

ABSTRACT
Host and viral factors influence the HIV-1 infection course. Reduced Nef function has been observed in HIV-1 controllers during the chronic phase, but the kinetics and mechanisms of Nef attenuation in such individuals remain unclear. We examined plasma RNA-derived Nef clones from 10 recently infected individuals who subsequently suppressed viremia to less than 2,000 RNA copies/ml within 1 year postinfection (acute controllers) and 50 recently infected individuals who did not control viremia (acute progressors). Nef clones from acute controllers displayed a lesser ability to downregulate CD4 and HLA class I from the cell surface and a reduced ability to enhance virion infectivity compared to those from acute progressors (all P < 0.01). HLA class I downregulation activity correlated inversely with days postinfection (Spearman's R = −0.85, P = 0.004) and positively with baseline plasma viral load (Spearman's R = 0.81, P = 0.007) in acute controllers but not in acute progressors. Nef polymorphisms associated with functional changes over time were identified in follow-up samples from six controllers. For one such individual, mutational analyses indicated that four polymorphisms selected by HLA-A*31 and B*37 acted in combination to reduce Nef steady-state protein levels and HLA class I downregulation activity. Our results demonstrate that relative control of initial HIV-1 viremia is associated with Nef clones that display reduced function, which in turn may influence the course of HIV-1 infection. Transmission of impaired Nef sequences likely contributed in part to this observation; however, accumulation of HLA-associated polymorphisms in Nef that impair function also suggests that CD8+ T-cell pressures play a role in this phenomenon.
IMPORTANCE Rare individuals can spontaneously control HIV-1 viremia in the absence of antiretroviral treatment. Understanding the host and viral factors that contribute to the controller phenotype may identify new strategies to design effective vaccines or therapeutics. The HIV-1 Nef protein enhances viral pathogenesis through multiple mechanisms. We examined the function of plasma HIV-1 RNA-derived Nef clones isolated from 10 recently infected individuals who subsequently controlled HIV viremia compared to the function of those from 50 individuals who failed to control viremia. Our results demonstrate that early Nef clones from HIV controllers displayed lower HLA class I and CD4 downregulation activity, as well as a reduced ability to enhance virion infectivity. The accumulation of HLA-associated polymorphisms in Nef during the first year postinfection was associated with impaired protein function in some controllers. This report highlights the potential for host immune responses to modulate HIV pathogenicity and disease outcome by targeting cytotoxic T lymphocyte (CTL) epitopes in Nef.
INTRODUCTION
Rare HIV-1-infected individuals who suppress plasma viral loads (pVL) to fewer than 50 RNA copies/ml (“elite controllers” [EC]) or to fewer than 2,000 RNA copies/ml (“viremic controllers”) in the absence of antiretroviral therapy provide an opportunity to identify host and viral determinants of spontaneous HIV-1 control that could aid development of vaccines or novel therapeutics. However, the mechanisms underlying the HIV-1 controller phenotype, particularly those acting at the acute/early infection stage, remain incompletely defined.
Host genetic and immune factors influence HIV-1 control. Protective human leukocyte antigen (HLA) class I alleles, notably B*57 and B*27, have been associated with lower pVL and delayed rates of disease progression in natural-history studies (1, 2) and genome-wide-association studies (3–5), and these alleles are enriched among HIV-1 controllers (6–8). Polyfunctional cytokine production and rapid perforin or granzyme expression are also frequently observed in CD8+ cytotoxic T lymphocytes (CTL) from controllers (9–11), suggesting that qualitative immune characteristics also influence viremia (12). While CTL responses targeting the HIV-1 Gag protein are likely to be central mediators of immune control (13), responses to Nef might also be beneficial (14, 15). Taking the data together, cellular immune responses recognizing mutationally constrained viral epitopes presented by certain HLA alleles are believed to be key to effective HIV-1 suppression (16).
Viral genetic factors also influence HIV-1 pathogenesis. In vitro viral replication capacity independently associates with early clinical markers of pathogenesis (17). Moreover, Gag, Pol, and Nef proteins from chronically infected EC have consistently displayed relative functional attenuation (18–21), suggesting that impaired viral function is a hallmark of this phenotype (22). In many cases, host expression of protective HLA alleles or the presence of specific HLA-associated escape mutations or both are associated with even lower viral protein function in these individuals, suggesting that adaptation to host HLA-restricted CTL can further attenuate HIV-1. Consistent with the observed “genetic fragility” (i.e., mutationally sensitive nature) of the HIV-1 p24 capsid protein as a result of its critical role in virion assembly (23), functional costs of CTL escape have been observed most readily in Gag (24–28), but immune-driven functional costs have also been demonstrated in Pol (18, 29) and Env (19, 30). The observation that compensatory mutations that offset the functional impact of escape arise more frequently in progressors than in controllers (28, 31) (likely due in part to severely reduced viral replication in the latter individuals) further complicates the study of HIV-1 adaptation to its host and its pathogenic consequences.
A major gap in our knowledge of HIV-1 controllers is a poor understanding of early events following infection that contribute to clinical outcome in these individuals (22). HIV-1 controller cohorts are generally comprised of individuals identified during the chronic phase; as such, sequence/function relationships in early controller viruses and the role of early host immune responses in modulating viral pathogenesis remain incompletely defined. A recent study by our group demonstrated reduced in vitro replication capacities of recombinant HIV-1 strains encoding gag and protease from 18 recently infected individuals who subsequently controlled pVL to less than 2,000 RNA copies/ml compared to those from 45 individuals who progressed with higher viremia, which was likely due to both transmission of less-fit strains and selection of CTL escape mutations by protective HLA alleles (32). It remains to be determined whether other viral proteins in controllers exhibit similar early evidence of relative attenuation. In particular, we hypothesized that HIV-1 Nef, an ∼27-kDa accessory protein that enhances viral pathogenesis, may play an early role in determining HIV-1 control. Nef interacts with a number of host proteins and performs multiple functions (33), including downregulation of cell surface CD4 (34) and HLA class I (35), upregulation of HLA class II invariant chain (CD74) (36), enhancement of virion infectivity (37), stimulation of viral replication in peripheral blood mononuclear cells (PBMC) (38), and alteration of T cell receptor (TCR) signaling (39, 40). Nef's relevance to pathogenesis is illustrated by the exceptionally slow disease progression exhibited by individuals infected with nef-deleted or nef-defective HIV-1 (41–44).
To enhance our understanding of early Nef function in HIV-1 controllers, we examined the sequence and in vitro function of plasma RNA-derived clonal nef sequences from 10 recently infected individuals who subsequently suppressed pVL to less than 2,000 RNA copies/ml in the absence of treatment (acute controllers; “AC”) and from 50 recently infected individuals who failed to suppress viremia (acute progressors, “AP”). Nef clones were assessed for their ability to downregulate CD4 and HLA class I and to enhance virion infectivity. Longitudinal assessments were also performed on Nef clones from six AC from whom follow-up samples were available, including one individual for whom the impact of HLA-associated polymorphisms on Nef function was explored in detail using site-directed mutagenesis. Overall, our results indicate that HIV-1 viremia control is associated with the presence of early Nef clones that display reduced in vitro function. This reduced function is likely a result of acquisition of partially attenuated viral strains at transmission, combined with the subsequent selection of escape mutations by host CTL responses that impair one or more Nef activities.
MATERIALS AND METHODS
Study subjects.
This study was approved by the Research Ethics Boards at Simon Fraser University (Burnaby, BC, Canada) and the Massachusetts General Hospital (Boston, MA). As described previously (32), participants were identified at sites in the United States, Australia, and Germany during acute/early HIV-1 infection as defined by the Acute Infection Early Disease Research Program (AIEDRP) criteria (45). Study subjects included n = 10 acute controllers (AC) who spontaneously suppressed HIV-1 plasma viremia to less than 2,000 RNA copies/ml during the first year of infection and n = 50 acute progressors (AP) who failed to suppress HIV-1 viremia to less than 2,000 RNA copies/ml during this time (Fig. 1). The earliest available plasma samples were studied. For AC and AP, these were collected an estimated median of 72 (interquartile range [IQR], 57 to 99) days and 56 (IQR, 39 to 75) days postinfection, respectively (Table 1). Additional follow-up analyses were conducted for six AC for whom samples were available (Table 1). All participants remained untreated for a minimum of 1 year postinfection.
FIG 1.

Plasma viral loads of AC and AP during the first year postinfection. Lines illustrate the plasma viral load (in HIV RNA copies/ml) for each participant. (A) Data are plotted for 10 AC who achieved viremic control (<2,000 RNA copies/ml) within the first year of infection. (B) Data are plotted for 50 AP who did not control viremia during this time. Dashed lines indicate the threshold value (2,000 RNA copies/ml) used to identify acute controllers.
TABLE 1.
Description of acute controllers (n = 10)a
| Participant | HLAs | eDPIb (pVLc) |
|
|---|---|---|---|
| Initial | Follow-up | ||
| AC01 | A*01:01/A*31:01; B*37:01/B*51:09; C*01:02/C*06:02 | 33 (25,000) | 383 (50) |
| AC02 | A*02:01/A*32:01; B*35/B*44; C*04:01/C*05 | 72 (15,200) | 469 (774) |
| AC03 | A*02:01/A*24:01; B*15:01/B*41:01; C*03/C*17 | 86 (1,490) | |
| AC04 | A*02:01/A*24:02; B*27:05/B*35:12; C*01:02/C*01:02 | 72 (1,140) | 251 (50) |
| AC05 | A*24:02/A*31:01; B*39:01/B*52:01; C*07:02/C*12:02 | 71 (7,160) | 587 (250) |
| AC06 | A*01:01/A*02:01; B*44:02/B*57:01; C*05/C*06:02 | 135 (440) | |
| AC07 | A*25:01/A*68:01; B*18:01/B*57:01; C*06:02/C*12:03 | 98 (23,900) | 204 (1,720) |
| AC08 | A*02:01/A*31:01; B*08:01/B*44:02; C*05/C*07 | 102 (572) | |
| AC09 | A*01:01/A*68:02; B*08:01/B*44:02; C*05/C*07 | 52 (45,700) | 268 (110) |
| AC10 | A*01:01/A*01:01; B*15:17/B*49:01; C*07:01/C*12:03 | 59 (22,200) | |
Abbreviations: HLA, human leukocyte antigen; eDPI, estimated number of days postinfection; pVL, plasma viral load at baseline.
