear DNA genome that is approximately 152 kbp in length. Both the replicating DNA and encapsidated viral genomes contain nicks and gaps (1–8). In DNA isolated from virions, gaps have been reported to number 3 to 13 per genome (9, 10). These gaps are randomly located and are present on both strands (3, 5, 11). Little is known about how nicks and gaps arise, and it is anticipated that the study of nicks and gaps in the HSV genome may provide insight into host responses to incoming viral genomes and the mechanism of HSV-1 replication.
At the earliest stages of HSV infection, the viral genome is released from the capsid into the nucleus, where it becomes associated with a combination of viral and cellular proteins. Some of these proteins are factors utilized by the virus to initiate a robust program of viral gene expression and DNA synthesis, while others are now recognized for their ability to mount an intrinsic antiviral response. Several intrinsically antiviral pathways that act to silence viral gene expression have been identified, including PML and other components of ND10 (12, 13), the interferon-inducible DNA sensor IFI16 (14–16), and components of the DNA damage response (DDR) (17–20). HSV must counteract these intrinsically antiviral pathways to establish an environment conducive to lytic infection.
The cellular DDR is mediated by three phosphatidylinositol 3-kinase-like serine/threonine protein kinases (PIKKs): DNA-PK (DNA-dependent protein kinase), ATM (ataxia telangiectasia mutated), and ATR (ATM and Rad3 related) (21–23). The DNA-PK complex, comprised of DNA-PKcs and the Ku70/Ku80 heterodimer, responds to double-strand breaks and stimulates repair via nonhomologous end joining (NHEJ) (22). ATM is activated by double-strand breaks and stimulates repair via homologous recombination (HR) and single-strand annealing (SSA). ATR is activated by stalled replication forks and stretches of single-stranded DNA (ssDNA) adjacent to double-stranded DNA (dsDNA) (22). DNA repair proteins play both positive and negative roles during HSV infection, and HSV manipulates components of these pathways, activating some and disabling others (18–20, 24–32).
The incoming viral genome with double-strand ends, nicks, and gaps may be expected to be recognized by the cellular DDR and activate one or more of the DNA damage-sensing kinases. Activation of a DNA damage response can initiate cell cycle arrest, gene silencing, and apoptosis, any of which could negatively impact the viral life cycle by suppressing viral gene expression and virus production. It is also possible that some DDR pathways may result in “repair” of DNA in a manner that is not consistent with lytic infection. In HSV-infected cells, several DNA damage-sensing elements are degraded, or their signaling is blocked. For instance, the immediate early protein ICP0 is a major player in counteracting cellular antiviral mechanisms by degrading antiviral proteins, such as DNA-PKcs (18, 19). Interestingly, ICP0 is also a component of the viral tegument and is thus present at the earliest stages of viral infection, even before immediate early protein synthesis (33–35). ATR-mediated phosphorylation of downstream targets, replication protein A (RPA) and Chk1, is also inhibited by 3 h postinfection (27, 30, 36). Thus, it appears that HSV has evolved to inactivate several DDR elements that may recognize the unusual structure of the incoming viral genome.
In this study, we have explored the role of virion DNA in the activation of DDR kinases in the absence of the tegument using transfection-based assays. Virion DNA was treated with enzymes that would be expected to fill in gaps, extend 3′ ends, ligate nicks, cleave regions of single-stranded DNA, or result in 5′ flaps. These treatments were used to confirm the presence of approximately 15 gaps per viral genome. We were also able to estimate the average gap length to be approximately 30 bases. Virion DNA that had been enzymatically modified was tested for infectivity by transfection. Although many of the treatments did not affect infectivity, strand displacement synthesis resulting in 5′ flaps and endonucleolytic digestion of ssDNA caused significant decreases in infectivity. Virion DNA containing 5′ flaps activated robust DNA-PKcs activity that also correlated with the dramatic loss of infectivity. This suppression could be rescued by genetic deletion of DNA-PKcs or cotransfection with ICP0. These results are consistent with previous reports that DNA-PKcs represents a cellular antiviral response to infection.
MATERIALS AND METHODS
Cell lines.
Vero cells and HCT-116 cells were obtained from American Type Culture Collection (ATCC), and the derivative DNA-PK−/− cell line was generously provided by Eric A. Hendrickson (University of Minnesota Medical School, Minneapolis, MN) and has been previously described (37). Vero cells were grown in Dulbecco's modified minimal essential medium (DMEM) (Gibco) containing 5% fetal bovine serum (FBS). HCT-116 cells and DNA-PK−/− cells were grown in McCoy's 5A medium (modified) (Gibco) containing 10% FBS.
Viruses and plasmids.
The KOS strain of HSV-1 was used as the wild-type virus. The recombinant virus KOS-CMVGFP contains the green fluorescent protein (GFP) gene under the control of the human cytomegalovirus (HCMV) promoter (2.0-kbp insertion) in the intergenic region between the UL26 and UL27 genes (38). Wild-type ICP0 (pCI-110) and ICP0 ΔRING-Finger (pCI-FXE) were generously provided by Roger Everett (MRC Virology Unit, Glasgow, Scotland).
Preparation of viral DNA.
Virion DNA was isolated as previously described (39). Approximately 1 × 108 Vero cells were infected with KOS at a low multiplicity of infection (MOI; 0.1 to 0.5). When maximum cytopathic effects were observed, infected cells were harvested by scraping and spun down at 1,500 rpm for 15 min at 4°C. The supernatant was removed and stored at 4°C for later use. The cell pellet was then resuspended in 3 ml of cold 1× reticulocyte standard buffer (RSB) and incubated on ice for 10 min. Cells were then disrupted by Dounce homogenization. Cell debris and nuclei were then removed by centrifugation at 1,500 rpm for 10 min at 4°C. The supernatant was combined with the first supernatant and centrifuged at 9,000 rpm for 60 min at 4°C. Virions were resuspended in 5 ml of TNE (10 mM Tris-Cl [pH 7.4], 10 mM NaCl, 30 mM MgCl2) and frozen at −80°C. Virions were thawed, and SDS and proteinase K were added to final concentrations of 1% and 100 μg/ml, respectively. The tube was gently inverted and then incubated for 5 h at 37°C. Then DNA was either dialyzed overnight at 4°C against 1 liter of TE (10 mM Tris-Cl [pH 7.5], 1 mM EDTA) or gently extracted by phenol-chloroform-isoamyl alcohol (25:24:1) and then precipitated either by addition of 0.6 volume of 20% polyethylene glycol 8000 (PEG 8000)–2 M NaCl or by ethanol precipitation. DNA was incubated 1 h on ice and then centrifuged for 15 min at 20,000 × g. The pellet was washed with 70% ethanol, dried briefly, and resuspended in TE. The DNA was aliquoted and stored at −80°C.
Enzymes.
The Klenow fragment of Escherichia coli DNA polymerase I was from Boehringer Mannheim or New England BioLabs (NEB). T4 DNA polymerase, terminal transferase (TdT), calf intestine alkaline phosphatase (CIP), and mung bean nuclease were from NEB. T4 DNA ligase was from NEB or Invitrogen.
In vitro modification of virion DNA.
Virion DNA (200 ng, 2 fmol) was incubated with polymerases and other enzymes in a 25-μl reaction volume for 30 min, unless otherwise indicated. For reactions with Klenow polymerase and T4 DNA ligase, a buffer containing 10 mM Tris-Cl (pH 7.5), 5 mM MgCl2, and 7.5 mM dithiothreitol (DTT) was used. The manufacturer-supplied buffer (NEB) was used for reaction mixtures containing T4 polymerase, terminal transferase, mung bean nuclease, and CIP. ATP was used at 1 mM with T4 DNA ligase and alkaline phosphatase, unless otherwise noted, and deoxynucleoside triphosphates (dNTPs) were used at 0.5 mM where indicated. When different buffers were used, no-enzyme controls were run using the same buffer.
