

ABSTRACT
Although recombination is a major source of genetic variability in retroviruses, no recombinant strain had been observed for human T-lymphotropic virus type 1 (HTLV-1), the first isolated human-pathogenic retrovirus. Different genotypes exist for HTLV-1: Genotypes b and d to g are restricted to central Africa, while genotype c is only endemic in Australo-Melanesia. In contrast, the cosmopolitan genotype a is widely distributed. We applied a combination of phylogenetics and recombination analysis approaches to a set of new HTLV-1 sequences, which we collected from 19 countries throughout Africa, the continent where the virus has the largest endemic presence. This led us to demonstrate the presence of recombinants in HTLV-1. Indeed, the HTLV-1 strains currently present in North Africa have originated from a recombinant event between strains from Senegal and West Africa. This recombination is estimated to have occurred around 4,000 years ago. This recombination seems to have been generated during reverse transcription. In conclusion, we demonstrate that, albeit rare, recombination can occur in HTLV-1 and may play a role in the evolution of this retrovirus.
IMPORTANCE A number of HTLV-1 subtypes have been described in different populations, but none of the genetic differences between these subtypes have been ascribed to recombination events. Here we report an HTLV-1 recombinant virus among infected individuals in North Africa. This demonstrates that, contrary to what was thought, recombination can occur and could play a role in the evolution of HTLV-1.
INTRODUCTION
Genetic recombination is a major characteristic of the human retrovirus HIV-1 and plays a crucial role in evolution and pathogenicity (1, 2). In contrast, no recombination event has been described, so far, for human T-lymphotropic virus type 1 (HTLV-1), the first described human-pathogenic retrovirus (3). This fitted well with the remarkable genetic stability of HTLV-1, probably linked to viral amplification via clonal expansion of infected cells (4). The absence of HTLV-1 recombination was also supported by the fact that no superinfection at the cellular level had been described (5).
HTLV-1 infects at least 5 to 10 million people worldwide, mainly in geographical foci of high endemicity (6). HTLV-1 is the etiologic agent of a malignant CD4 lymphoproliferation (adult T-cell leukemia [ATL]) (7) and of a chronic progressive neuromyelopathy (tropical spastic paraparesis/HTLV-1 associated myelopathy [TSP/HAM]) (8). Among the infected individuals, 3 to 7% will develop such severe diseases. Different HTLV-1 genotypes exist, some of which are geographically restricted: Genotypes b and d to g are restricted to central Africa, while genotype c is endemic in Australo-Melanesia. In contrast, the cosmopolitan genotype a is widely distributed, supposedly dispersed during the past centuries through migration of infected populations, as for instance during the Atlantic slave trade (6, 9–11). Within this large cosmopolitan a subtype, several molecular clades have been described (e.g., Japanese and West African). Here we applied a combination of phylogenetics and recombination analysis approaches to a set of new HTLV-1 sequences, which we collected from 19 countries throughout Africa, the continent where the virus has the largest endemicity. We demonstrate that recombination has occurred in natura in HTLV-1 and that strains currently present in North Africa originated from a recombination event.
MATERIALS AND METHODS
Study population and ethics statement.
Samples were obtained from 41 HTLV-1-infected individuals (of different clinical statuses: ATL, TSP/HAM, and asymptomatic carriers) originating from 19 African countries (Table 1). All blood samples were obtained according to French laws and regulations (Articles L.1211-2 and L.1243-3 from Code de la Santé Publique). The human sample collection has been declared to the Ministère de l'Enseignement Supérieur et de la Recherche (2010 DC-1197).
TABLE 1.
