

ABSTRACT
Human APOBEC3 (A3) restriction factors provide intrinsic immunity against zoonotic transmission of pathogenic viruses. A3D, A3F, A3G, and A3H haplotype II (A3H-hapII) can be packaged into virion infectivity factor (Vif)-deficient HIVs to inhibit viral replication. To overcome these restriction factors, Vif binds to the A3 proteins in viral producer cells to target them for ubiquitination and proteasomal degradation, thus preventing their packaging into assembling virions. Therefore, the Vif-A3 interactions are attractive targets for novel drug development. HIV-1 and HIV-2 arose via distinct zoonotic transmission events of simian immunodeficiency viruses from chimpanzees and sooty mangabeys, respectively, and Vifs from these viruses have limited homology. To gain insights into the evolution of virus-host interactions that led to successful cross-species transmission of lentiviruses, we characterized the determinants of the interaction between HIV-2 Vif (Vif2) with human A3 proteins and compared them to the previously identified HIV-1 Vif (Vif1) interactions with the A3 proteins. We found that A3G, A3F, and A3H-hapII, but not A3D, were susceptible to Vif2-induced degradation. Alanine-scanning mutational analysis of the first 62 amino acids of Vif2 indicated that Vif2 determinants important for degradation of A3G and A3F are completely distinct from these regions in Vif1, as are the determinants in A3G and A3F that are critical for Vif2-induced degradation. These observations suggest that distinct Vif-A3 interactions evolved independently in different SIVs and their nonhuman primate hosts and conservation of the A3 determinants targeted by the SIV Vif proteins resulted in successful zoonotic transmission into humans.
IMPORTANCE Primate APOBEC3 proteins provide innate immunity against invading pathogens, and Vif proteins of primate lentiviruses have evolved to overcome these host defenses by interacting with them and inducing their proteasomal degradation. HIV-1 and HIV-2 are two human pathogens that induce AIDS, and elucidating interactions between their Vif proteins and human A3 proteins could facilitate the development of novel antiviral drugs. Furthermore, understanding Vif-A3 interactions can provide novel insights into the cross-species transmission events that led to the HIV-1 and HIV-2 pandemics and evolution of host-virus interactions. We carried out mutational analysis of the N-terminal 62 amino acids of HIV-2 Vif (Vif2) and analyzed A3G/A3F chimeras that retained antiviral activity to identify the determinants of the Vif2 and A3 interaction. Our results show that the Vif2-A3 interactions are completely different from the Vif1-A3 interactions, suggesting that these interactions evolved independently and that conservation of the A3 determinants resulted in successful zoonotic transmission into humans.
INTRODUCTION
Antiviral host restriction factors present in human T cells can potentially inhibit human immunodeficiency virus type 1 and type 2 (HIV-1 and HIV-2, respectively); however, viral mechanisms to counteract these host defenses have evolved, thus leading to productive infections. Understanding the mechanisms involved in these host-virus interactions could lead to important insights into barriers to the zoonotic transmission of pathogenic viruses, mechanisms of virus-host coevolution, and novel antiviral treatment strategies with targeted efforts to restore the natural host defenses. Members of the human apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3 (APOBEC3 [A3]) family of proteins are potent cellular restriction factors that provide a defense against viral infection by incorporating into virions and targeting nascent viral DNA for deamination of cytidine residues to uridines (1–8); cytidine deamination of the viral DNA leads to lethal hypermutation of the targeted virus. However, both HIV-1 and HIV-2 have accessory proteins, called viral infectivity factors (Vifs), that can bind to the APOBEC3 proteins to promote their ubiquitination and subsequent proteosomal degradation (9–18), thereby suppressing their virion incorporation.
Cellular restriction factors, including APOBEC3 proteins, can potentially serve as barriers to cross-species transmission (19–23). HIV-2 and HIV-1 differ in many ways, including their zoonotic origins (24). HIV-1 originated by four separate transmission events from great apes: simian immunodeficiency virus (SIV) from chimpanzees (SIVcpz) gave rise to groups M and N, while SIV from gorillas gave rise to group P. For HIV-1 group O, it is less clear whether this group arose via transmission of SIV directly from chimpanzees or gorillas. In the case of HIV-2, eight distinct transmission events of SIV from the sooty mangabey (SIVsm) gave rise to HIV-2 groups A to H. SIVsm Vif was able to support the replication of HIV-1ΔVif in human H9 cells, indicating that it can induce the degradation of human APOBEC3 proteins (25). Because the evolutionary histories of HIV-1 and HIV-2 are different, there are distinct differences between the viral genomes, including the presence or absence of certain accessory proteins. These differences may be attributed in some cases to viral adaptations to new hosts during cross-species transmission events (such as the presence of Vpx in some SIVs, but not in SIVcpz or HIV-1) (23). In addition, functionally conserved proteins have little homology between their sequences, as is the case with the accessory protein Vif.
Understanding Vif-A3 interactions can provide insights into virus-host interactions. APOBEC3 proteins and other restriction factors exhibit positive selection at certain sites (19, 26–28); specifically, the rate of nonsynonymous changes exceeds the rate of synonymous changes, indicating that specific sites are under evolutionary pressure to select for substitutions. The positive selection is viewed as a result of an arms race between the virus and the host, where a deleterious interaction with a viral protein selects for substitutions at the site of interaction.
In addition to providing insights into virus-host interactions, defining A3-Vif interactions has the potential to lead to the development of novel antiviral agents that interfere with Vif-induced degradation and restore the APOBEC3 defenses of the cell. To date, most studies have focused on defining the interactions between APOBEC3 and Vif from HIV-1 (Vif1), which is responsible for most of the AIDS pandemic. Vif1 interacts with numerous host cell proteins to form a ubiquitin ligase complex consisting of cullin 5, elongin B, and elongin C (29), RING finger protein 2 (30, 31), and CBFβ (32, 33). Further, Vif1 has several domains through which it binds APOBEC3 proteins, with some regions being specific for certain APOBEC3s (34–39). For example, the 40YRHHY44 motif of Vif1 is known to play a critical role in inducing the degradation of A3G, while the 14DRMR17 region is known to be critical for inducing the degradation of A3C, A3D, and A3F (34).
Unlike APOBEC3-Vif1 interactions, very few studies have attempted to define the interactions between HIV-2 Vif (Vif2) and the APOBEC3 proteins (11, 13). There is only ∼30% amino acid identity between Vif1 from NL4-3 and Vif2 from HIV-2ROD12. We hypothesize that the SIVs that gave rise to HIV-1 and HIV-2 possessed mechanisms to evade the human cellular defenses, including the APOBEC3 proteins, in order to achieve successful transmission to the new host species. This hypothesis predicts that the SIV Vif proteins, which evolved to overcome the APOBEC3 proteins in their nonhuman primate hosts, were able to interact with the human APOBEC3 proteins, because the simian APOBEC3 determinants recognized by SIV Vif proteins were conserved in the human APOBEC3 proteins. Two critical questions for understanding the limits to cross-species transmission are how often viruses have evolved successful APOBEC3-Vif interactions and how conserved the APOBEC3 determinants recognized by Vif proteins are across primate species. We sought to define the important regions in Vif2 that are required for interaction with human APOBEC3 proteins to gain a better understanding of the evolution and adaptations acquired by HIV-2 and HIV-1 in their respective Vif proteins to successfully overcome this family of host restriction factors.
We performed alanine-scanning mutagenesis of the N-terminal region of Vif2 and found that some determinants were specifically required to overcome A3G, A3F, or A3H haplotype II (A3H-hapII), while other determinants were required for activity against two or all three APOBEC3 proteins. In addition, we found that A3G and A3F mutants that are resistant to Vif1 are sensitive to degradation by Vif2, and analysis of A3G-A3F chimeras mapped the Vif2-interacting domains to regions different from the regions for the Vif1-interacting domains. These observations suggest that distinct Vif-A3 interactions evolved independently in different SIVs in adaptation to their nonhuman primate hosts and conservation of the A3 determinants targeted by the SIV Vifs resulted in successful zoonotic transmission of HIV-1 and HIV-2 into humans.
MATERIALS AND METHODS
Plasmid construction and cell culture.
The pFLAG-A3C, -A3D, -A3F, -A3G, -A3H-hapII, -A3H-hapI, -A3G-D128K, -A3F-E289K, -GFG2, and -FGF4 plasmids were previously described (34, 40–42). Myc-A3A-HA was kindly provided by Gisela Heidecker and David Derse (National Cancer Institute—Frederick). The pFLAG-A3B plasmid was constructed by modification of pAPOBEC3B-HA, which was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, from Bryan R. Cullen (43). The pcDNA-hVif1 (44) and pCR3.1-Vif2 (45) plasmids were also previously described. Codon-optimized full-length Vif2 was synthesized with a hemagglutinin (HA) tag on the C terminus and cloned into the BamHI and EcoRI sites of pCR3.1 (Genewiz Inc.). Vif2 mutants were generated by site-directed mutagenesis using a QuikChange II site-directed mutagenesis kit (Stratagene), and APOBEC3 mutants were generated by either site-directed mutagenesis or overlapping PCR.
The modified human embryonic kidney 293T (HEK293T) cell line (46) used for transfections and the HeLa-derived HIV-1 reporter cell line TZM-bl (47, 48) used for infections were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum, 1% penicillin-streptomycin, and 1% glutamine.
APOBEC3 degradation assays.
Six-well plates were seeded with 8 × 105 HEK293T cells per well 1 day prior to transfection. Transfections were carried out using previously described methods (1, 29, 30). To assay for degradation of the APOBEC3 proteins, the following plasmids and amounts were used: 0.34 μg pFLAG-APOBEC3 plasmids or myc-A3A-HA plasmid, 4.5 μg Vif2 plasmid (∼0.1:1 molar ratio of A3/Vif2), and 0.2 μg pGreen Lantern-1 (pGL) (Gibco BRL) as a positive control for transfections. To maintain equivalent DNA amounts, pcR3.1noMCS or pcDNA3.1noMCS (34) empty vectors were used when needed. To assay for degradation of the A3G/A3F (GF) and A3F/A3G (FG) chimeras, the following plasmids and amounts were used: 0.34 μg pFLAG-APOBEC3 plasmids and 0.34 μg hVif2-HA plasmid (∼1:1 molar ratio of A3/Vif2-HA). To maintain equivalent DNA amounts, pcR3.1noMCS or pcDNA3.1noMCS empty vectors were used when needed. After 48 h, cell lysates were harvested by removing the medium and rinsing the cells twice with phosphate-buffered saline (PBS). For each well, 250 μl of lysis buffer was used. The total protein concentration was determined by the Bradford assay using the Quick Start Bradford 1× dye reagent (Bio-Rad), and samples were normalized with lysis buffer and then mixed with NuPAGE LDS sample buffer (Invitrogen) containing β-mercaptoethanol. Western blot assays were performed to detect steady-state expression levels in the absence and presence of Vif2. FLAG-APOBEC3 proteins were detected using a rabbit anti-FLAG polyclonal antibody (Sigma) at a 1:5,000 dilution, and myc-A3A-HA was detected using a rabbit anti-c-myc monoclonal antibody (Sigma) at a 1:5,000 dilution. An IRDye 800CW-labeled goat antirabbit secondary antibody (LI-COR) was used at a 1:10,000 dilution to detect rabbit primary antibodies. To detect α-tubulin, mouse anti-α-tubulin antibody (Sigma) was used at a 1:5,000 dilution, followed by an IRDye 680-labeled goat antimouse secondary antibody at a 1:10,000 dilution (LI-COR). Protein bands were visualized using an Odyssey infrared imaging system (LI-COR) and quantified as previously described (49).
