

ABSTRACT
Immune-mediated lung injury is a hallmark of lower respiratory tract illness caused by respiratory syncytial virus (RSV). STAT4 plays a critical role in CD4+ Th1 lineage differentiation and gamma interferon (IFN-γ) protein expression by CD4+ T cells. As CD4+ Th1 differentiation is associated with negative regulation of CD4+ Th2 and Th17 differentiation, we hypothesized that RSV infection of STAT4−/− mice would result in enhanced lung Th2 and Th17 inflammation and impaired lung Th1 inflammation compared to wild-type (WT) mice. We performed primary and secondary RSV challenges in WT and STAT4−/− mice and used STAT1−/− mice as a positive control for the development of RSV-specific lung Th2 and Th17 inflammation during primary challenge. Primary RSV challenge of STAT4−/− mice resulted in decreased T-bet and IFN-γ expression levels in CD4+ T cells compared to those of WT mice. Lung Th2 and Th17 inflammation did not develop in primary RSV-challenged STAT4−/− mice. Decreased IFN-γ expression by NK cells, CD4+ T cells, and CD8+ T cells was associated with attenuated weight loss and enhanced viral clearance with primary challenge in STAT4−/− mice compared to WT mice. Following secondary challenge, WT and STAT4−/− mice also did not develop lung Th2 or Th17 inflammation. In contrast to primary challenge, secondary RSV challenge of STAT4−/− mice resulted in enhanced weight loss, an increased lung IFN-γ expression level, and an increased lung RSV-specific CD8+ T cell response compared to those of WT mice. These data demonstrate that STAT4 regulates the RSV-specific CD8+ T cell response to secondary infection but does not independently regulate lung Th2 or Th17 immune responses to RSV challenge.
IMPORTANCE STAT4 is a protein critical for both innate and adaptive immune responses to viral infection. Our results show that STAT4 regulates the immune response to primary and secondary challenge with RSV but does not restrain RSV-induced lung Th2 or Th17 immune responses. These findings suggest that STAT4 expression may influence lung immunity and severity of illness following primary and secondary RSV infections.
INTRODUCTION
Respiratory syncytial virus (RSV) is a major cause of bronchiolitis and viral pneumonia in children, resulting in significant morbidity and mortality worldwide (1, 2). Despite the importance of this pathogen, there is no licensed RSV vaccine and, apart from passive immunoprophylaxis or the highly toxic antiviral ribavirin, no therapy for RSV-induced illness (3, 4). Immune-mediated lung injury is a hallmark of lower respiratory tract illness in the mouse model of RSV infection and may contribute to illness severity in human infections (5–7). Several cell types contribute to the lung immune response to RSV in mice. Gamma interferon (IFN-γ)-expressing NK cells and CD4+ and CD8+ T cells contribute to the clearance of RSV from the lung (8–12). However, in the course of viral clearance, this immune response causes significant immunopathology and lung damage (9–11). Depending on the virus strain and host immune context of challenge, lung immunopathology can be mediated by IFN-γ-expressing NK cells, CD4+ Th1 cells, and CD8+ T cells that enhance viral clearance or by aberrant CD4+ T cell immune responses, including interleukin-13 (IL-13)-predominant Th2 and/or IL-17A-predominant Th17 immune responses (8, 13–15).
STAT4 plays a critical role in the differentiation of naive CD4+ T cells into Th1 cells (16–20). IL-12 receptor engagement is the predominant cytokine signal that results in STAT4 phosphorylation, homodimerization, and translocation to the nucleus (16). STAT4 and T-bet, acting downstream of IL-12 and IFN-γ, induce Th1 differentiation and IFN-γ expression by CD4+ T cells (17–23). In the absence of STAT4, CD4+ Th1 differentiation and IFN-γ expression are impaired (16, 17, 19, 20), but complete differentiation of the CD4+ Th1 phenotype appears to require both STAT4 and T-bet (18, 21, 24, 25). In addition to its role in CD4+ Th1 differentiation, STAT4 is also critical for NK cell and CD8+ T cell effector functions (23, 26–31). In NK cells and CD8+ T cells, STAT4 acts downstream of IL-12 as well as type I interferons to induce cell proliferation, IFN-γ expression, and/or cytotoxicity.
In the course of CD4+ Th1 differentiation, both STAT4 and STAT1 are capable of inducing the expression of T-bet (20–22, 24, 25, 32, 33). The order and relative contribution of STAT4 and STAT1 to T-bet expression and Th1 cell differentiation have been a matter of considerable debate (20–22). Negative regulation of Th2 and Th17 differentiation pathways in Th1 cells appears to be primarily under the control of T-bet (25, 34–38). We previously reported that STAT1−/− mice challenged with RSV A2 have significantly increased IL-13 and IL-17A protein expression levels and airway mucus expression in their lungs compared to wild-type (WT) BALB/c mice (8, 13). This immune response is also characterized by eosinophilic and neutrophilic infiltration into the lung, in contrast to the predominantly lymphocytic inflammation in the lungs of RSV-challenged WT BALB/c mice. Thus, STAT1 signaling is required to inhibit Th2 (airway eosinophils; lung IL-13 expression) and Th17 (airway neutrophils; lung IL-17A expression) lung immune responses in the RSV primary challenge model.
In the present study, we sought to determine the role of STAT4 signaling in the inhibition of Th2 and Th17 and the promotion of Th1 immune responses to RSV challenge. As both STAT4 and STAT1 increase T-bet expression levels in the course of Th1 differentiation, and T-bet negatively regulates Th2 and Th17 immune responses, we hypothesized that primary RSV challenge of STAT4−/− mice results in enhanced lung Th2 and Th17 immune responses, similar to what we previously reported for STAT1−/− mice (13). We also hypothesized that primary RSV challenge of STAT4−/− mice results in significantly impaired IFN-γ expression by NK cells, CD4+ T cells, and CD8+ T cells and that this attenuates both postchallenge weight loss and viral clearance. While previous work showed that STAT4-deficient C57BL/6 mice develop increased airway mucus expression, airway hyperreactivity, and airway eosinophilia following RSV strain Line 19 challenge (39), that study did not measure lung Th2 or Th17 cytokine expression levels in STAT4−/− mice and used RSV strain Line 19, which was subsequently shown to induce significant airway mucus expression in WT BALB/c mice (14, 39, 40). Moreover, several studies using BALB/c mice in non-RSV models have shown that STAT4 deficiency does not promote lung Th2 immune responses (41–44). Thus, we undertook this study to address these discrepancies and to investigate the role of STAT4−/− in regulating the lung immune response to RSV A2 in BALB/c mice.
To investigate the first hypothesis, that STAT4−/− mice develop RSV-induced lung Th2 and Th17 immune responses, we challenged WT, STAT1−/−, and STAT4−/− mice with RSV A2 and measured airway inflammatory cell recruitment, lung cytokine expression, and CD4+ T cell expression of T-bet, GATA-3, and ROR-γt. To investigate the second hypothesis, that RSV-induced IFN-γ expression, illness, and viral clearance are impaired in STAT4−/− mice, we determined cell-specific IFN-γ expression, postchallenge weight loss, and viral clearance in WT and STAT4−/− mice. Previous work using mouse models showed that secondary RSV challenge resulted in Th2-skewed lung immune responses. We therefore performed experiments to determine the lung immune response to secondary RSV challenge in WT and STAT4−/− mice. Investigation of RSV challenge in the context of an altered host immune response is critical to an understanding of the balance between successful and pathological immune responses to RSV.
MATERIALS AND METHODS
Cells, virus, and mice.
HEp-2 cells were obtained from the ATCC. The A2 and Line 19 strains of RSV were propagated as previously described (14). Female, 6- to 8-week-old, BALB/cJ mice were purchased from Jackson Laboratories (Bar Harbor, ME). STAT4−/− mice (BALB/cJ background) were purchased from Jackson Laboratories. STAT1−/− mice were kindly provided by Joan Durbin (New York University School of Medicine). Age-matched female WT, STAT1−/−, and STAT4−/− mice were used for experiments. All mice were maintained under specific-pathogen-free conditions. In caring for the animals, investigators adhered to protocols outlined in the revised Guide for the Care and Use of Laboratory Animals (45).
RSV challenge.
Mice were anesthetized by intramuscular injection of a ketamine-xylazine solution and challenged intranasally with 107 PFU of RSV A2 in 100 μl Dulbecco modified Eagle medium (DMEM). In selected experiments, mice were challenged intranasally with 107 PFU of UV-inactivated RSV A2 (UVRSV). For experiments utilizing RSV Line 19, mice were challenged with UV-inactivated or live RSV A2 or RSV Line 19 at a titer of 105 PFU. Virus was UV inactivated by exposing the viral suspension in a glass vial on ice to a UV light source (UVC30, 254 nm, two 15-W bulbs, 120 V; Kendro Lab Products, Asheville, NC) for 40 min. The day of primary challenge was denoted day 0 for all experiments. In selected experiments, mice were challenged intranasally a second time with 107 PFU of RSV A2. Secondary challenge was performed on day 42 post-primary challenge. Post-secondary challenge days were denoted days 43, 44, 45, and 46 and correspond to days 1, 2, 3, and 4 following repeat challenge, respectively.
Quantitation of lung viral load.
The lung viral load from the left lungs of mice challenged with RSV was quantified by a plaque assay, as previously described (46). Briefly, left lungs were harvested and individually ground with precooled mortars and pestles in sterile ground glass. Lung homogenates were diluted immediately in a serial fashion and were used to inoculate subconfluent HEp-2 cells in 12-well dishes. Cells were overlaid with DMEM containing 10% fetal bovine serum (FBS) and 0.75% methylcellulose. At day 5 postinoculation, cells were formalin fixed and stained with hematoxylin and eosin (H&E). Plaques were quantified visually and normalized to lung weight for each individual mouse.
Analysis of inflammatory cell infiltration into bronchoalveolar lavage fluid.
To obtain bronchoalveolar lavage (BAL) fluid, saline was instilled through a tracheostomy tube and withdrawn via a syringe. Total cells were counted on a hemocytometer by using trypan blue exclusion. Cytologic examination was performed on cytospin preparations (Thermo Shandon), and cytospin slides were fixed and stained by using DiffQuik (American Scientific Products). Differential counts were based on counts of 200 cells, using standard morphological criteria to classify the cells as neutrophils, eosinophils, lymphocytes, or macrophages.
Cytokine ELISA.
Cytokine protein expression from the left lung homogenate was measured by using available DuoSet (IFN-γ), Quantikine (IL-13, IL-17A, and IL-27), or Verikine (IFN-α and IFN-β) enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN).
Serum RSV F-protein-specific antibody ELISA.
Sera collected at day 42 post-primary challenge were tested for the presence of RSV F-protein-specific antibodies (Abs) by an ELISA. Briefly, ectodomain F protein from RSV A2 was fused to a GCN4 trimerization domain and a His tag and expressed in mammalian cells, as previously described (47). One hundred fifty nanograms of soluble RSV F protein was adsorbed onto Immulon 2B (Thermo Scientific, Rochester, NY) plates overnight in phosphate-buffered saline (PBS) at 4°C. The plate was blocked with 1% bovine serum albumin (BSA) in PBS for 1 h at room temperature and washed with 0.05% Tween 20 in PBS. Seventy-five microliters of serial 2-fold dilutions of serum was added to the plate in duplicate and incubated for 1 h at room temperature. The plates were washed; horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (1:5,000), IgG1 (1:500), or IgG2a (1:500) was added; and plates were incubated at room temperature for another hour (Southern Biotech, Birmingham, AL). Following an additional wash, 75 μl of Ultra-TMB (Pierce, Rockford, IL) was added to each well. The reaction was stopped by adding 100 μl of 1 M HCl to the mixture. Absorbance was measured at 450 nm. The serum endpoint dilution at 0.2 absorbance units above background (PBS blank) was calculated for each antibody type.
