

ABSTRACT
A unique aspect of human monocytes, compared to monocytes from many other species, is that they express the CD4 molecule. However, the role of the CD4 molecule in human monocyte development and function is not known. We determined that the activation of CD4 via interaction with major histocompatibility complex class II (MHC-II) triggers cytokine expression and the differentiation of human monocytes into functional mature macrophages. Importantly, we determined that CD4 activation induces intracellular signaling in monocytes and that inhibition of the MAPK and Src family kinase pathways blocked the ability of CD4 ligation to trigger macrophage differentiation. We observed that ligation of CD4 by MHC-II on activated endothelial cells induced CD4-mediated macrophage differentiation of blood monocytes. Finally, CD4 ligation by MHC-II increases the susceptibility of blood-derived monocytes to HIV binding and subsequent infection. Altogether, our studies have identified a novel function for the CD4 molecule on peripheral monocytes and suggest that a unique set of events that lead to innate immune activation differ between humans and mice. Further, these events can have effects on HIV infection and persistence in the macrophage compartment.
IMPORTANCE The CD4 molecule, as the primary receptor for HIV, plays an important role in HIV pathogenesis. There are many cell types that express CD4 other than the primary HIV target, the CD4+ T cell. Other than allowing HIV infection, the role of the CD4 molecule on human monocytes or macrophages is not known. We were interested in determining the role of CD4 in human monocyte/macrophage development and function and the potential effects of this on HIV infection. We identified a role for the CD4 molecule in triggering the activation and development of a monocyte into a macrophage following its ligation. Activation of the monocyte through the CD4 molecule in this manner increases the ability of monocytes to bind to and become infected with HIV. Our studies have identified a novel function for the CD4 molecule on peripheral monocytes in triggering macrophage development that has direct consequences for HIV infection.
INTRODUCTION
Monocytes, which typically represent approximately 10% of human peripheral blood mononuclear cells (PBMCs), are mobile progenitor cells to tissue-embedded macrophages and dendritic cells (DC). Monocytes are a heterogeneous group of cells that are distinguished by the expression of several cell surface molecules, including CD14, CD16, CD64, and CD4 (1, 2). Due to their expression of pattern recognition receptors, monocytes represent an immediate, nonproliferating effector population of cells that play key roles in innate immunity through their inflammatory and phagocytic responses (3). Their primary functions include triggering antimicrobial responses, maintaining tissue homeostasis, performing tissue repair, and scavenging toxic or foreign material (1, 4). The ability of monocytes to produce a variety of proinflammatory cytokines following their activation and their ability to serve as antigen-presenting cells (APCs) bridge the innate immune response with the modulation of the adaptive immune response during inflammation (1). Importantly, peripheral monocytes rapidly differentiate into macrophages following activation with various immune signals, including Toll-like receptor (TLR) ligands, interleukin 10 (IL-10), and IL-15 (5–7). These rapidly differentiated macrophages are characterized by their functional activities, including phagocytosis and antimicrobial activity, as well as their expression of CD209 (5–7).
One significant, unique aspect of human monocytes and macrophages, compared to mouse macrophages, is that they express the CD4 molecule (8). While the function of CD4 on T cells is well characterized, the function of CD4 on human monocytes is not well understood. The CD4 molecule is a 55- to 58-kDa membrane-bound glycoprotein member of the immunoglobulin family of receptors and interacts with major histocompatibility complex class II (MHC-II), IL-16, and human immunodeficiency virus (HIV) gp120 (9–11). Neither the interaction of CD4 on non-T cells with MHC-II nor the signaling capability of CD4 in macrophage lineage cells is fully understood. For instance, while it is clear that the intracellular domain of CD4 interacts with the Src family kinase Lck in T cells, CD4+ monocytes lack expression of Lck (9, 12).
