

ABSTRACT
The orf47-orf46-orf45 gene cluster of Kaposi's sarcoma-associated herpesvirus (KSHV) is known to serially encode glycoprotein L (gL), uracil DNA glycosylase, and a viral tegument protein. Here, we identify two novel mRNA variants, orf47/45-A and orf47/45-B, alternatively spliced from a tricistronic orf47-orf46-orf45 mRNA that is expressed in the orf47-orf46-orf45 gene locus during the early stages of viral reactivation. The spliced gene products, ORF47/45-A and ORF47/45-B, consist of only a partial region of gL (ORF47), a unique 7-amino-acid motif, and the complete tegument protein ORF45. Like the ORF45 protein, ORF47/45-A and ORF47/45-B expressed in cells sufficiently activate the phosphorylation of p90 ribosomal S6 kinase (RSK) and extracellular signal-regulated protein kinase (ERK). However, unlike ORF45, both ORF47/45-A and ORF47/45-B contain a signal peptide sequence and are localized at the endoplasmic reticulum (ER). Additionally, we found that ORF47/45-A and ORF47/45-B have an extra function that mediates the upregulation of GRP78, a master regulator of ER homeostasis. The important event regarding GRP78 upregulation can be observed in all tested KSHV-positive cell lines after viral reactivation, and knockdown of GRP78 in cells significantly impairs viral lytic cycle progression, especially at late lytic stages. Compared with some other viral glycoproteins synthesized through the ER, our results strongly implicate that the ORF47/45 proteins may serve as key effectors for controlling GRP78 expression and ER homeostasis in cells. Taken together, our findings provide evidence showing the reciprocal association between the modulation of ER homeostasis and the progression of the KSHV lytic cycle.
IMPORTANCE Emerging evidence has shown that several viruses appear to use different strategies to control ER homeostasis for supporting their productive infections. The two parts of this study identify two aspects of the association between the regulation of ER homeostasis and the progression of the KSHV lytic cycle. The first part characterizes the function of two early lytic cycle proteins, ORF47/45-A and ORF47/45-B, on the activation of a major ER chaperone protein, GRP78. In addition to the ability to promote GRP78 upregulation, the ORF47/45 proteins also activate the phosphorylation of RSK and ERK. The second part reveals that upregulation of GRP78 is essential for the progression of the KSHV lytic cycle, especially at late stages. We therefore propose that activation of GRP78 expression by viral proteins at the early lytic stage may aid with the protection of host cells from severe ER stress and may directly involve the assembly or release of virions.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV), also referred to as human herpesvirus 8 (HHV8), is implicated in the pathogenesis of Kaposi's sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman's disease (1–3). Similar to other herpesviruses, KSHV displays two distinctive life cycles: latency and lytic replication (4, 5). Although the authentic physiological determinants for the latent-to-lytic cycle switch of KSHV are not fully understood, various chemical or biological stimuli have been reported to trigger viral reactivation in the latently infected cells. These lytic replication-inducing agents or conditions include sodium butyrate (SB), 12-O-tetradecanoylphorbol-13-acetate (TPA), hypoxia, reactive oxygen species, and proinflammatory cytokines, as well as superinfection with other viruses (4, 6–10). Upon viral reactivation, viral lytic genes are expressed in an orderly fashion, i.e., immediate early, early, and late gene expression, ultimately leading to the production of infectious virions (11, 12). Viral immediate early genes are the first class of viral genes expressed during the lytic cycle and usually encode regulatory proteins that control the later stages of the lytic cycle (13). After the expression of immediate early genes, early genes encoding enzymes or regulators for ori-Lyt-dependent DNA synthesis are subsequently induced. Following lytic DNA replication, abundant levels of late gene products, such as capsid, tegument, and envelope proteins, are produced for virus assembly and egress (13).
The endoplasmic reticulum (ER) is the main compartment for protein folding and trafficking, as well as a reservoir of intracellular calcium. Disruption of ER function, a condition known as ER stress, causes the accumulation of unfolded or misfolded proteins in the ER. To maintain ER homeostasis, eukaryotic cells have evolved highly conserved signaling pathways called the unfolded protein response (UPR) to cope with the accumulation of unfolded or misfolded proteins in the ER (14, 15). There are three ER transmembrane sensors that initiate UPR signaling cascades in cells, including protein kinase RNA-like ER kinase (PERK), inositol-requiring protein 1 (IRE1), and activating transcription factor 6 (ATF6) (14, 15). Under nonstressed conditions, these three ER transmembrane sensors are maintained in an inactive state by association with a 78-kDa glucose-regulated protein, GRP78, also known as BiP or HSPA5, in the lumen of the ER. GRP78 is an ER chaperone protein critical for serving as a central regulator of ER homeostasis (16). Upon accumulation of unfolded proteins in the ER, GRP78 preferentially binds to unfolded proteins and dissociates from the UPR sensor molecules, which allow each sensor to be activated. After disassociation from GRP78, PERK undergoes autophosphorylation and then phosphorylates eukaryotic translation initiation factor 2α (eIF2α) to prevent further protein synthesis. However, a small subset of mRNAs can be selectively translated under stress conditions. The best-studied example of phosphorylated eIF2α-dependent translation is activating transcription factor 4 (ATF4), which can induce UPR response genes. IRE1 is an unusual transmembrane protein kinase in that its activity is coupled with endoribonuclease activity. Activation of IRE1 triggers its endoribonuclease activity, which cleaves X box-binding protein 1 (XBP-1) mRNA. The spliced gene product, XBP-1s, is a potent transcription factor that induces the expression of many UPR chaperone genes, including grp78. After being released from GRP78, ATF6 transits from the ER to the Golgi body, where ATF6 undergoes sequential proteolytic cleavages. The cleaved ATF6 is an active transcriptional factor that upregulates ER folding enzymes and chaperone proteins (including GRP78). These adaptive responses aid with the alleviation of protein aggregation in the ER by global attenuation of protein synthesis and upregulation of chaperone proteins or folding enzymes. However, if the ER stress is too severe, cells undergo apoptosis (17). Due to the fact that a full panel of viral structural genes could be robustly induced at late stages of the KSHV lytic cycle, abundant viral proteins synthesized through the ER might be deleterious to host cells, which may result in the prevention of the lytic cycle from proceeding. Therefore, the homeostatic regulation of ER functions would be important for the progression of the KSHV lytic cycle. To date, the potential modulation of ER homeostasis during the KSHV lytic cycle is largely unknown.
Open reading frame 45 (ORF45) of KSHV is a multifunctional tegument protein expressed in the immediate early and early stages of the viral lytic cycle (18, 19). Evidence has shown that the ORF45 protein plays critical roles in viral infection to target cells and in viral lytic replication. During viral infection, the ORF45 released from the virion is known to block the phosphorylation and nuclear translocation of IRF-7 (20), which leads to the prevention of activation of type I interferon (alpha/beta interferon [IFN-α/β]) genes in response to viral infection. On the other hand, when KSHV is reactivated from latency, the ORF45 protein is expressed and forms a complex with p90 ribosomal S6 kinase (RSK) and extracellular signal-regulated kinase (ERK) (21–23). Interaction of ORF45 with RSK and ERK stimulates the phosphorylation of both kinases. The sustained activation of RSK and ERK by ORF45 is thought to be essential for the KSHV lytic cycle (22). Additionally, Sathish et al. reported that ORF45 interacts with kinesin-2, a microtubule-based motor protein (24), suggesting that ORF45 plays a role in the transport of viral capsid-tegument complexes along microtubule filaments. The importance of ORF45 in the KSHV lytic cycle is emphasized by the phenotypic characterization of orf45-null recombinant KSHV (25). Cells with the orf45-null KSHV genome produced fewer virions than cells harboring the wild-type viral genome after lytic induction (25).
The orf45 gene is located at the most downstream region of the orf47-orf46-orf45 gene cluster in the viral genome (26). Two upstream genes, orf47 and orf46, encode glycoprotein L (gL) and uracil DNA glycosylase, respectively. We have previously mapped three overlapping mRNA transcripts that are separately initiated from the orf47, orf46, and orf45 gene promoters in the gene locus (27–29). Among these mRNA transcripts, the transcript initiated from the orf47 gene promoter is a tricistronic orf47-orf46-orf45 mRNA (27, 29). In this study, we provide evidence showing that two alternatively spliced mRNAs are derived from the tricistronic orf47-orf46-orf45 transcript. These two newly identified mRNA variants encode gene products, designated ORF47/45-A and ORF47/45-B, consisting of an N-terminal portion of gL (114 amino acids [aa] and 56 aa for ORF47/45-A and ORF47/45-B, respectively), an extra unique 7-aa motif, and the entire ORF45 protein region. Most importantly, our functional analyses revealed that both ORF47/45-A and ORF47/45-B are localized at the ER and receive the ability to augment GRP78 expression. The potential associations between GRP78 expression and the progression of the KSHV lytic cycle are characterized and discussed.
MATERIALS AND METHODS
Cell cultures and transfections.