Measurement of nucleotide incorporation into DNA.
The incorporation of labeled nucleotides was performed using Klenow polymerase, T4 polymerase, and T4 DNA ligase (reactions described above) with [α32P]dCTP included in the dNTP mixture. Reaction mixtures were incubated at 37°C for the times indicated in the figures, and reactions were stopped by adding 5 μl of 6× gel loading buffer (50% glycerol, 1% SDS, 50 mM EDTA, 0.2% bromphenol blue). Half (15 μl) of each sample was loaded onto a 0.7% agarose TAE (0.04 M Tris-acetate, 0.001 M EDTA) gel. The gel was dried onto DE81 paper (Whatman) and exposed to phosphorimager screens (National Diagnostics). Dilutions of the dNTP mixture were spotted onto paper and exposed to the phosphorimager screen along with the gel in order to aid in the quantification of the amount of label incorporated. The ImageQuant version 5.0 software package was used for quantification of the results. Testing at each time point was repeated independently at least three times. Adobe Photoshop (v.7.0) and Adobe Illustrator (v. 10) were used in preparation of the figures.
Calculation of gap number and length.
The incorporation of nucleotides by Klenow-ligase into 1 fmol of HSV genomes in a 40-min incubation was compared to the incorporation into the same amount of DNA by Klenow fragment alone, 500 fmol and 3,500 fmol, respectively. The number of gaps per genome was calculated by dividing the difference in incorporation between Klenow alone and T4 polymerase by the average length of Klenow strand displacement (200 nucleotides [nt]):
| (1) |
| (2) |
Infectivity assays.
Reactions were prepared and incubated as described above, without the labeled nucleotide. All cell types were transfected using Lipofectamine Plus reagent (Invitrogen) according to the manufacturer's suggested protocol. Vero cells at 50 to 70% confluence were transfected with incorporation products at a concentration of 200 ng of DNA/60-mm dish. Samples were overlaid with DMEM containing 2% methylcellulose at 17 to 20 h posttransfection. Plaques were fixed and stained with crystal violet 4 to 5 days later, and plaques were counted. HCT-116 and DNA-PK−/− cells at 70% confluence were transfected with purified HSV DNA at a concentration of 0.5 μg of DNA/35-mm dish. Samples were harvested at 48 h following transfection and titrated on Vero cells. For cotransfection experiments, HCT-116 cells were plated on 35-mm dishes at 70% confluence and transfected with 0.5 μg of viral DNA and 1 μg of plasmid DNA. Virus was collected at 48 h following transfection, and viral yield was determined by titration on Vero cells.
Pulsed-field gel electrophoresis.
Agarose gels were prepared using 1% pulsed-field gel-grade agarose (Bio-Rad) in 0.5× TBE (45 mM Tris, 45 mM borate, 1 mM EDTA [pH 8.3]) buffer. Samples were diluted in 6× gel loading dye (NEB) and loaded directly into the wells. Electrophoresis was performed using a CHEF-DR III apparatus (Bio-Rad) with 0.5× TBE running buffer. Samples were separated using 6 V/cm (approximately 200 V) for 24 h at 14°C. Switch times were ramped from 1 to 25 s. Lambda Ladder PFG Marker (NEB) and Vero cells infected with HSV-1 were used as size standards. Gels were stained with ethidium bromide, photographed, and transferred by Southern blotting to GeneScreen Plus membranes (Dupont NEN) according to the manufacturer's protocols. Membranes were probed with CDP-Star biotin detection reagent (NEB) and imaged using a ChemiDoc MP imaging system (Bio-Rad).
Western blot analysis.
Vero cells were transfected with 2.5 μg of untreated, Klenow-treated, or Klenow-ligase-treated infectious DNA (prepared as described above) per dish. Lipofectamine 2000 reagent (Invitrogen) was used at a concentration of 10 μl per transfection, as described by the manufacturer. Samples were harvested 24 h following transfection and prepared for Western blot analysis as previously described (32). The primary antibodies used were mouse monoclonal anti-RPA32 (9H8) (1:1,000; GeneTex), polyclonal rabbit anti-phospho-RPA S33 (1:3,000; Bethyl), polyclonal rabbit anti-phospho-RPA S4/S8 (1:3,000; Bethyl), monoclonal mouse anti-ICP0 5H7 (1:5,000; EastCoast Bio), and monoclonal mouse anti-Ku70 Ab-4 (N3H10) (1:1,000; Neomarkers). Western blots were quantified using ImageJ software.
Gene expression assay.
Vero cells were transfected with modified KOS-GFP DNA. Samples were fixed with 4% paraformaldehyde 24 h posttransfection and resuspended in PBS-EDTA (5 mM). GFP-positive cells were measured using flow cytometry using a BD LSR II, and analyzed using FlowJo software.
IF.
Immunofluorescence (IF) analysis was performed as described previously (31, 40). Briefly, cells adhered to glass coverslips were washed with PBS, fixed with 4% paraformaldehyde, and permeabilized with 1% Triton X-100. Cells were blocked in 3% normal goat serum and reacted with antibodies as indicated below. Primary antibodies included monoclonal mouse anti-ICP0 5H7 (1:200; EastCoast Bio) and polyclonal rabbit anti-phospho-RPA32 S4/S8 (1:200; Bethyl Laboratories). Alexa Fluor secondary antibodies (1:200; Molecular Probes) were used with fluorophores excitable at wavelengths of 488 and 594 nm. Images were captured using a Zeiss LSM 510 confocal nonlinear optical (NLO) microscope equipped with argon and HeNe lasers and a Zeiss 63× objective lens (numerical aperture, 1.4). Images were processed and arranged using Adobe Photoshop CS3 and Illustrator CS3.
RESULTS
In vitro modification of HSV-1 DNA.
The HSV genome is packaged as a linear, double-stranded DNA molecule that contains nicks and gaps. This unusual structure prompted us to ask how the presence of nicks and gaps might influence early steps in viral infection, particularly with respect to host factors that respond to incoming DNA. HSV-1 DNA, purified from virions, is capable of initiating a productive infection in transfected cells. Transfection experiments thus permit us to explore the infectivity of virion DNA in the absence of tegument proteins that may act to promote or disable host factors responding to incoming viral genomes. To better understand how the structural attributes of virion DNA affect infectivity and host responses, DNA isolated from virions was enzymatically modified in vitro prior to transfection.
We first sought to confirm the presence of gaps in purified HSV DNA and to demonstrate that gaps could be repaired in vitro using a gap-filling polymerase. Purified DNA was treated with the Klenow fragment of E. coli DNA polymerase I, which was expected to recognize free 3′ termini and incorporate nucleotides, thereby filling in gaps. In a control reaction, virion DNA was denatured and primed with random hexamers. Figure 1A shows that labeled nucleotides incorporated into the boiled and random-primed DNA migrated as a smear (left side), whereas nucleotides incorporated into virion DNA migrated primarily as a single species. The extent of incorporation was quantified and displayed in the graph shown in Fig. 1B. Incorporation of label into nondenatured virion DNA reached a plateau after 5 to 10 min of incubation, indicating that the gaps in this DNA substrate had been filled. It is unlikely that the plateau is due to inactivation of the enzyme or an exhaustion of the dNTPs, as the incorporation into the random-primed sample continued to be linear and did not reach a plateau in the same time frame.
FIG 1.

Purified HSV-1 DNA contains gaps than can be filled by DNA polymerase. (A) The incorporation of labeled nucleotides by the Klenow fragment of E. coli DNA polymerase I into 200 ng of HSV-1 DNA was performed as described in Materials and Methods. For lanes 1 to 6, prior to incubation in the reaction mixture, the DNA was boiled for 2 min and quickly cooled, and random primers were added. Samples were analyzed by agarose gel electrophoresis. (B) Quantification of labeled nucleotides incorporated into the HSV-1 DNA.
Klenow polymerase strand displacement activity can be used to measure gap number and length.