Flow cytometry.
Lungs were harvested, minced, and digested for 40 min at 37°C in RPMI 1640 containing 5% FBS, collagenase type I (1.25 mg/ml), and DNase (0.2 mg/ml). The digestion was stopped with 50 μl of 0.5 M EDTA, and minced lung was passed through a 70-μm strainer. For intracellular cytokine staining (ICS), cells were stimulated in a solution containing RPMI 1640, 10% FBS, 1 μM ionomycin (Sigma-Aldrich, St. Louis, MO), 50 ng/ml phorbol myristate acetate (PMA) (Sigma-Aldrich), and 0.07% GolgiStop (BD Pharmingen, Franklin Lakes, NJ) for 6 h at 37°C in 5% CO2. In experiments to determine RSV-specific CD8+ T cell IFN-γ expression levels, cells were stimulated in a solution containing RPMI with FBS, GolgiStop, and 1 μM synthetic peptide representing the H-2Kd-restricted CD8+ T cell epitope M282–90 (SYIGSINNI) (Biosynthesis, Lewisville, TX). Cells were counted by using a hemocytometer, and the total number of cells from the lungs of each mouse was recorded. One million cells from the lungs of each mouse were stained for cell surface molecules and intracellular cytokines, as described previously (48). Cell viability was determined by using a Live/Dead blue staining kit (Invitrogen, Life Technologies, Grand Island, NY). We used the following antibodies: anti-CD3ε (clone 145-2C11), anti-CD4 (clone H129.19), anti-CD8a (clone 53-6.7), anti-IFN-γ (clone XMG1.2), anti-CD49b (DX5) (clone HMα2), and anti-CD19 (clone 1D3) from BD Pharmingen and anti-NKG2D (clone CX5), anti-GATA-3 (clone TWAJ), anti-T-bet (clone 4B10), and anti-ROR-γt (clone AFKJS-9) from eBioscience (San Diego, CA). IgG1κ (clone P3.6.2.8.1), IgG2aκ (clone eBR2a), and IgG2bκ (clone eB149/10H5) were used as isotype controls for anti-T-bet, anti-ROR-γt, and anti-GATA-3, respectively (eBioscience). Anti-CD16/32 Ab (BD Pharmingen) was used to prevent nonspecific staining. RSV-specific CD8+ T cells were quantified by using an H-2Kd–M282–90 tetramer purchased from Beckman-Coulter (Fullerton, CA). CD19 surface staining was performed to exclude B cells from tetramer flow, as previously described (49). Nonspecific background tetramer staining was determined by using an H-2Kd–HA204–212 influenza virus tetramer (Beckman-Coulter) (50, 51). Cell samples were analyzed by using an LSR II flow cytometer (BD Biosciences). Data were analyzed by using FlowJo software (Tree Star, Ashland, OR).
Lung fixation and histological examination.
Mice were sacrificed on day 8 postchallenge. Following perfusion of the right ventricle with PBS, the trachea was exposed and cannulated, and lungs were inflated with 10% neutral buffered formalin. Lungs were removed and fixed in 10% neutral buffered formalin for 24 h at 4°C prior to processing and embedding. Five-micrometer sections were cut and stained with H&E or periodic acid-Schiff (PAS) stain. Lungs were evaluated by light microscopy by an experienced veterinary pathologist (K.L.B.) blind to group assignment. A semiquantitative scoring system was used for four parameters: airway mucus, peribronchiolar inflammation, perivascular inflammation, and interstitial pneumonia. Inflammation and pneumonia were scored on a scale of 0 to 4, and airway mucus was scored on a scale of 0 to 3, with 0 denoting the absence of the parameter in the lung. Perivascular and peribronchiolar infiltrates in H&E sections were scored as follows: 1 for minimal infiltrates of leukocytes observed around airways or vessels, 2 for mild infiltration of leukocytes around airways or vessels, 3 for moderate infiltration of leukocytes around airways or vessels, and 4 for severe infiltration of leukocytes around airways or vessels. Interstitial pneumonia in H&E sections was scored as follows: 1 for few leukocytes observed in alveolar spaces with no inflammation in alveolar walls, 2 for few leukocytes in alveolar spaces with mild inflammation/thickening of alveolar walls; 3 for many leukocytes in alveolar spaces with moderate thickening of alveolar walls, and 4 for marked numbers of leukocytes in alveolar spaces with extensive inflammation/thickening of alveolar walls. Airway mucus in PAS sections was scored as follows: 1 for <10% PAS-positive cells, 2 for 10 to 30% PAS-positive cells, and 3 for >30% PAS-positive cells.
Statistical analyses.
Experimental groups were compared by Student's t test or one-way analysis of variance (ANOVA) with Bonferroni posttest correction in order to calculate P values. Comparison between weight curves was performed by repeated-measures ANOVA. Statistical analyses were performed by using GraphPad Prism 5.01 (GraphPad, San Diego, CA). ELISA and viral titer data values below the limit of detection were assigned a value of half the limit of detection.
RESULTS
STAT4 deficiency does not result in RSV-induced Th2 or Th17 lung immune responses to primary challenge.
WT BALB/c mice challenged with RSV A2 develop a lung CD4+ T cell response to challenge that is characterized by IFN-γ expression by CD4+ Th1 cells, CD8+ T cells, and NK cells (6). However, we and others have shown previously that RSV-challenged STAT1−/− mice developed airway eosinophilia, neutrophilia, and lung expression of IL-13 and IL-17A, which did not occur in WT BALB/c mice (8, 13). IL-17A expression in STAT1−/− mice occurred primarily in CD4+ T cells (48). These cellular and cytokine responses are characteristic of Th2 (airway eosinophilia; lung IL-13 expression) and Th17 (airway neutrophilia; lung IL-17A expression) immune responses. To test the hypothesis that STAT4 deficiency increases RSV-induced Th2 and Th17 lung immune responses, similar to those seen in STAT1−/− mice, we challenged WT, STAT1−/−, and STAT4−/− mice with RSV (A2). As we have previously shown, WT mice developed airway cellularity that was predominantly lymphocytic with no detectable airway eosinophils and minimal airway neutrophils following RSV challenge (Fig. 1A to E). STAT1−/− mice had significantly increased total airway cell numbers compared to WT mice at days 4, 6, and 8 postchallenge (Fig. 1A). The increased airway cellularity in STAT1−/− mice was accounted for almost entirely by the significant increase in numbers of airway eosinophils and neutrophils compared to WT mice (Fig. 1B and C). STAT4−/− mice had an airway cellular response to RSV challenge that was predominantly lymphocytic (Fig. 1A to E). RSV-challenged STAT4−/− mice had no airway eosinophils and minimal airway neutrophils (Fig. 1D and E).
FIG 1.

STAT1-deficient mice, but not STAT4-deficient mice, develop RSV-induced airway eosinophil and neutrophil recruitment. Bronchoalveolar lavage (BAL) was performed on RSV-challenged WT, STAT1−/−, and STAT4−/− mice. Total numbers of airway cells (A), eosinophils (B), neutrophils (C), macrophages (D), and lymphocytes (E) are shown (n = 2 to 3 mice per group on day 0 and 4 to 6 mice per group for all other days). Data are representative of two independent experiments. For panels A to C, * indicates a P value of <0.05 versus WT mice, ** indicates a P value of <0.01 versus WT and STAT4−/− mice, and ‡ indicates a P value of <0.001 versus WT and STAT4−/− mice; for panels D and E, * indicates a P value of <0.05 versus STAT1−/− mice.
To further characterize the lung immune response to RSV challenge, we next measured IL-13 and IL-17A protein levels in the lungs of WT, STAT1−/−, and STAT4−/− mice. As before, RSV-challenged STAT1−/− mice had significantly increased lung protein expression levels of IL-13 and IL-17A compared to WT mice (Fig. 2A and B) (13). In contrast to STAT1−/− mice and similar to WT mice, STAT4−/− mice had no detectable IL-13 or IL-17A protein expression postchallenge. Despite marked airway eosinophilia in STAT1−/− mice, we did not detect lung IL-5 protein expression in these mice at the chosen time points (data not shown). The lung protein expression level of CCL11 (eotaxin-1), however, was significantly increased in RSV-challenged STAT1−/− mice on days 4, 6, and 8 postchallenge compared to WT and STAT4−/− mice (Fig. 2C).
FIG 2.
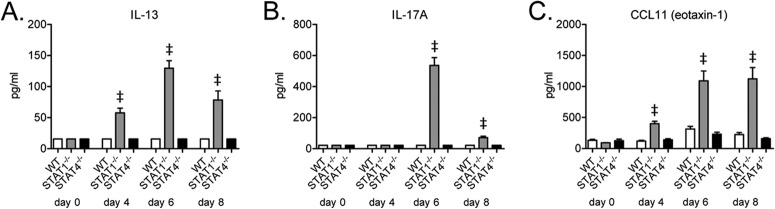
STAT4 deficiency does not result in RSV-induced lung protein IL-13 or IL-17A expression. Shown are lung protein expression levels of IL-13 (A), IL-17A (B), and CCL11 (eotaxin-1) (C) from RSV-challenged mice (n = 2 to 3 mice per group on day 0 and 4 to 6 mice per group for all other days). Limits of detection for IL-13, IL-17A, and CCL11 were 31.25 pg/ml, 43.75 pg/ml, and 15.6 pg/ml, respectively. Data are representative of two independent experiments. ‡, P < 0.001 versus WT and STAT4−/− mice.
STAT4 deficiency does not enhance RSV-induced airway mucus expression.
As STAT1−/− mice develop significant airway mucus expression following RSV A2 challenge (13), we next determined whether STAT4−/− mice develop RSV-induced airway mucus expression. Given the lack of detectable IL-13 expression in lungs of RSV-challenged STAT4−/− mice, we hypothesized that STAT4−/− mice do not develop airway mucus following RSV challenge. Lung sections from WT, STAT1−/−, and STAT4−/− mice challenged with UV-inactivated or live RSV A2 were stained with PAS stain or H&E, and inflammation and airway mucus scores were determined. In these experiments, both UVRSV- and RSV-challenged WT mice developed modest airway mucus expression that was primarily limited to the upper airways (Fig. 3). As we have shown previously (13), RSV-challenged STAT1−/− mice had significantly increased airway mucus expression, with mucus expression extending to the lower airways compared to WT mice (Fig. 3). In contrast, RSV-challenged STAT4−/− mice had no expression of airway mucus, with significantly decreased mucus expression compared to both RSV-challenged WT and STAT1−/− mice.
FIG 3.

STAT4 deficiency does not enhance RSV-induced airway mucus expression. Lung sections stained with PAS stain or H&E were prepared from UV-inactivated RSV (UVRSV)- or live-RSV-challenged WT, STAT1−/−, and STAT4−/− mice on day 6 postchallenge. (A) Representative PAS and H&E-stained lung sections shown at a ×200 magnification. (B) Histopathology scores for airway mucus expression and peribronchiolar, perivascular, and interstitial inflammation (n = 2 mice per group for UVRSV-challenged groups, and n = 3 mice per group for RSV-challenged groups). *, P < 0.05; **, P < 0.01; ‡, P < 0.001.
Similar to the airway mucus expression results, RSV-challenged STAT1−/− mice had significantly increased lung inflammation compared to WT and STAT4−/− mice (Fig. 3). In contrast, RSV-challenged STAT4−/− mice had significantly decreased peribronchiolar and interstitial inflammation compared to RSV-challenged WT mice. Thus, STAT4 deficiency decreased RSV-induced lung inflammation but did not result in enhanced airway mucus expression.
Following primary RSV challenge, the frequency of T-bet-expressing lung CD4+ T cells in STAT4−/− mice is intermediate compared to the frequency in both WT and STAT1−/− mice.