HH-B2 (11), BCBL1 (5), BC3 (30), and BCP1 (31) are PEL cell lines infected with KSHV. All PEL cell lines were cultured in RPMI 1640 medium supplemented with 15% fetal bovine serum (FBS). HEK293T (293T) is a human embryonic kidney cell line transformed with the E1 region of adenovirus and the simian virus 40 T antigen (32). HEK293T cells were grown in high-glucose Dulbecco modified Eagle medium (DMEM) supplemented with 10% FBS. 293T(BAC16) cells with the KSHV genome (on bacmid BAC16) were maintained in DMEM containing 10% FBS plus hygromycin at 200 μg/ml. For viral lytic induction, HH-B2, BC3, and 293T(BAC16) cells were treated with 3 mM SB, while BCBL1 and BCP1 cells were treated with TPA (30 ng/ml). The concentration of phosphonoacetic acid (PAA) used in the study was 200 μg/ml. Transfection experiments were performed using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen).
Rapid amplification of cDNA ends (RACE).
The orf47/orf45 mRNA variants expressed in HH-B2 cells were identified using a GeneRacer kit (Invitrogen). Total RNAs from HH-B2 cells treated with 3 mM SB for 48 h were extracted using an RNeasy minikit (Qiagen). Two micrograms of total RNAs was reverse transcribed into first-strand cDNAs using an oligo(dT) primer provided in the GeneRacer kit. The first-strand cDNAs were employed as the templates for PCR amplification. The primary and nested PCR amplifications were performed using specific forward primers RACE-A1 and RACE-A2, respectively, in combination with reverse primers provided in the GeneRacer kit. The nucleotide sequences of the RACE-A1 and RACE-A2 primers within the orf47 region are 5′-ATGGGGATCTTTGCGCTATTTGCC and 5′-TGTTGCGCAATTCAGGCATCGGCA, respectively. Two major PCR products consistently amplified in the gels were eluted, cloned, and sequenced.
Northern blot analysis.
HH-B2 cells were treated with 3 mM SB in the presence or absence of 200 μg/ml PAA for 12, 24, 36, and 48 h. Total RNAs from these treated cells were extracted, fractionated on 1% formaldehyde-agarose gels, and transferred to nylon membranes (Hybond-N). Probes were labeled by the random-primed method. DNA probes corresponding to nucleotides 69917 to 69412 and nucleotides 68576 to 68101 of the KSHV genome (GenBank accession no. U75698) were employed to detect the orf47- and orf45-containing mRNAs, respectively. Northern hybridization was carried out as described previously (29).
Reverse transcriptase PCR (RT-PCR).
Total RNAs were extracted from the PEL cell lines using an RNeasy minikit (Qiagen) in combination with an RNase-free DNase set (Qiagen). RNA samples were first converted into cDNAs by using SuperScript II reverse transcriptase (Invitrogen) and random priming. The cDNA samples were then amplified by PCR using primers RT-PCR-1 (5′-TACGTCGCCTTACCATGTTGCGCA) and RT-PCR-2 (5′-GCCTGCCAAATCCTGGAAGTGATG).
Plasmid construction.
The expression plasmids pCMV-F-ORF47+46+45, pCMV-F-ORF47/45-A, pCMV-F-ORF47/45-B, and pCMV-F-ORF45 were generated by insertion of PCR-amplified cDNA fragments into pFLAG-CMV-2 (Sigma). To construct plasmids carrying proteins with an added C-terminal triple FLAG (3F) tag, the coding region of orf47/45-A, orf47/45-B, orf45, orf47 (gL), orf8 (gB), orf22 (gH), orf39 (gM), orf53 (gN), or orf51 (K8.1) was individually amplified by PCR and cloned into pCMV-3Tag-3 (Stratagene). The grp78 gene promoter-driven reporter was constructed by inserting the human grp78 promoter element from positions −191 to +29 into pGL3-Basic (Promega) at XhoI and HindIII sites. This human grp78 promoter fragment encompasses three tandem copies of the ER stress response element (ERSE) (33).
Western blot analysis.
Protein extracts were mixed with 3× sodium dodecyl sulfate (SDS) gel loading buffer, and the mixture was boiled for 5 min before proteins were resolved on an 8% to 12% polyacrylamide gel. After electrophoresis, the proteins in the gel were transferred onto a polyvinylidene difluoride membrane (Bio-Rad), where they were probed with specific antibodies. The rabbit polyclonal antibody against ORF47/45-A was generated using chemically synthesized peptides as immunogens. Two immunogenic peptides, CIAKLRSKTGDITV and AYTRSTPYKA, were used to generate polyclonal antibodies. The anti-ORF50 antibody was obtained as described previously (34). Antibodies to FLAG (A8592; Sigma), K8 (sc-57889; Santa Cruz), ORF45 (sc-53883; Santa Cruz), K8.1 (sc-65446; Santa Cruz), GRP78 (sc-13968; Santa Cruz), RSK (9355; Cell Signaling), phospho-RSK (9344; Cell Signaling), ERK (sc-94; Santa Cruz), phospho-ERK (sc-7383; Santa Cruz), XBP-1s (619502; BioLegend), PERK (sc-13073; Santa Cruz), phospho-PERK (sc-32577; Santa Cruz), eIF2α (sc-133132; Santa Cruz), phospho-eIF2α (sc-293100; Santa Cruz), ATF6 (MAB6762; Abnova), and actin (sc-47778; Santa Cruz) were purchased commercially.
Confocal immunofluorescence analysis.
293T cells or A7 melanoma cells (4 × 104) were seeded on coverslips and transiently transfected with the expression plasmids. Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min and then washed with PBS three times for 5 min each time. The fixed cells were permeabilized with 0.1% Triton X-100 in PBS for 10 min. After the cells were incubated with blocking solution (CAS-Block; Invitrogen) at room temperature for 1 h, anti-ORF45 (1:200) and anti-protein disulfide isomerase (anti-PDI; 1:200) antibodies were added to treat the cells at 4°C overnight. After three further washing steps, appropriate secondary antibodies were added. DAPI (4′,6-diamidino-2-phenylindole) staining was performed at room temperature for 3 min. Cells were mounted for examination with a Leica confocal laser scanning system (TCS-SP5II). Image processing was carried out using LAS AF Lite software (Leica).
Luciferase assays.
Cells (7 × 105) were transfected with a fixed amount (0.8 μg) of plasmid DNA that included the firefly luciferase reporter, the pTK-Renilla reporter, and effector plasmids. At 24 h or 48 h posttransfection, the firefly luciferase activity in the cells was measured and normalized to the renilla luciferase activity. Luciferase reporter assays were performed according to the manufacturer's protocol for the luciferase reporter assay system (Promega). The fold activation was calculated as the normalized firefly luciferase activity in the presence of effector divided by that in the absence of effector. The value of the fold activation represents that from at least three independent experiments with duplicate samples.
Lentivirus-based knockdown.
All RNA interference (RNAi) reagents were obtained from the National RNAi Core Facility Platform at the Institute of Molecular Biology/Genomic Research Center, Academia Sinica, supported by the National Core Facility Program for Biotechnology Grants of the National Science Council in Taiwan. To prepare short hairpin RNA (shRNA) lentiviral particles, the pLKO.1 plasmid carrying grp78 shRNA or control shRNA was cotransfected with pCMV-ΔR8.91 and pMD.G into 293T cells. Cotransfection of the plasmid pCMV-ΔR8.91 provided the expression of the gag, pol, and rev genes of HIV-1, and cotransfection of pMD.G provided the expression of the vesicular stomatitis virus glycoprotein G (VSV-G) envelope. The target sequence of grp78 shRNA is 5′-AGATTCAGCAACTGGTTAAAG. At 24 h posttransfection, the culture medium was replaced with fresh DMEM. The lentivirus-containing media were then collected at day 1 and day 2. Lentivirus transduction into BC3, BCBL1, and 293T(BAC16) cells was performed for 24 h in the presence of Polybrene at a final concentration of 8 μg/ml.
Cell viability assay.
For determination of cell viability, cells were cultured and evaluated with a tetrazolium-based colorimetric assay {the 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]-2H-tetrazolium hydroxide (XTT) assay; Roche}. The absorbance of the converted dye was measured at a wavelength of 450 nm using a microtiter plate reader. A reference wavelength of 630 nm was used to assess nonspecific readings. Duplicate samples were analyzed for each cell line, and each experiment was repeated at least twice.
TaqMan PCR quantification.
The culture media of the BCBL1, BC3, and 293T(BAC16) cells were collected at day 3 after KSHV reactivation. The culture supernatants were supplemented with 2 mM MgCl2 and 0.5 mM CaCl2 and treated with DNase I (30 units/ml) for 1 h to remove contaminating DNA. After the reaction was stopped by EDTA at a final concentration of 5 mM, samples were further treated with 0.1% SDS and 100 μg/ml proteinase K at 37°C overnight. Viral DNA was extracted by phenol-chloroform (1:1) and precipitated with ethanol in the presence of glycogen. TaqMan PCR was used to quantify KSHV DNA levels. The nucleotide sequences of the TaqMan probe and orf65 primers used for the quantitative PCR were 5′-FAM-ACTCCACGGTTGTCCAATCG-TAM (where FAM is 6-carboxyfluorescein and TAM is 6-carboxytetramethylrhodamine), 5′-CGGTTGACAGAATAAAGAA (forward primer), and 5′-GTCAGACATTCTCACAAC (reverse primer).
Nucleotide sequence accession numbers.