Macrophages are an important cellular target and viral reservoir for HIV in infected individuals (13). Besides HIV infection of the CD4+ T helper cell population, macrophages are thought to constitute the second most-infected cellular subset and the longest-lived one. Infected macrophages are found distributed in every tissue in the body in HIV-infected individuals (13–16). Macrophages are believed to be important in the persistence and pathogenesis of HIV infection due to their widespread anatomical presence and are thought to contribute to long-lived reservoirs of virus-infected cells (16). Subsets of these cells, particularly more mature monocytes and macrophages, including the CD14+ CD16+ monocyte subset, are preferentially infected with HIV both in vivo and in vitro (15–19). Monocytes productively infected with HIV have been found in the peripheral blood and constitute a significant, continually evolving viral reservoir (20–24). These infected monocytes can further differentiate in the tissues into mature, virus-harboring macrophages that can disseminate HIV to other cell types, including T cells (15, 22, 25). Further, infected macrophages appear to be more resistant to the cytopathic effects of HIV than are CD4+ T helper cells (14, 26, 27), and HIV infection can alter macrophage function by affecting phagocytic activity and modulating the production and/or secretion of factors that influence antigen-presenting cell activity and change bystander cell responses (13, 14, 28). In addition, macrophages may have an important role in chronic inflammation and atherosclerosis in HIV-infected individuals (16). Thus, in these studies, we were interested in examining the functional outcome resulting from the interaction of CD4 on monocytes with proinflammatory MHC-II and the effects of this interaction on HIV infection of these cells (29). Since activation of macrophages can be triggered by a variety of stimuli, including pathogen-associated molecular patterns (PAMPs), Fc receptor binding, and cytokine stimulation (1, 3, 4), we hypothesized that activation of CD4 on human monocytes results in unique signaling pathways leading to monocyte activation and differentiation and that this affects the ability of these cells to be infected with or bound by HIV.
MATERIALS AND METHODS
Human peripheral blood.
Human peripheral blood cells were obtained at the University of California, Los Angeles, from anonymous donors using an Institutional Review Board (IRB)-approved written consent form in accordance with UCLA IRB-approved protocols by the UCLA Center for AIDS Research (CFAR) Virology Laboratory and were distributed for this study without personal identifying information.
Antibodies, cytokines, and soluble MHC.
The following antibodies were used in phenotypic assessment of cells by flow cytometry: CD11c, CD14, CD4, HLA-DR (Coulter), CD16, CD163, CD45, and IgG controls (eBioscience); phosphorylated p38 (pp38) mitogen-activated protein kinase (MAPK), phosphorylated extracellular signal-regulated kinase 1/2 (pERK1/2), pNF-κB, phosphorylated signal transducer and activator of transcription 3 (pSTAT3), CD36, and CD209 (BD Biosciences); and TLR8 (Invivogen). For CD4-blocking experiments, a previously identified (30, 31) cocktail of anti-CD4 blocking antibodies, including clones 13B8.2 and BL4 (Immunotech), RPA-T4 and OKT4 (eBioscience), SK3 (Leu3a) (BD Bioscience), and S3.5 (Invitrogen), was used to examine CD4 specificity. All were obtained in forms without azide and in tissue culture-optimized forms when available, and all were used at an initial staining concentration of 1 μg/ml. Mouse IgG1, IgG2a, and IgG2b (eBioscience) were used as controls. In CD36-blocking experiments, purified anti-CD36 antibodies (BD Bioscience) or mouse IgM isotype (eBioscience) was used as a control. Recombinant human gamma interferon (IFN-γ) (Peprotech), granulocyte-macrophage colony-stimulating factor (GM-CSF) (Invitrogen), IL-15 (Gibco), and IL-16 (R&D Systems) were used to examine their effects on monocytes. Unlabeled soluble MHC-II (sMHC-II) (HLA-DRB1*0401) with the human class II-associated invariant chain peptide (CLIP) (amino acids 103 to 117) tetramer or soluble MHC class I (sMHC-I) (HLA-A*0201) with an Epstein-Barr virus latent membrane protein 2A (LMP2A) (amino acids 426 to 434) peptide were obtained from the NIH Tetramer Core Facility. Endotoxin testing of these reagents determined them to be <0.26 IU/ml utilizing the ToxinSensor Gel-Clot Assay (GeneScript).
Purification and differentiation of human peripheral blood monocytes.