Virus production and titration.
To produce virus, the following plasmids and amounts were used for transfections: 3.33 μg of the HIV-1 vector genome pHDV-EGFP (50), 0.67 μg of vesicular stomatitis virus glycoprotein (VSV-G) expression plasmid pHCMV-G (51), 0.34 μg of pFLAG-A3G or pFLAG-A3F or 0.67 μg of pFLAG-A3H-hapII expression plasmid, and 4.5 μg of either Vif2 or human Vif1 (hVif1) or 0.34 μg of C-terminally HA-tagged and codon-optimized Vif2 (hVif2-HA). Molar ratios (HDV-EGFP/VSV-G/A3) were 1:0.4:0.2 (A3G or A3F), 0.4 (A3H-hapII), or 0.8 (the A3G mutant with five alanine substitutions [the A3G>5A mutant]) and either 2.7 (Vif2), 2.5 (hVif1), or 0.2 (hVif2-HA). For the FLAG-A3G>5A experiments, the expression of this protein was greatly reduced compared to that of wild-type (WT) A3G at equivalent DNA amounts (data not shown), so the amount of the FLAG-A3G>5A expression plasmid was increased to 1.36 μg. To maintain equal amounts of DNA, pCR3.1noMCS or pcDNA3.1noMCS empty vectors were used when needed. After 48 h, the virus-containing supernatant was clarified by low-speed centrifugation or filtered through a 0.45-μm-pore-size filter, and virus producer cell lysates were used for Western blot analyses, as described above, except that horseradish peroxidase-labeled goat antirabbit or goat antimouse secondary antibodies were used at a 1:10,000 dilution to detect the rabbit anti-FLAG or mouse anti-α-tubulin primary antibodies, respectively. For Vif1 detection, a rabbit anti-Vif polyclonal antibody (HIV-1HXB2 Vif antiserum from Dana Gabuzda, AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH) (52) was used at a 1:5,000 dilution. For the Vif2 experiments with mutants with multiple alanine mutations (multiple-alanine mutants), the integrated band intensities for the FLAG-A3 proteins were measured using Nikon Elements software, and the relative degradation was calculated with the value for the no-Vif2 control for each APOBEC3 set equal to 0% degradation. The average A3 degradation for three independent experiments was calculated (with any negative degradation values assigned to be 0% degradation). Virus production was quantitated using a p24 CA enzyme-linked immunosorbent assay. TZM-bl cells were seeded in 96-well plates (4 × 103 cells per well) 1 day prior to infection. Virus samples were normalized for their p24 CA contents prior to TZM-bl cell infections. The supernatant was removed 72 h later and replaced with an equal volume of PBS and Britelite luciferase solution (Perkin-Elmer). Luciferase enzyme activity was immediately measured using a LUMIstar Galaxy luminometer or a 1450 MicroBeta JET detector (PerkinElmer). Spearman correlation coefficients (rss) were calculated from the averages of the relative infectivity versus degraded APOBEC3 protein.
Virus production in CEM2n cells.
CEM2n cells (6 × 106 cells in 2 ml medium) were transfected using nucleofection (nucleofected) with HDV-GFP, VSV-G, and either control pCR3.1 DNA, WT Vif2-HA, or Vif2-HA mutant VPG>3A, KVG>3A, or WAW>3A using a Amaxa Nucleofector II kit, program 001. At 3 h after transfection using nucleofection, the medium was replaced with 750 ml of fresh medium; 48 h later, virus was harvested and used to infect TZM-bl cells, and luciferase activity was determined 48 h after infection of the TZM-bl cells.
Co-IP assays.
Anti-FLAG coimmunoprecipitation (co-IP) assays were done as previously described (34, 40). HEK293T cells were seeded in a 100-mm-diameter dish (4 × 106 cells per dish) and transfected. The following DNA amounts were used for the co-IP assays with A3B-A3H: 5 μg pFLAG-APOBEC3 plasmids, 0.5 μg hVif2-HA plasmid, and 1.8 μg pGL as a positive control for the transfections. To maintain equivalent DNA amounts, pCR3.1noMCS or pcDNA3.1noMCS empty vectors were used when needed. After 48 h, the medium was removed and the cells were rinsed twice with PBS. Lysis buffer (1 ml) was added, total cell extracts were harvested, and samples for co-IP were prepared as previously described (34). To detect eluted complexes as well as the input cell lysates, Western blotting was performed. The APOBEC3 proteins were detected using a rabbit anti-FLAG polyclonal antibody (Sigma) at a 1:5,000 dilution, the hVif2-HA proteins were detected using a mouse anti-HA monoclonal antibody (Sigma) at a 1:5,000 dilution, and α-tubulin was detected using mouse anti-α-tubulin antibody (Sigma) at a 1:5,000 dilution. Rabbit and mouse primary antibodies were detected using 1:10,000 dilutions of an IRDye 800CW-labeled goat antirabbit secondary antibody (LI-COR) or an IRDye 680-labeled goat antimouse secondary antibody (LI-COR). Protein bands were visualized using an Odyssey infrared imaging system (LI-COR).
RESULTS
APOBEC3 binding and sensitivity to Vif2.
To characterize APOBEC3-Vif2 interactions, we coexpressed human APOBEC3 and Vif2 proteins and used Western blotting to determine the sensitivity of each human APOBEC3 protein to Vif2-induced degradation (Fig. 1A and B). In this assay, we compared the level of APOBEC3 protein in the absence of Vif2 to that in the presence of Vif2. The results indicated that A3A is insensitive (no degradation was observed) and A3D has very little sensitivity to Vif2-induced degradation (>80% of A3D remained in the presence of Vif2). A3B and A3F had intermediate sensitivity (63% and 56% of A3B and A3F remained, respectively), while A3C, A3G, and A3H-hapII were all highly sensitive to Vif2-induced degradation (<25% of A3C, A3G, or A3H-hapII remained). We also examined A3H-hapI sensitivity, but because this protein is unstable in the absence of Vif2, very little of the protein was detectable even in the absence of Vif2, and it was not possible to reliably quantify its sensitivity.
FIG 1.
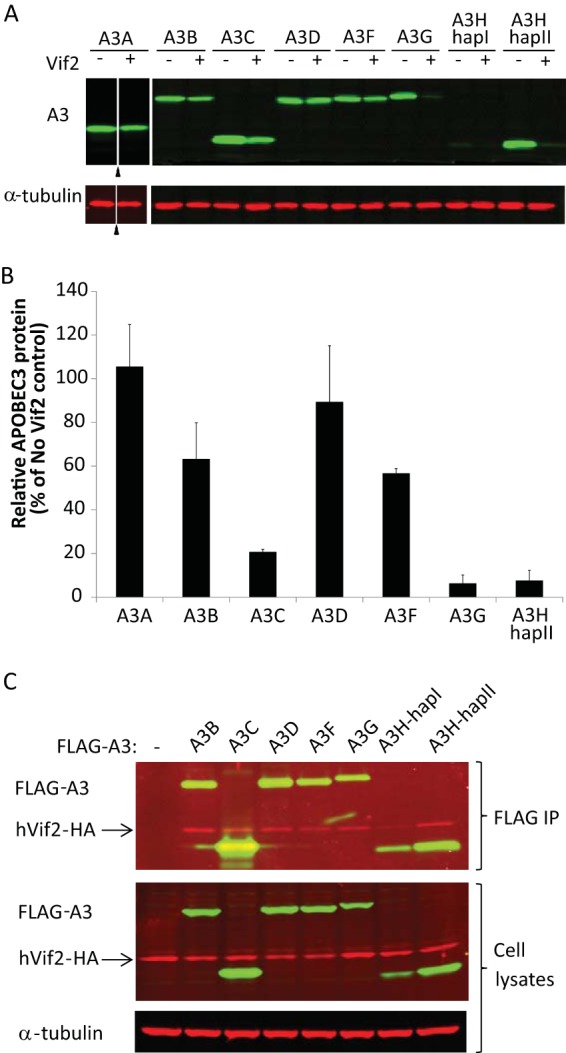
Interactions between human APOBEC3 proteins and HIV-2 Vif. HEK293T cells that were cotransfected with A3 expression plasmids and either empty vector or Vif2 expression plasmids were analyzed. (A) Sensitivity of human A3 proteins to Vif2. Cell lysates were analyzed by quantitative Western blotting using an anti-myc antibody to detect myc-A3A, an anti-FLAG antibody to detect FLAG-A3B, -A3C, -A3D, -A3F, -A3G, -A3H-hapI, and -A3H-hapII, or an anti-α-tubulin antibody to detect α-tubulin. Triangles, lanes derived from the same blot with extraneous lanes were removed; lanes +, with Vif2; lanes −, without Vif2. (B) The A3 band intensities were normalized by the α-tubulin intensities, and the relative A3 protein levels (FLAG or myc) in the presence of Vif2 were calculated as a percentage of the amount of A3 protein in the absence of Vif2 (set equal to 100% for each A3 protein). A3H-hapI was not quantitated due to the low signal intensity in the absence of Vif2. The average of two independent experiments is shown; error bars indicate standard deviations. (C) Co-IP assays to determine binding of Vif2 to A3 proteins. HEK293T cells were cotransfected with FLAG-A3 expression plasmids and hVif2-HA expression plasmids. Cell lysates and immunoprecipitated proteins were analyzed by Western blotting using anti-FLAG and anti-HA antibodies to detect FLAG-A3 and hVif2-HA proteins, respectively. α-Tubulin was also detected in the cell lysates as a loading control.