The GenBank accession numbers of the full-length orf47/45-A and orf47/45-B cDNAs from the study are KJ490393 and KJ490394, respectively.
RESULTS
Two mRNA variants spliced from the tricistronic orf47-orf46-orf45 mRNA are identified in KSHV-infected cells after lytic induction.
We have previously mapped the transcription start site and the 3′ polyadenylation cleavages site of the orf47 gene transcript using RACE analyses (27–29). The orf47 gene transcript is a tricistronic mRNA that includes orf47, orf46, and orf45 (Fig. 1A). Actually, when specific PCR products generated in 3′ RACE experiments were carefully analyzed (Fig. 1B), we unexpectedly found two spliced mRNA variants derived from the tricistronic orf47-orf46-orf45 mRNA. These two alternatively spliced mRNAs, orf47/45-A and orf47/45-B, consisted of only a part of the orf47 region and the downstream orf45 region (Fig. 1A). The orf47/45-A transcript has a longer orf47 portion than the orf47/45-B transcript. Like most eukaryotic spliced mRNAs, the splicing of both orf47/45-A and orf47/45-B variants from the orf47-orf46-orf45 mRNA follows the GU-AG rule, with a consensus donor site and an acceptor site (Fig. 1A and 2A). The estimated sizes of the orf47/45-A mRNA (1.7 kb) and orf47/45-B mRNA (1.5 kb) appeared to be close to the estimated size of the orf45 mRNA (1.6 kb) (Fig. 1A). In Northern blotting analyses, the corresponding orf47/45-A and orf47/45-B mRNAs were detected in HH-B2 cells after treatment with sodium butyrate (SB) using either the orf47 or the orf45 region as a probe (Fig. 1A and C). The newly identified orf47/45-A and orf47/45-B genes may belong to the members of the early lytic gene family because treatment with phosphonoacetic acid (PAA), an inhibitor of KSHV lytic DNA synthesis, did not significantly affect their relative mRNA levels (Fig. 1C). The expression of the orf47/45 transcripts in different KSHV-infected PEL cell lines was further confirmed by RT-PCR. As shown in Fig. 1D, the unspliced orf47-orf46-orf45 mRNA and the spliced orf47/45 variants could be simultaneously detected by RT-PCR in all tested PEL cell lines, including the HH-B2, BCBL1, BC3, and BCP1 cell lines, after treatment with appropriate chemical inducers. According to the predicted coding sequence of the orf47/45-A and orf47/45-B mRNAs, the gene products of these two spliced mRNAs, designated ORF47/45-A and ORF47/45-B, would encompass an N-terminal ORF47 portion (114 aa and 56 aa for ORF47/45-A and ORF47/45-B, respectively), an extra unique 7-aa motif that is adjacent to the ORF45-coding region, and the entire ORF45-coding region (Fig. 2A and B). On the basis of the prediction, ORF47/45-A, ORF47/45-B, and ORF45 would contain 528, 470, and 407 amino acids, respectively, and the calculated molecular masses would be 56.7, 50.1, and 43.3 kDa, respectively.
FIG 1.

Identification of orf47/45-A and orf47/45-B mRNAs expressed in cells after KSHV reactivation. (A) Schematic diagram of the spliced and unspliced orf45-containing transcripts expressed during the KSHV lytic cycle. The numbers in the diagram represent the nucleotide positions of the KSHV genome (GenBank accession no. U75698). The locations of probes for Northern blotting and specific primers for the RACE and RT-PCR assays are indicated. (B) Representative data from 3′ RACE analysis. Two major PCR products corresponding to the tricistronic orf47-orf46-orf45 mRNA and alternatively spliced orf47/45 mRNAs were amplified in the 3′ RACE experiment. (C) Northern blotting of the orf45-containing mRNAs in HH-B2 cells. Total RNAs were prepared from HH-B2 cells treated with SB for different times (12, 24, 36, and 48 h) in the presence or absence of PAA. The orf47- and orf45-specific DNA probes were used in Northern hybridization. (D) Detection of the tricistronic orf47-orf46-orf45 mRNA and spliced orf47/45 mRNAs by RT-PCR in different KSHV-infected cell lines treated with SB or TPA. RT(−), no reverse transcriptase; RT(+), addition of reverse transcriptase.
FIG 2.

Coding regions of the orf47, orf45, and spliced orf47/45 genes. (A) The nucleotide (nt) and amino acid sequences of the individual coding regions in the orf47-orf46-orf45 gene locus. The orf47 and orf45 coding regions are shown in boxes. The brackets represent either splicing donor sites (nucleotides 69748 and 69574) or the acceptor site (nucleotide 68597) of the orf47/45 transcripts. The circled “M” indicates the translational initiation codon for each gene. The poly(A) signal, AATAAA, is shown in a gray box, and the 3′ polyadenylated cleavage site (nucleotide 67325) of the orf45-containing transcript is indicated by an arrow. (B) The predicted open reading frames of the ORF47/45-A, ORF47/45-B, and ORF45 proteins are 528, 470, and 407 amino acids, respectively.
ORF47/45 proteins are expressed during the early stages of the KSHV lytic cycle.
To determine whether ORF47/45 proteins are indeed expressed during the KSHV lytic cycle, an antibody specific to the ORF47/45 proteins, but not ORF45, was generated by the use of chemically synthesized peptides. Since the short ORF47 portion (56 aa) in ORF47/45-B was poorly immunogenic, two specific peptides from ORF47/45-A were used as immunogens for the generation of a specific antibody in rabbit. These two immunogenic peptides included CIAKLRSKTGDITV, which is located within the ORF47 portion of ORF47/45-A, and AYTRSTPYKA, which is near the splice junction and overlaps the unique 7-aa motif. The specificity of the generated antibody for ORF47/45-A was verified by Western blotting using the lysates of 293T cells transfected with pCMV-3F, pCMV-ORF47/45-A(3F), or pCMV-ORF45(3F). As expected, the expressed ORF47/45-A and ORF45 with a C-terminal triple FLAG (3F) tag could be detected in Western blots using either anti-FLAG or anti-ORF45 antibody (Fig. 3A, left and middle). However, only ORF47/45-A and not ORF45 was specifically recognized by anti-ORF47/45-A antibody (Fig. 3A, right). A noteworthy finding was that although ORF47/45-A and ORF45 have predicted molecular masses of 56.7 and 43.3 kDa, respectively, both proteins aberrantly migrated on SDS-polyacrylamide gels to approximately similar positions of 70 to 75 kDa (Fig. 3A, left and middle).
FIG 3.

Endogenous ORF47/45 proteins are expressed during the KSHV lytic cycle. (A) Protein extracts of 293T cells transfected with pCMV, pCMV-ORF47/45-A(3F), and pCMV-ORF45(3F) were subjected to Western blot (WB) analysis using antibodies against FLAG, ORF45, and ORF47/45-A. CMV, cytomegalovirus. (B to D) Western blot analysis of viral lytic proteins in different KSHV-positive cell lines. The expression of viral lytic proteins, including ORF50, K8, ORF45, and ORF47/45, was examined in HH-B2, BC3, or 293T(BAC16) cells treated with SB for the indicated times.
Next, the expression of endogenous ORF47/45-A protein was examined in KSHV-infected cell lines by Western blotting using the generated antibody. As shown in Fig. 3B to D, the expression of ORF47/45-A was detected in HH-B2, BC3, and 293T(BAC16) cells after SB treatment. Furthermore, the expression kinetics of ORF47/45-A in these PEL cells appeared to be similar to that of the immediate early or early lytic proteins, such as ORF50, K8, and ORF45. These results strongly suggest that the ORF47/45 proteins are early lytic proteins.
Both ORF47/45-A and ORF47/45-B, but not ORF45, contain a signal peptide and are localized to the ER.
Since the ORF45 protein is critical in the KSHV lytic cycle, we attempted to compare the possible structural and functional differences between ORF47/45-A or ORF47/45-B and ORF45. Initially, plasmids that express proteins with an added N-terminal FLAG tag were constructed (Fig. 4A). In addition to ORF47/45-A-, ORF47/45-B-, and ORF45-expressing plasmids, we also constructed an expression plasmid encompassing a full-length tricistronic orf47-orf46-orf45 cDNA (Fig. 4A; pCMV-F-ORF47+46+45). Although the orf45 gene was located at the most downstream position of the orf47-orf46-orf45 region, transfection of pCMV-F-ORF47+46+45 into 293T cells still allowed the expression of the ORF45 protein, detected by immunoblotting with anti-ORF45 antibody (Fig. 4B, left, lane 5). Importantly, point mutations (mt-ATG) at the ORF45 initiation codon and its adjacent in-frame ATG in pCMV-F-ORF47+46+45 did not alter the expression of the ORF45 protein (Fig. 4B, right; compare lanes 2 and 3), suggesting that the ORF45 protein expressed from the pCMV-F-ORF47+46+45-transfected cells would be ORF47/45-A or ORF47/45-B, or both.
FIG 4.