Median (IQR) eDPI for AC (n = 10) at baseline, 72 (57 to 99). Median (IQR) eDPI for all AP (n = 50) at baseline, 56 (39 to 75) (P = 0.07). Median (IQR) eDPI for AP > 40 eDPI (n = 37) at baseline, 72 (53 to 88) (P = 0.4). P values represent Mann-Whitney comparisons to AC cohort.
Median (IQR) pVL in numbers of HIV RNA copies per milliliter for AC (n = 10) at baseline, 11,180 (998 to 24,175). Median (IQR) pVL for all AP (n = 50) at baseline, 337,500 (35,925 to 750,100) (P < 0.001). Median (IQR) pVL for AP > 40 eDPI (n = 37) at baseline, 186,000 (22,350 to 750,005) (P < 0.001). P values represent Mann-Whitney comparisons to AC cohort.
Viral (HIV-1 Nef) and Host (HLA class I) genotyping.
HIV-1 nef gene products were amplified from plasma RNA as described previously (32). Briefly, nested reverse transcription-PCR (RT-PCR) was performed using HIV-1-specific primers, where the second-round forward and reverse primers included EcoRI and SacII restriction sites, respectively, used for cloning. Amplicons were ligated into pIRES2-enhanced green fluorescent protein (EGFP) (Clontech), transformed into Escherichia cloni 10G cells (Lucigen), and selected on LB agar plates containing kanamycin. Colonies were screened by restriction enzyme digest to verify nef insertion. Nested RT-PCR amplicons and pIRES2-nef-EGFP clones were sequenced bidirectionally on a 3130xl or 3730xl automated DNA sequencer (Applied Biosystems, Inc.). Chromatograms were analyzed using Sequencher v5.0 (Genecodes) or RECall (46). In bulk sequences, nucleotide mixtures were called if the subdominant peak height exceeded 25% (Sequencher) or the subdominant peak area exceeded 20% (RECall) of the dominant peak. All sequences were confirmed to be HIV-1 subtype B using the Recombinant Identification Program (RIP; http://www.hiv.lanl.gov/content/sequence/RIP/RIP.html). nef sequences were aligned to HXB2 using an in-house tool based on the HyPhy platform (47), and phylogenetic analysis was conducted using PhyML (48, 49). Each clone sequence was also compared to the original bulk sequence to enumerate the number of amino acid differences between them. These steps ensured that each Nef clone encoded an intact open reading frame and was free of gross genetic defects (e.g., large deletions) and that clones chosen for functional analysis were representative of each individual's circulating viral quasispecies. HLA class I typing was performed using genomic DNA extracted from PBMC or plasma using sequence-based methods (50).
CD4 and HLA class I downregulation assays.
Nef-mediated CD4 and HLA class I downregulation function was assessed by flow cytometry following transient transfection, as described previously (21). Briefly, 3 × 105 CEM-A*02-positive (CEM-A*02+) cells in Opti-MEM (Life Technologies) were transfected with 5 μg of pIRES2-nef-EGFP by electroporation (Bio-Rad Gene Pulser MXcell) (square wave, 250 V, 2,000 μF, infinite Ω, 25 ms) and recovered in R10+ medium (RPMI 1640 containing 10% fetal calf serum, 2 mM l-glutamine, 100 U of penicillin/ml, and 100 μg of streptomycin/ml) (Sigma). After 24 h, unfixed transfected cells were stained with allophycocyanin (APC)-labeled anti-CD4 and phycoerythrin (PE)-labeled anti-HLA-A*02 antibodies (BD Biosciences), and surface expression of these molecules was detected using a Guava easyCyte 8HT flow cytometer (Millipore). For each Nef clone, the median fluorescence intensity (MFI) of CD4 or HLA class I on GFP-positive (Nef-producing) cells was normalized to that of cells transfected with a positive control (pIRES2-EGFP containing SF2 strain Nef; NefSF2) and a negative control (empty pIRES2-EGFP) using the following formula: (MFIpatient Nef − MFInegative control)/(MFIpositive control − MFInegative control). Normalized values less than 1.0 represent downregulation activities lower than those of the positive-control NefSF2, while values greater than 1.0 represent downregulation activities higher than those of NefSF2. The calculated value of the negative control is zero. Each clone was tested in a minimum of 3 replicates, and the results were averaged.
Virion infectivity assays.
nef clones were transferred into a pNL4.3 backbone plasmid as described previously (51) and confirmed by sequencing. Recombinant viruses carrying nef from HIV-1 strain SF2 (NL4.3-nefSF2) and those lacking nef (NL4.3Δnef) served as positive and negative controls, respectively. Infectious viruses were generated by transfection of HEK-293T cells with each proviral clone, and virus-containing supernatant was harvested at 48 h, as described previously (52). Viral stocks were quantified using a p24Gag enzyme-linked immunosorbent assay (ELISA) (ZeptoMetrix Corp.), and aliquots were stored at −80°C until use. Recombinant virus infectivity was determined by exposing 104 TZM-bl cells (catalog no. 8129; NIH AIDS Reagent Program) to 3 ng p24Gag virus stock followed by chemiluminescence detection 48 h later, as described previously (53). Infectivity values represent the means of the results of duplicate experiments, normalized to NL4.3-nefSF2, such that values less than 1.0 or greater than 1.0 indicated lower or higher activity than the positive-control strain, respectively.
Western blot analysis.
Steady-state Nef protein levels were measured by Western blotting for a subset of AC and AP clones. For this, 5 × 106 CEM-A*02+ cells were transfected with 10 μg of pIRES2-nef-EGFP using electroporation. After 24 h, cells were pelleted, lysed, and analyzed as described previously (52), with modifications as follows. Total cell lysates were subjected to SDS-PAGE using Mini-Protean TGX 4% to 20% gels (Bio-Rad Laboratories), and proteins were electroblotted onto a polyvinylidene difluoride (PVDF) membrane. Nef protein was detected using a polyclonal rabbit (catalog no. 2949; NIH AIDS Reagent Program) (1:4,000) (54) or sheep (catalog. no. ARP444; NIBSC Center for AIDS Reagents) (1:2,000) antibody followed by staining with secondary donkey anti-rabbit (GE Healthcare) (1:30,000) or anti-sheep (Jackson ImmunoResearch) (1:35,000) antibody. For all experiments, expression of beta-actin was assessed simultaneously using primary mouse anti-actin (Sigma) (1:20,000) and secondary goat anti-mouse (Jackson ImmunoResearch) (1:20,000) antibodies. Band intensities were quantified with ImageQuant LAS 400 (GE Healthcare) and compared to the positive-control SF2 Nef values.
Site-directed mutagenesis.
nef variants were generated using a QuikChange II site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions or using a PCR-based overlap extension method (55). Mutations were validated by bidirectional sequencing of the full-length nef insert prior to functional analysis.
Statistics.
Statistical analyses were conducted in Prism 5.0 (Graphpad Software, Inc.). The Mann-Whitney U test was used to compare Nef functions between groups. In patients with longitudinal (baseline versus follow-up) Nef functional assessments, the Wilcoxon paired test was employed. Spearman's correlation was used to investigate relationships between different Nef functions or samples with clinical HIV-1 parameters (e.g., CD4 T-cell count and plasma viral load). All tests were two-tailed, with P < 0.05 considered statistically significant.
Nucleotide sequence accession numbers.
All unique clonal nef sequences from AC have been deposited in GenBank (accession numbers KJ996014 to KJ996066).
RESULTS
Isolation of early Nef clones from HIV-1 controllers and progressors.
Clinical data from HIV-1-infected individuals enrolled at sites in the United States, Germany, and Australia were retrospectively screened to identify persons with acute/early infection who subsequently maintained viremia control below 2,000 RNA copies/ml during the first year postinfection and for whom a baseline sample within 150 days of their estimated date of infection was available for study (32) (Fig. 1). A comparison group of 50 acute progressors (AP) enrolled at the same sites who displayed pVL of greater than 2,000 RNA copies/ml over a similar period of time were also assessed. Consistent with a previous study undertaken in this cohort (32), no significant enrichment of protective HLA class I alleles was observed in AC compared to AP (e.g., HLA-B*57 prevalence was 20% in AC versus 10% in AP; Fisher's exact test P = 0.3) (data not shown).
The times of baseline sample collection differed slightly between cohorts: for AC, samples were collected a median of 72 (interquartile range [IQR], 57 to 99) days following the estimated infection date, while those for AP were collected a median of 56 (IQR, 39 to 75) days postinfection (Mann-Whitney test; P = 0.065) (Table 1). Baseline pVL was also significantly lower in AC, which is not surprising given that these patients were selected based on their ability to subsequently control viremia. Nevertheless, we were concerned that differences in baseline pVL could have been due in part to the earlier enrollment times for AP (who thus had a higher likelihood of being sampled closer to the time of peak viremia) and that this could potentially bias downstream analyses. Exclusion of AP with the earliest enrollment times (prior to 40 days postinfection) allowed us to identify a subset of 37 AP with baseline sample dates (median, 72 [IQR, 53 to 88] days) that were comparable to those of AC (P = 0.42) (Table 1). For the remainder of our study, all primary analyses were performed on the entire AP cohort, with secondary analyses performed using the AP subset matched for the enrollment date. Results were consistent in all cases, even when not explicitly indicated in the text.