Next, we examined the binding of Vif2 to the APOBEC3 proteins. To do so, we cotransfected C-terminally tagged Vif2 and FLAG-tagged APOBEC3 expression plasmids and performed co-IP assays using anti-FLAG antibody (Fig. 1C). Because there are currently no commercially available antibodies against native Vif2, it was necessary to use the C-terminally HA-tagged and codon-optimized Vif2 (hVif2-HA) for Western blotting (Fig. 1C). We determined that hVif2-HA was pulled down in the presence of each of the FLAG-tagged APOBEC3 proteins, indicating that hVif2-HA can bind to these APOBEC3 proteins.
Identification of distinct regions of Vif2 involved in inhibition of A3F and A3G.
We previously used alanine-scanning mutagenesis to identify two distinct regions in the first 60 amino acids of Vif1 that are critical for neutralization of A3F and A3G (34, 40, 42). An alignment of the N termini of Vif1 and Vif2 shows that these proteins are dissimilar (Fig. 2A). To determine how Vif2 counteracts the human APOBEC3 proteins, the first 60 amino acids in Vif2 were targeted for multiple-alanine-scanning mutagenesis; in addition, a mutant with double alanine substitutions of amino acids 61 and 62 was constructed. Amino acids that were not conserved between Vif2 and SIVsm Vif (Vif2 amino acids 6, 28, 32, 37, and 39) were excluded because residues critical for degradation of human APOBEC3 proteins were expected to be conserved between these two Vifs. In addition, Vif2 is known to functionally complement Vif-deficient HIV-1, so to evaluate the effects of alanine mutations on Vif2 function, the ability of the alanine substitution mutants to neutralize A3G and A3F was examined by determining their ability to rescue the infectivity of p24 CA-normalized HIV-1Δvif in TZM-bl cell infectivity assays (Fig. 2B). The average relative infectivity compared to that for the no-Vif/no-APOBEC3 control was 0.3% in the presence of A3G without Vif2 and 59.7% in the presence of A3G and Vif2. For A3F, the average relative infectivity was 8.0% in the presence of A3F without Vif2 and 40.8% in the presence of A3F and Vif2.
FIG 2.

Effect of Vif2 multiple-alanine substitutions on its activity against A3G, A3F, and A3H-hapII. (A) Alignment of Vif1 and Vif2 N-terminal 62 amino acids. The locations of amino acids replaced by alanines to generate multiple-alanine substitution mutants 1 to 19 are indicated with brackets below the sequence. (B) Effects of multiple alanine mutations in Vif2 on HIV-1Δvif infectivity in the presence of A3G, A3F, or A3H-hapII in HEK293T cells. Viruses were produced from HEK293T cells that were transfected with pHDV-EGFP, expression plasmids for VSV-G, A3G, A3F, and A3H-hapII, and either empty vector, WT Vif2, or mutant Vif2. Amino acid substitutions are shown for the multiple-alanine-substitution mutants, numbered 1 to 19. Dashed lines, inhibition of virus infectivity by A3G, A3F, or A3H-hapII in the absence of Vif2. Error bars indicate standard errors of the means (SEMs) of the results from three infectivity experiments. All comparisons were made on the basis of the results for the no-Vif2 controls for each A3 protein using two-tailed Student's t test. A P value of <0.05 was considered statistically significant (*). The F box labels indicate Vif2 mutants that had no activity against A3F but retained activity against A3G and/or A3H-hapII. The G box label indicates the Vif2 mutant that had no activity against A3G but retained activity against A3F and A3H-hapII. The H box label indicates the Vif2 mutant that had no activity against A3H-hapII but retained activity against A3G and A3F. Mutants 2 to 4, 7, 12, and 16 to 19 lost activity against all three A3 proteins. (C) Effects of multiple alanine mutations in Vif2 on HIV-1Δvif infectivity in the presence of A3G, A3F, or A3H-hapII in CEM2n cells. Viruses were produced from CEM2n cells that were nucleofected with pHDV-EGFP and either empty vector, WT Vif2, or the indicated multiple-alanine mutants of Vif2 that specifically blocked A3F, A3G, or A3H-hapII. Relative infectivity was determined for two independent virus stocks by infection of TZM-bl cells with p24 CA-normalized virus and quantitation of luciferase activity at 72 h postinfection. The infectivity in the absence of Vif2 was set equal to 1.0. All comparisons were made on the basis of the results for the Vif2 control for using two-tailed Student's t test. A P value of <0.05 was considered statistically significant (*).
In the presence of A3G, mutant 1 (E2-E3-D4>3A), mutant 5 (V15-P16-G17>3A), mutant 6 (R18-M19-E20>3A), and mutant 15 (W49-A50-W51>ASA) were able to rescue infectivity to a statistically significant level compared to that for the no-Vif2 control. In the presence of A3F, only mutants 14 (K46-V47-G48>3A) and 15 (W49-A50-W51>ASA) were able to significantly rescue infectivity compared to that for the no-Vif2 control. Overall, most Vif2 multiple-alanine mutants were severely dysfunctional in their activity against A3G and A3F.
Because most of the Vif2 multiple-alanine mutants lost function against A3G and A3F, it is possible that the Vif2 mutants have a nonspecific defect, such as folding or decreased expression. Because there is no commercially available antibody against native Vif2, the protein expression levels of these Vif2 mutants could not be detected. To overcome this issue, we tested the functionality of the Vif2 multiple-alanine mutants against A3H-hapII, a more distantly related member of the APOBEC3 family that is able to inhibit HIV-1 and is sensitive to wild-type Vif2. Mutant 1, mutant 5, mutant 8 (S24-L25-V26>3A), mutant 9 (K27-L29-K30>3A), mutant 10 (Y31-T33-K34>3A), mutant 11 (D35-L36-K38>3A), mutant 13 (P43-H44-H45>3A), and mutant 14 were able to significantly rescue infectivity in the presence of A3H-hapII compared to that for the no-Vif2 control (23.5%). These data suggest that at least one amino acid that was altered in each of the mutants 8, 9, 10, 11, and 13 is important for inducing the degradation of both A3G and A3F but not that of A3H-hapII. Interestingly, mutants 1 and 5 retained their function against A3G and A3H-hapII but did not retain their function against A3F, suggesting that there are two distinct regions in Vif2 that are important only for A3F. Also, statistical analysis indicated that mutant 6 retains significant function against A3G but not A3F. Mutant 6 had partial function against A3H-hapII, but its function was not statistically significantly different from that for the no-Vif2 control. Taken together, these data indicate that the residues that were mutated in mutants 1, 5, and 6 (labeled F box in Fig. 2B) are specifically required for A3F neutralization. Likewise, mutant 14 contained changes to residues that were important for A3G but not A3F, and the residues in these regions are labeled G box in Fig. 2B. Mutant 15, which retained its function against A3G and A3F, did not retain its function against A3H-hapII, suggesting that this region of Vif2 is specifically important for A3H-hapII (labeled H box in Fig. 2B). Together, these data indicate that, like Vif1, Vif2 evolved to recognize different APOBEC3 proteins through distinct regions. In addition, like Vif1, some determinants of Vif2 are important for inducing degradation of more than one APOBEC3 protein.
Because mutants 2 to 4, 7, 12, and 16 to 19 did not retain the ability to rescue infectivity above background levels, these mutants are either important for neutralization of all APOBEC3 proteins tested or have a general defect that abrogated their ability to neutralize A3F, A3G, and A3H-hapII. Further testing of the amino acids in these regions is required to distinguish between these possibilities.
CEM2n cells were previously reported to express A3G, A3F, and A3H (53). To determine whether the residues in the F, G, and H boxes of Vif2 are important for the infectivity of viruses produced in the presence of native levels of A3 proteins, CEM2n cells were nucleofected with pHDV-EGFP and either empty vector, WT Vif2, or multiple-alanine mutant VPG>3A, KVG>3A, or WAW>ASA. Viral infectivity was determined by p24 CA-normalized HIV-1Δvif in TZM-bl cell infectivity assays (Fig. 2C). The infectivity of the no-Vif control (empty vector) was significantly reduced compared to that of the WT Vif2 control, suggesting that native A3 protein expression in the nucleofected CEM2n cells was sufficient for inhibition of the viruses produced in the absence of Vif. Interestingly, rescue of viral infectivity was deficient when KVG>3A was used to complement Vif-deficient HIV. Because KVG>3A is nonfunctional against A3G, these data indicate that native A3G inhibited the virus. The viruses produced in the presence of either the VPG>3A or the WAW>3A mutant did not have severely reduced infectivity, strongly suggesting that the levels of A3F and A3H in CEM2n cells are too low to significantly inhibit HIV-1 in a single cycle of replication. These data agree with previously published results from us and other investigators indicating that A3G expression is sufficient to substantially inhibit HIV-1 replication in T cell lines, while A3F does not exert a strong antiviral effect in early infections (41, 54) but can inhibit an A3G-specific Vif mutant in multiple cycles of replication (41). However, results from another study suggest that A3F levels in CEM2n cells are sufficient to inhibit HIV-1 replication (53).
In addition to assessing the Vif2 mutant function in infectivity rescue experiments, Western blots were also performed on the producer cell lysates for the viruses used for the assay whose results are presented in Fig. 2 to detect the degradation of A3G, A3F, and A3H-hapII (Fig. 3A). These data indicate that nearly complete degradation of A3G was required for the rescue of HIV-1 infectivity by Vif2 or its mutants (e.g., see the lanes for Vif2 mutants 1, 5, 6, and 15 in Fig. 3A). The data also indicate that some Vif2 mutants could cause extensive, yet incomplete degradation of A3G (e.g., see the lanes for Vif2 mutants 10, 11, 13, and 14 in Fig. 3A); the amounts of A3G remaining were sufficient to inhibit HIV-1. To account for variation between experiments, the average APOBEC3 degradation from three independent experiments was determined and used in scatter plots of the average relative infectivity versus the average APOBEC3 degradation. These data were used to determine Spearman's rank correlation coefficient (rs = 0.86), indicating a strong positive relationship between relative infectivity and Vif2-induced A3G degradation (Fig. 3B). Likewise, cell lysates for virus production in the presence of A3F or A3H-hapII were analyzed by Western blotting. Nearly complete degradation of A3F and A3H-hapII was required for the statistically significant rescue of HIV-1 infectivity. Spearman's rank correlation coefficients indicated strong positive relationships between relative infectivity and Vif2-induced A3F degradation (rs = 0.85; Fig. 3C) or Vif2-induced A3H-hapII degradation (rs = 0.94; Fig. 3D).
FIG 3.