In order to measure the gap number and length of virion DNA, we compared nucleotide incorporation using DNA polymerases with different biochemical properties. The Klenow fragment of DNA polymerase I extends 3′ termini at gaps and is also capable of strand displacement synthesis. Because Klenow lacks 5′-to-3′ exonuclease activity, nucleotides are displaced from the template ahead of the growing chain but are not cleaved, resulting in a 5′ flap. T4 polymerase, on the other hand, does not possess strand displacement activity, and it dissociates from the DNA template when it reaches the end of a gap (41). In addition, treatment with Klenow and T4 DNA ligase together prevents strand displacement synthesis because under these conditions, once Klenow has filled in the gap, the 3′ terminus of the newly synthesized fragment is ligated to the 5′ terminus of the adjacent strand.
Figure 2 shows the incorporation of labeled nucleotides into HSV DNA by Klenow polymerase alone, Klenow and ligase together, or T4 DNA polymerase. While all three reactions reached a plateau, Klenow incorporated approximately 7-fold more label than the other two reactions. This is consistent with the known ability of Klenow to carry out strand displacement synthesis. On the other hand, the inability of T4 DNA polymerase and Klenow-ligase to perform strand displacement synthesis is consistent with the lower levels of incorporation observed for these conditions.
FIG 2.

Incorporation of labeled nucleotides into HSV-DNA by Klenow fragment polymerase alone, Klenow and ligase together, or T4 DNA polymerase. Purified HSV-1 DNA was incubated with labeled nucleotides and the following enzymes, as described in Materials and Methods: Klenow fragment, Klenow fragment together with T4 DNA ligase, and T4 DNA polymerase.
We took advantage of the known biochemical properties of T4 DNA ligase and Klenow polymerase to measure the number and length of gaps in HSV-1 DNA using equation 1, as described in Materials and Methods. The difference in incorporation between Klenow treatment and Klenow-ligase treatment (3,000 fmol) represents the “extra” incorporation by Klenow resulting from strand displacement. Klenow polymerase is known to displace approximately 200 bases before it dissociates from a DNA template (42, 43). To estimate the number of invasions and, consequently, the number of gaps, the value for extra incorporation was divided by 200. According to this calculation, the number of gaps in the HSV-1 genome is 15, which is comparable to previous determinations of 3 to 13 gaps per genome (9, 10). Furthermore, using equation 2, the average gap size is calculated to be 33 nucleotides.
The conditions under which the ligation was performed (low DNA concentration and high concentration of ligase at 37°C for 20 min) support complete sealing of the nicks but are unlikely to result in any end-to-end ligation of molecules. In order to verify that the ends of the HSV-1 DNA were not ligated during the reaction, products were separated by pulsed-field gel electrophoresis. The untreated HSV-1 DNA migrated as a linear 152-kb monomer on the gel (Fig. 3, lanes WT). After treatment with Klenow or Klenow and ligase together (Fig. 3, lanes K and KL, respectively), the DNA was still monomeric length, and no larger-molecular-size species were observed, indicating that the ligase did not produce concatemers or circles. Species that are shorter than monomeric length were observed in all lanes, indicating that a portion of the DNA was sheared. Since fragmentation is more noticeable for the untreated sample, it is likely that the nicked DNA was more vulnerable to breakage than DNA that had been repaired.
FIG 3.

Virion DNA before and after treatment with Klenow or Klenow and ligase. Biotinylated nucleotides were incorporated into HSV DNA using Klenow alone or Klenow and ligase. Pulsed-field gel electrophoresis was performed with untreated (WT), Klenow-treated (K), and Klenow-ligase-treated (KL) HSV virion DNA. The gel was probed with ethidium bromide (total DNA) and CDP-Star biotin detection reagent (incorporated nucleotides).
Filling in nicks and gaps did not affect infectivity.
The incorporation studies demonstrated that virion HSV-1 DNA contained gaps that were readily filled using purified DNA polymerases. We next asked whether the changes in DNA structure, made in vitro, affected the infectivity of the modified DNA, as measured by a plaque assay following transfection. Plaque numbers for the control DNA were in the range of 50 to 150 per 60-mm dish. Treatment of the DNA with T4 DNA ligase did not affect infectivity (Fig. 4). Likewise, DNA that was filled in and ligated by treatment with Klenow-ligase was fully infectious. This indicates that, at least in the context of transfection, gaps and nicks are not required for infectivity. Filling in nicks and gaps also did not potentiate the infection, as percent infectivity was similar to that of the untreated control.
FIG 4.

Expected structure and measured infectivity of HSV virion DNA following various in vitro treatments. Virion DNA was treated with the indicated enzymes and tested for infectivity. The numbers of plaques have been normalized, with the plaque number obtained in control reactions set at 100%.
Treatment with mung bean nuclease destroys infectivity, confirming the presence of gaps.
Infectivity was completely eliminated when HSV-1 DNA was incubated with mung bean nuclease, an endonuclease that degrades ssDNA (Fig. 4). This result confirms the presence of gaps in virion DNA, since digestion of the ssDNA at gaps would fragment the genome and abolish infectivity. On the other hand, virion DNA that had been treated with Klenow-ligase and then incubated with an excess of mung bean nuclease retained its infectivity. HSV has been reported to contain complementary single 3′ nucleotide overhangs at both ends (44, 45). Treatment with mung bean nuclease following repair with Klenow-ligase would be expected to blunt the ends of the HSV genome by removing overhangs. These overhangs have been suggested to facilitate end ligation and contribute to circularization of the genome prior to replication (46). The observation that HSV-1 DNA was still infectious after this treatment confirms that Klenow-ligase can fill in gaps and that 3′ overhangs are not required for infectivity.
Treatment with calf intestine alkaline phosphatase is tolerated.
End ligation and circularization of DNA would be expected to require the presence of a 5′ phosphate. In order to determine whether 5′ phosphates on viral DNA are required for infectivity, HSV-1 DNA was treated with calf intestine phosphatase. This treatment has no effect on infectivity, suggesting that incoming DNA does not need to be end ligated (Fig. 4). We cannot, however, rule out the possibility that cellular kinases could rephosphorylate viral DNA.
Virion DNA with 3′ flaps retains infectivity.
To further analyze the effects of in vitro modification on infectivity of virion DNA, we tested the effect of random 3′ tails added onto the DNA by terminal transferase (TdT). Treatment with TdT acts in a template-independent manner to catalyze the addition of deoxynucleotides to an available 3′ terminus. We used terminal transferase to add tails, which were approximately 100 to 300 nucleotides in length, onto the HSV-1 DNA (47–50). Despite the fact that the 3′ additions were random, and therefore nonhomologous, these additions had no effect on the infectivity of the DNA (Fig. 4).
Treatment with Klenow polymerase abolishes infectivity.
Interestingly, DNA treated with Klenow alone produced only 0 to 10% of the number of plaques seen with untreated DNA (Fig. 4). In the absence of nucleotides, Klenow treatment had no effect on infectivity, demonstrating that incorporation of nucleotides into viral DNA was necessary for the loss of infectivity. In addition, if Klenow was prevented from generating 5′ flaps by coincubation with T4 DNA ligase, infectivity was not impaired. Thus, the dramatic decrease in infectivity with Klenow-treated DNA was seen only when strand displacement synthesis was allowed. Since strand displacement synthesis is believed to result in the formation of 5′ flaps, these results suggest that 5′ flaps are responsible for the lack of infectivity of Klenow-treated DNA.