Participant-derived nef alleles were amplified from baseline plasma by nested RT-PCR and sequenced. As expected from an acutely infected population, the median number of amino acid mixtures observed in bulk Nef HIV sequences was low: a median of 0 (IQR, 0 to 1) for AC compared to 0 (IQR, 0 to 2) for AP (P = 0.3). Nef amplicons were cloned into a cytomegalovirus (CMV) expression plasmid, and individual clones were isolated and resequenced to confirm an intact Nef open reading frame. For each AC, a minimum of 5 baseline nef clones (median, 7; range, 5 to 13) were isolated and sequenced. For each AP, 1 to 13 baseline clones (median, 2) were isolated and sequenced. All clones were HIV-1 subtype B. Each Nef clone was verified to cluster closely with the original bulk plasma HIV RNA sequence (see Fig. S1 in the supplemental material). Overall, the median number of amino acid differences between the clonal and original bulk sequences was 0 (IQR, 0 to 1) for both AC and AP. For each patient, the first isolated clone was arbitrarily designated a “representative clone,” while the most frequently observed clone was designated the “predominant clone” (see Fig. S1). AC and AP nef sequences did not display substantial intracohort clustering in a combined phylogeny (Fig. 2A; see also Fig. S1), consistent with previous analyses of gag and pol genes from these persons (32). Together with previous results, our data indicate that early HIV-1 control in AC is unlikely to be due to gross HIV sequence defects or recent descent from a common attenuated viral ancestor. We confirmed Nef expression by Western blotting for the predominant clone isolated from all 10 AC and compared detection levels to those of a random subset of 10 AP. No significant differences in band intensity were observed between cohorts (representative data shown in Fig. 2B), suggesting that Nef proteins from AC exhibited in vitro stability comparable to the stability of those from AP.
FIG 2.

Nef sequences from AC and AP display no substantial phylogenetic clustering. (A) An unrooted maximum-likelihood phylogenetic tree depicts the representative Nef clone isolated from 10 AC (red) and 50 AP (blue), indicating that sequences did not exhibit substantial clustering by cohort. The HIV-1SF2 Nef sequence (black) is included for comparison. A phylogenetic tree that includes all clonal Nef sequences used for this study is shown in Fig. S1 in the supplemental material. (B) Western blot analysis was used to examine steady-state protein levels in AC and AP Nef clones, along with NefSF2 (positive control) and ΔNef (negative control). β-Actin was used as a cellular protein control.
Nef clones from AC display impaired CD4 and HLA class I downregulation function.
The ability of Nef clones to downregulate cell surface CD4 and HLA class I molecules was examined at 24 h following transfection using flow cytometry (Fig. 3). For each AC, the function of 2 to 5 unique clones was assessed. Results were normalized to those of a positive control (SF2Nef) that displays strong CD4 and HLA class I downregulation activities (21, 56) such that values less than 1.0 indicate lower Nef function and values greater than 1.0 indicate higher Nef function relative to SF2Nef. A negative-control plasmid lacking nef did not downregulate either surface protein (Fig. 3A and B); as such, its activity is 0 in this assay. Based on an assessment of a representative clone isolated from each individual, the ability of Nef to downregulate CD4 in AC (median, 0.94 [IQR, 0.87 to 0.98]) was modestly lower than that observed in AP (0.99 [0.97 to 1.02]) (P = 0.004) (Fig. 3C). In addition, HLA class I downregulation activity was markedly lower in AC (0.83 [0.65 to 0.93]) than in AP (1.00 [0.94 to 1.06]) (P < 0.001) (Fig. 3D). Secondary analyses using the “date-of-enrollment-matched” AP subset were consistent with a lower function of AC compared to that seen with AP Nefs (CD4, P = 0.01; HLA, P = 0.001; data not shown), indicating that our findings are not attributable simply to biases related to the sample collection date. Similarly, analyses comparing the median downregulation functions of all Nef clones isolated in AC versus AP also indicated significantly lower activities in the latter group (CD4, P = 0.01; HLA, P < 0.001; Fig. S2 in the supplemental material and data not shown), suggesting that our observations were not driven by biases related to analyses of only a single clone per patient. Together, these results suggest that Nef-mediated HLA downregulation (and, to a lesser extent, CD4 downregulation) may contribute to early viremia control.
FIG 3.

Reduced in vitro function is observed for AC Nef clones. Nef CD4 and HLA class I downregulation functions were assessed in ≥3 replicates, using flow cytometry. (A and B) Representative flow cytometry plots display relative levels of downregulation of CD4 (A) and HLA class I (B) for CEM-A*02+ cells transfected with NefSF2 (positive control) or ΔNef (negative control) or for one participant-derived Nef clone. The x axis (GFP+) indicates transfected cells, and the y axis indicates surface expression of CD4 or HLA class I molecules. (C and D) Scatter plots depict normalized CD4 (C) or HLA class I (D) downregulation activities in representative Nef clones from AC (n = 10, red) compared to downregulation activities in those from AP (n = 50, blue). A dashed line (---) in each panel illustrates normalized activity of 1.0, which represents the function of NefSF2. Values below 1.0 indicate lower function relative to NefSF2, while values above 1.0 indicate higher Nef function.
Nef clones from AC show reduced ability to enhance virion infectivity.
We next constructed recombinant NL4.3 viruses encoding a representative early clonal nef sequence from 10 AC and 41 AP and assessed virion infectivity. No difference in viral production between stocks containing AC and AP nef sequences was observed, as determined by p24Gag ELISA, although there was a trend toward higher values among AC-derived viruses (Fig. 4A). TZM-bl cells were inoculated with virus (3 ng p24Gag), and infectivity was measured at 48 h postinfection using chemiluminescence assays. Data were normalized to a positive-control virus, NL4.3-nefSF2, as described above. All recombinant viral stocks displayed infectivity values greater than those determined for the NL4.3Δnef negative control, whose mean activity was 0.02 (standard deviation, 0.009; n = 8 replicates) in this assay. Overall, viruses encoding AC-derived nef alleles displayed significantly lower infectivity values (median, 0.59 [IQR, 0.44 to 0.71]) compared to those from AP (1.16 [0.71 to 1.51]) (P = 0.003) (Fig. 4B). Results were consistent in a secondary analysis using “date-of-enrollment-matched” AP clones (P = 0.01; data not shown).
FIG 4.
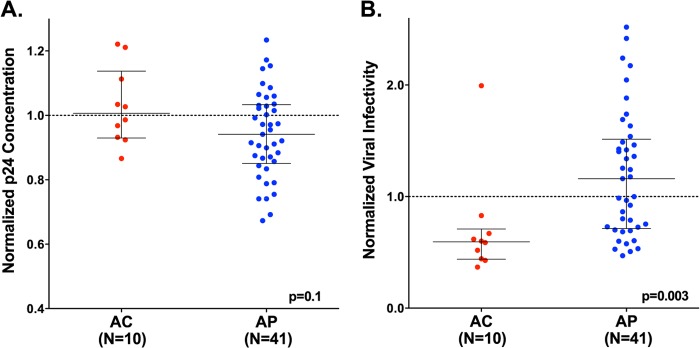
Reduced ability to enhance virion infectivity in AC Nef clones. Plots depict the normalized ability of recombinant viruses encoding representative Nef clones from AC (n = 10, red) and AP (n = 41, blue) to enhance viral production (p24Gag ng/ml) (A) and viral infectivity (B) in Tzm-bl cell assays. A dashed line (---) in each panel illustrates normalized activity of 1.0, which represents the function of positive-control NL4.3-nefSF2. Values below 1.0 indicate lower Nef function relative to NL4.3-nefSF2, while values above 1.0 indicate higher function. Each Nef-containing recombinant virus was tested in duplicate. Note that the negative-control NL4.3Δnef strain displayed a mean infectivity level of 0.02 (standard deviation [SD], 0.009; n = 8 replicates) in this assay.
HLA class I downregulation activity correlates with baseline clinical parameters in AC.
We next wanted to characterize the relationship between these three Nef functions and markers of clinical status in the AC and AP cohorts (Table 2 and Table 3, respectively; see also Fig. S3 in the supplemental material). As expected for samples obtained during early infection following peak viremia, pVL correlated inversely with the estimated number of days postinfection in both AC (Spearman R = −0.77, P = 0.01) and AP (R = −0.38, P = 0.007). Consistent with previous observations in other cohorts (21, 56, 57), Nef CD4 and HLA class I downregulation functions correlated positively in both AC (R = 0.74, P = 0.02) and AP (R = 0.56, P < 0.001). Given that these activities are largely genetically separable in mutational studies of HIV-1 reference strains (58, 59), this result supports the idea of the existence of secondary genetic determinants lying outside critical motifs in patient-derived Nef isolates that may modulate function and/or protein stability. Of note, the ability of Nef to enhance virion infectivity correlated significantly with baseline pVL in AP (R = 0.54, P < 0.001) but not AC. This may have been due to the generally low activity seen among AC clones for this function, but it warrants further investigation. Surprisingly, Nef's HLA class I downregulation activity correlated inversely with estimated numbers of days postinfection (R = −0.85, P = 0.004) and positively with pVL (R = 0.81, P = 0.007) in AC. Similar associations were not seen for AP, despite enhanced statistical power to observe differences in this group. Together, these results suggest that longitudinal alterations in Nef function may contribute to spontaneous HIV-1 control in AC but not in AP.