Effects of Vif2 and its multiple-alanine-substitution mutants on A3G, A3F, and A3H-hapII steady-state levels. (A) Cell lysates of the viral producers in Fig. 2 were analyzed by Western blotting using anti-FLAG or anti-α-tubulin antibodies. The numbers above the lanes correspond to the Vif2 mutant (as labeled in Fig. 2) that was present in the cotransfection. The results are representative of those from three independent experiments. Triangle, lanes derived from the same blot with extraneous lanes were removed. (B to D) Correlation between relative infectivity in the presence of A3G (B), A3F (C), or A3H-hapII (D) and Vif-mediated degradation by different multiple-alanine-substitution Vif2 mutants. The Western blot bands of FLAG-A3 and α-tubulin in the producer cell lysates for the viruses used in the infectivity assay whose results are shown in Fig. 2 were quantitated using Nikon Elements software. Spearman's rank correlation coefficients (rss) were calculated and are indicated in the correlation plots. All calculated rs values were greater than the critical value (0.576, n = 21) for a significance (α) level of 0.005, indicating a strong correlation between infectivity and A3 degradation.
Identification of specific Vif2 residues involved in inducing degradation of A3F and A3G.
After identification of the F box and G box regions, we performed single alanine substitutions in the residues substituted in mutants 5, 6, 13, and 14 to determine which residues contribute to the ability of Vif2 to neutralize A3F and/or A3G. We included mutant 13 (P43-H44-H45>3A) in this analysis because it was adjacent to the G box and aligned with the 40YRHHY44 motif in Vif1, which was previously shown to be important for degradation of A3G but not that of A3F. Again, we assessed the function of these single-alanine mutants by determining their ability to complement HIV-1Δvif (Fig. 4A). The average relative infectivity in the presence of A3G without Vif2 was <1% compared to that for the no-Vif/no-APOBEC3 control, and that in the presence of A3G with Vif2 was 77% compared to that for the control. For A3F, the average relative infectivity was 13% in the presence of A3F without Vif2 and 50% in the presence of A3F with Vif2. The rescue of infectivity for a given Vif2 mutant in the presence of A3F or A3G was compared to the inhibition of infectivity in the presence of these APOBEC3 proteins without any Vif2 (dotted lines in Fig. 4A).
FIG 4.

Effect of single alanine substitutions in APOBEC3-specific regions of Vif2 on its activity against A3G and A3F. (A) Vif2 mutant rescue of HIV-1Δvif infectivity in the presence of A3G and A3F. Viruses were produced and infectivity was determined as described in the legend to Fig. 2. Data are plotted as relative infectivity levels, with the level in the absence of any A3 protein being set equal to 100%. Dashed lines, inhibition by A3G or A3F in the absence of Vif2. Error bars indicate standard errors of the means (SEMs) of the results from three to four infectivity experiments performed with independent virus stocks. All comparisons were made on the basis of the results for no-Vif2 controls for each A3 protein using two-tailed Student's t test. A P value of <0.05 was considered statistically significant (*). Boxes indicate mutants which retained significant activity against A3G but not A3F (solid) or retained significant activity against A3F but very little activity against A3G (dotted). (B) Effects of Vif2 on the degradation of A3G and A3F. Cell lysates of the viral producers for which the results are presented in panel A were analyzed by Western blotting using anti-FLAG or anti-α-tubulin antibodies. The results are from one representative experiment of three independent experiments. (C) Summary of Vif2 mutational analysis. The alignment of the Vif1 and Vif2 N-terminal 60 amino acids is shown. Dots, amino acid identity; dashes, gaps; fuchsia boxes, multiple-alanine mutants or single-amino-acid-substitution mutants that lost activity against A3F but not A3G and/or A3H-hapII; green box labeled G, G48 single-amino-acid-substitution mutant that lost activity against A3G but not A3F; blue box, multiple-alanine-substitution mutant that lost activity against A3H-hapII but not A3G or A3F; gray box, multiple-alanine-substitution mutants that lost activity against A3G and A3F but not A3H-hapII; black boxes, multiple-alanine-substitution mutants that lost activity against A3G, A3F, and A3H-hapII.
All Vif2 mutants with single mutations in the F box rescued infectivity to statistically significant levels compared to that for the no-Vif2 control in the presence of A3G (Fig. 4A). Most of the F box mutants had at least some function against A3F compared to that of the no-Vif2 control; however, only the V15A, G17A, and R18A mutants rescued infectivity to statistically significant levels compared to that of the no-Vif2 control, while the P16A, M19A, and E20A mutants did not. Among these, the P16A mutant seemed to be especially important since it had the smallest amount of activity against A3F. Western blots were also performed on the lysates of cells that produced the viruses used for the assay whose results are presented in Fig. 4A to detect Vif2-induced degradation of A3G and A3F (Fig. 4B). Each Vif2 mutant with a single mutation in the F box induced degradation of A3G to some extent, and the V15A, G17A, and R18A mutants induced the degradation of A3F, while the P16A, M19A, and E20A mutants induced little to no A3F degradation. Together, these data suggest that P16, M19, and E20 are important for Vif2's ability to overcome the antiviral activity of A3F but are less important for blocking the antiviral activity of A3G.
Among the single-amino-acid mutants derived from mutants 13 and 14, all single mutants except H45A significantly rescued viral infectivity in the presence of A3G compared to that for the no-Vif2 control. The G48A mutant showed a very low level of activity against A3G but significant activity against A3F, indicating that this amino acid is important for Vif2's ability to overcome the antiviral activity of A3G but not that of A3F. The P43A, H44A, and V47A mutants significantly rescued virus infectivity in the presence of A3G but not A3F, indicating that these amino acids are specifically important for Vif2's ability to overcome the antiviral activity of A3F but not that of A3G. Western blot data for the viral producer cell lysates showed that each Vif2 single mutant with a mutation in the G box induced degradation of A3G to some extent, although the H45A and G48A mutants were the least efficient (Fig. 4B). Degradation of A3F was induced with the P43A, H44A, K46A, and G48A mutants, while the H45A and V47A mutants poorly induced the degradation of A3F, consistent with their poor ability to rescue infectivity compared to that of wild-type Vif2. Taken together, the P16A, M19A, E20A, P43A, H44A, and V47A mutants were defective in their ability to rescue virus infectivity in the presence of A3F; of these, the P16A mutant appears to be especially important since it had the least amount of activity against A3F. Only the G48A mutant was defective in its ability to rescue virus infectivity in the presence of A3G but not A3F.
A summary of the Vif2 mutational analysis is shown in Fig. 4C. The residues shown in fuchsia indicate a multiple-alanine mutant or single amino acids that are critical for the degradation of A3F, the G48 residue underlined in green is critical for degradation of A3G, and the multiple alanine mutation underlined in blue contains a determinant that is critical for the degradation of A3H-hapII. The multiple-alanine mutants and H45A underlined in gray contain determinants that are critical for the degradation of A3G and A3F but not that of A3H-hapII. Finally, the multiple-alanine mutants underlined in black have lost the ability to degrade A3G, A3F, and A3H-hapII and may be important for the overall structure and function of Vif2.
Vif2 can efficiently induce the degradation of Vif1-resistant A3G and A3F.
Previously, we and others found that the determinant in A3G for Vif1-induced degradation includes D128 and the surrounding residues (45, 55–57), and we found that the critical determinant in A3F for Vif1-induced degradation includes E289 and the surrounding residues (42, 58). A3G D128K and GFG2 mutants (9) and A3F E289K and FGF4 mutants (7), were previously shown to be resistant to Vif1-induced degradation (Fig. 5A). The GFG2 mutant has substitutions in A3G, with the corresponding residues in A3F being as follows: F126Y, D128E, P129R, and Q132R (31). The FGF4 mutant (29) has substitutions of nine amino acids of A3F, with the corresponding residues from A3G being as follows: G285Q, V287M, E289K, L291I, A292S, R293K, H294N, S295K, and N296H.
FIG 5.

Vif2 function against wild-type and Vif1-resistant A3G or A3F. (A) Schematic representation of Vif1 binding sites in A3G (left) and A3F (right). The Vif1 interaction determinants and surrounding residues in A3G and A3F are shown extending out from each protein. Important determinants for Vif1 interactions are shown in bold, and single residues critical for Vif1-induced degradation used in this study are underlined (D128 in A3G and E289 in A3F). Residues that are mutated in the GFG2, D128K, FGF4, and E289K mutants are shown below the wild-type residues. Dashes, residues that were not mutated. (B) Vif2 rescue of HIV-1Δvif infectivity in the presence of A3G and its mutants or A3F and its mutants. Viruses were produced and infectivity was determined as described in the legend to Fig. 2. Data are plotted as relative infectivity levels, with the level in the absence of any A3 protein being set equal to 100%. Error bars indicate standard errors of the means (SEMs) of the results from 2 to 6 infectivity experiments performed with independent virus stocks. All comparisons were made on the basis of the results for no-Vif controls for each A3 protein using two-tailed Student's t test. A P value of <0.05 was considered statistically significant (*). (C) Effects of Vif2 on the degradation of A3. Cell lysates of the viral producers for which the results are shown in panel B were analyzed by Western blotting using anti-FLAG or anti-α-tubulin antibodies.
Here, we wanted to assess whether mutations in these determinants of A3G and A3F that confer resistance to Vif1 also confer resistance to Vif2. Infectivity assays were used to compare the abilities of Vif1 and Vif2 to neutralize these A3G and A3F mutants (Fig. 5B). As expected, the A3G D128K and GFG2 mutants were resistant to Vif1, as were the A3F E289K and FGF4 mutants; in contrast, Vif2 could efficiently rescue virus infectivity in the presence of these Vif1-resistant mutants. Western blot analyses were also performed on the lysates of cells used to produce the viruses analyzed in the assay whose results are presented in Fig. 5B to detect the Vif1- or Vif2-induced degradation of A3G and A3F (Fig. 5C). Consistent with the infectivity data, A3G mutants D128K and GFG2 as well as A3F mutants E289K and FGF4 were resistant to Vif1-induced degradation, while each of these mutants was readily degraded by Vif2.
Because D128 and surrounding residues in A3G have been shown to play an important role in numerous A3G proteins from various primate species and it has been suggested that Vif2-induced degradation of A3G may be dependent on residues 128 to 130 (12), we wanted to further test the potential role of this region in Vif2 susceptibility. Because it is possible that the substitutions used in the GFG2 mutant either introduced A3F's Vif2-binding site or were too structurally conservative, we targeted D128 and the surrounding region in A3G by mutating five residues (D128, P129, D130, Q132, and E133) to alanines to create the A3G>5A mutant (Fig. 6A). The single-round infectivity assays whose results are shown in Fig. 6B and the Western blotting results shown in Fig. 6C indicated that the A3G>5A mutant was resistant to Vif1 but sensitive to Vif2, indicating that these amino acids of A3G did not play a role in the Vif2-mediated degradation of A3G.