Human peripheral blood was obtained from anonymous healthy adult donors in the form of leukopacks through the UCLA Center for AIDS Research Virology Core Laboratory. Monocytes were purified through magnetic activated cell sorting either through positive selection utilizing CD14 microbeads or by negative selection using the Monocyte Isolation Kit II (Miltenyi Biotech). Purity was verified by flow cytometry and was typically greater than 90% CD14+ monocytes with <0.5% CD3+ cells. The purified monocytes were stimulated with medium alone, IL-15 (200 ng/ml), GM-CSF (100 units [or 10 ng]/ml), lipopolysaccharide (LPS) (10 ng/ml), soluble MHC-II (1 μg/ml), macrophage colony-stimulating factor (M-CSF) (10 ng/ml) or soluble MHC-I (1 μg/ml) in RPMI 1640 medium containing 10% fetal calf serum (FCS). In cultures containing blocking CD4 monoclonal antibodies (MAbs), cells were pretreated for 30 min at 4°C prior to culture or treatment with a cocktail of the CD4-blocking antibodies or a cocktail of equivalent concentrations of mouse IgGs, with antibodies of each subclass (IgG1, IgG2a, and IgG2b) at the same concentration as the matching type antibody in the CD4-blocking cocktail. The cells were then cultured for 48 h at 37°C.
Flow cytometry.
Cell surface marker expression was analyzed by multiparametric flow cytometry utilizing antibodies conjugated to either fluorescein isothiocyanate (FITC), peridinin chlorophyll protein (PerCP)-Cy5.5, phycoerythrin (PE), electron-coupled dye (ECD), PE-Cy5, PE-Cy7, allophycocyanin (APC), APC-Alexa 750, APC-eFluor780 (eBioscience), Alexa 700, or eFluor405, Pacific Blue, or Pacific Orange (all from eBioscience) in appropriate combinations. Cells were acquired on an LSR II flow cytometer (BD Biosciences), part of the UCLA CFAR Flow Cytometry Core Laboratory, using FACSDiva software and were analyzed by FlowJo software. Intracellular TLR8 staining was performed using the Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (BD Biosciences) according to the manufacturer's instructions. All the cells were analyzed following fixation in a 1% formaldehyde solution.
Endocytosis and TLR8 functional assays.
Macrophages derived from 2-day treatment with medium alone, sMHC-II, IL-15, or GM-CSF were incubated at 37°C with one of the following for 1 h: Lucifer Yellow (LY) (0 to 1 mg/ml; Invitrogen), FITC-labeled dextran (0 to 1 mg/ml; Invitrogen), or green-fluorescent-protein-labeled Escherichia coli (multiplicity of infection [MOI] = 0 to 10). Some of these cells were incubated for 4 h at 37° with Dil (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate)-labeled CuSO4-oxidized low-density lipoprotein (Dil-oxLDL) (Intracel) (0 to 10 mg/ml). For CD36-blocking studies, cells differentiated for 2 days in sMHC-II were pretreated with 10 μg/ml of either mouse IgM as a control or anti-CD36 for 30 min at 37°C. The cells were then washed and incubated as described above with Dil-oxLDL. Cells were harvested and analyzed by flow cytometry. The intracellular uptake under each condition was determined by subtracting the mean fluorescence intensity of macrophages derived from controls treated with medium alone from that of macrophages derived from the sMHC-II-, IL-15-, or GM-CSF-treated cells. For TLR8 activation studies, macrophages derived from 2-day treatment with medium alone, sMHC-II, IL-15, or GM-CSF were placed in culture with the HIV-derived single-stranded RNA40 (ssRNA40) or a control RNA, ssRNA41 (0.5 μg/ml) (Invivogen), TLR8 ligand. Culture supernatants were harvested 24 h later and assessed for cytokine production with a Cytometric Bead Array using beads and antibodies specific for IL-1β, IL-6, and IL-12 (BD Biosciences), followed by flow cytometry, according to the manufacturer's instructions.
Cell signaling assays.
For measurement of intracellular calcium flux, freshly purified human peripheral blood monocytes were incubated in phosphate-buffered saline (PBS) containing 1.5 μM Fluo-4 AM (Invitrogen) at 37°C for 2 h, washed, and kept at 4°C. For CD4-cross-linking studies, the cells were divided and an aliquot was prestained with biotinylated CD4 MAb OKT4 for 20 min at 4°C (similar to the method described previously [32, 33]). The cells were then sequentially stimulated with prewarmed (37°) RPMI 1640 medium containing nothing, 5 μM ionomycin (Invitrogen), 2 μg/ml sMHC-II, 200 ng/ml IL-16, or 20 μg/ml NeutrAvidin (Invitrogen) to cross-link CD4. The calcium flux was measured by immediately placing cells on an FC500 flow cytometer (Coulter) and assessing fluorescence using a 488-nm laser. For measurement of p38 phosphorylation, freshly purified monocytes were stimulated in the above-described manner with either medium alone or sMHC-II. The cells were removed from culture and placed in cold (−20°C) methanol to fix and permeabilize the cells. The cells were then stained separately for phosphorylated (pT180/pY182) MAPK p38, phosphorylated (pT202/pY204) ERK1/2, phosphorylated (pY705) STAT3, and phosphorylated (pS529) NF-κB using the PhosFlow MAb system (BD Biosciences). The cells were subsequently analyzed by flow cytometry.