TABLE 2.
Correlation analyses for acute controllers (n = 10)a
| Parameter | Statistic | Baseline pVL | Nef CD4 downregulation | Nef HLA downregulation | Nef infectivity enhancement |
|---|---|---|---|---|---|
| Baseline eDPI | R | −0.77 | −0.52 | −0.85 | 0.19 |
| P | 0.007 | 0.1 | 0.004 | 0.6 | |
| Baseline pVL | R | 0.48 | 0.81 | 0.01 | |
| P | 0.2 | 0.01 | 1.0 | ||
| CD4 downregulation | R | 0.74 | −0.05 | ||
| P | 0.02 | 0.9 | |||
| HLA downregulation | R | 0.15 | |||
| P | 0.7 |
Abbreviations: pVL, plasma viral load; HLA, human leukocyte antigen; eDPI, estimated days postinfection. Data represent Spearman R and P values. Significant associations are highlighted in bold type.
TABLE 3.
Correlation analyses for acute progressors (n = 50)a
| Parameter | Statistic | Baseline pVL | Nef CD4 downregulation | Nef HLA downregulation | Nef infectivity enhancement |
|---|---|---|---|---|---|
| Baseline eDPI | R | −0.38 | −0.05 | 0.05 | −0.11 |
| P | 0.007 | 0.7 | 0.8 | 0.5 | |
| Baseline pVL | R | 0.17 | 0.15 | 0.54 | |
| P | 0.3 | 0.3 | <0.001 | ||
| CD4 downregulation | R | 0.56 | 0.20 | ||
| P | <0.001 | 0.2 | |||
| HLA downregulation | R | −0.10 | |||
| P | 0.6 |
Abbreviations: HLA, human leukocyte antigen; eDPI, estimated days postinfection; pVL, plasma viral load. Data represent Spearman R and P values; significant associations are highlighted in bold type.
Changes in Nef activity observed over time in some AC participants.
To directly explore changes in Nef function over time in AC, we cloned plasma RNA Nef sequences at a single follow-up time point, collected a median of 283 (IQR, 161 to 427) days after the date of baseline enrollment, from 6 AC individuals from whom a sample was available (Table 1). A median of 4 Nef clones (range, 1 to 11) were isolated per patient at the follow-up time point. CD4 and HLA class I downregulation activities were measured for all unique clones, while a single nef allele was used to generate recombinant NL4.3 stocks to assess viral infectivity enhancement (Fig. 5). In a paired analysis comparing the functions of a representative Nef clone from baseline and follow-up samples, we observed no overall differences in the ability of Nef to downregulate CD4 or HLA class I or to enhance viral infectivity (Wilcoxon paired test; all P > 0.5). This suggests that Nef functional alterations (whether they be increases or decreases over time) are not generalizable or consistent phenomena during the first year postinfection. However, we did observe substantial changes in these Nef functions in individual cases.
FIG 5.
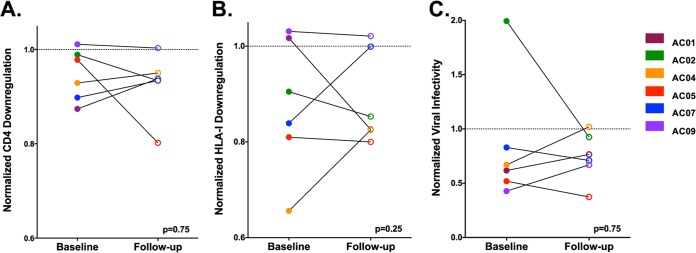
No consistent changes in Nef activity were observed in AC patients over time. The ability of Nef to downregulate CD4 (A) and HLA class I (B) or to enhance viral infectivity (C) is shown for representative clones derived from 6 AC patients at the baseline (closed circles) and follow-up (open circles) time points, as presented in Table 1. A dashed line (---) indicates normalized activity of 1.0, which represents the function of the positive-control NefSF2 in each assay. The Wilcoxon P value is shown; no significant differences in Nef function were observed between baseline and follow-up clones in a paired test. Nef downregulation activities were tested in ≥3 replicates, while viral infectivity was tested in duplicate.
To address this further, we analyzed clonal nef sequences from baseline and follow-up time points in 4 AC who displayed the greatest changes in Nef function over time to identify polymorphisms that might contribute to these observations. Notably, in all 4 individuals, most observed sequence changes were linked to an HLA class I allele expressed by the host or to reversion of transmitted viral polymorphisms associated with HLA alleles presumably expressed in previous a host, as defined by reference lists of HLA-associated polymorphisms derived from published statistical association studies (60, 61). For example, selection of 4 polymorphisms associated with HLA-A*31 and B*37 in subject AC01 (discussed below) was accompanied by a 19% reduction in HLA class I downregulation activity, while in subject AC02, reductions in Nef-mediated infectivity enhancement corresponded to the appearance of a B*35-associated polymorphism, H40Q, and reversion to the consensus sequence at D63E (located in the acidic E62EEE65 cluster that binds PACS-2 [62]). In AC05, an 18% reduction in CD4 downregulation activity was observed in conjunction with D54E and the B*39-associated N162S polymorphism. Finally, recovery of wild-type HLA class I downregulation function in follow-up clones from AC07 coincided with 10 polymorphisms in the C-terminal half of Nef, the most notable being reversion of a well-characterized A*23/A*24-associated escape mutation (F135Y) that is reported to alter this activity (63). These results indicate that certain host HLA-associated CTL pressures can select mutations that impact Nef function.
HLA-driven polymorphisms act in combination to reduce Nef function in AC01.
AC01 was of particular interest because this subject displayed the greatest reduction in HLA class I downregulation activity and also controlled pVL to less than 50 RNA copies/ml within the first year. Of the 11 clones isolated from this patient at 383 days postinfection, 10 were identical and encoded 4 amino acid polymorphisms (R19K, H40R, M182L, and V194A) that together resulted in a 19% reduction in HLA class I downregulation compared to baseline clone levels (Fig. 6A). The remaining follow-up clone contained a fifth polymorphism (E149K) and displayed 15% and 13% reductions in HLA class I and CD4 downregulation activities, respectively (data not shown). E149 is located at the base of a central loop in Nef (amino acids 149 to 179) that binds to AP-2 (64), suggesting that an E149K charge-reversal mutation may alter interactions with this clathrin adapter complex that are required for CD4 downregulation.
FIG 6.

Accumulation of HLA-associated polymorphisms is associated with reduced Nef function and expression. (A) An HXB2 alignment of unique Nef sequences derived from patient AC01 at the baseline (n = 5) and follow-up (n = 2) time points is shown. NefHXB2 data are indicated for reference. Sequences are labeled by participant identifier (ID) (AC01) followed by the time of sample collection (baseline, b; follow-up, f) and colony identifier (A, B, etc). A solid line distinguishes baseline clones (above the line) from follow-up clones (below the line). Four polymorphisms (R19K, H40R, M182L, and V194A; highlighted in yellow) were observed in all follow-up clones and are associated with host HLA-A*31 or HLA-B*37, as discussed in the text. (B) A model of the Nef protein structure illustrates the location of residues/motifs previously associated with HLA class I downregulation function (green), the four polymorphisms observed in AC01 (red and magenta), and an area of potential overlap near R19 (magenta). (C) A bar graph depicts the normalized HLA class I downregulation activities of baseline clone AC01b_F and follow-up clone AC01f_A (red) and their corresponding single-amino-acid mutants (yellow) at the four polymorphic sites; all samples were tested in ≥4 replicates. A dashed line (---) indicates normalized activity of 1.0, which represents the function of the positive-control NefSF2. (D) Steady-state protein expression for NefSF2 (positive control), ΔNef (negative control), AC01b_F, and AC01f_A and their corresponding single-amino-acid mutants was examined by Western blotting. β-Actin was used as a cellular protein control. The relative band intensity (%) for each Nef clone was calculated by normalization to β-actin and then NefSF2. As such, a number > 100% or < 100% represents relative Nef expression greater or less than that of NefSF2.
Published HLA-association studies (60, 61) indicate that mutations at Nef residues 19, 40, 182, and 194 are highly significantly associated with HLA-A*31 (codons 19 and 194) and B*37 (Nef codons 40 and 182), two alleles expressed by AC01. R19K is located in a putative A*31-restricted CTL epitope predicted on the basis of this individual's autologous baseline viral sequence (K9LAGWPTIR19 [the site of mutation is underlined]; prediction made using NetMHCpan2.8 [65, 66]), while V194A lies within a published A*31-restricted epitope (S188LAFRHVAR196), although the baseline nef sequence in AC01 is slightly different (TLAFHHVAR) (67). Likewise, H40R is found within a published CTL epitope (R35DLEKHGAI43) detected in B*37-expressing vaccine recipients (68), while M182L resides in a predicted A*31-restricted epitope (V180LEWRFDSR188, based on the nef consensus sequence) as well as a putative B*37-restricted epitope that is predicted in AC01's baseline viral sequence (K178EVLMKFDTRL189).
Viral adaptation to CTL in early infection commonly involves the appearance of transient polymorphisms in or near the epitope that is under immune pressure, from which the final escape form is ultimately selected (67, 69, 70). Baseline Nef clones from AC01 exhibited polymorphisms near codon 40, notably, D36N and E38K, that are consistent with this phenomenon (Fig. 6A). Furthermore, clones isolated at days 51, 65, and 93 encoded D36N, E38G, and K39T (adjacent to the final escape site at codon 40) and H192Y (adjacent to the final escape site at codon 194) (data not shown).