The spliced orf47/45-A and orf47/45-B gene products, ORF47/45-A and ORF47/45-B, contain a removable signal peptide. (A) Schematic diagram of plasmid constructs expressing proteins with an added N-terminal FLAG tag. The orf45-containing cDNAs, including orf47-orf46-orf45, orf47/45-A, orf47/45-B, and orf45, were cloned into pCMV-FLAG, an N-terminal FLAG-tagged vector. Point mutations at the initiation codon and the second in-frame ATG of the orf45 gene are indicated in the pCMV-F-ORF47+46+45 plasmid construct. (B) Expression of the ORF45 protein from the pCMV-F-ORF47+46+45 plasmid construct. 293T cells were transfected for 48 h with either empty vector or the indicated expression plasmids. Cell lysates were analyzed by Western blotting with anti-ORF45 antibody. (C) Effect of phosphorylation on the gel mobility of ORF47/45-A, ORF47/45-B, and ORF45. 293T cells were transfected with plasmids expressing F-ORF47/45-A, F-ORF47/45-B, and F-ORF45. At 48 h posttransfection, the cell lysates were prepared and incubated with or without bacteriophage lambda protein phosphatase (λPPase) for 2 h at 30°C. The ORF45-related isoforms were examined by Western blotting with anti-ORF45 antibody. (D) The cell extracts used in the assay whose results are presented in panel C were analyzed by immunoblotting with anti-FLAG antibody. The numbers to the left of the gels in panels C and D are molecular masses (in kilodaltons). (E) Schematic diagram of the plasmid constructs carrying wild-type or mutant proteins with a C-terminal triple FLAG tag. Point mutations at the initiation codon and the second in-frame ATG of the orf45 gene are indicated in the mutant constructs. (F) Western blot analysis of wild-type and mutant ORF47/45-A and ORF47/45-B proteins.
As mentioned earlier, ORF47/45-A, ORF47/45-B, and ORF45 would contain 528, 470, and 407 amino acids, respectively, and would have calculated molecular masses of 56.7, 50.1, and 43.3 kDa, respectively. However, all these proteins migrated at 70 to 75 kDa on SDS-polyacrylamide gels (Fig. 4C, lanes 3, 5, and 7). This discrepancy might be due to the acquisition of different sets of posttranslational modifications (e.g., phosphorylation) during their biosynthesis. Since ORF45 is known to be a phosphorylated protein, we investigated the possible involvement of phosphorylation in the inconsistency. Protein extracts of 293T cells transfected with pCMV-F-ORF47/45-A, pCMV-F-ORF47/45-B, and pCMV-F-ORF45 were prepared and then treated with bacteriophage lambda protein phosphatase (λPPase). Although we detected multiple fast-mobility ORF45-related isoforms after removing phosphate groups, we did not observe significant differences in the profiling of these fast-mobility isoforms among the ORF47/45-A, ORF47/45-B, and ORF45 proteins (Fig. 4C, lanes 4, 6, and 8). Intriguingly, when the same cell lysates were analyzed by Western blotting using anti-FLAG antibody, we failed to detect ORF47/45-A and ORF47/45-B (Fig. 4D, lanes 3 to 6), suggesting that the N-terminal FLAG tag has been lost from both the ORF47/45-A and the ORF47/45-B proteins. This hypothesis was supported by the prediction that a conserved signal peptide sequence is present at the N-terminal 20-aa region of both ORF47/45-A and ORF47/45-B, according to the SignalP server (35). Thus, the mobility inconsistency between the ORF47/45 proteins and ORF45 may be partly attributed to the presence of a removable signal peptide in ORF47/45-A and ORF47/45-B. On the other hand, we also constructed plasmids carrying ORF47/45-A, ORF47/45-B, and ORF45 with a C-terminal triple FLAG tag (Fig. 4E). The expressed ORF47/45-A and ORF47/45-B tagged with a C-terminal FLAG tag could be easily detected in Western blots using either anti-ORF45 or anti-FLAG antibody (Fig. 4F). To further verify the differences between the translated ORF47/45 proteins and ORF45, the possible ORF45 initiation codons were mutated in the ORF47/45-A and ORF47/45-B constructs (Fig. 4E). As shown in Fig. 4F, ORF47/45-A and ORF47/45-B point mutants still normally expressed their corresponding proteins, as detected by immunoblotting using either anti-ORF45 or anti-FLAG antibody. Although the ORF47/45-A, ORF47/45-B, and ORF45 proteins had similar mobilities on SDS-polyacrylamide gels, our results demonstrated that the translated ORF47/45 proteins are indeed different from the ORF45 protein.
The presence of a conserved signal peptide sequence in ORF47/45-A and ORF47/45-B prompted us to compare the subcellular localization between ORF47/45 proteins and ORF45 in cells. Both N-terminally FLAG-tagged and nontagged proteins were examined in 293T cells and in A7 melanoma cells. By confocal immunofluorescence examination, the FLAG-tagged ORF45 was distributed in both the nucleus and cytoplasm of 293T cells or A7 melanoma cells, whereas FLAG-tagged ORF47/45-A and ORF47/45-B were exclusively detected in the cytoplasm (Fig. 5A and B). In particular, we found that the FLAG-tagged ORF47/45 proteins, but not ORF45, were predominantly colocalized with protein disulfide isomerase (PDI), a cellular ER-resident protein (Fig. 5A and B). By expressing the nontagged ORF45 and ORF47/45 proteins in 293T or A7 cells, we got the same results that we did with FLAG-tagged proteins (Fig. 5C and D). The nontagged ORF47/45-A and ORF47/45-B proteins were specifically detected in the cytoplasm (Fig. 5C and D, panels ii and iii). On the basis of the intensity correlation analysis (Fig. 5B and C, panels xiii, xiv, and xv), the fluorescence intensities between the ORF47/45 proteins (green) and PDI (red) had a perfectly linear relationship, further suggesting that the ORF47/45 proteins are colocalized with PDI. However, the ORF45 protein in the cytoplasm did not show the same linear correlation with PDI (Fig. 5B and C, panels xiii). Due to the presence of a signal peptide sequence in the ORF47/45 proteins and their colocalization with PDI, we conclude that both ORF47/45-A and ORF47/45-B are ER-localized proteins.
FIG 5.

Both ORF47/45-A and ORF47/45-B are ER-localized proteins. (A and B) Subcellular localizations of N-terminally FLAG-tagged ORF45, ORF47/45-A, and ORF47/45-B in 293T cells and in melanoma A7 cells. At 24 h after transfection with the indicated expression constructs, cells were stained with anti-ORF45, anti-PDI, and DAPI and then analyzed using confocal microscopy. (C and D) Subcellular localizations of nontagged ORF45, ORF47/45-A, and ORF47/45-B in 293T cells and in A7 cells. Single-channel confocal images (i to ix) and merged images (x, xi, and xii) are shown as indicated. Arrows in merged images (x, xi, and xii), cells selected for the intensity correlation analysis (xiii, xiv, and xv). The fluorescence intensities of pixels along the black line in each selected cell were collected. Note that the fluorescence intensities between ORF47/45-A or ORF47/45-B (green) and PDI (red) in cells are linearly related.
Expression of ORF47/45-A or ORF47/45-B in cells induces the phosphorylation of RSK and ERK as well as the upregulation of GRP78.
The ORF45 protein is known to form a complex with p90 RSK and ERK and is able to activate both kinases (22). To determine whether the ORF47/45 proteins activate RSK and ERK, protein extracts of 293T cells transfected with pCMV-F-ORF47/45-A, pCMV-F-ORF47/45-B, or pCMV-F-ORF45 were used to evaluate the phosphorylation of RSK and ERK. Like ORF45, both ORF47/45-A and ORF47/45-B with an added N-terminal FLAG tag were able to induce the phosphorylation of RSK and ERK (Fig. 6A). Additionally, the nontagged or C-terminally FLAG-tagged ORF47/45 proteins also sufficiently activated the phosphorylation of RSK and ERK (Fig. 6B).
FIG 6.

Both ORF47/45-A and ORF47/45-B activate the phosphorylation of ERK and p90 RSK. 293T cells were transfected with plasmids expressing either N-terminally FLAG-tagged proteins (A) or C-terminally FLAG-tagged or nontagged proteins (B). At 48 h posttransfection, the levels of ERK, phospho-ERK (p-ERK), RSK, and phospho-RSK (p-RSK) induced by these effectors were determined by Western blotting using specific antibodies.
Due to the localization of the ORF47/45 proteins at the ER, we investigated whether the ORF47/45 proteins affect ER homeostasis. The expression of GRP78, a well-established marker in maintaining ER homeostasis, was examined in cells after transfection with effector plasmids (Fig. 7A). In Western blotting, the N-terminally FLAG-tagged ORF47/45-A and ORF47/45-B, but not ORF45, significantly enhanced the GRP78 levels in 293T cells (Fig. 7A). In a parallel experiment, treatment of the transfected cells with tunicamycin (Tm), an ER stress inducer, served as a positive control for GRP78 upregulation (Fig. 7A). To analyze whether the ORF47/45 proteins induce GRP78 at the transcription level, the grp78 gene promoter-driven luciferase reporter was cotransfected with effector plasmids in 293T cells or in HH-B2 cells. The luciferase reporter assay showed that both ORF47/45-A and ORF47/45-B activated the grp78 promoter by approximately 6.5-fold in 293T cells or in HH-B2 cells (Fig. 7B and C). In contrast, ORF45 or ORF50 in the cells did not induce grp78 promoter activation (Fig. 7B and C). In addition to the use of the N-terminal FLAG fusion proteins, activation of GRP78 expression was also examined using nontagged or C-terminally FLAG-tagged proteins. Similarly, both nontagged and C-terminally FLAG-tagged ORF47/45 proteins significantly induced the upregulation of GRP78 (Fig. 7D). Furthermore, point mutations (mt-ATG) in the ORF47/45-A and ORF47/45-B expression constructs, which completely eliminated the possible translational initiation of ORF45, did not influence the activation of GRP78 expression (Fig. 7E). Our results allow us to strongly conclude that only the ORF47/45 proteins and not ORF45 activate the expression of GRP78.