One explanation for the development of lung Th2 and Th17 immune responses following primary RSV challenge in STAT1−/− but not STAT4−/− mice might be the differential regulation of T-bet expression in CD4+ T cells of these mice. T-bet is a transcription factor critical for the differentiation of CD4+ Th1 cells in response to a variety of stimuli (32, 52). T-bet expression in CD4+ T cells is dependent on both STAT4 and STAT1 (20, 21). Importantly, T-bet negatively regulates CD4+ Th2 and Th17 differentiation through several mechanisms (25, 33, 36, 37). We therefore hypothesized that RSV-induced CD4+ T cell T-bet expression is impaired in STAT1−/− and STAT4−/− mice compared to WT mice and that the CD4+ T cell T-bet expression impairment in STAT1−/− mice is greater than that in STAT4−/− mice. To test this hypothesis, we determined the frequency and intensity of CD4+ T-bet expression in WT, STAT1−/−, and STAT4−/− mice following primary challenge by flow cytometry. The frequency of T-bet-expressing CD4+ T cells was significantly decreased in both STAT1−/− and STAT4−/− mice compared to WT mice (Fig. 4A and B). In addition, the frequency of T-bet-expressing CD4+ T cells in STAT1−/− mice was significantly decreased compared to STAT4−/− mice (Fig. 4A and B). Cell-specific expression of T-bet by CD4+ T cells was not decreased in either STAT4−/− or STAT1−/− mice compared to WT mice when measured by geometric mean fluorescence intensity (gMFI) (Fig. 4B).
FIG 4.

The frequency of T-bet-expressing lung CD4+ T cells in STAT4−/− mice is intermediate compared to frequencies in both WT and STAT1−/− mice. Lung mononuclear cells were harvested from RSV-challenged mice on day 6. Surface and intracellular flow staining was performed. (A) Representative dot plots for CD4+ T cells expressing T-bet, ROR-γt, or GATA-3. (B to D) Frequency of T-bet (B)-, ROR-γt (C)-, or GATA-3 (D)-expressing CD4+ T cells along with geometric mean fluorescence intensities (gMFI) for each transcription factor. Cells were gated on live CD3+ CD4+ cells. Gating was determined by using the respective isotype control antibodies (n = 5 to 7 per group). *, P < 0.05; **, P < 0.01; ‡, P < 0.001.
ROR-γt and GATA-3 are critical transcription factors for CD4+ Th17 and Th2 differentiation, respectively (53, 54). We next determined whether the differentially decreased frequency of expression of T-bet in STAT1−/− and STAT4−/− CD4+ T cells was associated with differential frequency or intensity of CD4+ T cell expression of ROR-γt and GATA-3. STAT1−/− CD4+ T cells had significantly increased frequency and intensity of expression of ROR-γt and GATA-3 compared to both WT and STAT4−/− CD4+ T cells (Fig. 4A, C, and D). Expression of ROR-γt and GATA-3 in STAT4−/− CD4+ T cells was not significantly different compared to that in WT CD4+ T cells (Fig. 4A, C, and D). Thus, STAT4−/− CD4+ T cells had an RSV-induced T-bet expression frequency that was intermediate compared to that seen in WT or STAT1−/− CD4+ T cells and was not associated with increased expression of ROR-γt or GATA-3.
STAT4 deficiency does not result in enhanced lung Th2 or Th17 immune responses following primary challenge with RSV strain Line 19.
As primary challenge of STAT4−/− mice with RSV A2 did not result in lung Th2 immune responses or Th17 immune responses, we next determined the ability of STAT4 deficiency to enhance lung Th2 immune responses in response to RSV Line 19. It was possible that the induction of lung Th2 immune responses by STAT4 deficiency was RSV strain dependent. RSV Line 19 challenge of WT BALB/c mice is well described to result in lung IL-13 protein expression and airway mucus expression (14, 15, 46). Although lung IL-13 protein expression following RSV Line 19 challenge is modest, airway mucus expression is robust and IL-13 dependent (14, 15). Because of the baseline ability of RSV Line 19 to induce lung IL-13 protein expression in WT mice, we hypothesized that STAT4−/− mice challenged with RSV Line 19 develop higher lung IL-13 expression levels than do WT mice. To test this hypothesis, we challenged WT, STAT1−/−, and STAT4−/− mice with UV-inactivated or live RSV A2 or RSV Line 19. STAT1−/− mice were included in this experiment as a positive control for the development of Th2 and Th17 immune responses to RSV infection. A lower-inoculum titer of RSV A2 (1 × 105 PFU/mouse) was used for this experiment, as this titer corresponded to the RSV Line 19 titer available to us. Mice were harvested on day 6 postchallenge, as this was the time of peak IL-13 protein expression in STAT1−/− mice in our previous experiments.
Mice challenged with UV-inactivated RSV (either A2 or Line 19) developed minimal airway cellularity that consisted primarily of macrophages (Fig. 5A and D). As in our previous experiments (Fig. 1 and 2), WT mice challenged with RSV A2 developed mild airway lymphocytosis but no airway eosinophilia, airway neutrophilia, or lung IL-13 or IL-17A protein expression (Fig. 5A to G). WT mice challenged with RSV Line 19 had greater airway lymphocytosis than did RSV A2-challenged mice but again did not have detectable airway eosinophils or airway neutrophils (Fig. 5B, C, and E). RSV Line 19-challenged WT mice did have detectable lung IL-13 protein expression that was similar to what was previously described in the literature (Fig. 5F) (15).
FIG 5.

STAT1−/− mice infected with RSV strain Line 19 develop enhanced airway eosinophilia and lung IL-13 expression but decreased airway neutrophilia and lung IL-17 expression compared to RSV A2-challenged STAT1−/− mice. WT, STAT1−/−, and STAT4−/− mice were infected with either UV-inactivated or live RSV A2 or RSV Line 19 (1 × 105 PFU/mouse). BAL and lung harvests were performed on day 6 postinfection. Total numbers of airway cells (A), eosinophils (B), neutrophils (C), macrophages (D), and lymphocytes (E) and lung IL-13 (F) and IL-17A (G) expression levels are shown. Lower limits of detection for IL-13 and IL-17A ELISAs were 31.25 pg/ml and 43.75 pg/ml, respectively (n = 5 mice per group). *, P < 0.05; ‡, P < 0.001.
STAT1−/− mice challenged with RSV Line 19 had significantly increased levels of airway eosinophils and lung IL-13 expression compared to RSV A2-challenged STAT1−/− mice (Fig. 5B and F). At the same time, RSV Line 19-challenged STAT1−/− mice had significantly fewer airway neutrophils and decreased lung IL-17A expression compared to RSV A2-challenged STAT1−/− mice (Fig. 5C and G). Similar to what we found previously in STAT4−/− mice challenged with RSV A2, STAT4−/− mice challenged with RSV Line 19 did not develop increased airway eosinophilia, airway neutrophilia, or lung IL-13 or IL-17A protein expression levels compared to WT mice challenged with RSV Line 19 (Fig. 5B, C, F, and G). Thus, the ability of STAT4 deficiency to promote Th2 or Th17 lung immune responses was not enhanced by challenge with RSV Line 19.
STAT4 deficiency does not attenuate RSV-induced lung IFN-γ expression but impairs RSV-induced cell-specific IFN-γ expression by CD4+ and CD8+ T cells and NK cells.
STAT4−/− mice did not develop Th2 or Th17 lung immune responses to RSV challenge; we therefore next focused on the role of STAT4 in lung IFN-γ protein expression following RSV challenge. We have shown previously that STAT1−/− mice demonstrate significantly increased lung IFN-γ protein expression levels following RSV challenge, and these mice were not used for the remainder of the study (8).
Because STAT4 has a well-described role in IFN-γ expression by CD4+ Th1 cells, CD8+ T cells, and NK cells (16, 55), we hypothesized that STAT4−/− mice would have significantly decreased whole-lung IFN-γ protein expression levels compared to WT mice. We therefore measured lung IFN-γ expression by an ELISA on day 6 postchallenge. Day 6 was selected as we have previously shown that it is the time of peak IFN-γ expression following RSV A2 challenge in WT BALB/c mice (13). Unexpectedly, on day 6 postchallenge, there was no significant difference in lung IFN-γ protein expression levels between WT and STAT4−/− mice (Fig. 6A).
FIG 6.

STAT4 deficiency does not attenuate RSV-induced lung IFN-γ expression but impairs RSV-induced cell-specific IFN-γ expression by CD4+ and CD8+ T cells and NK cells. (A) Lung IFN-γ protein expression from RSV-challenged mice on day 6 postchallenge. The lower limit of detection was 31.25 pg/ml. Data from three representative experiments were combined (n = 20 mice for the WT group, and n = 18 mice for the STAT4−/− group). (B to L) Lung mononuclear cells were harvested from RSV-challenged WT and STAT4−/− mice on days 4 and 6 or days 6 and 8 postchallenge. Restimulation was performed with PMA-ionomycin for CD4+ T cells and NK cells or with RSV M282–90 peptide for CD8+ T cells. Unrestimulated cell were surface stained for tetramer analysis. (B to D) Frequency (B), total number (C), and gMFI (D) of IFN-γ-expressing CD4+ T cells gated on live CD3+ CD4+ cells. (E and F) Frequency (E) and total number (F) of either influenza virus (Flu) or RSV M282–90 tetramer-expressing CD8+ cells, gated on live CD19− CD8+ cells. (G to I) Frequency (G), total number (H), and gMFI (I) of IFN-γ-expressing CD8+ T cells gated on live CD3+ CD8+ cells. (J to L) Frequency (J), total number (K), and gMFI (L) of IFN-γ-expressing NK cells gated on live CD3− DX5+ NKG2D+ cells (n = 5 mice per group; data are representative of two [J to L] or three [B to I] independent experiments). *, P < 0.05; **, P < 0.01; ‡, P < 0.001 (versus the WT).
We next determined the effect of STAT4 deficiency on IFN-γ expression by CD4+ T cells, CD8+ T cells, and NK cells. STAT4 and T-bet are critical for promoting CD4+ Th1 differentiation (17, 19–22, 32, 56). As we showed that CD4+ T cells from RSV-challenged STAT4−/− mice had decreased T-bet expression levels compared to WT CD4+ T cells (Fig. 4A and B), we anticipated that STAT4−/− CD4+ T cells would have impaired IFN-γ expression following RSV challenge. STAT4−/− mice had significantly decreased frequencies and total numbers of IFN-γ-expressing CD4+ T cells as well as decreased IFN-γ gMFI on days 6 and 8 following primary RSV challenge compared to WT mice (Fig. 6B to D). Despite these decreases, STAT4−/− CD4+ T cells had residual IFN-γ expression, with 14 and 17% of CD4+ T cells expressing IFN-γ on days 6 and 8, compared to 24 and 28% of WT CD4+ T cells expressing IFN-γ on these respective days.
STAT4 and T-bet are also critical for effector CD8+ T cell differentiation and IFN-γ expression (26, 56–58). We determined the effect of STAT4 deficiency on RSV-specific CD8+ T cell immune responses using the H2-Kd-restricted immunodominant M282–90 peptide and M282–90 tetramer (59, 60). STAT4 deficiency resulted in no significant differences in RSV-specific CD8+ T cell frequencies or numbers compared to WT mice (Fig. 6E and F). However, we found a modest but significant decrease in the frequency of IFN-γ-expressing CD8+ T cells in STAT4−/− compared to WT mice on day 8 postchallenge (Fig. 6G). While the total numbers of IFN-γ-expressing CD8+ T cells were not significantly different between groups, STAT4−/− CD8+ T cells had a significantly decreased IFN-γ gMFI compared to WT CD8+ T cells (Fig. 6H and I). Thus, although STAT4−/− mice did not develop Th2 or Th17 lung immune responses, IFN-γ expression by both CD4+ and CD8+ T cells was impaired in STAT4−/− compared to WT mice.