For signal transduction inhibition studies, freshly purified human monocytes were incubated with the indicated inhibitor for 30 min at 4°C. The concentrations of each inhibitor were based on previously published reports from our group and others (33, 34). The inhibitors used and their concentrations were as follows: PP2 {(4-amino-5-(4-chlorophenyl)-7-(dimethylethyl)pyrazolo[3,4-d]pyrimidine}, 10 μM; PD98059, 100 μM; U0126, 10 nM (Calbiochem); bisindolylmaleimide (BIM) I, 1 μM; and H89, 1 μM (Sigma). The cells were then treated either with medium only or with sMHC-II and analyzed as described above. Statistically significant differences were determined using the t test.
DNA microarray data and analysis.
Microarray data were analyzed by the UCLA CFAR Biostatistics Core. Fresh blood monocytes, obtained through negative selection from three separate individual donors, were treated with RPMI 1640 medium alone or with medium containing sMHC-II (1 μg/ml) for 3 h. The cells were harvested, and RNA was purified using the RNeasy procedure (Qiagen). RNA was then analyzed by the UCLA Clinical Microarray Core Laboratory using the Human U133 Plus 2.0 Array (Affymetrix). Data were analyzed using Dchip software (Cheng Li Lab, Harvard University). Visual inspection did not find any potential outliers or suspect areas. Data across all groups were normalized using the perfect match (PM)-mismatch (MM) difference. Genes were selected for further analysis if the paired t test of the difference between treatment and control was significant (P < 0.1) across all 3 patients. The genes were then analyzed using hierarchical clustering. For transcription factor association analysis, we used the Transcription Element Listening System (TELiS) (35). Data were imported into the algorithm, and transcription factor binding site association was output. The P values indicate whether a transcription factor binding motif (TFBM) is present in a greater fraction of differentially expressed genes than in the whole genome.
Cytokine array.
Monocytes were treated either with medium alone or with sMHC-II for 24 h, and the supernatants were harvested and examined for differential cytokine expression using the Human Cytokine Array G5 (RayBiotech) according to the manufacturer's instructions. Quantitation of array data and comparisons were performed using Imagequant TL software (GE Healthcare Life Sciences). Cytokines that were modulated in 3 out of 3 donors were identified.
HUVEC culture, activation, and macrophage differentiation.
Human cord blood endothelial cells (HUVECs) were purchased in viable frozen form from Lonza or Cell Applications Inc. The cells were thawed and cultured for two passages prior to use according to the manufacturer's instructions. Prior to each experiment, cells (cultured for 2 to 7 passages) were plated, allowed to grow to confluence, and maintained at confluence for at least 3 days. Some cultures were treated with IFN-γ (50 ng/ml) or with medium alone for 24 h. HUVECs were harvested using cold PBS containing 5 mM EDTA and were analyzed for expression of HLA-DR by flow cytometry. The cells were then washed 4 times with RPMI 1640 medium containing 10% FCS and freshly isolated human monocytes labeled with carboxyfluorescein succinimidyl ester (CFSE) (Invitrogen) and were placed in culture with the HUVECs. In some cultures, monocytes were pretreated with the cocktail of CD4-blocking MAbs or with a cocktail of mouse IgGs for 20 min at 4°C. The cells were then analyzed for phenotypic marker expression by flow cytometry 2 days following culture.
HIV stocks.
Virus was grown in 293T cells from a plasmid encoding the full-length molecular clone, and viral p24 levels in the supernatant of plasmid-transduced cells were quantitated by a standard enzyme-linked immunosorbent assay (ELISA) (Coulter, Hialeah, FL). Viral infectivity was determined by limiting-dilution titration on CEMx174 cells or phytohemagglutinin (PHA)-stimulated PBMCs.
HIV infection.
Primary monocytes were isolated using CD14 microbeads from human peripheral blood as described above. The cells were either left untreated (medium only) or treated with sMHC-II for 3 days. The cells were then washed and treated with medium alone or were infected with the dual-tropic molecular clone HIV-189.6 at an MOI of approximately 1. The cells were then cultured for 3 additional days and analyzed by flow cytometry for cell surface marker expression as described above and for intracellular expression of HIV-1 Gag (p24) by staining with the KC57 antibody (Becton, Dickinson, San Jose, CA).