FIG 6.

Vif2 function against A3G>5A, A3F>5A, and A3G single-amino-acid-substitution mutants D128K, P129D, and P129A. (A) Schematic representation of A3G and A3F mutants. Residues in the mutated sites in A3G and A3F are shown extending out from each protein, and the substitution mutations are indicated. (B) Vif2 rescue of HIV-1Δvif infectivity in the presence of A3G and its mutants or A3F and its mutants. Viruses were produced and infectivity was determined as described in the legend to Fig. 2. Data are plotted as relative infectivity levels, with the level in the absence of any A3 protein being set equal to 100%. Error bars indicate standard errors of the means (SEMs) of the results from 3 to 4 infectivity experiments performed with independent virus stocks. All comparisons were made on the basis of the results for the no-Vif controls for each A3 protein using two-tailed Student's t test. A P value of <0.05 was considered statistically significant (*). (C) Effects of Vif2 on the degradation of A3G and A3F mutants. Cell lysates of the viral producers for which the results are shown in panel B were analyzed by Western blotting using anti-FLAG antibody for detection of A3G or A3F, anti-HA antibody for detection of Vif1 or Vif2, and anti-α-tubulin antibody for detection of a-tubulin. (D) Effects of Vif2 and Vif1 on the degradation of A3G-W127A. Degradation was analyzed by Western blotting using anti-FLAG or anti-α-tubulin antibodies. All lanes shown are from the same Western blot. Triangles, extraneous and uninformative lanes were removed.
Analysis of A3G/A3F chimeras suggested that the region in A3F that mediates sensitivity to Vif2 is in the N-terminal half of A3F (the data are shown in Fig. 7 and discussed later). Therefore, we hypothesized that the region in A3F (127ERDYRR132) that is homologous to the region in A3G that contains D128 (128DPDYQE133) is critical for Vif2-mediated degradation of A3F. To test this hypothesis, we generated the mutant A3F>5A, in which the 127ERDYRR132 amino acids were replaced with AAAYAA (Fig. 6A). However, the single-round infectivity assays whose results are shown in Fig. 6B and the Western blotting assays whose results are shown in Fig. 6C indicated that the A3F>5A mutant was sensitive to Vif2 as well as Vif1, demonstrating that this region of A3F is not important for Vif2-mediated degradation.
FIG 7.

Sensitivity of A3G/A3F and A3F/A3G chimeras to hVif2-HA. (A) Schematic structures of A3G/A3F (GF) and A3F/A3G (FG) chimeras. Full-length A3G and the A3G portions of the GF and FG chimeras are shown in green, whereas the full-length A3F and A3F portions of the chimeras are shown in fuchsia. The numbers indicate the amino acid position from each A3 protein at which the A3G and A3F portions were fused to generate the chimeras; for example, the G99F99 chimera contains amino acids 1 to 99 from A3G and amino acids 99 to 373 from A3F. Quantitation of hVif2-HA-induced degradation of each chimera is shown in the bar graph to the right of the schematic structures of the chimeras. The relative amount of each A3 protein remaining in the presence of absence of hVif2-HA was determined by quantitative Western blotting, and the amount of each A3 protein in the absence of Vif2 was set equal to 100%. Data are plotted as the average of two independent experiments, and error bars indicate standard deviations. (B) Representative images of Western blots indicating sensitivity to Vif2-HA are shown. Cell lysates were analyzed by using an anti-FLAG antibody to detect FLAG-A3 or an anti-α-tubulin antibody to detect α-tubulin. Lanes +, with Vif2; lanes −, without Vif2. Comparison of the Vif2 sensitivity of the G120F120 and G149F145 chimeras indicates that the determinants in A3F that confer sensitivity to Vif2 are between amino acids 121 and 144; a comparison of the G163F159 and G321F315 chimeras indicates that the determinants in A3G that confer sensitivity to Vif2 are between amino acids 163 and 321.
Recently, Letko et al. reported that the P129D mutant of A3G is resistant to degradation by both Vif1 and Vif2 (11). However, our GFG2 and A3G>5A mutants, which contain the P129R and P129A mutations, respectively, were sensitive to Vif2 (Fig. 5 and 6, respectively). To further analyze the role of P129, we generated P129D and P129A mutants and analyzed their sensitivity to Vif1 and Vif2 (Fig. 6B). The single-round infectivity assays showed that P129D was resistant to Vif2 as well as Vif1, whereas P129A was resistant to Vif1 but sensitive to Vif2. The sensitivity of these mutants to Vif1- and Vif2-mediated degradation was also analyzed by Western blotting (Fig. 6C). The Western blotting results confirmed the findings of the infectivity assays and showed that the A3G>5A, D128K, and P129A mutants were all resistant to Vif1 and sensitive to Vif2-induced degradation; only the P129D mutant was resistant to both Vif1 and Vif2. Taken together, these results indicate that D128 and the surrounding region are not involved in Vif2-mediated degradation of A3G. However, the P129D mutation confers resistance to Vif2, perhaps by inducing a conformational change that alters the structure of another distal domain that interacts with Vif2.
To further analyze the region surrounding D128 in A3G, we determined the sensitivity of the A3G-W127A mutant, which has been shown to be deficient in virion incorporation (59–61). Because this mutant lacks antiviral activity, we tested the ability of Vif1 and Vif2 to induce the degradation of A3G-W127A by Western blotting (Fig. 6D). The results showed that the W127A mutant was depleted in the presence of Vif2 as well as Vif1, indicating that it is not involved in Vif2-mediated degradation of A3G.
Sensitivity of A3G/A3F chimeras to Vif2-mediated degradation.
We previously constructed and analyzed a series of A3G/A3F (GF) and A3F/A3G (FG) chimeras that retained their antiviral activity (40). The structures of the GF and FG chimeras and their sensitivities to Vif2-mediated degradation, determined by quantitative Western blotting, are shown in Fig. 7A and B. The results showed that the chimera G99F99 (a chimera that contains amino acids 1 to 99 from A3G and amino acids 99 to 373 from A3F) and the chimera G120F120 were sensitive to Vif2-mediated degradation, and the amounts of APOBEC3 proteins remaining were 18 and 23% of the amount for the no-Vif2 control, respectively; these amounts of APOBEC3 proteins remaining were similar to the 17% A3F remaining compared to the amount for the no-Vif2 control, indicating that G99F99 and G120F120 had sensitivity to Vif2 similar to that of wild-type A3F. In contrast, G149F145 was much more resistant to Vif2, since the amount of APOBEC3 protein remaining was 69% compared to the amount for the no-Vif2 control. A comparison of G120F120 and G149F145 indicated that A3F amino acids 120 to 145 contain determinants that confer sensitivity to Vif2.
The results in Fig. 7A and B show that the G163F159 chimera is also resistant to Vif2-mediated degradation and the amount of APOBEC3 protein remaining is 86% of the amount for the no-Vif2 control. The resistance to Vif2 conferred by G163F159 indicates that A3G residues 1 to 163 and A3F residues 159 to 373 are not sufficient to confer Vif2 sensitivity and therefore are unlikely to contain the Vif2 sensitivity determinants. In contrast, the chimeras G321F315, F248G257, F282G291, F300G309, and F314G322 were all sensitive to Vif2-mediated degradation. The F282G291 chimera is fully sensitive to Vif2 (4% of the A3 remains relative to the amount in the no-Vif2 control), whereas the F314G322 chimera is less sensitive to Vif2 (20% of the A3 protein remains relative to the amount in the no-Vif2 control), indicating that a determinant between G291 and G321 confers sensitivity to Vif2-mediated degradation. On the other hand, the F248G257 chimera is fully sensitive to Vif2 (3% of A3 remains relative to the amount in the no-Vif2 control), while the G163F159 chimera is quite resistant to Vif2 (86% of the A3 protein remains relative to the amount in the no-Vif2 control), indicating that other determinants between G163 and G257 are important for Vif2 sensitivity. Taken together, these results indicate that multiple determinants between G163 and G321 confer sensitivity to Vif2-mediated degradation.
Identification of an A3F determinant important for Vif2-mediated degradation.
To identify determinants in A3F that are important for Vif2-mediated degradation, we focused on A3F amino acids 120 to 145, since the G120F120 chimera was sensitive and the G149F145 chimera was resistant to Vif2. Homology alignment of A3F and A3G in this region identified several amino acids that are conserved and three regions, labeled box 1, box 2, and box 3, that are different between the two A3 proteins. As shown in Fig. 6, the A3F>5A mutant, in which A3F box 1 amino acids were replaced by alanines, was sensitive to Vif2. In addition, a chimera that we previously reported (42), in which residues 125 to 131 in box 1 of A3F were replaced by the corresponding residues of A3G, was Vif2 sensitive (Fig. 8B). These results indicated that box 1 amino acids are not involved in Vif2-mediated degradation. Next, we found that a chimera in which the box 2 amino acids CRLS were replaced by the equivalent amino acids RSLC in A3G was sensitive to Vif2, indicating that these A3F amino acids are also not involved in Vif2 sensitivity (Fig. 8AII and B). Finally, we generated a chimera in which the A3F box 3 amino acids AGARV were replaced by the equivalent amino acids PRATM in A3G. Western blotting indicated that the PRATM chimera was resistant to Vif2-mediated degradation (Fig. 8AI and C). Single-round infection assays indicated that this chimera retained some antiviral activity, although its antiviral activity was impaired compared to that of wild-type A3F (Fig. 8D). Importantly, the antiviral activity of the PRATM chimera was resistant to Vif2, indicating that box 3 amino acids constitute a determinant that confers sensitivity to Vif2. To further analyze the role of box 3 amino acids, single amino acid substitutions A140K, G141R, R143T, and V144M were constructed. We elected to generate an A140K substitution, because molecular modeling of A3F on the basis of the A3C structure suggested that the A140 residue is part of α-helix 4, and substitution with a proline would likely result in a shortening of α-helix 4 and a large disruption of this structure (Fig. 8AIII) (62–64). In addition, an alternative alignment of A3F and A3G (Fig. 8AI, alignment 2) suggested that A140 could be aligned with a lysine in A3G. The sensitivity of the A3F single-amino-acid mutants to Vif2-mediated degradation was determined by Western blotting. The results indicated that the A140K and G141R substitutions were as sensitive to Vif2-mediated degradation as wild-type A3F, whereas the R143T and V144M substitutions were partially resistant to Vif2. The single-round infectivity assays indicated that all four single-amino-acid-substitution mutants retained their antiviral activity, and infectivity was fully restored by Vif2 in the presence of the A140K and G141R mutations and only partly restored in the presence of the R143T and V144M mutations. Taken together, these results suggest that the box 3 amino acids AGARV in A3F constitute a determinant that confers Vif2 sensitivity, with amino acids R143 and V144 playing a more important role in Vif2 sensitivity than A140 and G141.