The drastic reduction in infectivity after Klenow treatment was not expected, and we were interested to determine its cause. To rule out the possibility that Klenow treatment caused a reduction in transfection efficiency and thus reduced the number of genomes entering the nucleus, we measured gene expression from DNA that had been treated with either Klenow or Klenow-ligase. Virion DNA was prepared from cells that had been infected with a GFP-expressing virus and used to transfect Vero cells in the presence of phosphonoacetic acid (PAA) to prevent viral replication. Thus, gene expression is expected to originate only from input genomes, allowing for a direct comparison between treated and untreated virion DNA. GFP-positive cells were analyzed by fluorescence-activated cell sorting (FACS). GFP expression was detected in cells transfected with untreated, Klenow-treated, or Klenow-ligase-treated KOS-GFP DNA. Figure 5 depicts histograms showing the distribution of GFP intensity plotted against the number of GFP-positive cells. These data indicate that transfection efficiencies are similar for treated and untreated samples and that the loss of infectivity associated with Klenow treatment is not caused by differences in transfection efficiency.
FIG 5.

Transfection efficiency of treated and untreated KOS-GFP DNA. Vero cells were transfected with untreated (black), Klenow-treated (red), or Klenow-ligase-treated (blue) KOS-GFP DNA. The graph depicts overlaid histograms of cells sorted by FACS and gated for GFP-positive cells.
Cotransfection of ICP0 with untreated and Klenow-treated HSV-1 DNA dramatically improves infectivity.
Cotransfection of ICP0 with infectious DNA has previously been shown to enhance plaque formation in Vero and U2OS cells (51). We were interested to see whether ICP0 could boost infectivity in cells transfected with Klenow-treated DNA. HCT-116 cells were cotransfected with wild-type or mutant ICP0 and purified HSV-1 DNA that was either left untreated or treated with Klenow. The ICP0 mutant lacked the RING finger domain (ICP0-FXE). Expression of wild-type ICP0 dramatically increased viral yield from Klenow-treated DNA, by more than 3 orders of magnitude, compared to cotransfection of an empty vector with Klenow-treated DNA (Table 1). Consistent with previous findings (51), cotransfection with wild-type ICP0 also improved the infectivity of untreated virion DNA, although to a lesser extent. There was also a modest effect on the infectivity of both the untreated and Klenow-treated DNA when cotransfected with ICP0-FXE. This may be due to other functions of ICP0 that are unrelated to its ubiquitin ligase activity. Similar results were also obtained with Vero cells (data not shown). These results confirm that ICP0 expression improves the infectivity of purified HSV-1 DNA. Interestingly, the presence of ICP0 is able to overcome the loss of infectivity exhibited by Klenow-treated virion DNA containing 5′ flaps. Although it is unknown whether 5′ flaps arise during HSV infection, it has been shown that replicating viral DNA adopts complex structures (52, 53), and these structures may include 5′ flaps.
TABLE 1.
Viral yields from cells cotransfected with HSV-1 virion DNA and plasmid DNA
| Viral DNA treatment | Plasmid cotransfected | Viral titer (PFU/ml), mean ± SE | % infectivity |
|---|---|---|---|
| None (wild type) | Empty vector | 2.9 × 103 ± 1.7 × 103 | 100 |
| ICP0 | 4.1 × 105 ± 8.1 × 104 | 14,000 | |
| ICP0 ΔFXE | 1.2 × 104 ± 3.7 × 103 | 420 | |
| Klenow | Empty vector | 3.0 × 101 ± 1.1 × 101 | 1 |
| ICP0 | 1.3 × 105 ± 4.4 × 104 | 4,600 | |
| ICP0 ΔFXE | 1.0 × 103 ± 1.9 × 102 | 35 |
HSV-1 DNA stimulates replication protein A (RPA) phosphorylation in transfected cells but not infected cells.
Since HSV virion DNA is linear, it has ends that may be recognized as double-strand breaks by DNA-PK. Furthermore, it is possible that gaps in HSV DNA or other unusual structures could mimic the substrates that activate ATR (ssDNA adjacent to dsDNA). Therefore, the structure of the viral genome might be expected to result in activation of both ATR and DNA-PK. We have, however, previously reported that RPA, which is a target for phosphorylation by both kinases, is not phosphorylated during HSV infection (24, 32). HSV inactivates components of both NHEJ and HR pathways through the action of ICP0. As an immediate early protein, ICP0 is expressed very early in infection, but it is also present in the tegument of the incoming virion (33–35), potentially entering the nucleus at the time of DNA entry. These observations led us to examine whether virion DNA could induce DNA-PK and ATR kinase activity in transfected cells in which ICP0 and other viral proteins are not present. The cellular ssDNA-binding protein RPA is a well-known downstream target of both DNA-PKcs and ATR in response to DNA damage (54). The S33 residue of the middle subunit, RPA32, is specifically phosphorylated by ATR (55), while the S4 and S8 residues of RPA32 are phosphorylated by DNA-PK (54, 56–58).
In uninfected Vero cells treated with UV, RPA32 is phosphorylated by DNA-PK and ATR, as detected using specific antibodies for phospho-RPA32 S4/S8 and phospho-RPA32 S33, respectively (Fig. 6). Consistent with our previous findings, no phosphorylated RPA was detected in HSV-infected cells (Fig. 6) (24, 32, 59). On the other hand, phospho-RPA32 S4/S8 was detected in cells transfected with purified virion DNA, demonstrating that DNA-PK is activated under these conditions (Fig. 6). Interestingly, phospho-RPA32 S33 was not detected, suggesting that incoming virion DNA is not a suitable substrate for ATR activation (Fig. 6).
FIG 6.

HSV-1 DNA stimulates RPA phosphorylation in transfected cells but not infected cells. Vero cells were either infected with HSV-1 at an MOI of 10 and harvested at 3 h postinfection (KOS) or transfected with 700 ng of virion DNA and harvested 3 h following serum addition (KOS DNA). As a positive control for pRPA32 S4/S8, Vero cells were treated with 50 J/m2 and allowed to recover for 1 h at 37°C (UV).
The observation that DNA-PK is not activated in infected cells is consistent with the known ability of ICP0 to degrade DNA-PKcs at least in some cell types (18, 19, 60, 61). Interestingly, however, DNA-PKcs is not degraded in HSV-infected Vero cells (24), raising the possibility that DNA-PK might be inhibited by a mechanism other than degradation. Vero cells were transfected with ICP0 or an ICP0 RING finger mutant (ICP0-FXE), which lacks ubiquitin ligase activity; they were then UV irradiated and analyzed by immunofluorescence with antibodies specific for ICP0 and pRPA-S4S8. Cells expressing ICP0 failed to induce RPA32 S4/S8 phosphorylation after UV damage, as there was no pRPA32 S4/S8 staining in cells that were positive for ICP0 expression (Fig. 7, top). Conversely, pRPA32 S4/S8 was observed in cells expressing ICP0-FXE, suggesting that the ubiquitin ligase activity of ICP0 is required for inhibition of RPA S4/S8 phosphorylation (Fig. 7, bottom). Thus, ICP0 is able to inhibit DNA-PK-dependent signaling, even in cell types in which DNA-PKcs is not degraded. These observations suggest either that ICP0 degrades another protein necessary for activation of DNA-PKcs or that ubiquitination of DNA-PKcs is inhibitory even without degradation.
FIG 7.

ICP0 prevents RPA32 S4/S8 phosphorylation in Vero cells. Vero cells were transfected with ICP0 wt or the FXE mutant and then irradiated with 100 J/m2 of UV. Cells were allowed to recover for 2 h and then fixed and stained as indicated. Arrows highlight cells transfected with ICP0.
Addition of 5′ flaps to virion DNA increases hyperphosphorylation of RPA32.
We were intrigued by the dramatic loss of infectivity observed with the Klenow-treated DNA and by the ability of ICP0 to rescue infectivity (Table 1). We hypothesized that DNA with 5′ flaps may activate a more robust cellular DNA damage response than wild-type HSV-1 DNA. Given previous reports that DNA-PK is antiviral, we wanted to test the possibility that loss of infectivity may correlate with a robust DNA damage response. Therefore, we asked whether modifications to virion DNA could alter the DDR following transfection. Vero cells were transfected with virion DNA that was either left untreated, treated with Klenow (containing 5′ flaps), or treated with Klenow-ligase (filled in and ligated). Untreated transfected DNA stimulated RPA32 S4/S8 phosphorylation, and Klenow-ligase-treated DNA exhibited a level of RPA32 S4/S8 phosphorylation similar to that of the untreated sample. Interestingly, Klenow-treated DNA elicited a greater pRPA S4/S8 signal than either the untreated or filled-in sample, perhaps due to the 5′ flaps produced by Klenow treatment (Fig. 8).