FIG 7.

Both ORF47/45-A and ORF47/45-B, but not ORF45, induce GRP78 expression. (A) Activation of GRP78 expression by the ORF47/45 proteins with an added N-terminal FLAG tag. 293T cells were transfected with the indicated expression plasmids for 48 h, and then GRP78 expression levels were determined by Western blotting. In parallel experiments, the transfected cells were treated with 20 μg/ml tunicamycin (Tm), an ER stress inducer, for 18 h before cell collection. (B and C) Activation of the grp78 promoter-driven reporter by FLAG-tagged ORF47/45 proteins in 293T and HH-B2 cells. The grp78 promoter-driven reporter plasmid was cotransfected with the effector plasmids into 293T cells for 48 h or into HH-B2 cells for 24 h. The fold activation of the grp78 promoter in the reporter assays was measured by comparing the normalized firefly luciferase activity in the presence of effectors to that in the presence of the empty control. (D) Activation of GRP78 expression by C-terminally FLAG-tagged or nontagged ORF47/45 proteins. (E) Effect of wild-type and mutated ORF47/45 proteins on GRP78 upregulation. The ORF47/45 mutant constructs contain point mutations at the potential translational initiation codons of ORF45. (F) Activation of GPR78 expression by ORF47 in 293T cells. (G and H) Effect of ORF47 on grp78 promoter activation in 293T and HH-B2 cells. Luciferase reporter assays were carried out in 293T cells or HH-B2 cells as described in the legends to panels B and C.
Since ORF47/45-A and ORF47/45-B contain an N-terminal portion of ORF47 (gL), we determined whether ORF47 has the ability to mediate GRP78 upregulation. Plasmids carrying C-terminally FLAG-tagged proteins were used for transient transfection (Fig. 7F). In Western blot analyses, we found that ORF47, like the ORF47/45 proteins, could mediate GRP78 upregulation in 293T cells (Fig. 7F). The luciferase reporter assay also demonstrated that ORF47 could activate the grp78 promoter in 293T cells (Fig. 7G). Interestingly, although ORF47 was sufficient to activate the grp78 promoter in 293T cells, we failed to detect the activation of the grp78 promoter by ORF47 in HH-B2 cells (Fig. 7H). The reason for the cell type-dependent activation of the grp78 promoter by ORF47 is currently unknown.
Different regions of the ORF47/45 proteins separately regulate ERK/RSK phosphorylation and GRP78 upregulation.
To determine the critical region responsible for GRP78 upregulation and ERK/RSK activation, a series of ORF47/45-A deletion mutants was constructed (Fig. 8A). Among these deletion mutants, the mutant with a deletion at the N terminus (ΔL38) that removed the conserved signal peptide sequence was defective in activating GRP78 expression, but it still retained the ability to activate ERK/RSK phosphorylation in 293T cells (Fig. 8A and B, lane 5). Conversely, two mutants with internal deletions, including a mutant with a deletion of amino acids 56 to 142 at the N terminus (the ΔL56-142 mutant) and a mutant with a deletion in the tegument protein at the C terminus (the ΔTegu-III mutant), were competent to induce GRP78 expression but lost their ability to activate ERK/RSK phosphorylation (Fig. 8B, lanes 6 and 9). It should be noted that when cells were transfected with the ΔL56-142 mutant (Fig. 8B, lane 6), an unusual RSK degradation was reproducibly observed in our experiments. The ORF47/45-B protein with an internal deletion (from aa 56 to 114) of ORF47/45-A was competent to regulate both GRP78 upregulation and ERK/RSK phosphorylation (Fig. 8B, lane 3). Additionally, two mutants with C-terminal deletions, ΔTegu-I and ΔTegu-II, and a mutant with an internal deletion from aa 236 to 307, ΔTegu-IV, still retained the complete ability to mediate GRP78 upregulation and ERK/RSK phosphorylation (Fig. 8B, lanes 7, 8, and 10). Therefore, the region of ORF47/45-A critical for GRP78 upregulation is located in the N-terminal 38-aa region, whereas the region critical for ERK/RSK phosphorylation is located mainly from aa 114 to 216, which corresponds to the spliced junction region and the ORF45 portion.
FIG 8.

Different regions of ORF47/45-A critically mediate GRP78 upregulation and RSK/ERK phosphorylation. (A) Summary of effects of ORF47/45-A deletions on GRP78 upregulation and ERK/RSK activation. All expressed protein constructs contain a triple FLAG tag at their C-terminal ends. (B) 293T cells were transfected with the indicated plasmids. The levels of GRP78, ERK, phospho-ERK (p-ERK), RSK, and phospho-RSK (p-RSK) in these transfected cells were determined 24 h after transfection. (C and D) Effects of ORF47/45-A deletion mutants on activation of the grp78 promoter-driven reporter in 293T cells and in HH-B2 cells.
Activation of the grp78 promoter by different ORF47/45-A deletions was also examined by reporter assays in 293T cells (Fig. 8C). Consistent with the results of Western blot analysis, all ORF47/45-A deletions, except ΔL38, were sufficient to activate the grp78 promoter in 293T cells (Fig. 8C). However, when the reporter assays were performed in HH-B2 cells (Fig. 8D), we found that specific ORF47/45-A internal deletion mutants (including the ΔL56-142, ΔTegu-III, and ΔTegu-IV mutants) displayed a 30 to 52% reduction in the ability to regulate the grp78 promoter. The deletion regions in these mutants were mainly located within aa 56 to 307 of ORF47/45-A (Fig. 8D), suggesting that the mapped region in ORF47/45-A is required for full activation of GRP78 expression in HH-B2 cells.
The ORF47/45 proteins selectively induce UPR signaling regulators.
In addition to GRP78, we attempted to assess the effect of the ORF47/45 proteins or ORF47 on other UPR-related signaling regulators. Before investigating the action of the ORF47/45 proteins or ORF47 on the expression of UPR signaling regulators, the time course of GRP78 activation in 293T cells was initially tested (Fig. 9A). At 16, 24, and 48 h posttransfection, we found that ORF47/45-mediated GRP78 expression increased gradually over time, whereas ORF47-mediated GRP78 activation peaked at 24 h after transfection. The modulation of UPR signaling regulators by the ORF47/45 proteins or ORF47 was therefore assessed 24 h after transfection (Fig. 9B). Besides GRP78 expression, the levels of XBP-1s, PERK, phospho-PERK, eIF2α, phospho-eIF2α, ATF6, and cleaved ATF6 were examined in the study. As shown in Fig. 9B, we found that ORF47/45-A and ORF47/45-B sufficiently induce the expression of GRP78 and XBP-1s but not the phosphorylation of PERK and eIF2α, and we also found that they do not induce the cleavage of ATF6. These results implicate that the ORF47/45 proteins selectively activate the IRE1-XBP-1s signaling pathway but not the PERK and ATF6 signaling networks.
FIG 9.

Not all viral lytic proteins synthesized through the ER activate GRP78 expression and UPR signaling regulators. (A) Kinetic examination of GRP78 activation in response to the ORF47/45 proteins and ORF47. At 16, 24, and 48 h posttransfection, the levels of GRP78 induced by the indicated effector proteins were determined by Western blotting. (B) Activation of GRP78 and UPR signaling regulators by viral lytic proteins. The plasmids carrying ORF47/45-A, ORF47/45-B, ORF45, gL, gB, and gH were individually transfected into 293T cells. The levels of GRP78, spliced XBP-1 (XBP-1s), PERK, phospho-PERK (p-PERK), eIF2α, phospho-eIF2α (p-eIF2), ATF6, and cleaved ATF6 were examined at 24 h after transfection. All expressed viral lytic proteins contained a C-terminal triple FLAG tag. (C) Effect of gM, gN, and K8.1 on GRP78 expression in 293T cells. (D and E) Activation of the grp78 promoter by viral lytic proteins in 293T cells and in HH-B2 cells. The fold induction of the grp78 promoter-driven reporter by effector proteins was examined and calculated as described in the legend to Fig. 7B and C.
Not all viral lytic proteins synthesized through ER upregulate GRP78 expression.