FIG 8.

Identification of a determinant in A3F that confers sensitivity to Vif2. (AI) Two alignments of A3F amino acids 120 to 144 with the corresponding amino acids in A3G are shown. Dots, identical amino acids; dashes, gaps; box 1, box 2, and box 3, regions that contain significant differences between A3F and A3G. (AII) Schematics of A3F, the box1 amino acids 125YWERDYR131 (light shaded box) and mutant FG125-131F, and box 3 amino acids 140AGARV144 (dark shaded box) and mutants PRATM, A140K, G141R, R143T, and V144M are shown. (AIII) The predicted structure for A3F N-terminal amino acids 1 to 189 was simulated by the SwissModel server using the APOBEC3C crystal structure as a template (73). The model shows that replacement of A140 with a proline, but not a lysine, results in shortening of helix 4. Amino acids R143 and V144, shown in fuchsia, are predicted to be part of β-sheet 5. (B) Sensitivity of FG125-131F and CRLS>RSLC mutants to Vif2-mediated degradation. Quantitative Western blot analyses of cell lysates were performed by using an anti-FLAG antibody to detect FLAG-A3F, FG125-131F, or the A3F CRLS>RSLC mutant, an anti-HA antibody to detect hVif2-HA, and an anti-α-tubulin antibody to detect α-tubulin. Lanes +, with Vif2; lanes −, without Vif2; triangle, lanes derived from the same blot with extraneous lanes were removed. (CI) Sensitivity of A3F box 3 mutants to Vif2-mediated degradation. Quantitative Western blots were performed as described in the legend to panel B. (CII) Quantitation of the amount of A3F remaining relative to the amount for the no-Vif2 control. The error bars indicate standard deviations from three independent experiments. (D) hVif2-HA rescue of HIV-1 ΔVif infectivity in the presence of A3F and its mutants. Viruses were produced and infectivity was determined as described in the legend to Fig. 2. Data are plotted as relative infectivity levels, with the level in the absence of any A3 protein being set equal to 100%. Error bars indicate standard errors of the means (SEMs) of the results from three independent experiments. A P value of <0.05 was considered statistically significant (*).
DISCUSSION
In these studies, we aimed to gain a better understanding of the host-pathogen interactions of HIV-2 by examining the Vif2-APOBEC3 interactions using mutational analyses combined with HEK293T cell cotransfections and TZM-bl cell infectivity assays to identify amino acids critical to these interactions. This robust system was used for identification of residues critical for Vif1-APOBEC3 interactions (34, 40) that we subsequently confirmed in both T cell lines and primary CD4+ T cells (41, 65). A summary of the Vif2 interactions with human A3G and A3F and their comparison to previously determined Vif1 interactions are shown in Fig. 9A. Vif2 amino acids P16, M19, E20, P44, H45, and V47 are important for interaction with A3F, whereas the 140AGARV144 motif in the N-terminal half of A3F is critical for interaction with Vif2. In contrast, previous studies have shown that the highly charged 14DRMR17 motif in Vif1 is critical for interaction with A3F, while the 289EFLARH294 motif in the C-terminal half of A3F is critical for interaction with Vif1. Our results showed that G48 of Vif2 was essential for specific interaction with A3G, and analysis of GF chimeras localized the Vif2 interaction domain to amino acids 163 to 321 of A3G. Additional studies are needed to identify the specific A3G determinants that are important for interaction with Vif2. Previous studies showed that the 40YRHHY44 motif of Vif1 is critical for interaction with A3G and the 128DPD130 motif of A3G is critical for interaction with Vif1. In addition, we identified a region important for A3H-hapII neutralization that is not required for A3F or A3G neutralization. Studies conducted by us and others have determined that Vif1 also has distinct regions for the neutralization of A3F, A3G, and A3H-hapII (34–39). It is important to point out that it cannot be concluded that these motifs that have been identified to be critical for interaction through mutational analyses are themselves directly involved in the protein-protein interactions. Structures of Vif-A3 complexes are needed to determine whether the same motifs directly interact or indirectly affect the interactions.
FIG 9.
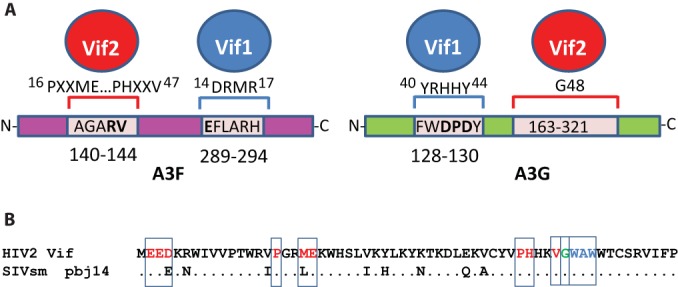
Summary of Vif2 and Vif1 interactions with human A3F and A3G (A) and alignment of the N-terminal 60 amino acids of HIV-2 Vif and SIVsm strain pbj14 Vif (B). Red letters, amino acids critical for specifically inducing the degradation of A3F; green letter G, G48, which is critical for inducing the degradation of A3G; blue letters, a multiple-alanine mutant (W49, A50, W51>ASA) that was defective in inducing degradation of A3H-hapII.
Although HIV-1 and HIV-2 are both human retroviruses that are capable of causing AIDS, each of these pathogens arose via distinct zoonotic sources, and these viruses differ greatly at the molecular level and in pathogenesis. A comparison of the N-terminal amino acid sequences of Vif2 and SIVsm Vif indicates that the amino acids identified to be critical for interactions with A3G, A3F, and A3H-hapII are highly conserved (Fig. 9B). The only exceptions are a conserved D-to-E change at amino acid 4 and an M-to-L change at amino acid 16. The high conservation of these amino acids suggests that the interactions that are critical for degradation of A3G and A3F are present in the SIVsm Vif. Consistent with this, Letko et al. recently showed that SIVsm Vif proteins can efficiently degrade human A3G (11). The conservation of these critical Vif2 amino acids suggests that the SIVsm Vif possessed the ability to degrade human A3G and A3F, which facilitated its zoonotic transmission to humans to form HIV-2. Interestingly, it was recently determined that Vif in rhesus macaque SIV (SIVmac) adapted to better antagonize one allelic A3G variant in rhesus macaques that is insensitive to SIVsm, while it retained its ability to antagonize other rhesus macaque A3G alleles and A3G from sooty mangabeys (19). Additional studies are needed to determine whether the amino acids that we have identified in HIV-2 Vif are also important for SIVsm Vif to induce degradation of human and sooty mangabey A3G and A3F. Finally, since the zoonotic transmission of SIVsm to rhesus macaques gave rise to SIVmac, it would be interesting to determine whether the same amino acids in SIVsm Vif are critical for inducing the degradation of rhesus macaque A3G and A3F.
Systematic studies of APOBEC3 family member expression patterns have demonstrated that in primary CD4+ T cells, the major target cells of HIV infection, A3B, A3D, A3F, A3G, and A3H consistently showed moderate to significant expression, but it is less clear whether A3A and A3C can be readily expressed (66, 67). In addition, while it seems that the levels of A3D, A3F, and A3G expression are sufficient to inhibit Vif-deficient HIV-1 in primary CD4+ T cells (41, 68), it is unknown which A3 proteins can inhibit replication of Vif-deficient HIV-2 in this cell type and whether Vif2 can counteract these antiviral proteins. The results of our studies showed that A3A was insensitive to Vif2 and A3B, which is Vif1 insensitive (43), displayed partial sensitivity to Vif2-induced degradation. While A3A expression may be induced in CD4+ T cells and dendritic cells under certain cell treatment conditions (67), other conditions indicate that induced A3A expression is poor (66) and A3B induction is only moderate (66, 67). Interestingly, although A3D is highly sensitive to Vif1 and is targeted by Vif1 through the same motif as A3C and A3F (the EFLARH domain) (42), it was not very sensitive to Vif2, which is consistent with previous findings (69). Although A3D expression is inducible in CD4+ T cells (66) and macrophages (67), human A3D cannot strongly inhibit Vif-deficient HIV-2 in single-cycle assays (70). Whether A3D plays a major role in HIV-2 replication remains to be clarified. Interestingly, Vif from SIVmac, which shares the SIVsm origins with HIV-2, can degrade rhesus macaque A3D (71). It would be interesting to determine whether human A3D and sooty mangabey A3D retained sensitivity to SIVsm Vif, as it has been previously shown that SIV Vifs can adapt to antagonize A3G variants in different hosts (19).
As with Vif1, A3C and A3G can be targeted by Vif2 for efficient degradation, while A3F is also sensitive, albeit to a lesser extent. Our results also indicate that A3H-hapII is efficiently degraded by the Vif2 used in our studies (derived from HIV-2ROD12). Because A3H-hapII sensitivity to Vif1 is dependent on the subtype from which Vif1 is derived (35, 72), it would be interesting to determine whether A3H-hapII is sensitive to other Vif2 variants as well.
Interestingly, Vif2 binding was readily detected by Western blotting for all APOBEC3 proteins except A3A, despite the differences in Vif2-induced degradation. It is possible that interaction with another host factor, such as CBFβ, is required for inducing degradation and that the interaction with a putative host factor occurs for some APOBEC3 proteins but not others. An alternative possibility is that after binding to Vif2, APOBEC3 proteins have to undergo a conformational change that induces ubiquitination and degradation, and this conformational change occurs with some APOBEC3 proteins but not others. These experiments provide insight into the A3 proteins that can be targeted by Vif2, and it would be interesting to determine which of these proteins can inhibit the replication of HIV-2 and induce hypermutation ex vivo.