FIG 8.
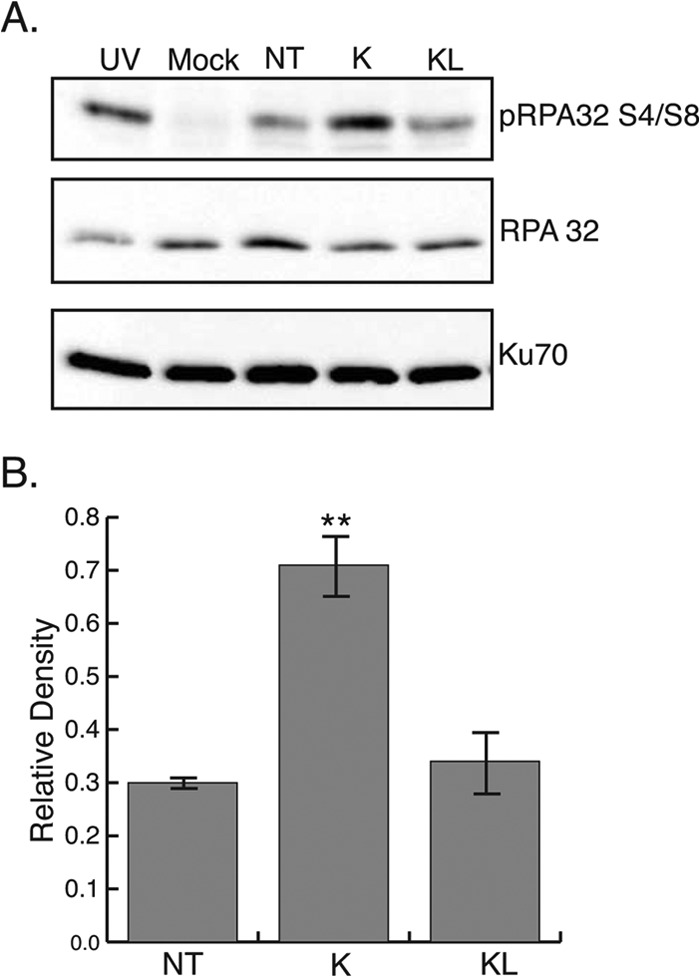
Addition of 5′ flaps to virion DNA increases hyperphosphorylation of RPA32. (A) Vero cells were transfected with purified KOS DNA that was either left untreated (NT) or treated with Klenow (K) or Klenow and ligase (KL). Samples were harvested at 3 h following serum addition. As a positive control for pRPA S4/S8, Vero cells were treated with 50 J/m2 and allowed to recover for 1 h at 37°C (UV). (B) Densitometry analysis of Western blot in panel A. Relative density was calculated by the ratio of the densities of pRPA S4/S8 and Ku70. **, P = 0.001.
Infectivity of Klenow-treated DNA is rescued in the absence of DNA-PK.
As described above, virion DNA treated with Klenow polymerase exhibited a marked decrease in infectivity and increased levels of pRPA32 S4/S8 phosphorylation. Since pRPA32 S4/S8 is a substrate of DNA-PKcs, we wanted to determine if DNA-PKcs itself was responsible for the reduced virus yield seen with Klenow-treated DNA. HCT-116 (wild-type) and DNA-PK−/− cells were transfected with untreated or Klenow-treated DNA, and infectivity was measured. Infectivity of Klenow-treated DNA in transfected HCT-116 cells was about 11% that of untreated DNA in transfected HCT-116 cells (Fig. 9). This is a decrease similar to that observed when Klenow-treated DNA was compared to untreated DNA in transfected Vero cells (Fig. 4). In DNA-PK−/− cells transfected with Klenow-treated DNA, however, infectivity was 100% compared with that of untreated DNA in transfected DNA-PK−/− cells (Fig. 9). This suggests that DNA-PKcs plays a role in the loss of infectivity phenotype observed in wild-type cells.
FIG 9.

Infectivity of Klenow-treated DNA is rescued in the absence of DNA-PK. HCT-116 cells and DNA-PK−/− cells were transfected with 500 ng of untreated or Klenow-treated virion DNA. Samples were harvested at 48 h following serum addition and titrated for virus yield on Vero cells. Infectivity is reported as percentage of the untreated DNA control. Viral titers were normalized to untreated DNA control set at 100%.
DISCUSSION
The results presented in this paper confirm that HSV-1 virion DNA has an unusual structure, containing multiple nicks and gaps. Virion DNA was treated with various combinations of enzymes that fill in gaps, ligate nicks, create 3′ and 5′ flaps, blunt the ends, and fragment the genome at sites of ssDNA. Untreated virion DNA was as infectious as DNA whose nicks and gaps were filled in and ligated, suggesting that nicks and gaps are not required for virion DNA to be infectious. Other treatments, including blunting of the termini, removal of 5′ phosphates, and production of 3′ flaps, are also tolerated. On the other hand, the formation of 5′ flaps in the DNA following treatment with Klenow alone resulted in a dramatic reduction (95 to 100%) in infectivity. Untreated virion DNA was able to activate DNA-PK activity in transfected cells, a somewhat surprising result considering that DNA-PK is not activated in HSV-infected cells, even at early times postinfection. Interestingly, virion DNA with 5′ flaps stimulated an even more robust activation of DNA-PK than did untreated virion DNA. Infectivity of Klenow-treated DNA was restored in cells that do not express DNA-PK and in cells cotransfected with the immediate early protein ICP0, which is known to degrade DNA-PKcs. Thus, we suggest that virion DNA with 5′ flaps induces an antiviral state that is dependent on DNA-PK and which can be suppressed by ICP0.
In this study, we have used DNA with 5′ flaps to explore how the cell responds to an unusual DNA structure. Although 5′ flaps are unlikely to be present in virion DNA, the mechanisms of HSV DNA replication are poorly understood. Furthermore, it is clear that unusual and complex structures containing X and Y junctions arise in infected cells, and it is thus possible that 5′ flaps may arise during replication. We have been particularly interested in whether cells respond to unusual viral DNA conformations by signaling a damage response that could be antiviral. For instance, the DDR response may silence or degrade viral DNA.
Using a novel assay based on the biochemical properties of the Klenow polymerase, we were also able to estimate that virion DNA contains, on average, 15 gaps per genome, consistent with published observations (9, 10). The presence of gaps in virion DNA might have been expected to activate the ATR kinase; however, ATR signaling was not activated in cells transfected with virion DNA. It has been reported that ATR activation requires a stretch of ssDNA capable of binding at least two consecutive RPA complexes (approximately 65 nucleotides) (62, 63). In this study, we estimated that the average size of gaps in virion DNA was approximately 33 nucleotides, long enough bind a single RPA complex but too short to activate ATR signaling. Thus, the incoming genome does not appear to be a suitable substrate for ATR activation.
The presence of nicks and gaps in HSV virion DNA has interesting implications for early stages of viral infection. Only a few other DNA viruses are known to package nicked and gapped genomes: pseudorabies virus (PRV) and Marek's disease virus (MDV) have nicks and gaps that are randomly distributed (64–66), while other DNA viruses contain nicks and gaps located at specific sites. For example, there are five major nicks in T5, which are thought to occur as a result of a virally encoded nicking enzyme and may play a role in the two-step transfer mechanism for ejecting DNA into its host (67–69). Like T5, T7 DNA also has single-strand interruptions at specific sites, but these are thought to be the product of premature terminase activity during packaging (70). Little is known, however, about whether the structure of viral genomes plays a role in infectivity or stimulation of host DDR.