STAT4 is critical for induction of IFN-γ expression by NK cells in several models (29–31, 61). NK cells promote illness and lung injury and prevent development of allergic airway inflammation following RSV challenge (9, 62–64). A role for STAT4 in NK cell IFN-γ expression during RSV challenge has not previously been determined. Therefore, we determined NK cell expression of IFN-γ following primary RSV challenge in our model. Peak IFN-γ expression by NK cells following RSV challenge has been shown to occur on day 4 (64). Following RSV challenge, STAT4−/− mice had a significant decrease in the frequency and total number of IFN-γ-expressing NK cells as well as decreased IFN-γ gMFI on both days 4 and 6 compared to WT mice (Fig. 6J to L). Taken together, these results show that STAT4 regulates IFN-γ expression in cells critical for the innate and adaptive immune responses to RSV.
STAT4 deficiency attenuates weight loss following primary RSV challenge.
RSV challenge of BALB/c mice induces weight loss that correlates with lung immunopathology (11, 12). CD4+ and CD8+ T cells and NK cells contribute to the development of this weight loss (9–12, 62, 65). As STAT4−/− mice demonstrated decreased inflammation, as measured by histopathology and both lung and cell-specific IFN-γ expression, we anticipated that STAT4−/− mice would have decreased weight loss following primary RSV challenge. Therefore, we challenged WT and STAT4−/− mice with live or UV-inactivated RSV and measured postchallenge weight loss. Following challenge with live RSV, WT mice developed biphasic weight loss, as previously described (6, 12). STAT4−/− mice developed postchallenge weight loss that was significantly attenuated at both early and later time points compared to WT mice (Fig. 7). Of note, WT mice challenged with RSV had significantly greater weight loss than did UVRSV-challenged WT mice. In contrast, postchallenge weight loss for STAT4−/− mice challenged with live RSV was not statistically different from weight loss in UVRSV-challenged STAT4−/− mice (Fig. 7). Thus, STAT4−/− mice had significantly attenuated post-RSV-challenge weight loss.
FIG 7.
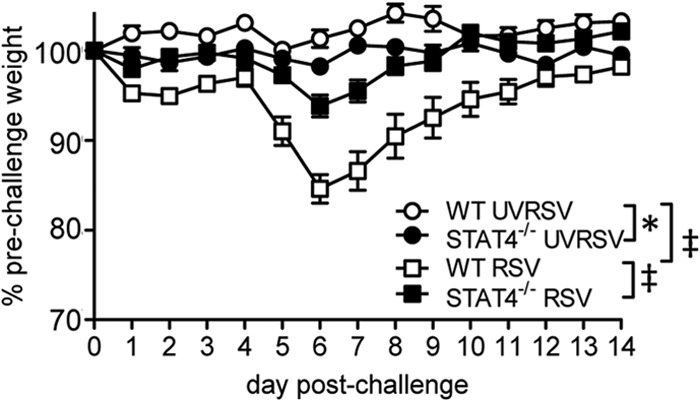
STAT4 deficiency attenuates RSV-induced weight loss following primary RSV challenge. WT and STAT4−/− mice were challenged with UVRSV or live RSV (RSV). Postchallenge weight loss as a percentage of prechallenge weight on day 0 is shown (n = 5 for WT mice challenged with UVRSV, n = 5 for STAT4−/− mice challenged with UVRSV, n = 15 for WT mice challenged with RSV, and n = 17 for STAT4−/− mice challenged with RSV). Statistical significance between curves was assessed by repeated-measures ANOVA across curves with a Bonferonni posttest. *, P < 0.05; ‡, P < 0.001. Data are representative of two independent experiments.
STAT4 deficiency significantly decreases RSV titers following primary challenge.
CD4+ T cells, CD8+ T cells, and NK cells are critical for viral clearance following RSV challenge (9–11). As decreased immunopathology and weight loss typically correlate with impaired viral clearance following RSV challenge, we hypothesized that STAT4−/− mice have increased lung RSV titers postinfection compared to WT mice. Lung viral titers in WT and STAT4−/− mice were quantified at day 4 postchallenge, which was previously shown to be the peak time of viral replication in this model (11, 12). Unexpectedly, the RSV titer was significantly decreased in the lungs of STAT4−/− mice compared to WT mice (Fig. 8). No virus was detectable in either group at day 6 or day 8 postchallenge (data not shown).
FIG 8.
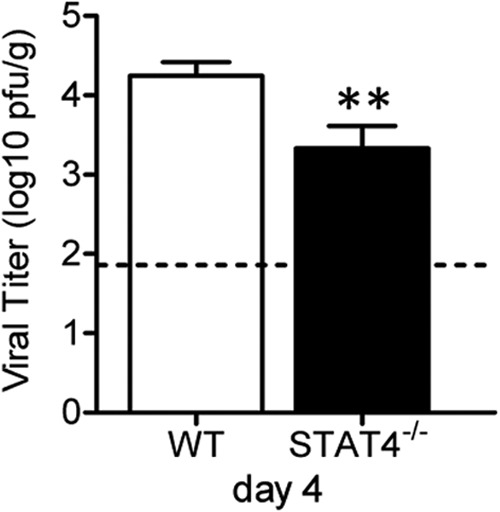
STAT4 deficiency enhances lung viral clearance following primary RSV challenge. WT and STAT4−/− mice were RSV challenged on day 0, and lungs were harvested on day 4. Lung RSV titers were determined by a plaque assay (lower limit of detection of 1.8 log10 PFU/g). Data from four independent, representative experiments were combined (n = 20 for WT mice, and n = 22 for STAT4−/− mice). **, P < 0.01 versus the WT.
The decrease in peak viral titers on day 4 postinfection in STAT4−/− compared to WT mice suggested that STAT4−/− mice might have enhanced innate defenses against viral infections. Type I interferons and IL-27 are STAT1-activating cytokines that promote antiviral host defense and regulate inflammation through a variety of pathways (66, 67). To determine whether STAT4−/− mice had increased expression levels of each of these cytokines at early time points following primary RSV challenge, we measured IFN-α, IFN-β, and IL-27 protein levels in lungs of RSV-challenged mice by ELISAs at 12 and 24 h postinfection. We found no significant differences in lung protein expression levels of these cytokines between WT and STAT4−/− mice (Fig. 9). Thus, STAT4−/− mice have decreased peak RSV titers despite having no differences in early lung expressions of several innate cytokines.
FIG 9.
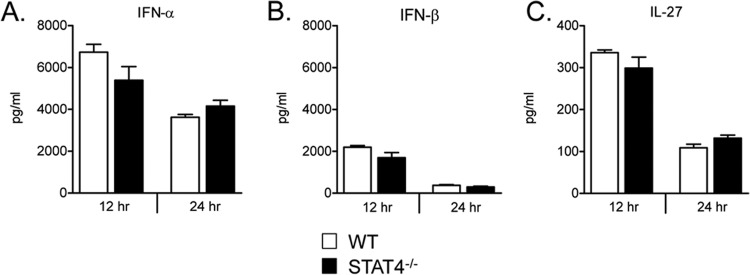
Lung expression of IFN-α, IFN-β, and IL-27 at early time points following RSV challenge. Shown are lung protein expression levels of IFN-α (A), IFN-β (B), and IL-27 (C) from RSV-challenged WT and STAT4−/− mice at 12 and 24 h post-primary infection. Lower limits of detection for IFN-α, IFN-β, and IL-27 were 250 pg/ml, 46.9 pg/ml, and 46.9 pg/ml, respectively (n = 4 to 5 mice per group).
STAT4 deficiency reduces RSV F-protein-specific antibody titers.
RSV challenge of BALB/c mice results in a prolonged elevation of levels of RSV F-protein-specific antibodies (68). Passive administration of antibody to F protein decreases viral replication in BALB/c mice (69). We determined the effect of STAT4 deficiency on F-specific antibody titers following challenge with RSV. Because IL-12–STAT4 signaling has been linked to T follicular helper cell function in humans (70), we hypothesized that STAT4−/− mice have impaired antibody responses following primary RSV challenge. We measured F-specific IgG, IgG1, and IgG2a levels in serum harvested from WT or STAT4−/− mice at day 42 postchallenge. In each case, STAT4−/− mice had significantly decreased antibody titers compared to WT mice (Fig. 10A to C). Thus, STAT4 deficiency alters both cellular and humoral immunity in response to RSV challenge.
FIG 10.
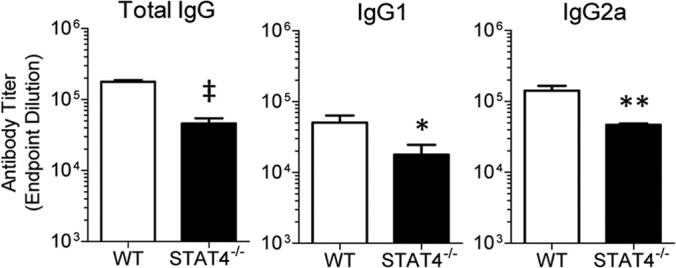
STAT4 deficiency decreases RSV F-protein-specific antibody titers. WT and STAT4−/− mice were challenged with RSV. On day 42 post-primary challenge, mice were bled, and serum was obtained. Endpoint dilution antibody titers were quantified by an ELISA for RSV F-protein-specific IgG (A), IgG1 (B), and IgG2a (C) (n = 6 to 7 mice per group). Data are representative of two independent experiments. *, P < 0.05; **, P < 0.01; ‡, P < 0.001 (versus the WT).
STAT4 deficiency increases weight loss and lung IFN-γ expression levels following secondary RSV challenge but does not enhance lung Th2 or Th17 immune responses.
Mouse models of secondary challenge with RSV have shown that WT mice first challenged with RSV as neonates and then rechallenged during adulthood develop Th2 immune responses following secondary challenge (71, 72). These rechallenge-induced Th2 immune responses correlated with impaired IFN-γ protein expression at primary neonatal challenge and were abrogated by exogenous IFN-γ given at primary neonatal challenge (73). Rechallenged WT mice first challenged with RSV in adulthood do not develop Th2 immune responses upon RSV secondary challenge (11, 71–73). Because we showed that STAT4−/− mice developed impaired cell-specific IFN-γ expression following primary RSV challenge, we hypothesized that secondary RSV challenge of STAT4−/− mice results in increased lung Th2 immune responses compared to WT mice.
To test this hypothesis, we challenged WT and STAT4−/− mice with RSV A2 at day 0 and again at day 42 with identical strains and titers of virus. In contrast to primary challenge, in which STAT4−/− mice had significantly decreased weight loss compared to WT mice, secondary RSV challenge of STAT4−/− mice caused a modest but significant increase in weight loss compared to rechallenged WT mice (Fig. 11A). Mice challenged with UVRSV at primary challenge and rechallenged with RSV did not develop weight loss following secondary challenge (Fig. 11A). To determine whether a Th2 bias developed in rechallenged STAT4−/− mice, we determined viral titers, numbers of airway inflammatory cells, and lung cytokine expression levels on days 44, 45, and 46. Previous work showed that rapid viral clearance occurs in mice following secondary RSV challenge, with virus being cleared from the lungs by day 2 post-secondary challenge (11, 68). Similarly, we found that WT and STAT4−/− mice had no detectable virus in the lungs at day 44, 45, or 46 (data not shown). Following secondary challenge, WT and STAT4−/− mice had gradually increasing total airway cell numbers, but neither group developed significant airway eosinophilia or neutrophilia (Fig. 11B to D). The majority of airway cells in both groups were lymphocytes and macrophages (Fig. 11E and F). The IFN-γ protein expression level in the lungs of STAT4−/− mice was significantly higher than that in WT mice on days 45 and 46 (Fig. 11G). While IL-13 was detected in the lungs of both groups following secondary challenge, the IL-13 protein expression level in STAT4−/− mice was not increased compared to WT mice (Fig. 11H). No IL-17A protein expression was detected in the lungs of WT or STAT4−/− mice (Fig. 11I). Thus, STAT4 deficiency enhanced illness and lung IFN-γ expression following secondary RSV challenge but did not result in enhanced Th2 or Th17 immune responses.