In addition to the ORF47/45 proteins and ORF47 (gL), several viral lytic glycoproteins, including ORF8 (gB), ORF22 (gH), ORF39 (gM), ORF53 (gN), and ORF51 (K8.1), were chosen to evaluate their ability to activate GRP78 expression. Expression plasmids that carry gB, gH, gM, gN, and K8.1 were constructed and were individually transfected into 293T cells. Among these viral glycoproteins, gH and K8.1 upregulated GRP78 expression, as detected by Western blotting (Fig. 9B and C). However, the expression of gB, gM, or gN in 293T cells did not induce GRP78 expression. The activation of GRP78 expression by gH and K8.1 in 293T cells was confirmed by a luciferase reporter assay using the grp78 promoter-driven reporter (Fig. 9D). In particular, although gH and K8.1 significantly activated the grp78 promoter-driven reporter in 293T cells, these two glycoproteins, like ORF47 (gL), failed to induce the grp78 promoter in HH-B2 cells (Fig. 9E). Under the conditions of the assay, only the ORF47/45 proteins sufficiently activated the grp78 promoter in both 293T and HH-B2 cells (Fig. 9D and E). Collectively, there are two important conclusions from these results. First, not all viral glycoproteins that are synthesized through the ER induce GRP78 expression. Second, the ORF47/45 proteins may be key effectors in controlling GRP78 expression and ER homeostasis in KSHV-infected cells.
GRP78 upregulation occurs during the KSHV lytic cycle.
To determine whether GRP78 upregulation occurs during the KSHV lytic cycle, different PEL cell lines were treated with appropriate lytic inducers. As expected, viral lytic proteins, including immediate early (ORF50 and ORF45), early (K8), and late (K8.1) proteins, were expressed in an orderly fashion after viral reactivation in HH-B2, BCBL1, or BC3 cells (Fig. 10). For example, the ORF50, ORF45, and K8 proteins were predominantly expressed at 24 h in HH-B2 and BC3 cells (Fig. 10A and C) or at 48 h in BCBL1 cells (Fig. 10B) after lytic induction, whereas the optimal expression of K8.1 occurred at later time points, at about 48 to 72 h. Under the conditions of the assay, we found that GRP78 expression was markedly enhanced at the early stage of lytic replication and then sustained during the whole lytic cycle in these PEL cell lines (Fig. 10).
FIG 10.

GRP78 expression is upregulated during the KSHV lytic cycle. The expression of GRP78 and viral lytic proteins, including ORF50, K8, ORF45, and K8.1, was determined in HH-B2 (A), BCBL1 (B), and BC3 (C) cells after treatment with lysis-inducing agents.
To determine the potential roles of viral late gene products on GRP78 upregulation during the KSHV lytic cycle, different PEL cells were treated with lytic inducers in combination with PAA. PAA treatment, as expected, abolished the expression of late lytic proteins, such as K8.1, but had only a modest effect on the expression of immediate early and early lytic proteins, such as ORF50, ORF45, and K8, in HH-B2, BCBL1, and BC3 cells (Fig. 11). When the levels of GRP78 in these cells during lytic replication were examined, GRP78 upregulation in these PEL cell lines remained almost unchanged in the presence of PAA (Fig. 11). These results suggest that activation of GRP78 expression is mainly dependent on the viral lytic proteins expressed at the immediate early or early stage.
FIG 11.
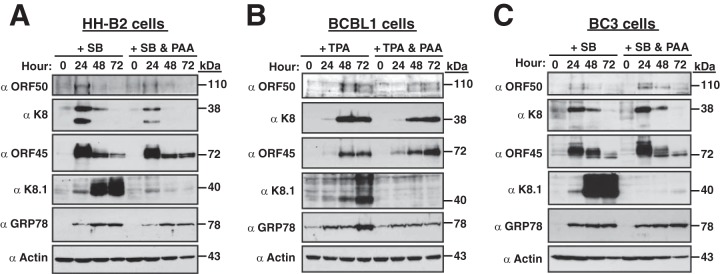
Blockade of viral late gene expression by PAA does not significantly affect sustained GRP78 upregulation during the KSHV lytic cycle. Different PEL cell lines, including HH-B2 (A), BCBL1 (B), and BC3 (C), were treated with a chemical inducing agent (SB or TPA) in the presence or absence of PAA (200 μg/ml). The expression of GRP78 and viral lytic proteins at different time points (0, 24, 48, and 72 h) after lytic induction was examined by Western blotting.
GRP78 upregulation is important for completion of the KSHV lytic cycle.
To determine the importance of GRP78 upregulation in the KSHV lytic cycle, lentiviral vector-based GRP78 knockdown was carried out in BCBL1, BC3, and 293T(BAC16) cells. Infection of lentivirus encoding grp78 shRNA in these cells resulted in more than 85% GRP78 knockdown compared with the level of GRP78 expression for the controls (Fig. 12A to C). When cell viability was examined in these cell lines, knockdown of GRP78 actually did not further affect the viability of BC3 and 293T(BAC16) cells during the lytic cycle (Fig. 12E and F). However, BCBL1 cells that received grp78 shRNA had significantly decreased viability at the late lytic stages (Fig. 12D). The expression of viral lytic proteins and the release of virus particles were also examined in these cell lines after GRP78 knockdown. As shown in Fig. 12G to I, knockdown of GRP78 markedly reduced the production of virions in these tested cell lines. Nevertheless, the effect of GRP78 knockdown on viral lytic gene expression varied in these cell lines (Fig. 12A to C). In BCBL1 cells, knockdown of GRP78 had only a modest effect on the expression of ORF50, K8, and ORF45. However, expression of the late gene product K8.1 was dramatically reduced in BCBL1 cells after GRP78 knockdown (Fig. 12A). In BC3 cells, the expression of viral lytic proteins, including ORF50, K8, ORF45, and K8.1, was only partially decreased after GRP78 knockdown (Fig. 12B). In 293T(BAC16) cells, knockdown of GRP78 expression actually did not alter the expression of viral lytic proteins, including ORF50, K8, ORF45, and K8.1 (Fig. 12C). A noteworthy finding was that 293T(BAC16) cells treated with grp78-specific shRNA led to an 82% reduction of viral particle release during lytic reactivation (Fig. 12I), suggesting that GRP78 may play critical roles in virion assembly or egress. Our results thus allow us to conclude that the control of GRP78 expression is critical for the completion of the KSHV lytic cycle.
FIG 12.

The controlled expression of GRP78 is important for the KSHV lytic cycle to go to completion. (A to C) Expression of viral lytic proteins in different PEL cells after GRP78 knockdown. The lentiviral vector-mediated GRP78 knockdown was performed in BCBL1, BC3, and 293T(BAC16) cells. The expression of ORF50, K8, ORF45, K8.1, and GRP78 at different time points after chemical treatment was determined by Western blotting. (D to F) Effects of GRP78 knockdown on the viability of BCBL1, BC3, and 293T(BAC16) cells during lytic cycle progression. Cell viability was analyzed by the XTT assay. (G to I) Effects of GRP78 knockdown on the release of virus particles from cultured BCBL1, BC3, and 293T(BAC16) cells. The KSHV loads in culture media on day 3 after TPA or SB treatment were evaluated using TaqMan quantitative PCR. siControl, control small interfering RNA; siGRP78, grp78-specific small interfering RNA.
DISCUSSION
In this study, we have identified two novel spliced genes located in the KSHV orf47-orf46-orf45 gene locus and have shown that these two spliced genes are expressed at the early stage of the viral lytic cycle (Fig. 1 and 3). The spliced gene products, ORF47/45-A and ORF47/45-B, are ER-localized proteins (Fig. 5) and can promote both RSK/ERK phosphorylation and GRP78 upregulation in cells (Fig. 6 and 7). The importance of RSK/ERK activation in the KSHV lytic cycle was previously well characterized by Kuang et al. (22). However, the possible association between GRP78 expression and the KSHV lytic cycle has remained elusive. As a result of our investigation, we showed that upregulation of GRP78 is observed in all tested KSHV-positive cell lines during viral lytic replication and knockdown of GRP78 markedly impairs the completion of the viral lytic cycle (Fig. 10 and 12). Due to the fact that the ORF47/45 proteins sufficiently activate GRP78 expression in either KSHV-negative or KSHV-positive cells, it is possible that the ORF47/45 proteins may be the key effectors responsible for GRP78 activation during viral lytic replication. To our knowledge, this is the first work to provide evidence showing the involvement of ER homeostatic regulation in the progression of the KSHV lytic cycle.
Structural and functional comparison of the ORF47/45 proteins, ORF45, and ORF47.
The two newly identified orf47/45-A and orf47/45-B transcripts, which consist of a part of the orf47 region and the full-length orf45 region, are believed to encode ORF47-ORF45 fusion proteins (ORF47/45-A and ORF47/45-B). According to their primary sequences, ORF47/45-A, ORF47/45-B, and ORF45 would encode 528, 470, and 407 amino acids, respectively, and would have calculated molecular masses of 56.7, 50.1, and 43.3 kDa, respectively. However, the translated ORF47/45-A, ORF47/45-B, and ORF45 proteins from their corresponding expression plasmids migrated to approximately similar positions (70 to 75 kDa) on SDS-polyacrylamide gels. At least four findings in our experiments support the suggestion that the translated the ORF47/45 proteins are indeed different from the well-characterized ORF45. First, the anti-ORF47/45-A antibody generated specifically recognizes the translated ORF47/45-A but not ORF45 (Fig. 3A). Second, point mutations at the potential ORF45 initiation codons in the ORF47-ORF46-ORF45 and ORF47/45 expression constructs, which block the possible translational initiation of ORF45 via leaky scanning, do not alter the expression of the translated ORF47/45 proteins (Fig. 4B and F). Third, the translated ORF47/45 proteins and ORF45 display different subcellular localizations in either 293T or A7 cells (Fig. 5). Lastly, the translated ORF47/45 proteins and ORF45 exhibit different abilities to activate GRP78 expression (Fig. 7). Since the translated ORF47/45 proteins and ORF45 have different subcellular localizations, we suggest that the mobility inconsistency between the ORF47/45 proteins and ORF45 is probably attributed to differences in posttranslational modifications or protein processing during their biosynthesis.