Some cellular DNA damage response pathways are inhibited by HSV infection.
Cells have several different DDR pathways that could be activated during infection, and it is possible that HSV has evolved to utilize those pathways that are conducive to productive infection while preventing pathways that inhibit lytic infection. Using GFP correction assays, we have recently determined that single-strand annealing (SSA) is increased in HSV-infected cells, while NHEJ and HR are inhibited (71). This “pathway choice” on the part of the virus may be facilitated by ICP0, consistent with its known ability to degrade components of the NHEJ and HR pathways. For instance, ICP0 is known to degrade DNA-PKcs, a component of the NHEJ pathway, as well as RNF8 and RNF168, which function in HR (17, 18). DNA-PKcs and Ku, which are both essential components of the NHEJ pathway, are inhibitory to HSV infection: replication is more efficient in cells lacking DNA-PKcs (19), and viral yields are increased almost 50-fold in Ku-deficient murine embryonic fibroblasts (25). In this study, we showed that ICP0 is able to inhibit DNA-PKcs-specific phosphorylation of RPA32 even in a cell type in which DNA-PKcs is not degraded (Table 1). The observation that HSV may utilize at least two different mechanisms to inhibit the activity of DNA-PKcs underscores the apparent necessity for the virus to inactivate NHEJ.
Although the direct relationship between DNA-PK activation and loss of infectivity is not known, the observation that loss of infectivity of virion DNA with 5′ flaps correlates with the robust activation of DNA-PK (Fig. 8) is consistent with the notion that DNA-PK is antiviral. Furthermore, infectivity of Klenow-treated DNA was restored in DNA-PK−/− cells (Fig. 9), suggesting that DNA-PKcs plays a role in the loss of infectivity phenotype observed in wild-type cells. Taken together, these results suggest that the activation of DNA-PK is associated with the drop in infectivity, and we are intrigued by the mechanism by which this occurs. It is possible that DNA with 5′ flaps can recruit cellular proteins, such as nucleases, that are able to process the 5′ flaps, resulting in extreme fragmentation of the viral genome and loss of infectivity. This model, however, does not explain the observation that the absence of DNA-PK itself can rescue infectivity. Another model posits that 5′ flaps either directly or indirectly activate DNA-PK, and this activation is antiviral. Support for this model comes from reports that DNA-PKcs kinase activity is potentiated by dsDNA with ssDNA ends compared to blunt-ended dsDNA (72–75). Additionally, DNA with a 5′ overhang exhibits a greater increase in DNA-PK kinase activity than a 3′ overhang (76). The observation that in the absence of DNA-PK the 5′ flap structure is tolerated supports the notion that the 5′-flapped genome represents a substrate for direct DNA-PK activation.
DNA-PK plays an important role in a variety of cellular processes, and the specific mechanism by which DNA-PK inhibits lytic infection is not understood. It is possible that DNA-PK, acting as part of the NHEJ pathway, promotes circularization of the viral genome, which has been correlated with establishment of latent or quiescent infection (77–79). The report by Jackson and DeLuca that the presence of ICP0 can inhibit circularization (77) may be consistent with this suggestion; however, further experimentation will be required to elucidate the relationship between NHEJ and circularization of viral genomes. Regardless of whether circularization is the mechanism by which NHEJ exerts its antiviral effects, the fate of the viral genome and the choice of repair/recombination pathway activated during infection appear to have important consequences for the establishment of lytic infection.
Although the mechanism of recombination in HSV infection is not well understood, recent experiments from our laboratory suggest that HSV-1 activates the SSA pathway. During infection, SSA is stimulated in a manner that appears to be dependent on the viral 5′-to-3′ exonuclease UL12 (71). UL12 interacts with ICP8 to form a two-component recombinase capable of strand exchange (80, 81). We are intrigued by the possibility that UL12 and ICP8 work together to promote recombination-dependent replication by SSA and that this pathway leads to the production of concatemeric DNA that can be packaged into infectious virus (82). In addition, we are currently exploring the possible involvement of cellular proteins in the stimulation of SSA in HSV-infected cells.
Taken together, these observations suggest that HSV has evolved mechanisms of “pathway choice” that promote lytic replication by inhibiting NHEJ. Our findings suggest that the presence of nicks and gaps in incoming DNA may result in the recruitment of a combination of cellular and viral proteins that stimulates a repair pathway that is beneficial to lytic replication, such as SSA. This process underscores the complex evolutionary relationships between HSV and its host.
ACKNOWLEDGMENTS
We thank members of our laboratory for discussions and suggestions on the manuscript.
This project was supported by National Institutes of Health grants AI069136 and AI021747, awarded to S.K.W.
Footnotes
Published ahead of print 25 June 2014
REFERENCES
- 1.Sheldrick P, Laithier M, Lando D, Ryhiner ML. 1973. Infectious DNA from herpes simplex virus: infectivity of double-stranded and single-stranded molecules. Proc. Natl. Acad. Sci. U. S. A. 70:3621–3625. 10.1073/pnas.70.12.3621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacob RJ, Roizman B. 1977. Anatomy of herpes simplex virus DNA. VIII. Properties of the replicating DNA. J. Virol. 23:394–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kieff ED, Bachenheimer SL, Roizman B. 1971. Size, composition, and structure of the deoxyribonucleic acid of herpes simplex virus subtypes 1 and 2. J. Virol. 8:125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frenkel N, Roizman B. 1972. Separation of the herpesvirus deoxyribonucleic acid duplex into unique fragments and intact strand on sedimentation in alkaline gradients. J. Virol. 10:565–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilkie NM. 1973. The synthesis and substructure of herpesvirus DNA: the distribution of alkali-labile single strand interruptions in HSV-1 DNA. J. Gen. Virol. 21:453–467. 10.1099/0022-1317-21-3-453 [DOI] [PubMed] [Google Scholar]
- 6.Hirsch I, Roubal J, Vonka V. 1976. Replicating DNA of herpes simplex virus type 1. Intervirology 7:155–175 [DOI] [PubMed] [Google Scholar]
- 7.Biswal N, Murray BK, Benyesh-Melnick M. 1974. Ribonucleotides in newly synthesized DNA of herpes simplex virus. Virology 61:87–99. 10.1016/0042-6822(74)90244-X [DOI] [PubMed] [Google Scholar]
- 8.Gordin M, Olshevsky U, Rosenkranz HS, Becker Y. 1973. Studies on herpes simplex virus DNA: denaturation properties. Virology 55:280–284. 10.1016/S0042-6822(73)81031-1 [DOI] [PubMed] [Google Scholar]
- 9.Sinden RR, Pettijohn DE, Francke B. 1982. Organization of herpes simplex virus type 1 deoxyribonucleic acid during replication probed in living cells with 4,5′,8-trimethylpsoralen. Biochemistry 21:4484–4490. 10.1021/bi00261a045 [DOI] [PubMed] [Google Scholar]
- 10.Ben-Porat T, Rixon FJ. 1979. Replication of herpesvirus DNA. IV: analysis of concatemers. Virology 94:61–70 [DOI] [PubMed] [Google Scholar]
- 11.Hyman RW, Oakes JE, Kudler L. 1977. In vitro repair of the preexisting nicks and gaps in herpes simplex virus DNA. Virology 76:286–294. 10.1016/0042-6822(77)90303-8 [DOI] [PubMed] [Google Scholar]