HIV attachment assay.
Peripheral blood monocytes were purified from healthy PBMCs by negative selection as described above. The cells were treated with sMHC-II or with medium alone for 3 days and were then washed and incubated with CCR5-tropic HIV-1NFN-SX for 3 h at 37°C. The cells were then washed three times with RPMI 1640 medium containing 10% FCS, and the pellets were lysed with PBS containing 1% Triton X-100 and analyzed for viral Gag (p24) by ELISA. Experiments were done in triplicate. For antibody blockade experiments, cells were treated as described above and preincubated with mouse IgG (10 μg/ml) (Sigma) or mouse anti-CD209 (10 μg/ml; clone DCN46; eBioscience) on ice for 10 min before HIV infection and p24 assay (described above). Experiments were done in triplicate.
Microarray data accession number.
The data from this study have been submitted to the Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo) under accession number GSE32939.
RESULTS
Ligation of CD4 on monocytes by MHC-II induces differentiation into macrophages.
CD4 is expressed by peripheral human monocytes, yet its function on these cells is not fully understood. To investigate the function of CD4 on human blood monocytes, we activated cells with either medium alone; the CD4 ligand sMHC-II (HLA-DR*0401); or known inducers of rapid monocyte differentiation, IL-15 or GM-CSF (5, 6, 36). The typical purity of isolated CD14+ monocytes was greater than 97%, with the following phenotype: CD45+ CD4+ CD11c+ TLR8lo CD16lo CD163− CD209− (Fig. 1A). Activation of monocytes via CD4 ligation for 48 h induced the expression of a variety of cell surface markers typically expressed by rapidly differentiated macrophages (5–7), including CD163, a scavenger receptor for hemoglobin; CD209 (DC-SIGN), a C-type lectin involved in phagocytosis; and CD16 (Fig. 1B). Treatment of cells with IL-15 induced the development of CD163lo CD209+ and CD163+ CD209− cells, and treatment with GM-CSF primarily induced CD163+ CD209− cells, similar to previous reports (5, 37). In contrast to activation via the CD4–sMHC-II interaction, we did not observe differentiation of monocytes in response to treatment with sMHC-I (HLA-A*0201) (Fig. 1C). This indicates that the CD4-mediated differentiation of monocytes into macrophages was not due to nonspecific effects of the MHC-II tetramer reagent and did not occur through CD4–MHC-I tetramer interaction. Activation of CD4 with sMHC-II also resulted in a dramatic increase in the expression of the pattern recognition receptor TLR8 compared to the medium control (53% versus 10%) (Fig. 1D). Meanwhile, levels of CD11c, CD4, and CD45 were not significantly different between sMHC-II- and medium-treated cells (Fig. 1D and E). Expression of these molecules remained constant regardless of the type of stimulation.
FIG 1.