FIG 11.

STAT4 deficiency enhances RSV-induced weight loss and lung IFN-γ protein expression following secondary challenge. WT and STAT4−/− mice were challenged with RSV on day 0 and rechallenged on day 42 with identical strains and titers of virus. (A) Post-secondary challenge weight loss as a percentage of day 42 weight is shown for mice challenged at day 0 and day 42 with RSV (WT-RSV-RSV and STAT4−/−-RSV-RSV) (n = 22 mice per group; data from four representative, independent experiments were combined) and for mice challenged with UVRSV at day 0 and RSV at day 42 (WT-UVRSV-RSV and STAT4−/−-UVRSV-RSV) (n = 5 mice per group). **, P < 0.01. (B to I) WT and STAT4−/− mice that underwent RSV challenge at day 0 and day 42 were harvested on days 44, 45, and 46. Total numbers of lymphocytes (B), neutrophils (C), eosinophils (D), lymphocytes (E), and macrophages (F) in BAL fluid are shown. (G to I) Lung protein expression levels of IFN-γ (G), IL-13 (H), and IL-17A (I) are shown, with lower limits of detection of 31.25, 31.25, and 43.75 pg/ml, respectively (for panels B to I, n = 10 to 11 mice per group, with data combined from two representative, independent experiments). *, P < 0.05; **, P < 0.01 (versus the WT).
NK cell and CD4+ T cell IFN-γ expression levels in STAT4−/− mice are impaired following secondary RSV challenge.
Because we found increased lung IFN-γ expression levels in STAT4−/− mice compared to WT mice following secondary RSV challenge, we next determined cell-specific IFN-γ expression levels in lungs of rechallenged mice. We hypothesized that following secondary challenge, IFN-γ expression levels from one or more cell types, including NK cells, CD4+ T cells, and/or CD8+ T cells, would be increased in STAT4−/− mice compared to WT mice to account for the increase in whole-lung IFN-γ expression levels. Lung lymphocytes were obtained on day 44 and day 46 and restimulated with PMA-ionomycin, and surface and intracellular staining was performed. STAT4−/− mice had significantly decreased frequencies and total numbers of IFN-γ-expressing NK cells and CD4+ T cells compared to WT mice (Fig. 12A to D). In addition, the IFN-γ gMFIs for NK cells and CD4+ T cells were significantly decreased on days 44 and 46, respectively, in STAT4−/− compared to WT mice (Fig. 12C and D). Thus, NK cells and CD4+ T cells do not appear to be the source of increased lung IFN-γ expression levels in STAT4−/− mice following secondary challenge.
FIG 12.

STAT4 deficiency impairs RSV-induced IFN-γ expression by NK cells and CD4+ T cells following secondary challenge. WT and STAT4−/− mice challenged with RSV on day 0 and day 42 were euthanized on days 44 and 46. Lung mononuclear cells were obtained and restimulated with PMA-ionomycin, and surface and intracellular staining was performed. (A and B) Representative dot plots for IFN-γ-expressing NK cells on day 4 postchallenge (A) and CD4+ T cells on day 8 postchallenge (B). (C and D) Frequency, total number, and gMFI of IFN-γ-expressing NK cells (C) and IFN-γ-expressing CD4+ T cells (D). NK cells were gated on live CD3− DX5+ NKG2D+ cells, and CD4+ T cells were gated on live CD3+ CD4+ cells (n = 11 to 12 mice per group for day 44, and n = 15 to 20 mice per group for day 46). *, P < 0.05; **, P < 0.01; ‡, P < 0.001 (versus the WT). Data from three representative, independent experiments were combined.
STAT4 deficiency increases the frequency and total number of RSV-specific CD8+ T cells following secondary challenge.
We next determined the RSV-specific CD8+ T cell response to secondary challenge in both WT and STAT4−/− mice. As CD8+ T cells are critical for illness following RSV rechallenge (68), and STAT4−/− mice had increased rechallenge weight loss compared to WT mice in our model, we hypothesized that the RSV-specific CD8+ T cell response to rechallenge is enhanced in STAT4−/− compared to WT mice. To test this hypothesis, we challenged mice with RSV on day 42 following primary challenge. Lung mononuclear cells were obtained either prior to rechallenge on day 42 or following rechallenge on days 44 and 46.
On day 42, both WT and STAT4−/− mice had increased frequencies and total numbers of RSV-specific CD8+ T cells compared to negative-control influenza virus-specific CD8+ T cells (Fig. 13B). The frequencies and total numbers of RSV-specific CD8+ T cells were not significantly different between WT and STAT4−/− mice. On day 44, the total numbers of RSV-specific CD8+ T cells in both WT and STAT4−/− mice were again significantly higher than the numbers of the respective influenza virus-specific CD8+ T cells. No significant difference in frequency or number of RSV-specific CD8+ T cells between WT and STAT4−/− mice was present on day 44. On day 46, a robust RSV-specific CD8+ T cell response in the lungs was noted for both WT and STAT4−/− mice (Fig. 13B). Notably, STAT4−/− mice had a 39% increase in the frequency and an 80% increase in the total number of lung RSV-specific CD8+ T cells compared to WT mice (Fig. 13A and B). Despite this increase in the frequency of RSV-specific CD8+ T cells, STAT4−/− mice had only a trend toward an increased frequency of IFN-γ expression by CD8+ T cells (Fig. 13C and D).
FIG 13.

STAT4 deficiency enhances the lung RSV-specific CD8+ T cell response to secondary challenge. Lung mononuclear cells were obtained from WT and STAT4−/− mice that underwent secondary RSV challenge on day 42 after primary RSV challenge. Surface staining for tetramer analysis was performed (A and B), and both surface and intracellular staining were performed following M2 peptide restimulation (C and D). (A and B) Representative dot plots from day 46 (A) and frequency and total number (B) of either RSV or influenza virus (Flu) tetramer-expressing CD8+ cells gated on live CD19− CD8+ cells. (C and D) Representative dot plots from day 46 (C) and frequency, total number, and gMFI (D) of IFN-γ-expressing CD8+ T cells gated on live CD3+ CD8+ cells (n = 11 to 12 mice per group for days 42 and 44, and n = 15 to 20 mice per group for day 46). Data were combined from three representative, independent experiments. *, P < 0.05; **, P < 0.01; ‡, P < 0.001.
To determine whether altered expression of innate cytokines in STAT4−/− mice might contribute to enhanced weight loss or RSV-specific CD8+ T cell responses following secondary challenge in STAT4−/− mice, we measured lung protein levels of type I interferons and IL-27 following secondary challenge in WT and STAT4−/− mice. No significant differences were present at 6 or 12 h postchallenge (Fig. 14). STAT4−/− mice did have significantly higher lung IFN-α protein expression levels than did WT mice at 24 h postchallenge (Fig. 14). No differences in lung IFN-β or IL-27 protein expression levels between WT and STAT4−/− mice were noted at 24 h post-secondary challenge.
FIG 14.
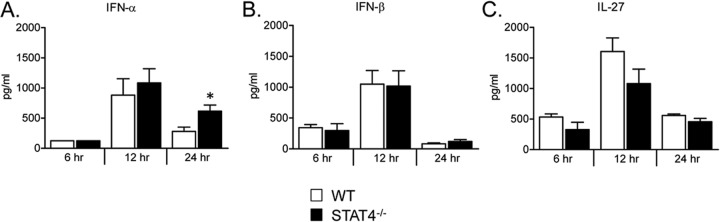
Lung protein expression of IFN-α, IFN-β, and IL-27 at early time points following secondary RSV challenge. Shown are lung protein expression levels of IFN-α (A), IFN-β (B), and IL-27 (C) from WT and STAT4−/− mice RSV challenged on days 0 and 42 and harvested at 6, 12, and 24 h post-secondary RSV challenge. Lower limits of detection for IFN-α, IFN-β, and IL-27 were 250 pg/ml, 62.5 pg/ml, and 62.5 pg/ml, respectively (n = 4 to 5 mice per group). *, P < 0.05 versus the WT.
Thus, STAT4 deficiency enhanced the lung RSV-specific CD8+ T cell immune response to secondary infection without resulting in enhanced Th2 or Th17 immune responses.
DISCUSSION
Immune-mediated lung injury is critical for RSV pathogenesis. Depending on host and viral factors, this injury can be mediated by IFN-γ-predominant lymphocytic inflammation, IL-13-predominant allergic inflammation with airway mucus expression, and/or IL-17A-predominant neutrophilic inflammation (6, 8, 13, 14, 46). We undertook the present study to determine (i) the role of STAT4 in cross-regulation between RSV-induced Th1 and Th2 or Th17 CD4+ T cell immune responses and (ii) the role of STAT4 in RSV-induced NK cell, CD4+ T cell, and CD8+ T cell IFN-γ expression, postchallenge weight loss, and viral clearance following both primary and secondary RSV challenge.
We demonstrated that following primary RSV challenge, STAT4−/− mice did not develop Th2 or Th17 immune responses. This finding was in marked contrast to the findings for STAT1−/− mice, which developed airway neutrophilia, eosinophilia, and mucus expression and significantly increased lung IL-17A and IL-13 expression levels compared to WT or STAT4−/− mice (13). As both STAT4 and STAT1 have been shown to increase T-bet expression levels in CD4+ T cells (20, 21), and T-bet is a negative regulator of Th2 and Th17 cell differentiation (33–37), we had anticipated that STAT4−/− mice would develop Th2 and/or Th17 immune responses to primary challenge with RSV. In our model, both STAT4−/− and STAT1−/− mice had decreased frequencies of T-bet-expressing CD4+ T cells compared to WT mice. However, STAT1−/− CD4+ T cells had a significantly decreased frequency of T-bet expression compared to STAT4−/− CD4+ T cells. In STAT1−/− mice, the decrease in the frequency of CD4+ T cell T-bet expression was accompanied by increases in the frequency and intensity of expression of ROR-γt and GATA-3, master regulators of Th2 and Th17 differentiation pathways, respectively, in CD4+ T cells. Despite the decreased frequency of T-bet expression in STAT4−/− CD4+ T cells compared to WT cells, neither the frequency nor the intensity of ROR-γt or GATA-3 expression increased in STAT4−/− CD4+ T cells. These results suggest that the residual frequency of CD4+ T cell T-bet expression in STAT4−/− mice may account for the lack of Th2 or Th17 immune responses to RSV in STAT4−/− mice.