Specimen identification, geographic anddemographic data, and clinical status of HTLV-1-infected individuals from northern,western, central, and austral African countriesa
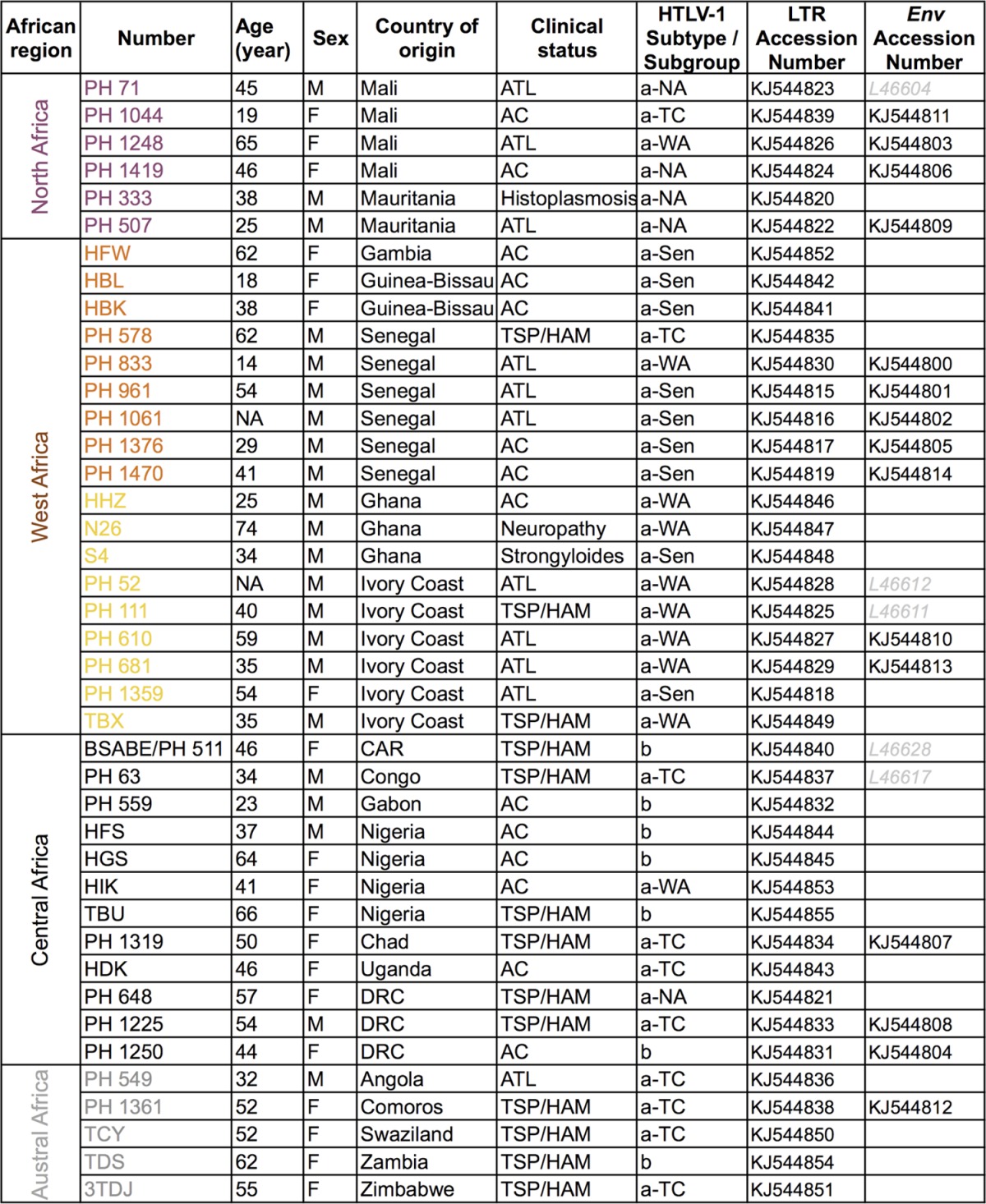
Provided are the HTLV-1 subtype and subgroup confirmed by phylogenetic analysis and new LTR and env African sequences characterized in this study. Abbreviations: AC, asymptomatic carrier; ATL, adult T-cell leukemia; TSP/HAM, tropical spastic paraparesis/HTLV-1-associated myelopathy; a-NA, a-WA, a-Sen, and a-TC, North African, West African, Senegalese, and transcontinental clades of the a subtype, respectively.
Viral sequencing.
For the purpose of this study, high-molecular-weight DNA was extracted from peripheral blood buffy coat using the QIAamp DNA blood minikit (Qiagen, Hilden, Germany). The samples were first subjected to PCR using human β-globin-specific primers to ensure that DNA was amplifiable.