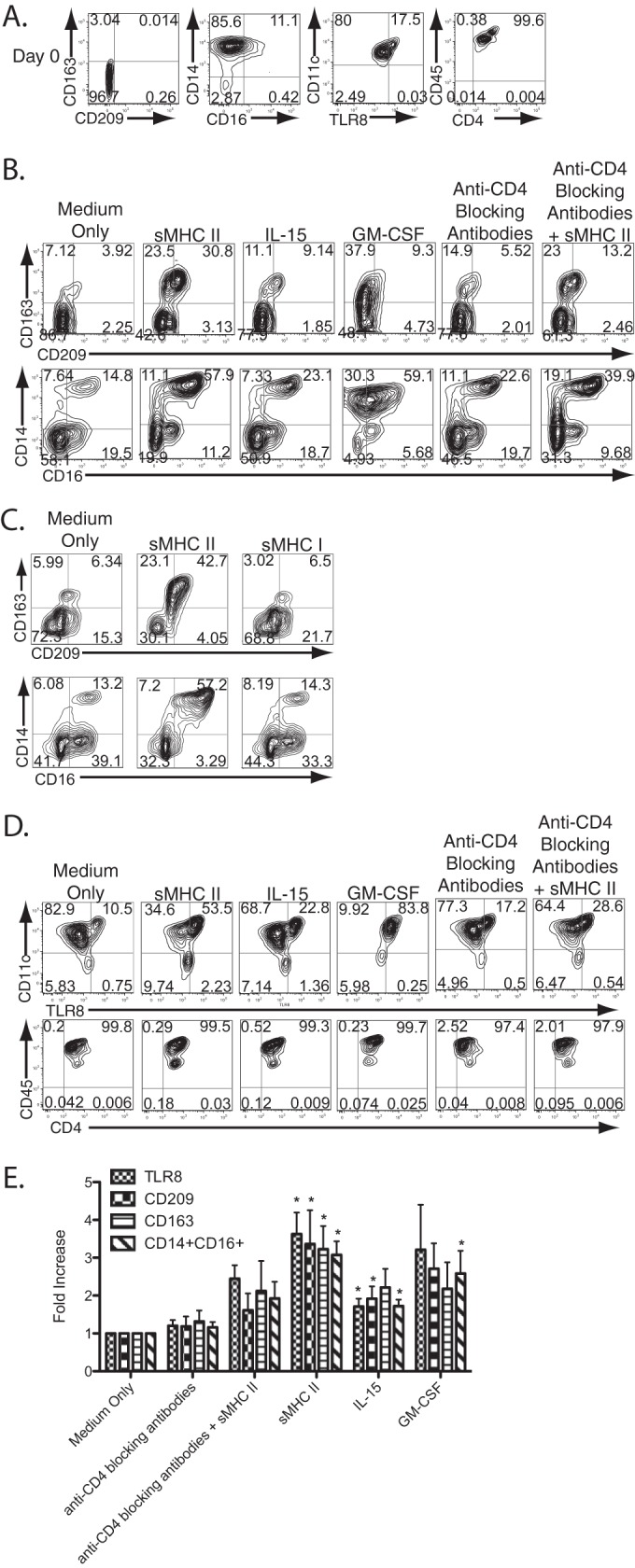
Ligation of CD4 expressed on human peripheral blood monocytes by MHC-II triggers macrophage differentiation. (A) Peripheral blood monocytes were purified by positive selection for CD14 and were examined by flow cytometry immediately following purification prior to stimulation. The numbers in each contour plot represent the percentage of cells in each quadrant. (B) Monocytes were pretreated with mouse IgG antibodies (medium only) or anti-CD4 blocking antibodies or were left untreated and then stimulated with medium, sMHC-II, IL-15, or GM-CSF, as indicated, for 2 days. The contour plots represent flow cytometry analysis of markers of macrophage differentiation. (C) Stimulation with MHC-I does not induce macrophage differentiation. Monocytes were stimulated with medium only (left column), sMHC-II tetramers (middle column), or sMHC-I tetramers (both obtained from the NIH Tetramer Core Facility) and analyzed by flow cytometry for markers of macrophage differentiation 2 days following stimulation. The numbers represent the percentage of cells in each quadrant. (D) Two-day-stimulated monocytes were further analyzed for TLR8 expression and stable expression of myeloid developmental markers. (E) Summary of the changes in phenotypic marker expression profiles, as determined by flow cytometry and staining for each indicated marker and compared to monocytes treated with medium only. The data are a summation of experiments with monocytes derived from at least seven separate donors. The asterisks indicate changes with statistically significant differences (P ≤ 0.05; t test) from medium-only controls. The error bars represent the standard deviations of the mean.
To check the specificity of this interaction, we utilized blocking monoclonal antibodies to CD4 (30, 31, 38). Addition of the CD4-blocking antibodies dramatically abrogated the induction of CD209 and TLR8 following sMHC-II activation (Fig. 1B). This CD4-specific blocking was dependent on the initial concentration of blocking antibodies and was decreased with serial dilution of the blocking antibodies (see Fig. S1 in the supplemental material). However, blocking CD4 did not change CD4 surface levels (Fig. 1D), nor did it inhibit activation via the TLR4 ligand LPS or alter the phenotype of previously activated cells (see Fig. S2A and B in the supplemental material). In addition, we detected no expression of MHC-II ligand lymphocyte activation gene 3 (Lag-3, or CD223), indicating that the effects observed are not due to MHC-II–Lag3 interaction (see Fig. S3 in the supplemental material). Thus, these data strongly suggest that the interaction of CD4 on the surfaces of monocytes with MHC-II induces phenotypic differentiation into macrophages.
MHC-II ligation of monocyte CD4 triggers differentiation into functional macrophages.