These observations motivated us to undertake a more in-depth analysis of Nef-mediated HLA class I downregulation activity in AC01. Of these 4 polymorphisms, only R19K is located near a residue or motif (namely, M20) that has been associated with this function (Fig. 6B) (58, 63, 71–77). To directly examine their impact on Nef, forward mutations corresponding to each of the 4 polymorphisms were introduced individually into the baseline clone (AC01b_F) whose sequence most closely resembled the B/HXB2 nef consensus sequence and backward mutations were introduced into the predominant follow-up clone (AC01f_A) to revert each of these polymorphisms individually. Single-amino-acid changes in the baseline clone had only a modest impact on HLA class I downregulation function (∼3% reduction in each case; Fig. 6C). None of the forward mutations altered baseline Nef protein stability appreciably, but each backwards mutation restored expression of the follow-up clone to a level that was comparable to that seen with the baseline sequence (Fig. 6D). Since none of these polymorphisms appeared to have a major effect on Nef function individually, and since Nef expression levels and in vitro function could be largely restored by reversion of any one of these mutations alone, our results suggest that all 4 HLA-associated polymorphisms acted in combination to reduce Nef stability, yielding reduced HLA class I downregulation activity in later clones from AC01. In summary, the emergence of HLA-associated mutations that additively impaired Nef function over time in subject AC01 strongly suggests that Nef functional impairment in this individual is not solely attributable to transmission of an attenuated virus but is rather a direct consequence of HLA immunogenetics and CD8+ T cell selection in vivo.
DISCUSSION
In this study, we examined nef alleles from 10 recently infected individuals who controlled HIV-1 plasma viremia to less than 2,000 RNA copies/ml in the absence of antiretroviral therapy (acute controllers [AC]) and 50 noncontrollers (acute progressors [AP]). While baseline Nef clones from both cohorts were generally functional (all patient-derived clones exhibited activities greater than that of the ΔNef control), those from AC displayed a median of a 5%-reduced ability to downregulate surface CD4, 17%-reduced ability to downregulate HLA class I molecules, and 57% poorer enhancement of virion infectivity compared to those from AP. Although we cannot draw definitive conclusions regarding the clinical significance of these observations, even small changes at the level of each virus-infected cell could be amplified through multiple cycles of infection, thereby influencing pathogenesis.
We previously observed that multiple Nef functions were impaired in chronically HIV-1-infected EC (21), and results presented here demonstrate that similar impairments can be seen at early times postinfection. We therefore conclude that modest functional attenuation of Nef is a relatively common occurrence in individuals who spontaneously control HIV-1. Our results indicate that this may be attributed to transmission of less-fit HIV-1 strains as well as to the acquisition of HLA-associated mutations that compromise Nef function at early times postinfection—and that the precise mechanism may be unique to each individual. Notably, the in vitro replication capacity of gag and protease recombinant viruses is also reduced in this AC cohort through apparently similar means (32). Together, our data support a model where the early function of major HIV-1 structural and accessory proteins has a long-term impact on viremia control and disease progression, which is consistent with studies highlighting the impact of a transmitted/founder viral sequence on clinical outcome (78–80).
In a cross-sectional analysis, baseline HLA class I downregulation function correlated inversely with the baseline sampling date in Nef clones from AC (Table 2). This is consistent with early attenuation of Nef following infection as the transmitted/founder virus adapts to its new host through selection of CTL escape mutations. A similar correlation was not observed for AP (Table 3), suggesting that early changes in Nef sequence in these individuals were not associated with reduced function. Intriguingly, Nef-mediated enhancement of virion infectivity correlated directly with pVL in AP but not in AC; however, additional studies will be necessary to confirm a potential link with this Nef function in the establishment of the viral load set point in HIV-infected persons whose disease progresses normally.
Longitudinal analyses of Nef clones from six AC revealed substantial changes in Nef function in individual patients over time, though neither the direction nor magnitude of these changes was consistent across patients (possibly due to the small number of individuals examined). However, among the four AC who exhibited substantial functional increases or decreases over time, these could be attributed to unique polymorphisms that arose in the first year of infection. Many were consistent with HLA-driven adaptations in the current host or with reversion of HLA-associated polymorphisms present in the baseline sequence; however, only one (F135Y) had been previously described to affect Nef function (63).
Our detailed forward and reverse mutagenesis of clones from AC01 demonstrated that multiple substitutions acted synergistically to affect Nef function in this individual. In particular, four polymorphisms selected by HLA-A*31 and B*37 were required to reduce the ability of Nef to downregulate HLA class I. Together, these results indicate that host CTL pressure can select for escape mutations that directly reduce Nef function over the infection course, indicating that, in at least a subset of controllers, functional impairments are not solely attributable to acquisition of attenuated strains but are rather a direct consequence of HLA-restricted CD8+ T cell selection in vivo. Reduced HLA class I downregulation function in follow-up clones from AC01 correlated with lower steady-state Nef protein levels in cells (Fig. 5), and site-directed mutagenesis confirmed that all four HLA-associated polymorphisms were necessary to see both effects. Intriguingly, we observed a 42% reduction in protein levels for the quadruple Nef mutant but only a 19% reduction in its HLA class I downregulation function. This suggests that there may be a threshold effect such that modest reductions in Nef expression are tolerated without a significant loss of function. It is well established that a greater amount of Nef protein is required to promote efficient downregulation of HLA class I (compared to CD4) (81); as such, HLA class I downregulation activity is likely to be more sensitive to alterations in protein expression or stability. Impaired HLA class I downregulation was thus likely due to reduced Nef expression in this case rather than to disruption of a critical functional motif. Further studies will be necessary to determine if this is a consistent observation for patient-derived Nef clones in other contexts.
Some limitations of this study merit discussion. First, the AC cohort represents a rare group of individuals enrolled during acute/early infection; therefore, the number of individuals and the number of specimens available were limited. When possible, we tested the downregulation activity of multiple Nef clones from each AC, allowing a better assessment of functional diversity within each individual. Similar results for CD4 or HLA class I downregulation were obtained when we analyzed a representative clone, a predominant Nef clone, and the median function of all clones, indicating that our conclusions are not biased by the isolate used. Second, Nef performs multiple roles in virus-infected cells and we have examined only three in vitro functions in this study. Our results demonstrate significant impairments in CD4 and HLA class I downregulation function as well as infectivity enhancement for early Nef clones from AC. We have not assessed the ability of Nef to modulate other cellular proteins or functions (including TCR signaling) or potential differences in viral replication capacity using multicycle assays. Further investigation would therefore extend the observations of this study; however, we believe that such work is unlikely to alter our conclusions significantly. Similarly, it has been reported that the effects of Nef on host HLA expression may differ between HLA alleles (82). While we assessed only downregulation of HLA-A*02 here, a previous study by our group observed a strong correlation between the abilities of patient-derived Nef clones to downregulate A*02 and B*07 in this same assay system (56). Changes in Nef function between baseline and follow-up clones sampled were observed up to 516 days later. Thus, it is not clear when these changes occurred. Even in the case of AC01, from whom some intermediate specimens were available, the 4 polymorphisms examined became fixed between days 93 and 383. Finally, even though acute controllers were recruited, on average, less than 2.5 months following their estimated date of infection, we cannot rule out the possibility of very early selection and fixation of immune-driven mutations prior to sampling. Despite these limitations, we believe that the data presented here provide a unique window into early events during natural HIV-1 infection that can help us to better understand the pathways and mechanisms of viral attenuation that may contribute to the controller phenotype.
We conclude that impaired early Nef function is associated with spontaneous control of HIV-1 viremia. This observation is consistent with previous data showing reduced Nef activity in an independent cohort of chronically HIV-1-infected EC (21) and suggests that early functional deficits in Nef—via acquisition of modestly attenuated sequences at transmission or very rapid fixation of host-driven viral mutations—contribute to spontaneous viral control. Importantly, changes in Nef function during the first year postinfection frequently coincided with the appearance or reversion of HLA-associated polymorphisms, indicating that within-host CTL pressures can drive the selection of mutations that modulate Nef activity in at least some individuals. These results highlight the potential for host immune responses to modulate HIV pathogenicity and disease outcome by targeting epitopes in Nef.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the Canadian Institutes for Health Research (CIHR), operating grants MOP-93536 (to Z.L.B.) and THA-118569 (to M.A.B.), and by grants-in-aid from the Global COE Program (Ministry of Education, Science, Sports, and Culture of Japan) and the Ministry of Health, Labor, and Welfare of Japan (to T.U.). X.T.K. is supported by a Master's Scholarship from the Canadian Association for HIV Research in partnership with Bristol-Myers Squibb Canada and ViiV Healthcare. P.M. is supported by Postdoctoral Fellowships from the Michael Smith Foundation for Health Research (MSFHR) and CIHR. A.Q.L. is supported by a CIHR Frederick Banting and Charles Best Masters Award. Z.L.B. is the recipient of a CIHR New Investigator Award and a Scholar Award from MSFHR. M.A.B. holds a Canada Research Chair (Tier 2) in Viral Pathogenesis and Immunity from the Canada Research Chairs Program.
The funders of this study played no role in determining the content of the manuscript or in our decision to publish.
We thank the clinicians, staff, and participants of the Acute Infection Early Disease Research Program (AIEDRP) for their critical contributions to this project. The following reagents were obtained from the NIH AIDS Research and Reference Reagents Program, Division of AIDS, NIAID: TZM-bl, catalog no. 8129, from John C. Kappes, Xiaoyun Wu and Tranzyme Inc.; and HIV-1 Nef antiserum (rabbit), no. 2949, from Ronald Swanstrom. Sheep antiserum to HIV-1 Nef (catalog no. ARP444) was obtained from the Centre for AIDS Reagents, NIBSC (United Kingdom) and was kindly donated by M. Harris.