For long terminal repeat (LTR) sequencing (774 bp long for the HHZ sequence), we followed the protocol previously described (12). Briefly, complete LTR sequences were obtained through 2 PCRs, generating LTR-gag (with primers Enh280 and R2380) and Tax-LTR (with primers F6501 and 3VLTRext) segments. The former was sequenced using the Enh280/R1021 pair of primers and the latter using the 8200LA/Rev3 pair of primers. The forward and reverse sequences were aligned using the ClustalW algorithm (MacVector 6.5 software; Oxford Molecular) and were found identical. Then the 2 fragments were concatenated. There was a 248-bp-long overlapping region. In this region, the sequences were identical. Of note, the recombination site is found in this region, and 4 out of 8 informative mutations were also present. This rules out the possibility of the recombination being an artifact consecutive to our sequencing strategy.
Concerning the env segment (522-bp-long fragment), we used the previously described protocol (13). A seminested PCR was performed: a first amplification with the env1/env22 pair of primers was followed by amplification with the env1/env2 pair.
Phylogenetic analyses.
Multiple sequence alignments were performed with the DAMBE program (v4.2.13) (14). For the studies on the env segment, no gaps or stop codons were observed. Absence of saturation of the alignment was confirmed by the test of Xia and Xie in the DAMBE program.
The most appropriate nucleotide substitution model was selected in the Modeltest v3.6 program (15), based on the Akaike information criterion (AIC). The best-fitting models were GTR-Γ and Tamura-Nei-Γ for the LTR and env sequences, respectively. Phylogenetic reconstructions were conducted in PAUP* v4.0b10 using the neighbor-joining method with 1,000 bootstrap replicates performed to test the robustness of the tree topology. Phylogenetic topologies were also confirmed using the maximum likelihood method (on the PAUP program).
Recombinant search.
The recombinant search and breakpoint detection were performed by boot scanning in Simplot v3.5.1 (16). This program compares inferred clusters of sequences to each other. Phylogenetic relationships of these clusters are estimated for successive overlapping subregions (window, 180 bp; step, 20 bp). For each subregion, the bootstrap value of the query and the references are calculated (according to the Kimura two-parameter model with 1,000 replicates). Bootstrap values are then plotted along the genome on an x/y plot, so that x values reflect the genome position at the midpoint of the analyzed windows and y values reflect the bootstrap value calculated from the windows. The recombination breakpoint determined is then verified by phylogenetic analyses of the 2 fragments.
Quartet mapping was performed on the 2 LTR segments of interest (17). Quartet mapping consists of a likelihood mapping in which 4 groups of interest are initially inferred. The methods evaluated the maximum likelihood values for each of three possible trees when considering a quartet consisting of a sequence from each defined group. The result is plotted inside an equilateral triangle, where each corner represents a specific tree topology. The center represents sets where trees are poorly supported, named the “starlike area.” From the biological standpoint, a likelihood mapping showing more than 20% of dots in the starlike area suggests that the data are noisy. Quartet mapping was performed with the Tree-Puzzle v5.2 program.
Molecular clock.
Molecular clock analysis was performed on the HTLV-1 LTR alignment using 2 external times (divergence of the subtype c Autralo-Melanesian strains and radiation of the HTLV-1a subtype, as defined previously [18]). The evolutionary rate was estimated by using a Bayesian Markov chain Monte Carlo (MCMC) molecular clock method using BEAST v1.8.0 (19). A strict molecular clock hypothesis was inferred. The GTR-Γ substitution model with 8 gamma categories was used. The MCMC chain was run for 20 million steps and sampled every 10,000 steps. Convergence and effective sampling sizes (ESS) of the MCMC chain were examined in Tracer v1.6 (http://beast.bio.ed.ac.uk). All parameter estimates showed significant ESSs (>150).
RESULTS
Complete LTR sequences were obtained from 41 HTLV-1-infected individuals originating from various African regions: 6 from North Africa, 18 from West Africa, 12 from central Africa, and 5 from austral Africa (Table 1). Phylogenetic analyses were performed on the 774-bp-long LTR fragment. The topologies of the phylogenetic tree were comparable for the neighbor-joining (Fig. 1) and maximum likelihood methods (data not shown).
FIG 1.