- 12.Everett RD, Parada C, Gripon P, Sirma H, Orr A. 2008. Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J. Virol. 82:2661–2672. 10.1128/JVI.02308-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lukashchuk V, Everett RD. 2010. Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J. Virol. 84:4026–4040. 10.1128/JVI.02597-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson KE, Chikoti L, Chandran B. 2013. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J. Virol. 87:5005–5018. 10.1128/JVI.00082-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orzalli MH, DeLuca NA, Knipe DM. 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. U. S. A. 109:E3008–E3017. 10.1073/pnas.1211302109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pham TH, Kwon KM, Kim Y-E, Kim KK, Ahn J-H. 2013. DNA sensing-independent inhibition of herpes simplex virus 1 replication by DAI/ZBP1. J. Virol. 87:3076–3086. 10.1128/JVI.02860-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lilley CE, Chaurushiya MS, Boutell C, Landry S, Suh J, Panier S, Everett RD, Stewart GS, Durocher D, Weitzman MD. 2010. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 29:943–955. 10.1038/emboj.2009.400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lees-Miller SP, Long MC, Kilvert MA, Lam V, Rice SA, Spencer CA. 1996. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J. Virol. 70:7471–7477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parkinson J, Lees-Miller SP, Everett RD. 1999. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J. Virol. 73:650–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lilley CE, Chaurushiya MS, Boutell C, Everett RD, Weitzman MD. 2011. The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog. 7:e1002084. 10.1371/journal.ppat.1002084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abraham RT. 2004. PI 3-kinase related kinases: ‘big' players in stress-induced signaling pathways. DNA Repair (Amst.) 3:883–887. 10.1016/j.dnarep.2004.04.002 [DOI] [PubMed] [Google Scholar]
- 22.Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol. Cell 40:179–204. 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cimprich KA, Cortez D. 2008. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9:616–627. 10.1038/nrm2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilkinson DE, Weller SK. 2004. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J. Virol. 78:4783–4796. 10.1128/JVI.78.9.4783-4796.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor TJ, Knipe DM. 2004. Proteomics of herpes simplex virus replication compartments: association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J. Virol. 78:5856–5866. 10.1128/JVI.78.11.5856-5866.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD. 2005. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A. 102:5844–5849. 10.1073/pnas.0501916102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shirata N, Kudoh A, Daikoku T, Tatsumi Y, Fujita M, Kiyono T, Sugaya Y, Isomura H, Ishizaki K, Tsurumi T. 2005. Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elicited by herpes simplex virus infection. J. Biol. Chem. 280:30336–30341. 10.1074/jbc.M500976200 [DOI] [PubMed] [Google Scholar]
- 28.Muylaert I, Elias P. 2007. Knockdown of DNA ligase IV/XRCC4 by RNA interference inhibits herpes simplex virus type I DNA replication. J. Biol. Chem. 282:10865–10872. 10.1074/jbc.M611834200 [DOI] [PubMed] [Google Scholar]
- 29.Gregory DA, Bachenheimer SL. 2008. Characterization of mre11 loss following HSV-1 infection. Virology 373:124–136. 10.1016/j.virol.2007.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohni KN, Livingston CM, Cortez D, Weller SK. 2010. ATR and ATRIP are recruited to herpes simplex virus type 1 replication compartments even though ATR signaling is disabled. J. Virol. 84:12152–12164. 10.1128/JVI.01643-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohni KN, Mastrocola AS, Bai P, Weller SK, Heinen CD. 2011. DNA mismatch repair proteins are required for efficient herpes simplex virus 1 replication. J. Virol. 85:12241–12253. 10.1128/JVI.05487-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohni KN, Dee AR, Smith S, Schumacher AJ, Weller SK. 2013. Efficient herpes simplex virus 1 replication requires cellular ATR pathway proteins. J. Virol. 87:531–542. 10.1128/JVI.02504-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 82:8605–8618. 10.1128/JVI.00904-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao F, Courtney RJ. 1992. Association of ICP0 but not ICP27 with purified virions of herpes simplex virus type 1. J. Virol. 66:2709–2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delboy MG, Siekavizza-Robles CR, Nicola AV. 2010. Herpes simplex virus tegument ICP0 is capsid associated, and its E3 ubiquitin ligase domain is important for incorporation into virions. J. Virol. 84:1637–1640. 10.1128/JVI.02041-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilkinson DE, Weller SK. 2006. Herpes simplex virus type I disrupts the ATR-dependent DNA-damage response during lytic infection. J. Cell Sci. 119:2695–2703. 10.1242/jcs.02981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruis BL, Fattah KR, Hendrickson EA. 2008. The catalytic subunit of DNA-dependent protein kinase regulates proliferation, telomere length, and genomic stability in human somatic cells. Mol. Cell. Biol. 28:6182–6195. 10.1128/MCB.00355-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balliet JW, Kushnir AS, Schaffer PA. 2007. Construction and characterization of a herpes simplex virus type I recombinant expressing green fluorescent protein: acute phase replication and reactivation in mice. Virology 361:372–383. 10.1016/j.virol.2006.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldstein DJ, Weller SK. 1988. Factor(s) present in herpes simplex virus type 1-infected cells can compensate for the loss of the large subunit of the viral ribonucleotide reductase: characterization of an ICP6 deletion mutant. Virology 166:41–51. 10.1016/0042-6822(88)90144-4 [DOI] [PubMed] [Google Scholar]
- 40.Livingston CM, DeLuca NA, Wilkinson DE, Weller SK. 2008. Oligomerization of ICP4 and rearrangement of heat shock proteins may be important for herpes simplex virus type 1 prereplicative site formation. J. Virol. 82:6324–6336. 10.1128/JVI.00455-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Panet A, van de Sande JH, Loewen PC, Khorana HG, Raae AJ, Lillehaug JR, Kleppe K. 1973. Physical characterization and simultaneous purification of bacteriophage T4 induced polynucleotide kinase, polynucleotide ligase, and deoxyribonucleic acid polymerase. Biochemistry 12:5045–5050. 10.1021/bi00749a003 [DOI] [PubMed] [Google Scholar]
- 42.Davey SK, Faust EA. 1990. Murine DNA polymerase alpha fills gaps to completion in a direct assay. Altered kinetics of de novo DNA synthesis at single nucleotide gaps. J. Biol. Chem. 265:4098–4104 [PubMed] [Google Scholar]
- 43.Kong H, Kucera RB, Jack WE. 1993. Characterization of a DNA polymerase from the hyperthermophile archaea Thermococcus litoralis. Vent DNA polymerase, steady state kinetics, thermal stability, processivity, strand displacement, and exonuclease activities. J. Biol. Chem. 268:1965–1975 [PubMed] [Google Scholar]
- 44.Davison A, Rixon F. 1985. Cloning of the DNA of Alphaherpesvirinae, p 103–124 In Yechiel Becker JH. (ed), Recombinant DNA research and viruses, vol 5 Springer US, Boston, MA [Google Scholar]
- 45.Mocarski ES, Roizman B. 1982. Structure and role of the herpes simplex virus DNA termini in inversion, circularization and generation of virion DNA. Cell 31:89–97. 10.1016/0092-8674(82)90408-1 [DOI] [PubMed] [Google Scholar]
- 46.Strang BL, Stow ND. 2005. Circularization of the herpes simplex virus type 1 genome upon lytic infection. J. Virol. 79:12487–12494. 10.1128/JVI.79.19.12487-12494.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roychoudhury R, Jay E, Wu R. 1976. Terminal labeling and addition of homopolymer tracts to duplex DNA fragments by terminal deoxynudeotidyl transferase. Nucleic Acids Res. 3:863–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang L, Bollum FJ. 1986. Molecular biology of terminal transferase. CRC Crit. Rev. Biochem. 21:27–52. 10.3109/10409238609113608 [DOI] [PubMed] [Google Scholar]