Footnotes
Published ahead of print 25 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01334-14.
REFERENCES
- 1.Kaslow RA, Carrington M, Apple R, Park L, Munoz A, Saah AJ, Goedert JJ, Winkler C, O'Brien SJ, Rinaldo C, Detels R, Blattner W, Phair J, Erlich H, Mann DL. 1996. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat. Med. 2:405–411. 10.1038/nm0496-405 [DOI] [PubMed] [Google Scholar]
- 2.Gao X, Nelson GW, Karacki P, Martin MP, Phair J, Kaslow R, Goedert JJ, Buchbinder S, Hoots K, Vlahov D, O'Brien SJ, Carrington M. 2001. Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. N. Engl. J. Med. 344:1668–1675. 10.1056/NEJM200105313442203 [DOI] [PubMed] [Google Scholar]
- 3.International HIV Controllers Study Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, Ripke S, Brumme CJ, Pulit SL, Carrington M, Kadie CM, Carlson JM, Heckerman D, Graham RR, Plenge RM, Deeks SG, Gianniny L, Crawford G, Sullivan J, Gonzalez E, Davies L, Camargo A, Moore JM, Beattie N, Gupta S, Crenshaw A, Burtt NP, Guiducci C, Gupta N, Gao X, Qi Y, Yuki Y, Piechocka-Trocha A, Cutrell E, Rosenberg R, Moss KL, Lemay P, O'Leary J, Schaefer T, Verma P, Toth I, Block B, Baker B, Rothchild A, Lian J, Proudfoot J, Alvino DM, Vine S, Addo MM, et al. 2010. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330:1551–1557. 10.1126/science.1195271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, Zhang K, Gumbs C, Castagna A, Cossarizza A, Cozzi-Lepri A, De Luca A, Easterbrook P, Francioli P, Mallal S, Martinez-Picado J, Miro JM, Obel N, Smith JP, Wyniger J, Descombes P, Antonarakis SE, Letvin NL, McMichael AJ, Haynes BF, Telenti A, Goldstein DB. 2007. A whole-genome association study of major determinants for host control of HIV-1. Science 317:944–947. 10.1126/science.1143767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLaren PJ, Coulonges C, Ripke S, van den Berg L, Buchbinder S, Carrington M, Cossarizza A, Dalmau J, Deeks SG, Delaneau O, De Luca A, Goedert JJ, Haas D, Herbeck JT, Kathiresan S, Kirk GD, Lambotte O, Luo M, Mallal S, van Manen D, Martinez-Picado J, Meyer L, Miro JM, Mullins JI, Obel N, O'Brien SJ, Pereyra F, Plummer FA, Poli G, Qi Y, Rucart P, Sandhu MS, Shea PR, Schuitemaker H, Theodorou I, Vannberg F, Veldink J, Walker BD, Weintrob A, Winkler CA, Wolinsky S, Telenti A, Goldstein DB, de Bakker PI, Zagury JF, Fellay J. 2013. Association study of common genetic variants and HIV-1 acquisition in 6,300 infected cases and 7,200 controls. PLoS Pathog. 9:e1003515. 10.1371/journal.ppat.1003515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pereyra F, Addo MM, Kaufmann DE, Liu Y, Miura T, Rathod A, Baker B, Trocha A, Rosenberg R, Mackey E, Ueda P, Lu Z, Cohen D, Wrin T, Petropoulos CJ, Rosenberg ES, Walker BD. 2008. Genetic and immunologic heterogeneity among persons who control HIV infection in the absence of therapy. J. Infect. Dis. 197:563–571. 10.1086/526786 [DOI] [PubMed] [Google Scholar]
- 7.Deeks SG, Walker BD. 2007. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity 27:406–416. 10.1016/j.immuni.2007.08.010 [DOI] [PubMed] [Google Scholar]
- 8.Migueles SA, Connors M. 2010. Long-term nonprogressive disease among untreated HIV-infected individuals: clinical implications of understanding immune control of HIV. JAMA 304:194–201. 10.1001/jama.2010.925 [DOI] [PubMed] [Google Scholar]
- 9.Ndhlovu ZM, Proudfoot J, Cesa K, Alvino DM, McMullen A, Vine S, Stampouloglou E, Piechocka-Trocha A, Walker BD, Pereyra F. 2012. Elite controllers with low to absent effector CD8+ T cell responses maintain highly functional, broadly directed central memory responses. J. Virol. 86:6959–6969. 10.1128/JVI.00531-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hersperger AR, Pereyra F, Nason M, Demers K, Sheth P, Shin LY, Kovacs CM, Rodriguez B, Sieg SF, Teixeira-Johnson L, Gudonis D, Goepfert PA, Lederman MM, Frank I, Makedonas G, Kaul R, Walker BD, Betts MR. 2010. Perforin expression directly ex vivo by HIV-specific CD8 T-cells is a correlate of HIV elite control. PLoS Pathog. 6:e1000917. 10.1371/journal.ppat.1000917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Migueles SA, Laborico AC, Shupert WL, Sabbaghian MS, Rabin R, Hallahan CW, Van Baarle D, Kostense S, Miedema F, McLaughlin M, Ehler L, Metcalf J, Liu S, Connors M. 2002. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat. Immunol. 3:1061–1068. 10.1038/ni845 [DOI] [PubMed] [Google Scholar]
- 12.Hersperger AR, Migueles SA, Betts MR, Connors M. 2011. Qualitative features of the HIV-specific CD8+ T-cell response associated with immunologic control. Curr. Opin. HIV AIDS 6:169–173. 10.1097/COH.0b013e3283454c39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, Bishop K, van der Stok M, Nair K, Khan N, Crawford H, Payne R, Leslie A, Prado J, Prendergast A, Frater J, McCarthy N, Brander C, Learn GH, Nickle D, Rousseau C, Coovadia H, Mullins JI, Heckerman D, Walker BD, Goulder P. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 13:46–53. 10.1038/nm1520 [DOI] [PubMed] [Google Scholar]
- 14.Adland E, Carlson JM, Paioni P, Kloverpris H, Shapiro R, Ogwu A, Riddell L, Luzzi G, Chen F, Balachandran T, Heckerman D, Stryhn A, Edwards A, Ndung'u T, Walker BD, Buus S, Goulder P, Matthews PC. 2013. Nef-specific CD8+ T cell responses contribute to HIV-1 immune control. PLoS One 8:e73117. 10.1371/journal.pone.0073117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riou C, Burgers WA, Mlisana K, Koup RA, Roederer M, Abdool Karim SS, Williamson C, Gray CM. 2014. Differential impact of magnitude, polyfunctional capacity, and specificity of HIV-specific CD8+ T cell responses on HIV set point. J. Virol. 88:1819–1824. 10.1128/JVI.02968-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allen TM, Altfeld M. 2008. Crippling HIV one mutation at a time. J. Exp. Med. 205:1003–1007. 10.1084/jem.20080569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prince JL, Claiborne DT, Carlson JM, Schaefer M, Yu T, Lahki S, Prentice HA, Yue L, Vishwanathan SA, Kilembe W, Goepfert P, Price MA, Gilmour J, Mulenga J, Farmer P, Derdeyn CA, Tang J, Heckerman D, Kaslow RA, Allen SA, Hunter E. 2012. Role of transmitted Gag CTL polymorphisms in defining replicative capacity and early HIV-1 pathogenesis. PLoS Pathog. 8:e1003041. 10.1371/journal.ppat.1003041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brumme ZL, Li C, Miura T, Sela J, Rosato PC, Brumme CJ, Markle TJ, Martin E, Block BL, Trocha A, Kadie CM, Allen TM, Pereyra F, Heckerman D, Walker BD, Brockman MA. 2011. Reduced replication capacity of NL4-3 recombinant viruses encoding reverse transcriptase-integrase sequences from HIV-1 elite controllers. J. Acquir. Immune Defic. Syndr. 56:100–108. 10.1097/QAI.0b013e3181fe9450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lassen KG, Lobritz MA, Bailey JR, Johnston S, Nguyen S, Lee B, Chou T, Siliciano RF, Markowitz M, Arts EJ. 2009. Elite suppressor-derived HIV-1 envelope glycoproteins exhibit reduced entry efficiency and kinetics. PLoS Pathog. 5:e1000377. 10.1371/journal.ppat.1000377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miura T, Brockman MA, Brumme ZL, Brumme CJ, Pereyra F, Trocha A, Block BL, Schneidewind A, Allen TM, Heckerman D, Walker BD. 2009. HLA-associated alterations in replication capacity of chimeric NL4-3 viruses carrying gag-protease from elite controllers of human immunodeficiency virus type 1. J. Virol. 83:140–149. 10.1128/JVI.01471-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mwimanzi P, Markle TJ, Martin E, Ogata Y, Kuang XT, Tokunaga M, Mahiti M, Pereyra F, Miura T, Walker BD, Brumme ZL, Brockman MA, Ueno T. 2013. Attenuation of multiple Nef functions in HIV-1 elite controllers. Retrovirology 10:1. 10.1186/1742-4690-10-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lobritz MA, Lassen KG, Arts EJ. 2011. HIV-1 replicative fitness in elite controllers. Curr. Opin. HIV AIDS 6:214–220. 10.1097/COH.0b013e3283454cf5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rihn SJ, Wilson SJ, Loman NJ, Alim M, Bakker SE, Bhella D, Gifford RJ, Rixon FJ, Bieniasz PD. 2013. Extreme genetic fragility of the HIV-1 capsid. PLoS Pathog. 9:e1003461. 10.1371/journal.ppat.1003461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crawford H, Prado JG, Leslie A, Hue S, Honeyborne I, Reddy S, van der Stok M, Mncube Z, Brander C, Rousseau C, Mullins JI, Kaslow R, Goepfert P, Allen S, Hunter E, Mulenga J, Kiepiela P, Walker BD, Goulder PJ. 2007. Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic human immunodeficiency virus type 1 infection. J. Virol. 81:8346–8351. 