It is possible that the residual frequency of T-bet expression in STAT4−/− CD4+ T cells in our model is induced by STAT1-activating cytokines such as IFN-γ, type I interferons, or IL-27 (66, 74). Interestingly, in an in vitro CD4+ T cell differentiation system, the addition of IL-27, but not IFN-γ, under Th1-polarizing conditions resulted in a partial rescue of tbx21 mRNA expression in STAT4−/− CD4+ T cells (20). Moreover, in a T-bet reporter mouse system, expression of T-bet was markedly impaired in IFN-γR−/− STAT4−/− CD4+ T cells during in vitro differentiation but not during in vivo differentiation, suggesting the presence of additional T-bet-activating cytokines in vivo (38). The addition of type I interferons or IL-27 to culture media rescued in vitro IFN-γR−/− STAT4−/− CD4+ T cell T-bet expression in this model (38). Notably, in our model, WT and STAT4−/− mice had similar expression levels of type I interferons and IL-27 in the lungs at early time points post-primary challenge. Thus, type I interferon, IL-27, and/or IFN-γ expression may account for the maintained CD4+ T cell T-bet expression frequency in STAT4−/− mice and thereby prevent development of Th2 or Th17 immune responses to RSV in these mice. Importantly, differential development of Th17 immune responses between STAT4−/− and STAT1−/− mice may not be entirely accounted for by differential T-bet expression. STAT4 signaling is activated by IL-23, which is critical for maintenance of the CD4+ Th17 lineage (75, 76). Additionally, STAT1-activating cytokines can impair Th17 immune responses through both T-bet-dependent and -independent mechanisms (77). We are currently investigating the role of STAT1/T-bet-activating cytokines in preventing Th2 and Th17 immune responses to RSV in STAT4-deficient mice. To this end, we attempted to create STAT1−/− STAT4−/− mice as well as IFNAR−/− STAT4−/− mice. No viable pups were generated from these breeding attempts, and these combinations appear to be embryonic lethal.
The lack of RSV-induced Th2 immune responses in STAT4-deficient BALB/c mice in our study contradicts previously reported data in which STAT4-deficient C57BL/6 mice developed increased airway hyperreactivity, lung eosinophilia, and airway mucus expression compared to WT mice challenged with RSV (39). Importantly, that previous study was performed by using an RSV A strain (Line 19) that was subsequently shown to induce airway mucus expression and lung IL-13 expression in BALB/c mice, while the RSV A2 strain, which we used in this study, does not (14, 39, 40, 46). To determine whether STAT4 deficiency enhances lung Th2 immune responses to RSV Line 19 in our model, we challenged WT, STAT1−/−, and STAT4−/− mice with RSV A2 or RSV Line 19. Unexpectedly, STAT4−/− mice challenged with RSV Line 19 did not develop increased lung Th2 immune responses compared to RSV Line 19-challenged WT mice. In contrast, STAT1−/− mice challenged with RSV Line 19 developed significantly greater lung Th2 immune responses than did both RSV Line 19-challenged WT and RSV A2-challenged STAT1−/− mice. These results further support the importance of STAT1, but not STAT4, in restraining RSV-induced lung Th2 immune responses in BALB/c mice. Importantly, the previously reported study demonstrating that STAT4−/− mice develop increased Th2 immune responses was performed by using C57BL/6 STAT4−/− mice, and our study used BALB/c STAT4−/− mice (39). Taken together, these data suggest a critical role for mouse genetic background in the development of Th2 immune responses following RSV challenge of STAT4−/− mice.
Previous studies utilized STAT4−/− mice to investigate the role of STAT4 in regulating the lung immune response to allergens and viral infection. Using an ovalbumin model of allergic airway inflammation in STAT4−/− mice on a BALB/c background, one group showed that STAT4−/− mice had significantly decreased airway eosinophilia, airway neutrophilia, and airway mucus expression compared to WT mice (41). Another group, using a similar model, demonstrated that STAT4−/− mice have decreased airway cellularity and no increase in lung IL-13 expression levels compared to WT mice (44). Using a distinct cockroach antigen model of allergic airway inflammation in BALB/c STAT4−/− mice, another group showed that STAT4−/− mice had significantly decreased peribronchial inflammation and lung IL-5 expression levels, with no increase in lung IL-13 expression levels (43). Finally, using a primary influenza virus challenge model in WT and STAT4−/− BALB/c mice, another study showed that influenza virus-challenged STAT4−/− mice did not develop increased Th2 immune responses, as quantified by IL-4 expression in the spleen or lungs postchallenge (42). Thus, those previous studies support our conclusions that STAT4, in the context of the BALB/c genetic background, is not critical for restraining lung Th2 immune responses.
NK cells, CD4+ T cells, and CD8+ T cells each mediate immunopathogenesis, weight loss, and viral clearance following primary RSV challenge (9, 11, 62, 64). STAT4 has a well-described role in promoting the expression of IFN-γ by each of these cell types (17, 19, 20, 28–30, 56, 78). In our model, WT and STAT4−/− mice had similar whole-lung IFN-γ expression levels on day 6 postchallenge. However, cell-specific IFN-γ expression levels were significantly decreased in STAT4−/− NK cells, CD4+ T cells, and CD8+ T cells compared to the respective WT cells. While this discrepancy was unexpected, it may be partially explained by a differential importance among IFN-γ-expressing cells for whole-lung IFN-γ expression depending on the day post-RSV challenge. As the frequency and number of IFN-γ-expressing CD8+ T cells were not significantly different between WT and STAT4−/− mice on day 6 postchallenge, it is possible that CD8+ T cells account for the lack of difference in whole-lung IFN-γ protein expression levels between WT and STAT4−/− mice.
Despite decreased weight loss and immunopathology in RSV-challenged STAT4−/− versus WT mice, peak RSV titers in the lungs of STAT4−/− mice were significantly decreased compared to WT mice. This finding was unexpected, as NK cells, CD4+ T cells, and CD8+ T cells are critical for RSV clearance. Mice depleted of each of these cell types develop significantly decreased weight loss that correlates with increased peak viral titers and delayed viral clearance from the lungs (9, 11). The etiology of decreased peak viral titers in STAT4−/− mice in our model is not clear. STAT4 is expressed in antigen-presenting cells, and it is therefore possible that STAT4 deficiency alters the innate immune response to RSV, resulting in either impaired viral replication or enhanced viral clearance (78–80). To this end, we measured lung protein expression levels of type I interferons and IL-27 at early time points post-RSV challenge in WT and STAT4−/− mice and found no significant between-group differences. We are currently further investigating the mechanisms that cause reduced viral titers in the lungs of STAT4−/− mice following primary RSV challenge.
Immune memory following primary RSV infection in people is incomplete, and reinfection can occur throughout life. In one study of infants and young children, RSV reinfection resulted in lower respiratory tract infection in 26% of cases (81). We demonstrated that STAT4−/− mice had significantly increased weight loss, lung IFN-γ protein expression levels, and RSV-specific CD8+ T cell responses following secondary RSV challenge. Modestly increased rechallenge weight loss in STAT4−/− mice contrasted with the attenuated weight loss that STAT4−/− mice developed following primary RSV challenge. Both STAT4−/− NK cells and CD4+ T cells had decreased cell-specific IFN-γ protein expression levels upon secondary challenge compared to the respective WT NK cells and CD4+ T cells. In contrast, RSV-specific CD8+ T cells showed a trend toward increased IFN-γ expression levels. It is possible that CD8+ T cells represent the source of increased IFN-γ levels in this secondary challenge model. Alternatively, other potential IFN-γ-expressing cells, such as γδ T cells or NKT cells, could account for the increased whole-lung IFN-γ protein expression levels upon secondary challenge in STAT4−/− mice in this model. Importantly, it is possible that the increased IFN-γ protein expression level in the lungs of STAT4−/− mice is not solely responsible for increased post-secondary challenge weight loss. We therefore measured the lung protein expression levels of type I interferons and IL-27 to determine a possible role for innate pathways in the enhanced weight loss seen in STAT4−/− mice. Notably, at 24 h post-secondary challenge, STAT4−/− mice had a modest but significant increase in lung IFN-α protein expression levels compared to WT mice. The significance of this result is unclear, but enhanced IFN-α protein expression may suggest dysregulated innate host defenses in STAT4−/− mice following secondary infection. Taken together, these secondary RSV challenge results suggest that STAT4 signaling may play a role in determining the severity of illness following RSV reinfection.
While RSV-specific CD8+ T cells were detectable in the lungs of both WT and STAT4−/− mice before (day 42) and following (day 44) secondary challenge, expansion of this CD8+ T cell population was not noted until 4 days following secondary challenge (day 46). These findings fit with those of a previous study in which BALB/c mice were challenged with RSV after prechallenge adoptive transfer of previously antigen-exposed RSV-specific CD8+ T cells (82). Following RSV challenge of these primed mice, proliferation of RSV-specific CD8+ T cells was higher in lymph nodes than in lungs at day 3 and day 4 post-RSV challenge (82). RSV-specific CD8+ T cells in the lungs were not detected until day 4 post-RSV challenge. The similarly delayed increase in the number of lung RSV-specific CD8+ T cells following rechallenge in our model supports these previously reported findings and may reflect an inherent CD8+ T cell lymph node expansion phase following RSV rechallenge.
Several mechanisms may account for the increased frequency and number of RSV-specific CD8+ T cells following secondary RSV challenge in STAT4−/− mice compared to WT mice. First, we have shown that F-protein-specific antibody titers at day 42 post-primary challenge are modestly decreased in STAT4−/− mice compared to those in WT mice. Mice depleted of B cells at primary challenge have increased illness and enhanced viral replication upon rechallenge with RSV compared to non-B cell-depleted mice (65). While RSV was not detected in the lungs of WT or STAT4−/− mice at day 44, 45, or 46 in our model, it is possible that differences in viral titers between the groups at earlier time points resulted in increased antigen-specific stimulation and expansion of RSV-specific CD8+ T cells in STAT4−/− mice compared to WT mice. Second, we demonstrated impaired IFN-γ expression by CD4+ T cells and NK cells following RSV rechallenge in STAT4−/− compared to WT mice. Decreased IFN-γ expression by these cells may have resulted in a compensatory increase in the number of RSV-specific CD8+ T cells. Third, the absence of STAT4 may have altered RSV-specific CD8+ T cell memory through CD8+ T cell-intrinsic effects. Previous work using IL-12−/− mice showed that IL-12 deficiency resulted in an increased contribution of antigen-specific CD8+ T cells to the CD8+ T cell memory pool (83). As IL-12 signals through STAT4, it is possible that STAT4 deficiency in CD8+ T cells in our model resulted in enhanced RSV-specific CD8+ T cell memory development.
Secondary challenge models of RSV have been important for understanding the development of Th2 immune responses and T cell memory in response to RSV (11, 68, 71–73). Several groups have reported that age at primary challenge significantly influences the Th1 versus Th2 skewing of the immune response to secondary RSV challenge. Secondary RSV challenge of mice previously RSV challenged as neonates results in the development of Th2 immune responses upon rechallenge, whereas rechallenge of mice first challenged as adults does not (71, 72). This phenotype was recapitulated in adult IFN-γ−/− mice, which developed lung Th2 immune responses following RSV rechallenge. Notably, provision of exogenous IFN-γ at primary challenge to neonatal mice or to adult IFN-γ−/− mice abrogated rechallenge Th2 immune responses in both groups (73). Because cell-specific IFN-γ expression levels were decreased in STAT4−/− compared to WT mice following primary challenge in our model, we hypothesized that secondary challenge of STAT4−/− mice results in lung Th2 immune responses in STAT4−/− but not WT mice. In contrast, we found that STAT4 deficiency in adult mice did not result in enhanced lung Th2 immune responses following secondary RSV challenge. These findings suggest that impaired IFN-γ expression at primary challenge may not be the sole determinant in the development of a Th2-skewed response to secondary RSV challenge.
In summary, we show a significant role for STAT4 in regulating the immune response to primary and secondary challenge with RSV A2 in BALB/c mice. STAT4-deficient mice had increased weight loss and numbers of lung RSV-specific CD8+ T cells following secondary infection compared to WT mice but did not develop lung Th2 or Th17 immune responses to primary or secondary challenge. These findings have important implications for an understanding of the development of RSV-induced immunopathology in response to primary and secondary RSV infection. Our findings suggest that potential future treatment approaches to inhibit STAT4 will not enhance RSV-induced Th2 or Th17 immune responses. However, STAT4 inhibition during RSV infection may contribute to enhanced CD8+ T cell-dependent immunopathology during subsequent infection.