Phylogenetic analysis of African LTR sequences. Phylogenetic comparison was performed on 774-nucleotide-long LTR fragments of 97 HTLV-1 African isolates, including the 41 sequences generated in this study (in boldface) and the 56 previously published sequences (in italics). The Melanesian sequence Mel5 was used as the outgroup. The sequence name was colored according to the localization origin of the individuals, as presented in Table 1. Purple, orange, and yellow sequences are from North Africa, the Senegalese area, and West Africa, respectively. The phylogeny was derived by the neighbor-joining method using the GTR model (gamma = 0.5325). Horizontal branch lengths are drawn to scale, with the bar indicating 0.01 nucleotide replacement per site. Numbers on each node indicate the percentage of bootstrap samples (of 1,000) in which the cluster to the right is supported.
The main HTLV-1 subtypes (a, b, and d) were identifiable (Fig. 1) (20–22). The sequences belonged to either the central African b subtype, with strains isolated in Nigeria, Democratic Republic of Congo (former Zaïre), Zambia, Central African Republic, and Gabon, or the cosmopolitan a subtype. Within the latter subtype, one large transcontinental clade (a-TC) and three geographically restricted clades were identified: we named these the Senegalese (a-Sen), North African (a-NA), and West African (a-WA) clades. Of note, the a-Sen group has previously been described and named “trans-Saharan” or “clade N” (within the subgroup HTLV-1a D) (23, 24).
Interestingly, unlike the other groups, the a-NA clade was not supported by a strong bootstrap value and had an intermediate position between the a-WA and a-Sen groups. These characteristics were reminiscent of a clade emerging from a recombination event. To test this hypothesis, we studied the phylogenetic relationship of the different groups through bootstrap resampling by the boot-scanning method (Fig. 2A) (16). The LTR sequences of the a-NA group could be decomposed into 3 distinct regions. (i) The 5′ region (positions 1 to 375), which corresponds to the U3 region of the LTR, is closely related to the a-Sen strains, (ii) the central region (positions 375 to 526) corresponds to a portion of the R region of the LTR and is conserved among strains of the different groups, and (iii) the 3′ region (positions 526 to 774), corresponding to an R-U5 region of the LTR, segregates with the a-WA strains. Thus, we concluded that the a-NA strains might have resulted from a recombination event between strains from the a-Sen and a-WA groups that occurred within the R region.
FIG 2.

a-NA strains derive from a recombination event. (A) The a-NA subgroup was compared by boot-scanning (Simplot program) to different clades (a-TC, a-WA, a-Sen, b, d, e, and c). The analysis used a 180-bp-long window and a 20 bp-long step. The x values reflect the genome position at the midpoint of the analyzed windows, and the y values reflect the bootstrap value calculated from the windows. (B and C) Phylogenetic trees corresponding to the first 375 nucleotides and the 399 last nucleotides, respectively, were derived from the neighbor-joining method (GTR; gamma = 0.5325). The groups of interest are colored as follows: red, green, and blue sequences belong to a-Sen, a-NA, and a-WA, respectively. In both trees, 4 informative sites determine the topology between the 3 clades of interest. The quartet mapping of each sequence fragment is presented.
When focusing on the nucleotide sequences, we observed that nucleotides at positions 153, 182, 263, and 319 are conserved between a-NA and a-Sen strains (but not a-WA strains), while nucleotides at positions 377, 557, 624, and 631 are identical between a-NA and a-WA strains (but not a-Sen strains). This suggests that the recombination event has occurred at the U3-R junction, around nucleotide position 375. Separate analysis of the U3 segment (positions 1 to 375) and the R-U5 segment (positions 376 to 774) of the LTR confirmed that the a-NA sequences are more closely related to a-Sen on the U3 region and close to a-WA on the RU5 segment (Fig. 2B and C). Topologies of the phylogenetic trees were consistent between neighbor-joining (Fig. 2B and C) and maximum likelihood (data not shown) studies.