The confocal microscopic analyses revealed that the ORF47/45 proteins are perfectly colocalized with PDI, an ER-resident protein. Based on the presence of a conserved N-terminal signal peptide sequence in the ORF47/45 proteins and their colocalization with PDI, we have concluded that the ORF47/45 proteins are ER-resident proteins. For the localization of ORF45 in cells, we consistently observed that either FLAG-tagged or nontagged ORF45 was distributed in both the nucleus and the cytoplasm. Our results seem to conflict with previous reports (19, 20, 24, 36, 37) showing that ORF45 is exclusively localized in the cytoplasm of cells (including 293T cells). This discrepancy may be due to the use of different plasmid constructs or variations in the cells or culture conditions tested. The plasmid constructs used in our confocal immunofluorescence carried nontagged or FLAG-tagged ORF45 protein, whereas constructs carrying green fluorescent protein (GFP)-tagged ORF45 were exclusively applied in previous studies. Additionally, since the ORF45 protein contains both a nuclear localization signal (NLS) and a nuclear export signal (NES) (36), it is possible that the different cells or culture conditions tested may alter cell behavior and allow the nuclear localization of ORF45. Although the ORF47/45 proteins also contain an NLS, we did not detect the nuclear localization of the ORF47/45 proteins in cells. It may be because the nascent peptide with a conserved signal peptide sequence in the ORF47/45 proteins is sufficient to initiate ER translocation before the completion of protein synthesis. On the basis of the cotranslational translocation, the ER-targeting signal-mediated translocation would be dominant over the NLS-mediated translocation. For the subcellular localization of ORF47, Hahn et al. (38) have shown that ORF47 is a soluble ER luminal protein, whereas coexpression of gH, an ORF47-interacting partner, evidently shifts ORF47 from the ER toward the cell membrane. Although ORF47 is an ER luminal protein, we suspect that the ORF47/45 proteins may be ER transmembrane proteins. Due to the fact that the ORF47/45 proteins have the ability to activate RSK/ERK phosphorylation, the possibility that the ORF47/45 proteins may reach the ER membrane with a C-terminal ORF45 portion lying on the cytosolic side is raised. According to this hypothesis, the ORF47/45 proteins may complex with cellular cytoplasmic proteins (e.g., RSK and ERK) through their cytosolic portions.
Our studies have demonstrated that the ORF47/45 proteins confer at least two functions in cells, including activation of RSK/ERK phosphorylation and GRP78 expression. The critical region of the ORF47/45 proteins responsible for RSK/ERK activation is mapped to the region from aa 114 to 216 (corresponding to the ORF45 portion), whereas the critical region for GRP78 upregulation is localized to the N-terminal 38-aa region (corresponding to the ORF47 portion) (Fig. 8). It is not surprising that ORF45 activates only RSK/ERK phosphorylation and ORF47 induces only GRP78 expression in cells. Unexpectedly, although ORF47 activated the grp78 promoter in 293T cells, we noticed that ORF47 failed to induce the grp78 promoter in KSHV-positive HH-B2 cells (Fig. 7H, 8D, and 9E). Besides ORF47 (gL), we also found that other viral glycoproteins, such as gH and K8.1, also displayed cell type-specific activation of the grp78 promoter (Fig. 9D and E). The reasons for the cell type-specific activation of the grp78 promoter by these viral glycoproteins and the regulatory mechanism underlying the activation of GRP78 by the ORF47/45 proteins are under investigation.
Reciprocal association between modulation of ER homeostasis and progression of the KSHV lytic cycle.
Emerging evidence has suggested that several viruses appear to use different ways to modulate ER homeostasis to support their productive infection. For example, both glycoprotein B (gB) and ICP34.5 of herpes simplex virus 1 (HSV-1) selectively block activation of the PERK-mediated pathway in the ER (39). Inactivation of PERK-mediated signaling thus supports efficient viral protein synthesis and prevents cells from ER stress-mediated apoptosis. In contrast, productive infection with human cytomegalovirus (HCMV) differentially induces UPR signaling pathways and activates GRP78 expression (40). Studies on the role of the controlled expression of GRP78 in HCMV infection have suggested that GRP78 is important for virion assembly and maturation (41–43). With respect to KSHV, we show here that homeostatic regulation of the ER is also critical for the KSHV lytic cycle.
Three important results regarding the association between homeostatic regulation of the ER and the KSHV lytic cycle have been identified in the study. First, two early lytic proteins, ORF47/45-A and ORF47/45-B, promote a specific UPR signaling cascade (IRE1-XBP-1s signaling) and activate GRP78 expression in cells (Fig. 7 and 9B). Second, GRP78 expression is initially upregulated at the early stage of lytic replication and PAA treatment does not alter sustained GRP78 activation (Fig. 10 and 11). Third, knockdown of GRP78 dramatically impairs the KSHV lytic cycle from going to completion (Fig. 12). Although different PEL cell lines displayed variable responses to GRP78 knockdown during viral lytic replication, a marked reduction in the release of viral particles is observed as a common feature for different grp78-specific shRNA-treated PEL cells. However, the expression of viral immediate early and early lytic genes was not significantly affected by GRP78 knockdown in these PEL cell lines. When cell viability and viral late gene expression were analyzed in these PEL cells, we found that GRP78 knockdown did not apparently affect cell viability and late gene expression in either BC3 or 293T(BAC16) cells. However, BCBL1 cells treated with grp78-specific shRNA showed significantly decreased viability and the complete loss of viral late gene expression (Fig. 12A and D). Thus, controlled GRP78 expression may partly contribute to the prevention of cell death and benefit the biosynthesis of viral late genes in BCBL1 cells during the KSHV lytic cycle. Based on these findings, we propose that the activation of GRP78 or specific UPR signaling regulators is necessary for the progression of lytic replication (Fig. 13). In the proposed model, the ORF47/45 proteins or specific viral proteins expressed at the early lytic stage may act as effectors for controlling GRP78 expression. GRP78 activation is probably mediated through UPR-dependent or UPR-independent signaling pathways. The increased GRP78 expression during lytic replication may function to protect host cells from severe ER stress and may directly involve the assembly and release of virions (Fig. 13). We believe that understanding the reciprocal association between homeostatic regulation in the ER and KSHV lytic cycle progression not only is relevant to virus-host interactions but also may be helpful to provide a conceptual basis for blocking virion maturation.
FIG 13.

Proposed model for the relationship between the KSHV lytic cycle and ER homeostatic regulation. After KSHV reactivation, viral lytic proteins, including the ORF47/45 proteins expressed at the immediate early or early stage, may modulate specific UPR signaling pathways and GRP78 upregulation. The increased amount of GRP78 may aid with the protection of host cells from severe ER stress and support the progression of the KSHV lytic cycle.
ACKNOWLEDGMENTS
This work was supported by grant NSC102-2628-B-182-006-MY3 from the National Science Council of Taiwan (to P.-J.C.) and by medical research grants CMRPD6B0012 (to P.-J.C.), CMRPD6C0032 (to P.-J.C.), CMRPF6C0012 (to L.-W.C.), and CMRPG6C0291 (to S.-S.W.) from the Chang-Gung Memorial Hospital at Chaiyi, Taiwan.
We gratefully thank George Miller at Yale University for his support at the initial stage of the study. We also thank Fanxiu Zhu at Florida State University for providing the BAC16 bacmid and Chi-Ting Tsai in the Conjoint Laboratory of Chang-Gung Memorial Hospital at Chaiyi, Taiwan, for excellent technical assistance with confocal microscopy.