Our results showed that the residues in A3F and A3G that are critical for inducing degradation by Vif1 are dispensable for inducing degradation by Vif2. Letko et al. recently reported that A3G amino acid P129 is an important determinant for degradation by SIVsm Vif, which is the ancestor of Vif2, on the basis of the observation that a P129D mutant of A3G is resistant to SIVsm Vif-mediated degradation, and its association with SIVsm Vif as well as Vif2 is reduced (11). Our results confirmed that the P129D mutant is resistant to Vif2 and inhibits viral replication in the presence of Vif2. However, we also observed that P129A was sensitive to Vif2, as were other Vif1-resistant mutants, D128K, GFG2, and A3G>5A. Furthermore, analysis of GF chimeras showed that the Vif2 sensitivity domain of A3G resides between residues 163 and 321. Taken together, these results suggest that Vif2 can readily induce degradation when the Vif1 interaction domain is mutated in A3G and that Vif2 interacts with A3G in a manner distinct from the interaction with Vif1. The observation by Letko et al. (11) and our studies indicate that the P129D mutant is indeed partially resistant to Vif2-induced degradation. The mechanism by which the P129D residue confers partial Vif2 resistance is not clear and needs to be investigated further. Alternative possibilities are that addition of another aspartic acid within this negatively charged motif either alters the dimerization of A3G proteins or alters their conformation, which affects Vif2 binding at a distal site. In this regard, Compton and Emerman (12) recently reported that the P129 residue is conserved in numerous species of Old World monkeys (OWMs); despite this conservation, the A3G proteins from the African green monkey (Chlorocebus sabaeus) and the mantled colobus monkey (Colobus guerza) are resistant to degradation by Vif2. Furthermore, Vifs from SIVs of the same lineage as HIV-2, SIVsm and SIVmac, had variable activity against A3Gs from diverse OWM species, despite the ubiquitous P at position 129. These results, along with our results with the P129A mutant and the GFG2 mutant in which P129 was replaced by R, strongly suggest that the identity of the P129 residue is not critical for A3G's sensitivity to Vif2.
Similar to the interactions of A3G with Vif1 and Vif2, the interactions of A3F with Vif1 and Vif2 are also distinct. Our results with the A3F-E289K and FGF4 mutants indicate that Vif2 does not recognize the Vif1 interaction site in A3F (289EFLARH294). In addition, the A3F>5A mutant data indicate that the 127ERDYRR132 region in A3F does not play a role in the neutralization of A3F by Vif2. Furthermore, our observation that the G120F120 chimera is sensitive to Vif2 but the G149F145 and G163F159 chimeras are resistant to Vif2 localized the Vif2 interaction site in A3F to amino acids 121 to 144. Additional mutational analysis indicates that amino acids R143 and V144 in A3F constitute a determinant that is critical for Vif2-mediated degradation of A3F. Finally, the observation that the F248G257 and F282G291 chimeras are sensitive to Vif2 but resistant to Vif1 supports the view that the A3F determinants that interact with Vif1 and Vif2 are distinct (40).
In summary, HIV-1 and HIV-2 can both target APOBEC3s via their Vif proteins. Although each Vif protein has distinct recognition sites to specifically target A3F and A3G, the sites in A3F and A3G recognized by these two dissimilar Vifs are distinct. Although it is likely to require future studies with additional extensive mutational analysis, it will be interesting to determine the exact site(s) in A3G that is targeted by Vif2.
ACKNOWLEDGMENTS
We thank Hibiki Doi and Rebecca Russell for technical assistance and Wei-Shau Hu for valuable discussion throughout the studies and manuscript preparation. We also thank Viviana Simon for valuable reagents and discussions.
This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
The content of this publication does not necessarily reflect the views or policies of the U.S. Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Published ahead of print 18 June 2014
REFERENCES
- 1.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113:803–809. 10.1016/S0092-8674(03)00423-9 [DOI] [PubMed] [Google Scholar]
- 2.Lecossier D, Bouchonnet F, Clavel F, Hance AJ. 2003. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science 300:1112. 10.1126/science.1083338 [DOI] [PubMed] [Google Scholar]
- 3.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424:99–103. 10.1038/nature01709 [DOI] [PubMed] [Google Scholar]
- 4.Suspene R, Sommer P, Henry M, Ferris S, Guetard D, Pochet S, Chester A, Navaratnam N, Wain-Hobson S, Vartanian JP. 2004. APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res. 32:2421–2429. 10.1093/nar/gkh554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu Q, Konig R, Pillai S, Chiles K, Kearney M, Palmer S, Richman D, Coffin JM, Landau NR. 2004. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 11:435–442. 10.1038/nsmb758 [DOI] [PubMed] [Google Scholar]
- 6.Zhang H, Yang B, Pomerantz RJ, Zhang CM, Arunachalam SC, Gao L. 2003. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424:94–98. 10.1038/nature01707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Izumi T, Shirakawa K, Takaori-Kondo A. 2008. Cytidine deaminases as a weapon against retroviruses and a new target for antiviral therapy. Mini Rev. Med. Chem. 8:231–238. 10.2174/138955708783744047 [DOI] [PubMed] [Google Scholar]
- 8.Izumi T, Burdick R, Shigemi M, Plisov S, Hu WS, Pathak VK. 2013. Mov10 and APOBEC3G localization to processing bodies is not required for virion incorporation and antiviral activity. J. Virol. 87:11047–11062. 10.1128/JVI.02070-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Izumi T, Takaori-Kondo A, Shirakawa K, Higashitsuji H, Itoh K, Io K, Matsui M, Iwai K, Kondoh H, Sato T, Tomonaga M, Ikeda S, Akari H, Koyanagi Y, Fujita J, Uchiyama T. 2009. MDM2 is a novel E3 ligase for HIV-1 Vif. Retrovirology 6:1. 10.1186/1742-4690-6-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Izumi T, Io K, Matsui M, Shirakawa K, Shinohara M, Nagai Y, Kawahara M, Kobayashi M, Kondoh H, Misawa N, Koyanagi Y, Uchiyama T, Takaori-Kondo A. 2010. HIV-1 viral infectivity factor interacts with TP53 to induce G2 cell cycle arrest and positively regulate viral replication. Proc. Natl. Acad. Sci. U. S. A. 107:20798–20803. 10.1073/pnas.1008076107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Letko M, Silvestri G, Hahn BH, Bibollet-Ruche F, Gokcumen O, Simon V, Ooms M. 2013. Vif proteins from diverse primate lentiviral lineages use the same binding site in APOBEC3G. J. Virol. 87:11861–11871. 10.1128/JVI.01944-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Compton AA, Emerman M. 2013. Convergence and divergence in the evolution of the APOBEC3G-Vif interaction reveal ancient origins of simian immunodeficiency viruses. PLoS Pathog. 9:e1003135. 10.1371/journal.ppat.1003135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Compton AA, Malik HS, Emerman M. 2013. Host gene evolution traces the evolutionary history of ancient primate lentiviruses. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368:20120496. 10.1098/rstb.2012.0496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu XH, Yu YK, Liu BD, Luo K, Kong W, Mao PY, Yu XF. 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302:1056–1060. 10.1126/science.1089591 [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi M, Takaori-Kondo A, Miyauchi Y, Iwai K, Uchiyama T. 2005. Ubiquitination of APOBEC3G by an HIV-1 Vif-Cullin5-Elongin B-Elongin C complex is essential for Vif function. J. Biol. Chem. 280:18573–18578. 10.1074/jbc.C500082200 [DOI] [PubMed] [Google Scholar]
- 16.Shindo K, Takaori-Kondo A, Kobayashi M, Abudu A, Fukunaga K, Uchiyama T. 2003. The enzymatic activity of CEM15/Apobec-3G is essential for the regulation of the infectivity of HIV-1 virion but not a sole determinant of its antiviral activity. J. Biol. Chem. 278:44412–44416. 10.1074/jbc.C300376200 [DOI] [PubMed] [Google Scholar]
- 17.Marin M, Rose KM, Kozak SL, Kabat D. 2003. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 9:1398–1403. 10.1038/nm946 [DOI] [PubMed] [Google Scholar]
- 18.Sheehy AM, Gaddis NC, Malim MH. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9:1404–1407. 10.1038/nm945 [DOI] [PubMed] [Google Scholar]
- 19.Krupp A, McCarthy KR, Ooms M, Letko M, Morgan JS, Simon V, Johnson WE. 2013. APOBEC3G polymorphism as a selective barrier to cross-species transmission and emergence of pathogenic SIV and AIDS in a primate host. PLoS Pathog. 9:e1003641. 10.1371/journal.ppat.1003641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bieniasz PD. 2004. Intrinsic immunity: a front-line defense against viral attack. Nat. Immunol. 5:1109–1115. 10.1038/ni1125 [DOI] [PubMed] [Google Scholar]
- 21.Yan N, Chen ZJJ. 2012. Intrinsic antiviral immunity. Nat. Immunol. 13:214–222. 10.1038/ni.2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirmaier A, Wu F, Newman RM, Hall LR, Morgan JS, O'Connor S, Marx PA, Meythaler M, Goldstein S, Buckler-White A, Kaur A, Hirsch VM, Johnson WE. 2010. TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS Biol. 8:e1000462. 10.1371/journal.pbio.1000462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Etienne L, Hahn BH, Sharp PM, Matsen FA, Emerman M. 2013. Gene loss and adaptation to hominids underlie the ancient origin of HIV-1. Cell Host Microbe 14:85–92. 10.1016/j.chom.2013.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharp PM, Hahn BH. 2011. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 1:a006841. 10.1101/cshperspect.a006841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaddis NC, Sheehy AM, Ahmad KM, Swanson CM, Bishop KN, Beer BE, Marx PA, Gao F, Bibollet-Ruche F, Hahn BH, Malim MH. 2004. Further investigation of simian immunodeficiency virus Vif function in human cells. J. Virol. 78:12041–12046. 10.1128/JVI.78.21.12041-12046.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyerson NR, Sawyer SL. 2011. Two-stepping through time: mammals and viruses. Trends Microbiol. 19:286–294. 10.1016/j.tim.2011.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel MR, Emerman M, Malik HS. 2011. Paleovirology—ghosts and gifts of viruses past. Curr. Opin. Virol. 1:304–309. 10.1016/j.coviro.2011.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson WE. 2013. Rapid adversarial co-evolution of viruses and cellular restriction factors. Curr. Top. Microbiol. Immunol. 371:123–151. 10.1007/978-3-642-37765-5_5 [DOI] [PubMed] [Google Scholar]
- 29.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302:1056–1060. 10.1126/science.1089591 [DOI] [PubMed] [Google Scholar]