As the viral diversity is low (a characteristic of HTLV-1), the groups defined in these analyses are not supported by strong bootstraps. Therefore, to strengthen our observation, quartet mapping was performed on the 2 LTR fragments (17). We considered 4 phylogenetic groups (the a-Sen, a-NA, and a-WA subgroups and the b subtype). For a given quartet (i.e., a group of 4 sequences, each originating from a different phylogenetic group), 3 topologies are possible: the a-NA strain is closely related to the a-Sen, a-WA, or b strain. The probabilities of each topology are reported as barycentric coordinates in a triangle map. Therefore, if the quartet is undefined (i.e., the quartet has a perfect starlike shape), the sequence is reported in the center of the triangle. In contrast, if the a-NA strain is closely related to the a-Sen strain, the sequence is reported close to the angle corresponding to the a-Sen group. The analyses confirm that indeed a-NA strains are related to the a-Sen and a-WA groups on the U3 and RU5 fragments, respectively. Together these data confirm that a-NA strains are recombinants of a-Sen and a-WA sequences.
This recombination site at the U3-R junction suggests that the recombination event could have occurred during reverse transcription (RT). Indeed, retrovirus particles incorporate 2 strains of genomic RNA, and during RT, the newly synthesized R-U5 single-strand DNA is transferred to the 5′ end of genomic RNA (25). If both genomic RNAs were from different HTLV-1 genotypes (considering that a single cell can be infected with 2 different HTLV-1 strains), such a jump occurring between viral genomes would result in a virus with a recombined LTR. Based on this model, we predicted that the env sequence from a-NA strains should be related to the env sequence from the a-Sen strains. Phylogenetic analysis of env sequences confirmed that while a-WA strains form a separate clade, the a-NA and a-Sen strains are virtually undistinguishable (Fig. 3).
FIG 3.
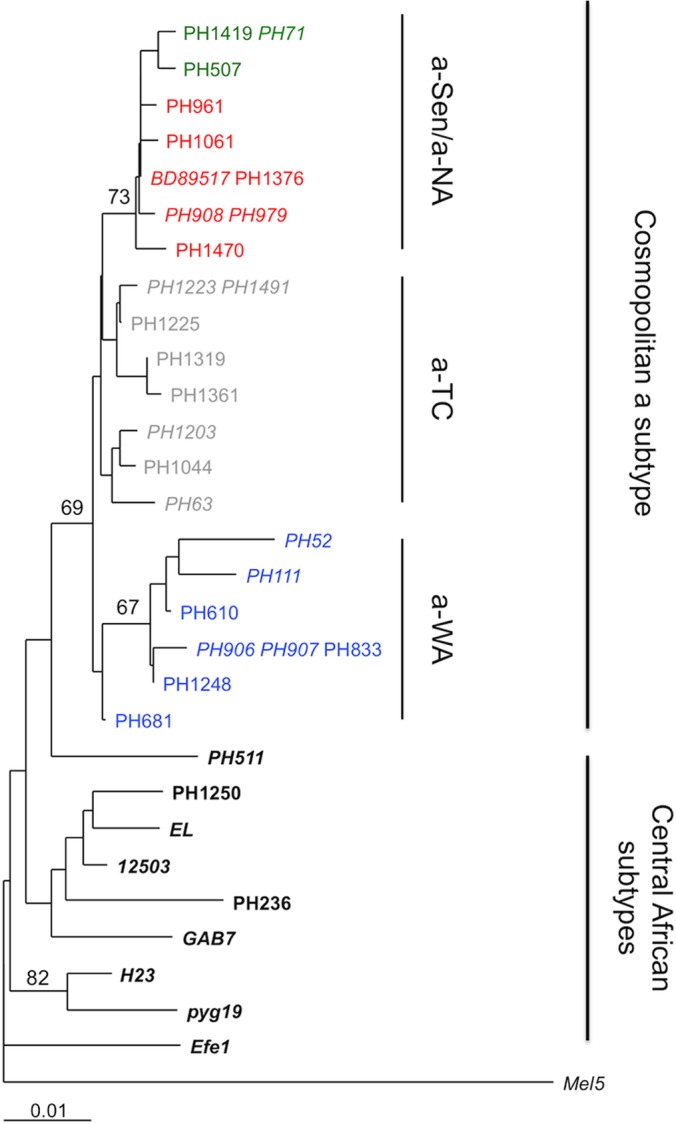
Phylogenetic analysis of African env sequences. Phylogenetic comparison was performed on 522-nucleotide-long env (gp21) fragments of 35 HTLV-1 African isolates. (The previously published sequences are in italics.) The Melanesian sequence Mel5 was used as outgroup. The phylogeny was derived by the neighbor-joining method using the Tamura-Nei model (gamma = 0.1382). Horizontal branch lengths are drawn to scale, with the bar indicating 0.01 nucleotide replacement per site. Numbers on each node indicate the percentage of bootstrap samples (of 1,000 replicated) in which the cluster to the right is supported.