ACKNOWLEDGMENTS
This work was supported by NIH grants RO1AI111820, R01HL090664, and U19AI095227 and Department of Veterans Affairs grant I01BX000624 to R.S.P. D.E.D. was supported by grants T32HD060554 and T32HL094296.
We thank Barney Graham for helpful discussions.
Footnotes
Published ahead of print 11 June 2014
REFERENCES
- 1.Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, Auinger P, Griffin MR, Poehling KA, Erdman D, Grijalva CG, Zhu Y, Szilagyi P. 2009. The burden of respiratory syncytial virus infection in young children. N. Engl. J. Med. 360:588–598. 10.1056/NEJMoa0804877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. 2005. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 352:1749–1759. 10.1056/NEJMoa043951 [DOI] [PubMed] [Google Scholar]
- 3.Collins PL, Melero JA. 2011. Progress in understanding and controlling respiratory syncytial virus: still crazy after all these years. Virus Res. 162:80–99. 10.1016/j.virusres.2011.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graham BS. 2011. Biological challenges and technological opportunities for respiratory syncytial virus vaccine development. Immunol. Rev. 239:149–166. 10.1111/j.1600-065X.2010.00972.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aherne W, Bird T, Court SD, Gardner PS, McQuillin J. 1970. Pathological changes in virus infections of the lower respiratory tract in children. J. Clin. Pathol. 23:7–18. 10.1136/jcp.23.1.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peebles RS, Jr, Graham BS. 2005. Pathogenesis of respiratory syncytial virus infection in the murine model. Proc. Am. Thorac. Soc. 2:110–115. 10.1513/pats.200501-002AW [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson JE, Gonzales RA, Olson SJ, Wright PF, Graham BS. 2007. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod. Pathol. 20:108–119. 10.1038/modpathol.3800725 [DOI] [PubMed] [Google Scholar]
- 8.Durbin JE, Johnson TR, Durbin RK, Mertz SE, Morotti RA, Peebles RS, Graham BS. 2002. The role of IFN in respiratory syncytial virus pathogenesis. J. Immunol. 168:2944–2952. 10.4049/jimmunol.168.6.2944 [DOI] [PubMed] [Google Scholar]
- 9.Li F, Zhu H, Sun R, Wei H, Tian Z. 2012. Natural killer cells are involved in acute lung immune injury caused by respiratory syncytial virus infection. J. Virol. 86:2251–2258. 10.1128/JVI.06209-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cannon MJ, Openshaw PJ, Askonas BA. 1988. Cytotoxic T cells clear virus but augment lung pathology in mice infected with respiratory syncytial virus. J. Exp. Med. 168:1163–1168. 10.1084/jem.168.3.1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graham BS, Bunton LA, Wright PF, Karzon DT. 1991. Role of T lymphocyte subsets in the pathogenesis of primary infection and rechallenge with respiratory syncytial virus in mice. J. Clin. Invest. 88:1026–1033. 10.1172/JCI115362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graham BS, Perkins MD, Wright PF, Karzon DT. 1988. Primary respiratory syncytial virus infection in mice. J. Med. Virol. 26:153–162. 10.1002/jmv.1890260207 [DOI] [PubMed] [Google Scholar]
- 13.Hashimoto K, Durbin JE, Zhou W, Collins RD, Ho SB, Kolls JK, Dubin PJ, Sheller JR, Goleniewska K, O'Neal JF, Olson SJ, Mitchell D, Graham BS, Peebles RS., Jr 2005. Respiratory syncytial virus infection in the absence of STAT 1 results in airway dysfunction, airway mucus, and augmented IL-17 levels. J. Allergy Clin. Immunol. 116:550–557. 10.1016/j.jaci.2005.03.051 [DOI] [PubMed] [Google Scholar]
- 14.Lukacs NW, Moore ML, Rudd BD, Berlin AA, Collins RD, Olson SJ, Ho SB, Peebles RS., Jr 2006. Differential immune responses and pulmonary pathophysiology are induced by two different strains of respiratory syncytial virus. Am. J. Pathol. 169:977–986. 10.2353/ajpath.2006.051055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stokes KL, Chi MH, Sakamoto K, Newcomb DC, Currier MG, Huckabee MM, Lee S, Goleniewska K, Pretto C, Williams JV, Hotard A, Sherrill TP, Peebles RS, Jr, Moore ML. 2011. Differential pathogenesis of respiratory syncytial virus clinical isolates in BALB/c mice. J. Virol. 85:5782–5793. 10.1128/JVI.01693-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaplan MH. 2005. STAT4: a critical regulator of inflammation in vivo. Immunol. Res. 31:231–242. 10.1385/IR:31:3:231 [DOI] [PubMed] [Google Scholar]
- 17.Kaplan MH, Sun YL, Hoey T, Grusby MJ. 1996. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature 382:174–177. 10.1038/382174a0 [DOI] [PubMed] [Google Scholar]
- 18.Schulz EG, Mariani L, Radbruch A, Hofer T. 2009. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-gamma and interleukin-12. Immunity 30:673–683. 10.1016/j.immuni.2009.03.013 [DOI] [PubMed] [Google Scholar]
- 19.Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, Sangster MY, Vignali DA, Doherty PC, Grosveld GC, Ihle JN. 1996. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature 382:171–174. 10.1038/382171a0 [DOI] [PubMed] [Google Scholar]
- 20.Thieu VT, Yu Q, Chang HC, Yeh N, Nguyen ET, Sehra S, Kaplan MH. 2008. Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity 29:679–690. 10.1016/j.immuni.2008.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, Murphy TL, Murphy KM. 2002. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat. Immunol. 3:549–557. 10.1038/ni794 [DOI] [PubMed] [Google Scholar]
- 22.Mullen AC, High FA, Hutchins AS, Lee HW, Villarino AV, Livingston DM, Kung AL, Cereb N, Yao TP, Yang SY, Reiner SL. 2001. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science 292:1907–1910. 10.1126/science.1059835 [DOI] [PubMed] [Google Scholar]
- 23.Nguyen KB, Watford WT, Salomon R, Hofmann SR, Pien GC, Morinobu A, Gadina M, O'Shea JJ, Biron CA. 2002. Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science 297:2063–2066. 10.1126/science.1074900 [DOI] [PubMed] [Google Scholar]
- 24.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. 2002. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science 295:338–342. 10.1126/science.1065543 [DOI] [PubMed] [Google Scholar]
- 25.Usui T, Preiss JC, Kanno Y, Yao ZJ, Bream JH, O'Shea JJ, Strober W. 2006. T-bet regulates Th1 responses through essential effects on GATA-3 function rather than on IFNG gene acetylation and transcription. J. Exp. Med. 203:755–766. 10.1084/jem.20052165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gil MP, Ploquin MJ, Watford WT, Lee SH, Kim K, Wang X, Kanno Y, O'Shea JJ, Biron CA. 2012. Regulating type 1 IFN effects in CD8 T cells during viral infections: changing STAT4 and STAT1 expression for function. Blood 120:3718–3728. 10.1182/blood-2012-05-428672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agarwal P, Raghavan A, Nandiwada SL, Curtsinger JM, Bohjanen PR, Mueller DL, Mescher MF. 2009. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J. Immunol. 183:1695–1704. 10.4049/jimmunol.0900592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. 2005. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 174:4465–4469. 10.4049/jimmunol.174.8.4465 [DOI] [PubMed] [Google Scholar]
- 29.Mack EA, Kallal LE, Demers DA, Biron CA. 2011. Type 1 interferon induction of natural killer cell gamma interferon production for defense during lymphocytic choriomeningitis virus infection. mBio 2(4):e00169-11. 10.1128/mBio.00169-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyagi T, Gil MP, Wang X, Louten J, Chu WM, Biron CA. 2007. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J. Exp. Med. 204:2383–2396. 10.1084/jem.20070401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen KB, Salazar-Mather TP, Dalod MY, Van Deusen JB, Wei XQ, Liew FY, Caligiuri MA, Durbin JE, Biron CA. 2002. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J. Immunol. 169:4279–4287. 10.4049/jimmunol.169.8.4279 [DOI] [PubMed] [Google Scholar]
- 32.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. 2000. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100:655–669. 10.1016/S0092-8674(00)80702-3 [DOI] [PubMed] [Google Scholar]
- 33.Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM. 2007. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat. Immunol. 8:145–153. 10.1038/ni1424 [DOI] [PubMed] [Google Scholar]
- 34.Hwang ES, Szabo SJ, Schwartzberg PL, Glimcher LH. 2005. T helper cell fate specified by kinase-mediated interaction of T-bet with GATA-3. Science 307:430–433. 10.1126/science.1103336 [DOI] [PubMed] [Google Scholar]
- 35.Fujiwara M, Hirose K, Kagami S, Takatori H, Wakashin H, Tamachi T, Watanabe N, Saito Y, Iwamoto I, Nakajima H. 2007. T-bet inhibits both TH2 cell-mediated eosinophil recruitment and TH17 cell-mediated neutrophil recruitment into the airways. J. Allergy Clin. Immunol. 119:662–670. 10.1016/j.jaci.2006.12.643 [DOI] [PubMed] [Google Scholar]
- 36.Lazarevic V, Chen X, Shim JH, Hwang ES, Jang E, Bolm AN, Oukka M, Kuchroo VK, Glimcher LH. 2011. T-bet represses T(H)17 differentiation by preventing Runx1-mediated activation of the gene encoding RORgammat. Nat. Immunol. 12:96–104. 10.1038/ni.1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oestreich KJ, Weinmann AS. 2012. T-bet employs diverse regulatory mechanisms to repress transcription. Trends Immunol. 33:78–83. 10.1016/j.it.2011.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu J, Jankovic D, Oler AJ, Wei G, Sharma S, Hu G, Guo L, Yagi R, Yamane H, Punkosdy G, Feigenbaum L, Zhao K, Paul WE. 2012. The transcription factor T-bet is induced by multiple pathways and prevents an endogenous Th2 cell program during Th1 cell responses. Immunity 37:660–673. 10.1016/j.immuni.2012.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tekkanat KK, Maassab H, Berlin AA, Lincoln PM, Evanoff HL, Kaplan MH, Lukacs NW. 2001. Role of interleukin-12 and stat-4 in the regulation of airway inflammation and hyperreactivity in respiratory syncytial virus infection. Am. J. Pathol. 159:631–638. 10.1016/S0002-9440(10)61734-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tekkanat KK, Maassab HF, Cho DS, Lai JJ, John A, Berlin A, Kaplan MH, Lukacs NW. 2001. IL-13-induced airway hyperreactivity during respiratory syncytial virus infection is STAT6 dependent. J. Immunol. 166:3542–3548. 10.4049/jimmunol.166.5.3542 [DOI] [PubMed] [Google Scholar]
- 41.Furuta S, Kagami S, Tamachi T, Ikeda K, Fujiwara M, Suto A, Hirose K, Watanabe N, Saito Y, Iwamoto I, Nakajima H. 2008. Overlapping and distinct roles of STAT4 and T-bet in the regulation of T cell differentiation and allergic airway inflammation. J. Immunol. 180:6656–6662. 10.4049/jimmunol.180.10.6656 [DOI] [PubMed] [Google Scholar]
- 42.Bot A, Rodrigo E, Wolfe T, Bot S, Von Herrath MG. 2003. Infection-triggered regulatory mechanisms override the role of STAT 4 in control of the immune response to influenza virus antigens. J. Virol. 77:5794–5800. 10.1128/JVI.77.10.5794-5800.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raman K, Kaplan MH, Hogaboam CM, Berlin A, Lukacs NW. 2003. STAT4 signal pathways regulate inflammation and airway physiology changes in allergic airway inflammation locally via alteration of chemokines. J. Immunol. 170:3859–3865. 10.4049/jimmunol.170.7.3859 [DOI] [PubMed] [Google Scholar]