- 49.Tu CP, Cohen SN. 1980. 3′-end labeling of DNA with [alpha-32P]cordycepin-5′-triphosphate. Gene 10:177–183. 10.1016/0378-1119(80)90135-3 [DOI] [PubMed] [Google Scholar]
- 50.Boulé JB, Rougeon F, Papanicolaou C. 2001. Terminal deoxynucleotidyl transferase indiscriminately incorporates ribonucleotides and deoxyribonucleotides. J. Biol. Chem. 276:31388–31393. 10.1074/jbc.M105272200 [DOI] [PubMed] [Google Scholar]
- 51.Yao F, Schaffer PA. 1995. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. J. Virol. 69:6249–6258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Severini A, Morgan AR, Tovell DR, Tyrrell DL. 1994. Study of the structure of replicative intermediates of HSV-1 DNA by pulsed-field gel electrophoresis. Virology 200:428–435. 10.1006/viro.1994.1206 [DOI] [PubMed] [Google Scholar]
- 53.Severini A, Scraba DG, Tyrrell DL. 1996. Branched structures in the intracellular DNA of herpes simplex virus type 1. J. Virol. 70:3169–3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anantha RW, Vassin VM, Borowiec JA. 2007. Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. J. Biol. Chem. 282:35910–35923. 10.1074/jbc.M704645200 [DOI] [PubMed] [Google Scholar]
- 55.Vassin VM, Anantha RW, Sokolova E, Kanner S, Borowiec JA. 2009. Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J. Cell Sci. 122:4070–4080. 10.1242/jcs.053702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shao RG, Cao CX, Zhang H, Kohn KW, Wold MS, Pommier Y. 1999. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J. 18:1397–1406. 10.1093/emboj/18.5.1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zernik-Kobak M, Vasunia K, Connelly M, Anderson CW, Dixon K. 1997. Sites of UV-induced phosphorylation of the p34 subunit of replication protein A from HeLa cells. J. Biol. Chem. 272:23896–23904. 10.1074/jbc.272.38.23896 [DOI] [PubMed] [Google Scholar]
- 58.Wang H, Guan J, Wang H, Perrault AR, Wang Y, Iliakis G. 2001. Replication protein A2 phosphorylation after DNA damage by the coordinated action of ataxia telangiectasia-mutated and DNA-dependent protein kinase. Cancer Res. 61:8554–8563 [PubMed] [Google Scholar]
- 59.Wilkinson DE, Weller SK. 2005. Inhibition of the herpes simplex virus type 1 DNA polymerase induces hyperphosphorylation of replication protein A and its accumulation at S-phase-specific sites of DNA damage during infection. J. Virol. 79:7162–7171. 10.1128/JVI.79.11.7162-7171.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davido DJ, Von Zagorski WF, Maul GG, Schaffer PA. 2003. The differential requirement for cyclin-dependent kinase activities distinguishes two functions of herpes simplex virus type 1 ICP0. J. Virol. 77:12603–12616. 10.1128/JVI.77.23.12603-12616.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin R, Noyce RS, Collins SE, Everett RD, Mossman KL. 2004. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J. Virol. 78:1675–1684. 10.1128/JVI.78.4.1675-1684.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wold MS. 1997. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 66:61–92. 10.1146/annurev.biochem.66.1.61 [DOI] [PubMed] [Google Scholar]
- 63.Blackwell LJ, Borowiec JA. 1994. Human replication protein A binds single-stranded DNA in two distinct complexes. Mol. Cell. Biol. 14:3993–4001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reznikoff WS, Thomas JCA. 1969. The anatomy of the SP50 bacteriophage DNA molecule. Virology 37:309–317. 10.1016/0042-6822(69)90214-1 [DOI] [PubMed] [Google Scholar]
- 65.Ben-Porat T, Stehn B, Kaplan AS. 1976. Fate of parental herpesvirus DNA. Virology 71:412–422. 10.1016/0042-6822(76)90369-X [DOI] [PubMed] [Google Scholar]
- 66.Lee LF, Kieff ED, Bachenheimer SL, Roizman B, Spear PG, Burmester BR, Nazerian K. 1971. Size and composition of Marek's disease virus deoxyribonucleic acid. J. Virol. 7:289–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rogers SG, Rhoades M. 1976. Bacteriophage T5-induced endonucleases that introduce site-specific single-chain interruptions in duplex DNA. Proc. Natl. Acad. Sci. U. S. A. 73:1576–1580. 10.1073/pnas.73.5.1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scheible PP, Rhoades EA, Rhoades M. 1977. Localization of single-chain interruptions in bacteriophage T5 DNA. I. Electron microscopic studies. J. Virol. 23:725–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang J, Jiang Y, Vincent M, Sun Y, Yu H, Wang J, Bao Q, Kong H, Hu S. 2005. Complete genome sequence of bacteriophage T5. Virology 332:45–65. 10.1016/j.virol.2004.10.049 [DOI] [PubMed] [Google Scholar]
- 70.Khan SA, Hayes SJ, Wright ET, Watson RH, Serwer P. 1995. Specific single-stranded breaks in mature bacteriophage T7 DNA. Virology 211:329–331. 10.1006/viro.1995.1411 [DOI] [PubMed] [Google Scholar]
- 71.Schumacher AJ, Mohni KN, Kan Y, Hendrickson EA, Stark JM, Weller SK. 2012. The HSV-1 exonuclease, UL12, stimulates recombination by a single strand annealing mechanism. PLoS Pathog. 8:e1002862. 10.1371/journal.ppat.1002862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martensson S, Hammarsten O. 2002. DNA-dependent protein kinase catalytic subunit. Structural requirements for kinase activation by DNA ends. J. Biol. Chem. 277:3020–3029. 10.1074/jbc.M106711200 [DOI] [PubMed] [Google Scholar]
- 73.DeFazio LG, Stansel RM, Griffith JD, Chu G. 2002. Synapsis of DNA ends by DNA-dependent protein kinase. EMBO J. 21:3192–3200. 10.1093/emboj/cdf299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rivera-Calzada A, Maman JD, Spagnolo L, Pearl LH, Llorca O. 2005. Three-dimensional structure and regulation of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs). Structure 13:243–255. 10.1016/j.str.2004.12.006 [DOI] [PubMed] [Google Scholar]
- 75.Llorca O, Pearl LH. 2004. Electron microscopy studies on DNA recognition by DNA-PK. Micron 35:625–633. 10.1016/j.micron.2004.05.004 [DOI] [PubMed] [Google Scholar]
- 76.Pawelczak KS, Turchi JJ. 2008. A mechanism for DNA-PK activation requiring unique contributions from each strand of a DNA terminus and implications for microhomology-mediated nonhomologous DNA end joining. Nucleic Acids Res. 36:4022–4031. 10.1093/nar/gkn344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jackson SA, DeLuca NA. 2003. Relationship of herpes simplex virus genome configuration to productive and persistent infections. Proc. Natl. Acad. Sci. U. S. A. 100:7871–7876. 10.1073/pnas.1230643100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Efstathiou S, Minson AC, Field HJ, Anderson JR, Wildy P. 1986. Detection of herpes simplex virus-specific DNA sequences in latently infected mice and in humans. J. Virol. 57:446–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rock DL, Fraser NW. 1983. Detection of HSV-1 genome in central nervous system of latently infected mice. Nature 302:523–525. 10.1038/302523a0 [DOI] [PubMed] [Google Scholar]
- 80.Reuven NB, Staire AE, Myers RS, Weller SK. 2003. The herpes simplex virus type 1 alkaline nuclease and single-stranded DNA binding protein mediate strand exchange in vitro. J. Virol. 77:7425–7433. 10.1128/JVI.77.13.7425-7433.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Reuven NB, Willcox S, Griffith JD, Weller SK. 2004. Catalysis of strand exchange by the HSV-1 UL12 and ICP8 proteins: potent ICP8 recombinase activity is revealed upon resection of dsDNA substrate by nuclease. J. Mol. Biol. 342:57–71. 10.1016/j.jmb.2004.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weller SK, Sawitzke JA. Homologous recombination promoted by DNA viruses: from phage λ to herpes simplex virus. Ann. Rev. Microbiol., in press. 10.1146/annurev-micro-091313-103424 [DOI] [PMC free article] [PubMed] [Google Scholar]