Finally molecular clock analyses were performed in order to date the recombination event; we hypothesized that a single event is at the origin of the a-NA group. Strict molecular clock analyses were performed using 2 different calibration dates: the divergence of the Australo-Melanesian strain at 50 ± 10 kiloyears and the subtype Ia radiation date (12,700 ± 2,000 years) (18). From the two hypotheses, the origins of the a-NA group were estimated to be 4,650 years before present (BP) (95% highest posterior density [HPD], 2,850 to 6,450; ESS, 255) and 3,740 years BP (95% HPD, 2,610 to 4,870; ESS, 283), respectively.
DISCUSSION
Here we demonstrate that albeit rare, recombination in HTLV-1 can occur and may play a role in the evolution of this virus, as for the other retroviruses (26). Indeed, the strains we sampled from North Africa are a result from a recombination between HTLV-1 strains currently found in Senegal and West Africa, which is estimated to having occurred around 4,000 years ago. This recombination seems to have been generated during reverse transcription.
The existence of this recombinant clade suggests that, in vivo, some cells can be infected by multiple HTLV-1 strains. Indeed, superinfection is a required condition for recombination to occur—a cell has to be infected with two viruses to generate a “hybrid” viral particle that would generate the recombinant virus we describe. The possibility of multiple HTLV-1 infections at the cellular level has been a matter of debate. Lymphocytes from patients with adult T-cell leukemia with multiple integrated viral genomes have been reported. However, it was unclear whether this was the consequence of multiple infections or HTLV-1 secondary amplification by retrotransposition (27, 28). While HTLV-1 receptors are still expressed at the cell surface of infected lymphocytes (29, 30), a recent report suggested that naturally infected T cells contain a single integrated HTLV-1 provirus (5). One can therefore speculate that the target cell for multiple infections might be another cell type (dendritic cells are sensitive to HTLV-1 infection [31]) or that recombination could occur upon formation of syncytia in vivo.
Considering that the a-NA clade was derived from a unique recombination event, we estimated that this recombinant might have originated from the intermixing of African populations infected with strains related to the ones currently present in West Africa and the Senegalese area: this event occurred around 4,000 years ago. The origins of such a recombination are, however, still unclear. Based on the current data, the recombination is unlikely to have occurred in a simian host. Indeed, no simian T-lymphotropic virus type 1 (STLV-1) subtype a strain has been described so far. However, STLV-1 genetic diversity in the Sahel and West Africa needs to be documented. Considering that recombination might have occurred during human intermixing, the place of recombination remains unknown. Either recombination occurred in West Africa (or the Senegal area) and was followed by migration of populations infected with the recombined virus, or populations infected with a-Sen and a-WA viral strains mixed in North Africa prior to recombination. As no a-NA strains were found in regions other than North Africa, it is more likely that the recombination occurred in North Africa, after migrations of infected individuals from West Africa and Senegal.
ACKNOWLEDGMENTS
We thank Peter Markov for discussions of this study and critical reading of the manuscript.
This work received funding from the French Government's Investissement d'Avenir Program, Laboratoire d'Excellence Integrative Biology of Emerging Infectious Diseases (grant no. ANR-10-LABX-62-IBEID).
A.G. directed the study. A.G., O.C., and P.V.A. designed the experiments. A.D. and O.C. performed the experiments. A.D., O.C., P.V.A., and A.G. analyzed and interpreted the data. O.G., O.H., and G.P.T. provided essential samples. G.P.T. edited the manuscript. A.D., O.C., P.V.A., and A.G. wrote the manuscript.