10.1128/JVI.00465-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez-Picado J, Prado JG, Fry EE, Pfafferott K, Leslie A, Chetty S, Thobakgale C, Honeyborne I, Crawford H, Matthews P, Pillay T, Rousseau C, Mullins JI, Brander C, Walker BD, Stuart DI, Kiepiela P, Goulder P. 2006. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J. Virol. 80:3617–3623. 10.1128/JVI.80.7.3617-3623.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brockman MA, Schneidewind A, Lahaie M, Schmidt A, Miura T, Desouza I, Ryvkin F, Derdeyn CA, Allen S, Hunter E, Mulenga J, Goepfert PA, Walker BD, Allen TM. 2007. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J. Virol. 81:12608–12618. 10.1128/JVI.01369-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rolland M, Manocheewa S, Swain JV, Lanxon-Cookson EC, Kim M, Westfall DH, Larsen BB, Gilbert PB, Mullins JI. 2013. HIV-1 conserved-element vaccines: relationship between sequence conservation and replicative capacity. J. Virol. 87:5461–5467. 10.1128/JVI.03033-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brockman MA, Brumme ZL, Brumme CJ, Miura T, Sela J, Rosato PC, Kadie CM, Carlson JM, Markle TJ, Streeck H, Kelleher AD, Markowitz M, Jessen H, Rosenberg E, Altfeld M, Harrigan PR, Heckerman D, Walker BD, Allen TM. 2010. Early selection in Gag by protective HLA alleles contributes to reduced HIV-1 replication capacity that may be largely compensated for in chronic infection. J. Virol. 84:11937–11949. 10.1128/JVI.01086-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brockman MA, Chopera DR, Olvera A, Brumme CJ, Sela J, Markle TJ, Martin E, Carlson JM, Le AQ, McGovern R, Cheung PK, Kelleher AD, Jessen H, Markowitz M, Rosenberg E, Frahm N, Sanchez J, Mallal S, John M, Harrigan PR, Heckerman D, Brander C, Walker BD, Brumme ZL. 2012. Uncommon pathways of immune escape attenuate HIV-1 integrase replication capacity. J. Virol. 86:6913–6923. 10.1128/JVI.07133-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Troyer RM, McNevin J, Liu Y, Zhang SC, Krizan RW, Abraha A, Tebit DM, Zhao H, Avila S, Lobritz MA, McElrath MJ, Le Gall S, Mullins JI, Arts EJ. 2009. Variable fitness impact of HIV-1 escape mutations to cytotoxic T lymphocyte (CTL) response. PLoS Pathog. 5:e1000365. 10.1371/journal.ppat.1000365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miura T, Brumme CJ, Brockman MA, Brumme ZL, Pereyra F, Block BL, Trocha A, John M, Mallal S, Harrigan PR, Walker BD. 2009. HLA-associated viral mutations are common in human immunodeficiency virus type 1 elite controllers. J. Virol. 83:3407–3412. 10.1128/JVI.02459-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miura T, Brumme ZL, Brockman MA, Rosato P, Sela J, Brumme CJ, Pereyra F, Kaufmann DE, Trocha A, Block BL, Daar ES, Connick E, Jessen H, Kelleher AD, Rosenberg E, Markowitz M, Schafer K, Vaida F, Iwamoto A, Little S, Walker BD. 2010. Impaired replication capacity of acute/early viruses in persons who become HIV controllers. J. Virol. 84:7581–7591. 10.1128/JVI.00286-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mwimanzi P, Markle TJ, Ueno T, Brockman MA. 2012. Human leukocyte antigen (HLA) class I down-regulation by human immunodeficiency virus type 1 negative factor (HIV-1 Nef): what might we learn from natural sequence variants? Viruses 4:1711–1730. 10.3390/v4091711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia JV, Miller AD. 1991. Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature 350:508–511. 10.1038/350508a0 [DOI] [PubMed] [Google Scholar]
- 35.Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard JM. 1996. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 2:338–342. 10.1038/nm0396-338 [DOI] [PubMed] [Google Scholar]
- 36.Schindler M, Wurfl S, Benaroch P, Greenough TC, Daniels R, Easterbrook P, Brenner M, Munch J, Kirchhoff F. 2003. Down-modulation of mature major histocompatibility complex class II and up-regulation of invariant chain cell surface expression are well-conserved functions of human and simian immunodeficiency virus nef alleles. J. Virol. 77:10548–10556. 10.1128/JVI.77.19.10548-10556.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Münch J, Rajan D, Schindler M, Specht A, Rücker E, Novembre FJ, Nerrienet E, Müller-Trutwin MC, Peeters M, Hahn BH, Kirchhoff F. 2007. Nef-mediated enhancement of virion infectivity and stimulation of viral replication are fundamental properties of primate lentiviruses. J. Virol. 81:13852–13864. 10.1128/JVI.00904-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller MD, Warmerdam MT, Gaston I, Greene WC, Feinberg MB. 1994. The human immunodeficiency virus-1 nef gene product: a positive factor for viral infection and replication in primary lymphocytes and macrophages. J. Exp. Med. 179:101–113. 10.1084/jem.179.1.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abraham L, Fackler OT. 2012. HIV-1 Nef: a multifaceted modulator of T cell receptor signaling. Cell Commun. Signal. 10:39. 10.1186/1478-811X-10-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Markle TJ, Philip M, Brockman MA. 2013. HIV-1 Nef and T-cell activation: a history of contradictions. Future Virol. 2013:8. 10.2217/fvl.13.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Daniel MD, Kirchhoff F, Czajak SC, Sehgal PK, Desrosiers RC. 1992. Protective effects of a live attenuated SIV vaccine with a deletion in the nef gene. Science 258:1938–1941. 10.1126/science.1470917 [DOI] [PubMed] [Google Scholar]
- 42.Deacon NJ, Tsykin A, Solomon A, Smith K, Ludford-Menting M, Hooker DJ, McPhee DA, Greenway AL, Ellett A, Chatfield C, Lawson VA, Crowe S, Maerz A, Sonza S, Learmont J, Sullivan JS, Cunningham A, Dwyer D, Dowton D, Mills J. 1995. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 270:988–991. 10.1126/science.270.5238.988 [DOI] [PubMed] [Google Scholar]
- 43.Kirchhoff F, Greenough TC, Brettler DB, Sullivan JL, Desrosiers RC. 1995. Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N. Engl. J. Med. 332:228–232. 10.1056/NEJM199501263320405 [DOI] [PubMed] [Google Scholar]
- 44.Zaunders JJ, Geczy AF, Dyer WB, McIntyre LB, Cooley MA, Ashton LJ, Raynes-Greenow CH, Learmont J, Cooper DA, Sullivan JS. 1999. Effect of long-term infection with nef-defective attenuated HIV type 1 on CD4+ and CD8+ T lymphocytes: increased CD45RO+CD4+ T lymphocytes and limited activation of CD8+ T lymphocytes. AIDS Res. Hum. Retroviruses 15:1519–1527. 10.1089/088922299309801 [DOI] [PubMed] [Google Scholar]
- 45.Little SJ, Frost SD, Wong JK, Smith DM, Pond SL, Ignacio CC, Parkin NT, Petropoulos CJ, Richman DD. 2008. Persistence of transmitted drug resistance among subjects with primary human immunodeficiency virus infection. J. Virol. 82:5510–5518. 10.1128/JVI.02579-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woods CK, Brumme CJ, Liu TF, Chui CK, Chu AL, Wynhoven B, Hall TA, Trevino C, Shafer RW, Harrigan PR. 2012. Automating HIV drug resistance genotyping with RECall, a freely accessible sequence analysis tool. J. Clin. Microbiol. 50:1936–1942. 10.1128/JCM.06689-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pond SL, Frost SD, Muse SV. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679. 10.1093/bioinformatics/bti079 [DOI] [PubMed] [Google Scholar]
- 48.Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52:696–704. 10.1080/10635150390235520 [DOI] [PubMed] [Google Scholar]
- 49.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59:307–321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- 50.Cotton LA, Abdur Rahman M, Ng C, Le AQ, Milloy MJ, Mo T, Brumme ZL. 2012. HLA class I sequence-based typing using DNA recovered from frozen plasma. J. Immunol. Methods 382:40–47. 10.1016/j.jim.2012.05.003 [DOI] [PubMed] [Google Scholar]
- 51.Ueno T, Motozono C, Dohki S, Mwimanzi P, Rauch S, Fackler OT, Oka S, Takiguchi M. 2008. CTL-mediated selective pressure influences dynamic evolution and pathogenic functions of HIV-1 Nef. J. Immunol. 180:1107–1116. 10.4049/jimmunol.180.2.1107 [DOI] [PubMed] [Google Scholar]
- 52.Mwimanzi P, Hasan Z, Hassan R, Suzu S, Takiguchi M, Ueno T. 2011. Effects of naturally-arising HIV Nef mutations on cytotoxic T lymphocyte recognition and Nef's functionality in primary macrophages. Retrovirology 8:50. 10.1186/1742-4690-8-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46:1896–1905. 10.1128/AAC.46.6.1896-1905.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shugars DC, Smith MS, Glueck DH, Nantermet PV, Seillier-Moiseiwitsch F, Swanstrom R. 1993. Analysis of human immunodeficiency virus type 1 nef gene sequences present in vivo. J. Virol. 67:4639–4650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. 10.1016/0378-1119(89)90358-2 [DOI] [PubMed] [Google Scholar]