- 30.Jager S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, Hernandez H, Jang GM, Roth SL, Akiva E, Marlett J, Stephens M, D'Orso I, Fernandes J, Fahey M, Mahon C, O'Donoghue AJ, Todorovic A, Morris JH, Maltby DA, Alber T, Cagney G, Bushman FD, Young JA, Chanda SK, Sundquist WI, Kortemme T, Hernandez RD, Craik CS, Burlingame A, Sali A, Frankel AD, Krogan NJ. 2012. Global landscape of HIV-human protein complexes. Nature 481:365–370. 10.1038/nature10719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stanley DJ, Bartholomeeusen K, Crosby DC, Kim DY, Kwon E, Yen L, Cartozo NC, Li M, Jager S, Mason-Herr J, Hayashi F, Yokoyama S, Krogan NJ, Harris RS, Peterlin BM, Gross JD. 2012. Inhibition of a NEDD8 cascade restores restriction of HIV by APOBEC3G. PLoS Pathog. 8:e1003085. 10.1371/journal.ppat.1003085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jager S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, Mahon C, Kane J, Franks-Skiba K, Cimermancic P, Burlingame A, Sali A, Craik CS, Harris RS, Gross JD, Krogan NJ. 2012. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 481:371–375. 10.1038/nature10693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang W, Du J, Evans SL, Yu Y, Yu XF. 2012. T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature 481:376–379. 10.1038/nature10718 [DOI] [PubMed] [Google Scholar]
- 34.Russell RA, Pathak VK. 2007. Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J. Virol. 81:8201–8210. 10.1128/JVI.00395-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Binka M, Ooms M, Steward M, Simon V. 2012. The activity spectrum of Vif from multiple HIV-1 subtypes against APOBEC3G, APOBEC3F, and APOBEC3H. J. Virol. 86:49–59. 10.1128/JVI.06082-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simon V, Zennou V, Murray D, Huang Y, Ho DD, Bieniasz PD. 2005. Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog. 1:e6. 10.1371/journal.ppat.0010006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen G, He Z, Wang T, Xu R, Yu XF. 2009. A patch of positively charged amino acids surrounding the human immunodeficiency virus type 1 Vif SLVx4Yx9Y motif influences its interaction with APOBEC3G. J. Virol. 83:8674–8682. 10.1128/JVI.00653-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He Z, Zhang W, Chen G, Xu R, Yu XF. 2008. Characterization of conserved motifs in HIV-1 Vif required for APOBEC3G and APOBEC3F interaction. J. Mol. Biol. 381:1000–1011. 10.1016/j.jmb.2008.06.061 [DOI] [PubMed] [Google Scholar]
- 39.Tian C, Yu X, Zhang W, Wang T, Xu R, Yu XF. 2006. Differential requirement for conserved tryptophans in human immunodeficiency virus type 1 Vif for the selective suppression of APOBEC3G and APOBEC3F. J. Virol. 80:3112–3115. 10.1128/JVI.80.6.3112-3115.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Russell RA, Smith J, Barr R, Bhattacharyya D, Pathak VK. 2009. Distinct domains within APOBEC3G and APOBEC3F interact with separate regions of human immunodeficiency virus type 1 Vif. J. Virol. 83:1992–2003. 10.1128/JVI.01621-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chaipan C, Smith JL, Hu WS, Pathak VK. 2013. APOBEC3G restricts HIV-1 to a greater extent than APOBEC3F and APOBEC3DE in human primary CD4+ T cells and macrophages. J. Virol. 87:444–453. 10.1128/JVI.00676-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith JL, Pathak VK. 2010. Identification of specific determinants of human APOBEC3F, APOBEC3C, and APOBEC3DE and African green monkey APOBEC3F that interact with HIV-1 Vif. J. Virol. 84:12599–12608. 10.1128/JVI.01437-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doehle BP, Schafer A, Cullen BR. 2005. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology 339:281–288. 10.1016/j.virol.2005.06.005 [DOI] [PubMed] [Google Scholar]
- 44.Nguyen KL, Llano M, Akari H, Miyagi E, Poeschla EM, Strebel K, Bour S. 2004. Codon optimization of the HIV-1 vpu and vif genes stabilizes their mRNA and allows for highly efficient Rev-independent expression. Virology 319:163–175. 10.1016/j.virol.2003.11.021 [DOI] [PubMed] [Google Scholar]
- 45.Xu HZ, Svarovskaia ES, Barr R, Zhang YJ, Khan MA, Strebel K, Pathak VK. 2004. A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc. Natl. Acad. Sci. U. S. A. 101:5652–5657. 10.1073/pnas.0400830101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang SL, Delgado R, King SR, Woffendin C, Barker CS, Yang ZY, Xu L, Nolan GP, Nabel GJ. 1999. Generation of retroviral vector for clinical studies using transient transfection. Hum. Gene Ther. 10:123–132. 10.1089/10430349950019255 [DOI] [PubMed] [Google Scholar]
- 47.Derdeyn CA, Decker JM, Sfakianos JN, Wu XY, O'Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E. 2000. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J. Virol. 74:8358–8367. 10.1128/JVI.74.18.8358-8367.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei XP, Decker JM, Liu HM, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu XY, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46:1896–1905. 10.1128/AAC.46.6.1896-1905.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burdick R, Smith JL, Chaipan C, Friew Y, Chen J, Venkatachari NJ, Delviks-Frankenberry KA, Hu WS, Pathak VK. 2010. P body-associated protein Mov10 inhibits HIV-1 replication at multiple stages. J. Virol. 84:10241–10253. 10.1128/JVI.00585-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Unutmaz D, KewalRamani VN, Marmon S, Littman DR. 1999. Cytokine signals are sufficient for HIV-1 infection of resting human T lymphocytes. J. Exp. Med. 189:1735–1746. 10.1084/jem.189.11.1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yee JK, Friedmann T, Burns JC. 1994. Generation of high-titer pseudotyped retroviral vectors with very broad-host-range. Methods Cell Biol. 43:99–112. 10.1016/S0091-679X(08)60600-7 [DOI] [PubMed] [Google Scholar]
- 52.Goncalves J, Jallepalli P, Gabuzda DH. 1994. Subcellular localization of the Vif protein of human immunodeficiency virus type 1. J. Virol. 68:704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Refsland EW, Hultquist JF, Harris RS. 2012. Endogenous origins of HIV-1 G-to-A hypermutation and restriction in the nonpermissive T cell line CEM2n. PLoS Pathog. 8:e1002800. 10.1371/journal.ppat.1002800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miyagi E, Brown CR, Opi S, Khan M, Goila-Gaur R, Kao S, Walker RC, Jr, Hirsch V, Strebel K. 2010. Stably expressed APOBEC3F has negligible antiviral activity. J. Virol. 84:11067–11075. 10.1128/JVI.01249-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schrofelbauer B, Chen D, Landau NR. 2004. A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif). Proc. Natl. Acad. Sci. U. S. A. 101:3927–3932. 10.1073/pnas.0307132101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mangeat B, Turelli P, Liao S, Trono D. 2004. A single amino acid determinant governs the species-specific sensitivity of APOBEC3G to Vif action. J. Biol. Chem. 279:14481–14483. 10.1074/jbc.C400060200 [DOI] [PubMed] [Google Scholar]
- 57.Bogerd HP, Doehle BP, Wiegand HL, Cullen BR. 2004. A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV type 1 virion infectivity factor. Proc. Natl. Acad. Sci. U. S. A. 101:3770–3774. 10.1073/pnas.0307713101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Albin JS, LaRue RS, Weaver JA, Brown WL, Shindo K, Harjes E, Matsuo H, Harris RS. 2010. A single amino acid in human APOBEC3F alters susceptibility to HIV-1 Vif. J. Biol. Chem. 285:40785–40792. 10.1074/jbc.M110.173161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bach D, Peddi S, Mangeat B, Lakkaraju A, Strub K, Trono D. 2008. Characterization of APOBEC3G binding to 7SL RNA. Retrovirology 5:54. 10.1186/1742-4690-5-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang T, Tian C, Zhang W, Luo K, Sarkis PT, Yu L, Liu B, Yu Y, Yu XF. 2007. 7SL RNA mediates virion packaging of the antiviral cytidine deaminase APOBEC3G. J. Virol. 81:13112–13124. 10.1128/JVI.00892-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huthoff H, Autore F, Gallois-Montbrun S, Fraternali F, Malim MH. 2009. RNA-dependent oligomerization of APOBEC3G is required for restriction of HIV-1. PLoS Pathog. 5:e1000330. 10.1371/journal.ppat.1000330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arnold K, Bordoli L, Kopp J, Schwede T. 2006. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. 10.1093/bioinformatics/bti770 [DOI] [PubMed] [Google Scholar]
- 63.Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. 2009. The SWISS-MODEL repository and associated resources. Nucleic Acids Res. 37:D387–D392. 10.1093/nar/gkn750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peitsch MC, Wells TN, Stampf DR, Sussman JL. 1995. The Swiss-3DImage collection and PDB-Browser on the World-Wide Web. Trends Biochem. Sci. 20:82–84. 10.1016/S0968-0004(00)88963-X [DOI] [PubMed] [Google Scholar]
- 65.Russell RA, Moore MD, Hu WS, Pathak VK. 2009. APOBEC3G induces a hypermutation gradient: purifying selection at multiple steps during HIV-1 replication results in levels of G-to-A mutations that are high in DNA, intermediate in cellular viral RNA, and low in virion RNA. Retrovirology 6:16. 10.1186/1742-4690-6-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. 2010. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 38:4274–4284. 10.1093/nar/gkq174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koning FA, Newman EN, Kim EY, Kunstman KJ, Wolinsky SM, Malim MH. 2009. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 83:9474–9485. 10.1128/JVI.01089-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gillick K, Pollpeter D, Phalora P, Kim EY, Wolinsky SM, Malim MH. 2013. Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. J. Virol. 87:1508–1517. 10.1128/JVI.02587-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Duggal NK, Fu W, Akey JM, Emerman M. 2013. Identification and antiviral activity of common polymorphisms in the APOBEC3 locus in human populations. Virology 443:329–337. 10.1016/j.virol.2013.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Duggal NK, Malik HS, Emerman M. 2011. The breadth of antiviral activity of Apobec3DE in chimpanzees has been driven by positive selection. J. Virol. 85:11361–11371. 10.1128/JVI.05046-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Virgen CA, Hatziioannou T. 2007. Antiretroviral activity and Vif sensitivity of rhesus macaque APOBEC3 proteins. J. Virol. 81:13932–13937. 10.1128/JVI.01760-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ooms M, Letko M, Binka M, Simon V. 2013. The resistance of human APOBEC3H to HIV-1 NL4-3 molecular clone is determined by a single amino acid in Vif. PLoS One 8:e57744. 10.1371/journal.pone.0057744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kitamura S, Ode H, Nakashima M, Imahashi M, Naganawa Y, Kurosawa T, Yokomaku Y, Yamane T, Watanabe N, Suzuki A, Sugiura W, Iwatani Y. 2012. The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat. Struct. Mol. Biol. 19:1005–1010. 10.1038/nsmb.2378 [DOI] [PubMed] [Google Scholar]