Footnotes
Published ahead of print 18 June 2014
REFERENCES
- 1.Negroni M, Buc H. 2001. Mechanisms of retroviral recombination. Annu. Rev. Genet. 35:275–302. 10.1146/annurev.genet.35.102401.090551 [DOI] [PubMed] [Google Scholar]
- 2.Schlub TE, Grimm AJ, Smyth RP, Cromer D, Chopra A, Mallal S, Venturi V, Waugh C, Mak J, Davenport MP. 2014. Fifteen to twenty percent of HIV substitution mutations are associated with recombination. J. Virol. 88:3837–3849. 10.1128/JVI.03136-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. 1980. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. U. S. A. 77:7415–7419. 10.1073/pnas.77.12.7415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wattel E, Vartanian JP, Pannetier C, Wain-Hobson S. 1995. Clonal expansion of human T-cell leukemia virus type I-infected cells in asymptomatic and symptomatic carriers without malignancy. J. Virol. 69:2863–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cook LB, Rowan AG, Melamed A, Taylor GP, Bangham CR. 2012. HTLV-1-infected T cells contain a single integrated provirus in natural infection. Blood 120:3488–3490. 10.1182/blood-2012-07-445593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gessain A, Cassar O. 2012. Epidemiological aspects and world distribution of HTLV-1 infection. Front. Microbiol. 3:388. 10.3389/fmicb.2012.00388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshida M, Miyoshi I, Hinuma Y. 1982. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc. Natl. Acad. Sci. U. S. A. 79:2031–2035. 10.1073/pnas.79.6.2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gessain A, Barin F, Vernant JC, Gout O, Maurs L, Calender A, de The G. 1985. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet ii:407–410 [DOI] [PubMed] [Google Scholar]
- 9.Gessain A, Gallo RC, Franchini G. 1992. Low degree of human T-cell leukemia/lymphoma virus type I genetic drift in vivo as a means of monitoring viral transmission and movement of ancient human populations. J. Virol. 66:2288–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alcantara LC, de Oliveira T, Gordon M, Pybus O, Mascarenhas RE, Seixas MO, Goncalves M, Hlela C, Cassol S, Galvao-Castro B. 2006. Tracing the origin of Brazilian HTLV-1 as determined by analysis of host and viral genes. AIDS 20:780–782. 10.1097/01.aids.0000216383.14808.13 [DOI] [PubMed] [Google Scholar]
- 11.Mota AC, Van Dooren S, Fernandes FM, Pereira SA, Queiroz AT, Gallazzi VO, Vandamme AM, Galvao-Castro B, Alcantara LC. 2007. The close relationship between South African and Latin American HTLV type 1 strains corroborated in a molecular epidemiological study of the HTLV type 1 isolates from a blood donor cohort. AIDS Res. Hum. Retroviruses 23:503–507. 10.1089/aid.2006.0203 [DOI] [PubMed] [Google Scholar]
- 12.Cassar O, Einsiedel L, Afonso PV, Gessain A. 2013. Human T-cell lymphotropic virus type 1 subtype C molecular variants among indigenous Australians: new insights into the molecular epidemiology of HTLV-1 in Australo-Melanesia. PLoS Negl. Trop. Dis. 7:e2418. 10.1371/journal.pntd.0002418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meertens L, Rigoulet J, Mauclere P, Van Beveren M, Chen GM, Diop O, Dubreuil G, Georges-Goubot MC, Berthier JL, Lewis J, Gessain A. 2001. Molecular and phylogenetic analyses of 16 novel simian T cell leukemia virus type 1 from Africa: close relationship of STLV-1 from Allenopithecus nigroviridis to HTLV-1 subtype B strains. Virology 287:275–285. 10.1006/viro.2001.1018 [DOI] [PubMed] [Google Scholar]
- 14.Xia X. 2013. DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 30:1720–1728. 10.1093/molbev/mst064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Posada D, Crandall KA. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818. 10.1093/bioinformatics/14.9.817 [DOI] [PubMed] [Google Scholar]