- 56.Mann JK, Byakwaga H, Kuang XT, Le AQ, Brumme CJ, Mwimanzi P, Omarjee S, Martin E, Lee GQ, Baraki B, Danroth R, McCloskey R, Muzoora C, Bangsberg DR, Hunt PW, Goulder PJ, Walker BD, Harrigan PR, Martin JN, Ndung'u T, Brockman MA, Brumme ZL. 2013. Ability of HIV-1 Nef to downregulate CD4 and HLA class I differs among viral subtypes. Retrovirology 10:100. 10.1186/1742-4690-10-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mwimanzi P, Markle TJ, Ogata Y, Martin E, Tokunaga M, Mahiti M, Kuang XT, Walker BD, Brockman MA, Brumme ZL, Ueno T. 2013. Dynamic range of Nef functions in chronic HIV-1 infection. Virology 439:74–80. 10.1016/j.virol.2013.02.005 [DOI] [PubMed] [Google Scholar]
- 58.Foster JL, Denial SJ, Temple BR, Garcia JV. 2011. Mechanisms of HIV-1 Nef function and intracellular signaling. J. Neuroimmune Pharmacol. 6:230–246. 10.1007/s11481-011-9262-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Landi A, Iannucci V, Nuffel AV, Meuwissen P, Verhasselt B. 2011. One protein to rule them all: modulation of cell surface receptors and molecules by HIV Nef. Curr. HIV Res. 9:496–504. 10.2174/157016211798842116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brumme ZL, Brumme CJ, Chui C, Mo T, Wynhoven B, Woods CK, Henrick BM, Hogg RS, Montaner JS, Harrigan PR. 2007. Effects of human leukocyte antigen class I genetic parameters on clinical outcomes and survival after initiation of highly active antiretroviral therapy. J. Infect. Dis. 195:1694–1704. 10.1086/516789 [DOI] [PubMed] [Google Scholar]
- 61.Brumme ZL, John M, Carlson JM, Brumme CJ, Chan D, Brockman MA, Swenson LC, Tao I, Szeto S, Rosato P, Sela J, Kadie CM, Frahm N, Brander C, Haas DW, Riddler SA, Haubrich R, Walker BD, Harrigan PR, Heckerman D, Mallal S. 2009. HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PLoS One 4:e6687. 10.1371/journal.pone.0006687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Atkins KM, Thomas L, Youker RT, Harriff MJ, Pissani F, You H, Thomas G. 2008. HIV-1 Nef binds PACS-2 to assemble a multikinase cascade that triggers major histocompatibility complex class I (MHC-I) down-regulation: analysis using short interfering RNA and knock-out mice. J. Biol. Chem. 283:11772–11784. 10.1074/jbc.M707572200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lewis MJ, Lee P, Ng HL, Yang OO. 2012. Immune selection in vitro reveals human immunodeficiency virus type 1 Nef sequence motifs important for its immune evasion function in vivo. J. Virol. 86:7126–7135. 10.1128/JVI.00878-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ren X, Park SY, Bonifacino JS, Hurley JH. 2014. How HIV-1 Nef hijacks the AP-2 clathrin adaptor to downregulate CD4. eLife 3:e01754. 10.7554/eLife.01754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. 2009. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 61:1–13. 10.1007/s00251-008-0341-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, Roder G, Peters B, Sette A, Lund O, Buus S. 2007. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One 2:e796. 10.1371/journal.pone.0000796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goonetilleke N, Liu MK, Salazar-Gonzalez JF, Ferrari G, Giorgi E, Ganusov VV, Keele BF, Learn GH, Turnbull EL, Salazar MG, Weinhold KJ, Moore S, CHAVI Clinical Core B. Letvin N, Haynes BF, Cohen MS, Hraber P, Bhattacharya T, Borrow P, Perelson AS, Hahn BH, Shaw GM, Korber BT, McMichael AJ. 2009. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J. Exp. Med. 206:1253–1272. 10.1084/jem.20090365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li F, Finnefrock AC, Dubey SA, Korber BT, Szinger J, Cole S, McElrath MJ, Shiver JW, Casimiro DR, Corey L, Self SG. 2011. Mapping HIV-1 vaccine induced T-cell responses: bias towards less-conserved regions and potential impact on vaccine efficacy in the Step study. PLoS One 6:e20479. 10.1371/journal.pone.0020479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fischer W, Ganusov VV, Giorgi EE, Hraber PT, Keele BF, Leitner T, Han CS, Gleasner CD, Green L, Lo CC, Nag A, Wallstrom TC, Wang S, McMichael AJ, Haynes BF, Hahn BH, Perelson AS, Borrow P, Shaw GM, Bhattacharya T, Korber BT. 2010. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS One 5:e12303. 10.1371/journal.pone.0012303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Henn MR, Boutwell CL, Charlebois P, Lennon NJ, Power KA, Macalalad AR, Berlin AM, Malboeuf CM, Ryan EM, Gnerre S, Zody MC, Erlich RL, Green LM, Berical A, Wang Y, Casali M, Streeck H, Bloom AK, Dudek T, Tully D, Newman R, Axten KL, Gladden AD, Battis L, Kemper M, Zeng Q, Shea TP, Gujja S, Zedlack C, Gasser O, Brander C, Hess C, Gunthard HF, Brumme ZL, Brumme CJ, Bazner S, Rychert J, Tinsley JP, Mayer KH, Rosenberg E, Pereyra F, Levin JZ, Young SK, Jessen H, Altfeld M, Birren BW, Walker BD, Allen TM. 2012. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 8:e1002529. 10.1371/journal.ppat.1002529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akari H, Arold S, Fukumori T, Okazaki T, Strebel K, Adachi A. 2000. Nef-induced major histocompatibility complex class I down-regulation is functionally dissociated from its virion incorporation, enhancement of viral infectivity, and CD4 down-regulation. J. Virol. 74:2907–2912. 10.1128/JVI.74.6.2907-2912.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Geyer M, Fackler OT, Peterlin BM. 2001. Structure-function relationships in HIV-1 Nef. EMBO Rep. 2:580–585. 10.1093/embo-reports/kve141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jia X, Singh R, Homann S, Yang H, Guatelli J, Xiong Y. 2012. Structural basis of evasion of cellular adaptive immunity by HIV-1 Nef. Nat. Struct. Mol. Biol. 19:701–706. 10.1038/nsmb.2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kuo LS, Baugh LL, Denial SJ, Watkins RL, Liu M, Garcia JV, Foster JL. 2012. Overlapping effector interfaces define the multiple functions of the HIV-1 Nef polyproline helix. Retrovirology 9:47. 10.1186/1742-4690-9-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mangasarian A, Piguet V, Wang JK, Chen YL, Trono D. 1999. Nef-induced CD4 and major histocompatibility complex class I (MHC-I) down-regulation are governed by distinct determinants: N-terminal alpha helix and proline repeat of Nef selectively regulate MHC-I trafficking. J. Virol. 73:1964–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schaefer MR, Wonderlich ER, Roeth JF, Leonard JA, Collins KL. 2008. HIV-1 Nef targets MHC-I and CD4 for degradation via a final common beta-COP-dependent pathway in T cells. PLoS Pathog. 4:e1000131. 10.1371/journal.ppat.1000131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamada T, Kaji N, Odawara T, Chiba J, Iwamoto A, Kitamura Y. 2003. Proline 78 is crucial for human immunodeficiency virus type 1 Nef to down-regulate class I human leukocyte antigen. J. Virol. 77:1589–1594. 10.1128/JVI.77.2.1589-1594.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chopera DR, Woodman Z, Mlisana K, Mlotshwa M, Martin DP, Seoighe C, Treurnicht F, de Rosa DA, Hide W, Karim SA, Gray CM, Williamson C, Team CS. 2008. Transmission of HIV-1 CTL escape variants provides HLA-mismatched recipients with a survival advantage. PLoS Pathog. 4:e1000033. 10.1371/journal.ppat.1000033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Goepfert PA, Lumm W, Farmer P, Matthews P, Prendergast A, Carlson JM, Derdeyn CA, Tang J, Kaslow RA, Bansal A, Yusim K, Heckerman D, Mulenga J, Allen S, Goulder PJ, Hunter E. 2008. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J. Exp. Med. 205:1009–1017. 10.1084/jem.20072457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fraser C, Lythgoe K, Leventhal GE, Shirreff G, Hollingsworth TD, Alizon S, Bonhoeffer S. 2014. Virulence and pathogenesis of HIV-1 infection: an evolutionary perspective. Science 343:1243727. 10.1126/science.1243727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu X, Schrager JA, Lange GD, Marsh JW. 2001. HIV Nef-mediated cellular phenotypes are differentially expressed as a function of intracellular Nef concentrations. J. Biol. Chem. 276:32763–32770. 10.1074/jbc.M101025200 [DOI] [PubMed] [Google Scholar]
- 82.Rajapaksa US, Li D, Peng YC, McMichael AJ, Dong T, Xu XN. 2012. HLA-B may be more protective against HIV-1 than HLA-A because it resists negative regulatory factor (Nef) mediated down-regulation. Proc. Natl. Acad. Sci. U. S. A. 109:13353–13358. 10.1073/pnas.1204199109 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.