- 44.Kim YS, Choi SJ, Choi JP, Jeon SG, Oh S, Lee BJ, Gho YS, Lee CG, Zhu Z, Elias JA, Kim YK. 2010. IL-12-STAT4-IFN-gamma axis is a key downstream pathway in the development of IL-13-mediated asthma phenotypes in a Th2 type asthma model. Exp. Mol. Med. 42:533–546. 10.3858/emm.2010.42.8.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.National Research Council. 1996. Guide for the care and use of laboratory animals. National Academies Press, Washington, DC [Google Scholar]
- 46.Moore ML, Chi MH, Luongo C, Lukacs NW, Polosukhin VV, Huckabee MM, Newcomb DC, Buchholz UJ, Crowe JE, Jr, Goleniewska K, Williams JV, Collins PL, Peebles RS., Jr 2009. A chimeric A2 strain of respiratory syncytial virus (RSV) with the fusion protein of RSV strain line 19 exhibits enhanced viral load, mucus, and airway dysfunction. J. Virol. 83:4185–4194. 10.1128/JVI.01853-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bates JT, Keefer CJ, Utley TJ, Correia BE, Schief WR, Crowe JE., Jr 2013. Reversion of somatic mutations of the respiratory syncytial virus-specific human monoclonal antibody Fab19 reveal a direct relationship between association rate and neutralizing potency. J. Immunol. 190:3732–3739. 10.4049/jimmunol.1202964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Newcomb DC, Boswell MG, Huckabee MM, Goleniewska K, Dulek DE, Reiss S, Lukacs NW, Kolls JK, Peebles RS., Jr 2012. IL-13 regulates Th17 secretion of IL-17A in an IL-10-dependent manner. J. Immunol. 188:1027–1035. 10.4049/jimmunol.1102216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Erickson JJ, Gilchuk P, Hastings AK, Tollefson SJ, Johnson M, Downing MB, Boyd KL, Johnson JE, Kim AS, Joyce S, Williams JV. 2012. Viral acute lower respiratory infections impair CD8+ T cells through PD-1. J. Clin. Invest. 122:2967–2982. 10.1172/JCI62860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hahn YS, Hahn CS, Braciale VL, Braciale TJ, Rice CM. 1992. CD8+ T cell recognition of an endogenously processed epitope is regulated primarily by residues within the epitope. J. Exp. Med. 176:1335–1341. 10.1084/jem.176.5.1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McGill J, Van Rooijen N, Legge KL. 2008. Protective influenza-specific CD8 T cell responses require interactions with dendritic cells in the lungs. J. Exp. Med. 205:1635–1646. 10.1084/jem.20080314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lazarevic V, Glimcher LH. 2011. T-bet in disease. Nat. Immunol. 12:597–606. 10.1038/ni.2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng W, Flavell RA. 1997. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89:587–596. 10.1016/S0092-8674(00)80240-8 [DOI] [PubMed] [Google Scholar]
- 54.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. 2006. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126:1121–1133. 10.1016/j.cell.2006.07.035 [DOI] [PubMed] [Google Scholar]
- 55.Oestreich KJ, Weinmann AS. 2012. Transcriptional mechanisms that regulate T helper 1 cell differentiation. Curr. Opin. Immunol. 24:191–195. 10.1016/j.coi.2011.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carter LL, Murphy KM. 1999. Lineage-specific requirement for signal transducer and activator of transcription (Stat)4 in interferon gamma production from CD4(+) versus CD8(+) T cells. J. Exp. Med. 189:1355–1360. 10.1084/jem.189.8.1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Q, Eppolito C, Odunsi K, Shrikant PA. 2006. IL-12-programmed long-term CD8+ T cell responses require STAT4. J. Immunol. 177:7618–7625. 10.4049/jimmunol.177.11.7618 [DOI] [PubMed] [Google Scholar]
- 58.Sullivan BM, Juedes A, Szabo SJ, von Herrath M, Glimcher LH. 2003. Antigen-driven effector CD8 T cell function regulated by T-bet. Proc. Natl. Acad. Sci. U. S. A. 100:15818–15823. 10.1073/pnas.2636938100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kulkarni AB, Connors M, Firestone CY, Morse HC, III, Murphy BR. 1993. The cytolytic activity of pulmonary CD8+ lymphocytes, induced by infection with a vaccinia virus recombinant expressing the M2 protein of respiratory syncytial virus (RSV), correlates with resistance to RSV infection in mice. J. Virol. 67:1044–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kulkarni AB, Collins PL, Bacik I, Yewdell JW, Bennink JR, Crowe JE, Jr, Murphy BR. 1995. Cytotoxic T cells specific for a single peptide on the M2 protein of respiratory syncytial virus are the sole mediators of resistance induced by immunization with M2 encoded by a recombinant vaccinia virus. J. Virol. 69:1261–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hwang I, Scott JM, Kakarla T, Duriancik DM, Choi S, Cho C, Lee T, Park H, French AR, Beli E, Gardner E, Kim S. 2012. Activation mechanisms of natural killer cells during influenza virus infection. PLoS One 7:e51858. 10.1371/journal.pone.0051858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harker JA, Godlee A, Wahlsten JL, Lee DC, Thorne LG, Sawant D, Tregoning JS, Caspi RR, Bukreyev A, Collins PL, Openshaw PJ. 2010. Interleukin 18 coexpression during respiratory syncytial virus infection results in enhanced disease mediated by natural killer cells. J. Virol. 84:4073–4082. 10.1128/JVI.02014-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kaiko GE, Phipps S, Angkasekwinai P, Dong C, Foster PS. 2010. NK cell deficiency predisposes to viral-induced Th2-type allergic inflammation via epithelial-derived IL-25. J. Immunol. 185:4681–4690. 10.4049/jimmunol.1001758 [DOI] [PubMed] [Google Scholar]
- 64.Hussell T, Openshaw PJ. 1998. Intracellular IFN-gamma expression in natural killer cells precedes lung CD8+ T cell recruitment during respiratory syncytial virus infection. J. Gen. Virol. 79(Part 11):2593–2601 [DOI] [PubMed] [Google Scholar]
- 65.Graham BS, Bunton LA, Rowland J, Wright PF, Karzon DT. 1991. Respiratory syncytial virus infection in anti-mu-treated mice. J. Virol. 65:4936–4942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hunter CA, Kastelein R. 2012. Interleukin-27: balancing protective and pathological immunity. Immunity 37:960–969. 10.1016/j.immuni.2012.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yoo JK, Kim TS, Hufford MM, Braciale TJ. 2013. Viral infection of the lung: host response and sequelae. J. Allergy Clin. Immunol. 132:1263–1276, quiz 1277. 10.1016/j.jaci.2013.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Graham BS, Bunton LA, Wright PF, Karzon DT. 1991. Reinfection of mice with respiratory syncytial virus. J. Med. Virol. 34:7–13. 10.1002/jmv.1890340103 [DOI] [PubMed] [Google Scholar]
- 69.Haynes LM, Tonkin J, Anderson LJ, Tripp RA. 2002. Neutralizing anti-F glycoprotein and anti-substance P antibody treatment effectively reduces infection and inflammation associated with respiratory syncytial virus infection. J. Virol. 76:6873–6881. 10.1128/JVI.76.14.6873-6881.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schmitt N, Bustamante J, Bourdery L, Bentebibel SE, Boisson-Dupuis S, Hamlin F, Tran MV, Blankenship D, Pascual V, Savino DA, Banchereau J, Casanova JL, Ueno H. 2013. IL-12 receptor beta1 deficiency alters in vivo T follicular helper cell response in humans. Blood 121:3375–3385. 10.1182/blood-2012-08-448902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Culley FJ, Pollott J, Openshaw PJ. 2002. Age at first viral infection determines the pattern of T cell-mediated disease during reinfection in adulthood. J. Exp. Med. 196:1381–1386. 10.1084/jem.20020943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dakhama A, Park JW, Taube C, Joetham A, Balhorn A, Miyahara N, Takeda K, Gelfand EW. 2005. The enhancement or prevention of airway hyperresponsiveness during reinfection with respiratory syncytial virus is critically dependent on the age at first infection and IL-13 production. J. Immunol. 175:1876–1883. 10.4049/jimmunol.175.3.1876 [DOI] [PubMed] [Google Scholar]
- 73.Lee YM, Miyahara N, Takeda K, Prpich J, Oh A, Balhorn A, Joetham A, Gelfand EW, Dakhama A. 2008. IFN-gamma production during initial infection determines the outcome of reinfection with respiratory syncytial virus. Am. J. Respir. Crit. Care Med. 177:208–218. 10.1164/rccm.200612-1890OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rauch I, Muller M, Decker T. 2013. The regulation of inflammation by interferons and their STATs. JAKSTAT 2:e23820. 10.4161/jkst.23820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Watford WT, Hissong BD, Bream JH, Kanno Y, Muul L, O'Shea JJ. 2004. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol. Rev. 202:139–156. 10.1111/j.0105-2896.2004.00211.x [DOI] [PubMed] [Google Scholar]
- 76.Mathur AN, Chang HC, Zisoulis DG, Stritesky GL, Yu Q, O'Malley JT, Kapur R, Levy DE, Kansas GS, Kaplan MH. 2007. Stat3 and Stat4 direct development of IL-17-secreting Th cells. J. Immunol. 178:4901–4907. 10.4049/jimmunol.178.8.4901 [DOI] [PubMed] [Google Scholar]
- 77.Villarino AV, Gallo E, Abbas AK. 2010. STAT1-activating cytokines limit Th17 responses through both T-bet-dependent and -independent mechanisms. J. Immunol. 185:6461–6471. 10.4049/jimmunol.1001343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fukao T, Frucht DM, Yap G, Gadina M, O'Shea JJ, Koyasu S. 2001. Inducible expression of Stat4 in dendritic cells and macrophages and its critical role in innate and adaptive immune responses. J. Immunol. 166:4446–4455. 10.4049/jimmunol.166.7.4446 [DOI] [PubMed] [Google Scholar]
- 79.Frucht DM, Aringer M, Galon J, Danning C, Brown M, Fan S, Centola M, Wu CY, Yamada N, El Gabalawy H, O'Shea JJ. 2000. Stat4 is expressed in activated peripheral blood monocytes, dendritic cells, and macrophages at sites of Th1-mediated inflammation. J. Immunol. 164:4659–4664. 10.4049/jimmunol.164.9.4659 [DOI] [PubMed] [Google Scholar]
- 80.Remoli ME, Ragimbeau J, Giacomini E, Gafa V, Severa M, Lande R, Pellegrini S, Coccia EM. 2007. NF-kappaB is required for STAT-4 expression during dendritic cell maturation. J. Leukoc. Biol. 81:355–363. 10.1189/jlb.0506319 [DOI] [PubMed] [Google Scholar]
- 81.Nokes DJ, Okiro EA, Ngama M, Ochola R, White LJ, Scott PD, English M, Cane PA, Medley GF. 2008. Respiratory syncytial virus infection and disease in infants and young children observed from birth in Kilifi District, Kenya. Clin. Infect. Dis. 46:50–57. 10.1086/524019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ostler T, Hussell T, Surh CD, Openshaw P, Ehl S. 2001. Long-term persistence and reactivation of T cell memory in the lung of mice infected with respiratory syncytial virus. Eur. J. Immunol. 31:2574–2582. 10.1002/1521-4141(200109)31:9<2574::AID-IMMU2574>3.0.CO;2-V [DOI] [PubMed] [Google Scholar]
- 83.Takemoto N, Intlekofer AM, Northrup JT, Wherry EJ, Reiner SL. 2006. Cutting edge: IL-12 inversely regulates T-bet and eomesodermin expression during pathogen-induced CD8+ T cell differentiation. J. Immunol. 177:7515–7519. 10.4049/jimmunol.177.11.7515 [DOI] [PubMed] [Google Scholar]