- 16.Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73:152–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nieselt-Struwe K, von Haeseler A. 2001. Quartet-mapping, a generalization of the likelihood-mapping procedure. Mol. Biol. Evol. 18:1204–1219. 10.1093/oxfordjournals.molbev.a003907 [DOI] [PubMed] [Google Scholar]
- 18.Salemi M, Desmyter J, Vandamme AM. 2000. Tempo and mode of human and simian T-lymphotropic virus (HTLV/STLV) evolution revealed by analyses of full-genome sequences. Mol. Biol. Evol. 17:374–386. 10.1093/oxfordjournals.molbev.a026317 [DOI] [PubMed] [Google Scholar]
- 19.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. 10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahieux R, Ibrahim F, Mauclere P, Herve V, Michel P, Tekaia F, Chappey C, Garin B, Van Der Ryst E, Guillemain B, Ledru E, Delaporte E, de The G, Gessain A. 1997. Molecular epidemiology of 58 new African human T-cell leukemia virus type 1 (HTLV-1) strains: identification of a new and distinct HTLV-1 molecular subtype in Central Africa and in pygmies. J. Virol. 71:1317–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salemi M, Van Dooren S, Audenaert E, Delaporte E, Goubau P, Desmyter J, Vandamme AM. 1998. Two new human T-lymphotropic virus type I phylogenetic subtypes in seroindeterminates, a Mbuti pygmy and a Gabonese, have closest relatives among African STLV-I strains. Virology 246:277–287. 10.1006/viro.1998.9215 [DOI] [PubMed] [Google Scholar]
- 22.Slattery JP, Franchini G, Gessain A. 1999. Genomic evolution, patterns of global dissemination, and interspecies transmission of human and simian T-cell leukemia/lymphotropic viruses. Genome Res. 9:525–540 [PubMed] [Google Scholar]
- 23.van Tienen C, de Silva TI, Alcantara LC, Onyango CO, Jarju S, Goncalves N, Vincent T, Aaby P, Whittle H, Schim van der Loeff M, Cotten M. 2012. Molecular epidemiology of endemic human T-lymphotropic virus type 1 in a rural community in Guinea-Bissau. PLoS Negl. Trop. Dis. 6:e1690. 10.1371/journal.pntd.0001690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zehender G, Ebranati E, De Maddalena C, Gianelli E, Riva A, Rusconi S, Massetto B, Rankin F, Acurie M, Galli M. 2008. Description of a “trans-Saharan” strain of human T-lymphotropic virus type 1 in West Africa. J. Acquir. Immune Defic. Syndr. 47:269–273. 10.1097/QAI.0b013e31816649a4 [DOI] [PubMed] [Google Scholar]
- 25.Hu WS, Hughes SH. 2012. HIV-1 reverse transcription. Cold Spring Harb. Perspect. Med. 2:a006882. 10.1101/cshperspect.a006882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robertson DL, Sharp PM, McCutchan FE, Hahn BH. 1995. Recombination in HIV-1. Nature 374:124–126. 10.1038/374124b0 [DOI] [PubMed] [Google Scholar]
- 27.Cook LB, Melamed A, Niederer H, Valganon M, Laydon D, Foroni L, Taylor GP, Matsuoka M, Bangham CR. 15 April 2014. The role of HTLV-1 clonality, proviral structure and genomic integration site in adult T cell leukemia/lymphoma. Blood. 10.1182/blood-2014-02-553602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bangham CR, Cook LB, Melamed A. 2013. HTLV-1 clonality in adult T-cell leukaemia and non-malignant HTLV-1 infection. Semin. Cancer Biol. 26C:89–98. 10.1016/j.semcancer.2013.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jain P, Manuel SL, Khan ZK, Ahuja J, Quann K, Wigdahl B. 2009. DC-SIGN mediates cell-free infection and transmission of human T-cell lymphotropic virus type 1 by dendritic cells. J. Virol. 83:10908–10921. 10.1128/JVI.01054-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghez D, Lepelletier Y, Lambert S, Fourneau JM, Blot V, Janvier S, Arnulf B, van Endert PM, Heveker N, Pique C, Hermine O. 2006. Neuropilin-1 is involved in human T-cell lymphotropic virus type 1 entry. J. Virol. 80:6844–6854. 10.1128/JVI.02719-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones KS, Petrow-Sadowski C, Huang YK, Bertolette DC, Ruscetti FW. 2008. Cell-free HTLV-1 infects dendritic cells leading to transmission and transformation of CD4(+) T cells. Nat. Med. 14:429–436. 10.1038/nm1745 [DOI] [PubMed] [Google Scholar]