Footnotes
Published ahead of print 25 June 2014
REFERENCES
- 1.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191. 10.1056/NEJM199505043321802 [DOI] [PubMed] [Google Scholar]
- 2.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. 10.1126/science.7997879 [DOI] [PubMed] [Google Scholar]
- 3.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280 [PubMed] [Google Scholar]
- 4.Miller G, Heston L, Grogan E, Gradoville L, Rigsby M, Sun R, Shedd D, Kushnaryov VM, Grossberg S, Chang Y. 1997. Selective switch between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J. Virol. 71:314–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2:342–346. 10.1038/nm0396-342 [DOI] [PubMed] [Google Scholar]
- 6.Chang J, Renne R, Dittmer D, Ganem D. 2000. Inflammatory cytokines and the reactivation of Kaposi's sarcoma-associated herpesvirus lytic replication. Virology 266:17–25. 10.1006/viro.1999.0077 [DOI] [PubMed] [Google Scholar]
- 7.Davis DA, Rinderknecht AS, Zoeteweij JP, Aoki Y, Read-Connole EL, Tosato G, Blauvelt A, Yarchoan R. 2001. Hypoxia induces lytic replication of Kaposi sarcoma-associated herpesvirus. Blood 97:3244–3250. 10.1182/blood.V97.10.3244 [DOI] [PubMed] [Google Scholar]
- 8.Lu C, Zeng Y, Huang Z, Huang L, Qian C, Tang G, Qin D. 2005. Human herpesvirus 6 activates lytic cycle replication of Kaposi's sarcoma-associated herpesvirus. Am. J. Pathol. 166:173–183. 10.1016/S0002-9440(10)62242-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lukac DM, Kirshner JR, Ganem D. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73:9348–9361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ye F, Zhou F, Bedolla RG, Jones T, Lei X, Kang T, Guadalupe M, Gao SJ. 2011. Reactive oxygen species hydrogen peroxide mediates Kaposi's sarcoma-associated herpesvirus reactivation from latency. PLoS Pathog. 7:e1002054. 10.1371/journal.ppat.1002054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gradoville L, Gerlach J, Grogan E, Shedd D, Nikiforow S, Metroka C, Miller G. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol. 74:6207–6212. 10.1128/JVI.74.13.6207-6212.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun R, Lin SF, Staskus K, Gradoville L, Grogan E, Haase A, Miller G. 1999. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J. Virol. 73:2232–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dourmishev LA, Dourmishev AL, Palmeri D, Schwartz RA, Lukac DM. 2003. Molecular genetics of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) epidemiology and pathogenesis. Microbiol. Mol. Biol. Rev. 67:175–212. 10.1128/MMBR.67.2.175-212.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samali A, Fitzgerald U, Deegan S, Gupta S. 2010. Methods for monitoring endoplasmic reticulum stress and the unfolded protein response. Int. J. Cell Biol. 2010:830307. 10.1155/2010/830307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang G, Yang ZQ, Zhang K. 2010. Endoplasmic reticulum stress response in cancer: molecular mechanism and therapeutic potential. Am. J. Transl. Res. 2:65–74 [PMC free article] [PubMed] [Google Scholar]
- 16.Wang M, Wey S, Zhang Y, Ye R, Lee AS. 2009. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 11:2307–2316. 10.1089/ars.2009.2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu J, Kaufman RJ. 2006. From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 13:374–384. 10.1038/sj.cdd.4401840 [DOI] [PubMed] [Google Scholar]
- 18.Zhu FX, Cusano T, Yuan Y. 1999. Identification of the immediate-early transcripts of Kaposi's sarcoma-associated herpesvirus. J. Virol. 73:5556–5567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu FX, Yuan Y. 2003. The ORF45 protein of Kaposi's sarcoma-associated herpesvirus is associated with purified virions. J. Virol. 77:4221–4230. 10.1128/JVI.77.7.4221-4230.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu FX, King SM, Smith EJ, Levy DE, Yuan Y. 2002. A Kaposi's sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc. Natl. Acad. Sci. U. S. A. 99:5573–5578. 10.1073/pnas.082420599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuang E, Fu B, Liang Q, Myoung J, Zhu F. 2011. Phosphorylation of eukaryotic translation initiation factor 4B (EIF4B) by open reading frame 45/p90 ribosomal S6 kinase (ORF45/RSK) signaling axis facilitates protein translation during Kaposi sarcoma-associated herpesvirus (KSHV) lytic replication. J. Biol. Chem. 286:41171–41182. 10.1074/jbc.M111.280982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuang E, Tang Q, Maul GG, Zhu F. 2008. Activation of p90 ribosomal S6 kinase by ORF45 of Kaposi's sarcoma-associated herpesvirus and its role in viral lytic replication. J. Virol. 82:1838–1850. 10.1128/JVI.02119-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuang E, Wu F, Zhu F. 2009. Mechanism of sustained activation of ribosomal S6 kinase (RSK) and ERK by Kaposi sarcoma-associated herpesvirus ORF45: multiprotein complexes retain active phosphorylated ERK and RSK and protect them from dephosphorylation. J. Biol. Chem. 284:13958–13968. 10.1074/jbc.M900025200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sathish N, Zhu FX, Yuan Y. 2009. Kaposi's sarcoma-associated herpesvirus ORF45 interacts with kinesin-2 transporting viral capsid-tegument complexes along microtubules. PLoS Pathog. 5:e1000332. 10.1371/journal.ppat.1000332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu FX, Li X, Zhou F, Gao SJ, Yuan Y. 2006. Functional characterization of Kaposi's sarcoma-associated herpesvirus ORF45 by bacterial artificial chromosome-based mutagenesis. J. Virol. 80:12187–12196. 10.1128/JVI.01275-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U. S. A. 93:14862–14867. 10.1073/pnas.93.25.14862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang PJ, Boonsiri J, Wang SS, Chen LY, Miller G. 2010. Binding of RBP-Jkappa (CSL) protein to the promoter of the Kaposi's sarcoma-associated herpesvirus ORF47 (gL) gene is a critical but not sufficient determinant of transactivation by ORF50 protein. Virology 398:38–48. 10.1016/j.virol.2009.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang PJ, Wang SS, Chen LY, Hung CH, Huang HY, Shih YJ, Yen JB, Liou JY, Chen LW. 2013. ORF50-dependent and ORF50-independent activation of the ORF45 gene of Kaposi's sarcoma-associated herpesvirus. Virology 442:38–50. 10.1016/j.virol.2013.03.023 [DOI] [PubMed] [Google Scholar]
- 29.Wang SS, Chang PJ, Chen LW, Chen LY, Hung CH, Liou JY, Yen JB. 2012. Positive and negative regulation in the promoter of the ORF46 gene of Kaposi's sarcoma-associated herpesvirus. Virus Res. 165:157–169. 10.1016/j.virusres.2012.02.010 [DOI] [PubMed] [Google Scholar]
- 30.Arvanitakis L, Mesri EA, Nador RG, Said JW, Asch AS, Knowles DM, Cesarman E. 1996. Establishment and characterization of a primary effusion (body cavity-based) lymphoma cell line (BC-3) harboring Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in the absence of Epstein-Barr virus. Blood 88:2648–2654 [PubMed] [Google Scholar]
- 31.Boshoff C, Gao SJ, Healy LE, Matthews S, Thomas AJ, Coignet L, Warnke RA, Strauchen JA, Matutes E, Kamel OW, Moore PS, Weiss RA, Chang Y. 1998. Establishing a KSHV+ cell line (BCP-1) from peripheral blood and characterizing its growth in Nod/SCID mice. Blood 91:1671–1679 [PubMed] [Google Scholar]
- 32.Graham FL, Smiley J, Russell WC, Nairn R. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36:59–74. 10.1099/0022-1317-36-1-59 [DOI] [PubMed] [Google Scholar]
- 33.Chao CC, Lin-Chao S. 1992. A direct-repeat sequence of the human BiP gene is required for A23187-mediated inducibility and an inducible nuclear factor binding. Nucleic Acids Res. 20:6481–6485. 10.1093/nar/20.24.6481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang SS, Chen LW, Chen LY, Tsai HH, Shih YC, Yang CT, Chang PJ. 2010. Transcriptional regulation of the ORF61 and ORF60 genes of Kaposi's sarcoma-associated herpesvirus. Virology 397:311–321. 10.1016/j.virol.2009.11.031 [DOI] [PubMed] [Google Scholar]
- 35.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8:785–786. 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- 36.Li X, Zhu F. 2009. Identification of the nuclear export and adjacent nuclear localization signals for ORF45 of Kaposi's sarcoma-associated herpesvirus. J. Virol. 83:2531–2539. 10.1128/JVI.02209-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abada R, Dreyfuss-Grossman T, Herman-Bachinsky Y, Geva H, Masa SR, Sarid R. 2008. SIAH-1 interacts with Kaposi's sarcoma-associated herpesvirus-encoded ORF45 protein and promotes its ubiquitylation and proteasomal degradation. J. Virol. 82:2230–2240. 10.1128/JVI.02285-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hahn A, Birkmann A, Wies E, Dorer D, Mahr K, Sturzl M, Titgemeyer F, Neipel F. 2009. Kaposi's sarcoma-associated herpesvirus gH/gL: glycoprotein export and interaction with cellular receptors. J. Virol. 83:396–407. 10.1128/JVI.01170-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mulvey M, Arias C, Mohr I. 2007. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J. Virol. 81:3377–3390. 10.1128/JVI.02191-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Isler JA, Skalet AH, Alwine JC. 2005. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 79:6890–6899. 10.1128/JVI.79.11.6890-6899.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buchkovich NJ, Maguire TG, Paton AW, Paton JC, Alwine JC. 2009. The endoplasmic reticulum chaperone BiP/GRP78 is important in the structure and function of the human cytomegalovirus assembly compartment. J. Virol. 83:11421–11428. 10.1128/JVI.00762-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buchkovich NJ, Maguire TG, Yu Y, Paton AW, Paton JC, Alwine JC. 2008. Human cytomegalovirus specifically controls the levels of the endoplasmic reticulum chaperone BiP/GRP78, which is required for virion assembly. J. Virol. 82:31–39. 10.1128/JVI.01881-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi-Chen Ou D, Lee SB, Chu CS, Chang LH, Chung BC, Juan LJ. 2011. Transcriptional activation of endoplasmic reticulum chaperone GRP78 by HCMV IE1-72 protein. Cell Res. 21:642–653. 10.1038/cr.2011.10 [DOI] [PMC free article] [PubMed] [Google Scholar]