An important innate immune function of macrophages is their ability to efficiently phagocytose pathogens and cellular debris. We were interested in determining whether the rapid phenotypic changes induced by CD4 activation correlated with macrophage functional responses. We initially compared the pinocytic abilities of macrophages differentiated through CD4 ligation or through treatment with either IL-15 or GM-CSF. CD4-mediated differentiation produced macrophages that had intermediate levels of uptake of LY (Fig. 2A; see Fig. S4A in the supplemental material). Meanwhile, phagocytosis of fluorescently labeled E. coli was similar under all three activation conditions (Fig. 2B; see Fig. S4B in the supplemental material). Uptake of fluorescently labeled dextran, which occurs through pinocytosis and receptor-mediated endocytosis, primarily through lectin-type receptors (39), was similar for CD4− and IL-15-differentiated macrophages (Fig. 2C; see Fig. S4C in the supplemental material). Finally, we compared the abilities of cells to take up oxidized lipids, a critical function of macrophages in maintaining cellular and tissue homeostasis through the scavenging of metabolic by-products. sMHC-II-differentiated macrophages efficiently took up Dil-oxLDL, and were similar to IL-15- or GM-CSF-differentiated monocytes (Fig. 2D; see Fig. S4D in the supplemental material).
FIG 2.

MHC-II ligation of CD4 on human monocytes differentiates them into functional macrophages. (A to D) Fresh human monocytes, purified by negative selection, were differentiated for 2 days in the presence of medium only, sMHC-II, IL-15, or GM-CSF and assessed for phagocytic activity, separately, of Lucifer Yellow (A), fluorescent E. coli (B), dextran-FITC (C), and Dil-oxLDL (D) at the indicated concentrations. The graphs depict the change in the mean fluorescence intensity (MFI) over cells differentiated in the presence of medium only. (E) CD36 expression levels on CD209+ macrophages differentiated for 2 days in medium alone or with sMHC-II, performed in triplicate. The error bars represent the standard deviations of the mean, and differences were statistically significant (P < 0.05; t test). (F) Dil-oxLDL uptake by macrophages differentiated for 2 days in sMHC-II or medium only and pretreated with either mouse isotype or with anti-CD36 blocking antibody was assayed. The graph represents the change in the MFI over gated CD209+ cells differentiated in the presence of medium only and is representative of three separate experiments. A second representative experiment is shown in Fig. S4 in the supplemental material.
We next examined whether the class B scavenger receptor CD36 was involved in the uptake of Dil-oxLDL (5, 40). We found that CD36 was more highly expressed on CD4-differentiated macrophages (Fig. 2E; see Fig. S4E in the supplemental material) and that addition of a CD36-specific monoclonal antibody reduced the uptake of Dil-oxLDL by approximately 50 to 60% (Fig. 2F; see Fig. S4F in the supplemental material). Altogether, CD4-mediated differentiation of human monocytes produced functional macrophages with increased phagocytic activity and scavenger receptor expression.
Increased TLR8 expression following CD4-mediated differentiation modulates innate immune responses.
We next wanted to test the functional relevance of the upregulation of TLR8 during CD4-mediated differentiation of monocytes. Given that TLR8 is important for the innate immune system to sense ssRNA, including that of HIV (41, 42), we determined whether CD4–MHC-II-mediated differentiation of human monocytes enhanced responses to a TLR8 ligand. CD4–MHC-II-differentiated macrophages were activated with a GU-rich sequence of HIV RNA (ssRNA40) or a control ssRNA (ssRNA41) (41). In response to activation with ssRNA40, CD4-MHC-II-differentiated macrophages produced higher levels of IL-1β and IL-6 and lower levels of IL-12 than monocytes cultured in medium alone (Fig. 3). In contrast, activation with the control ssRNA41 did not result in any significant changes in cytokine production. The increase in TLR8 expression following CD4-mediated differentiation results in cells with an enhanced ability to modulate cytokines involved in innate immune responses to HIV following TLR8 activation. These experiments demonstrate that the newly developed macrophage is functionally capable of responding to TLR8 ligands like HIV. However, CD4–MHC-II-mediated differentiation could have additional effects in the presence of HIV by allowing virus attachment through increased expression of CD209 and increased infection of the newly differentiated macrophage (see below). Thus, despite the cell's increased ability to respond to viral RNA through TLR8, HIV could actively subvert the engagement of innate antiviral responses following macrophage differentiation.
FIG 3.
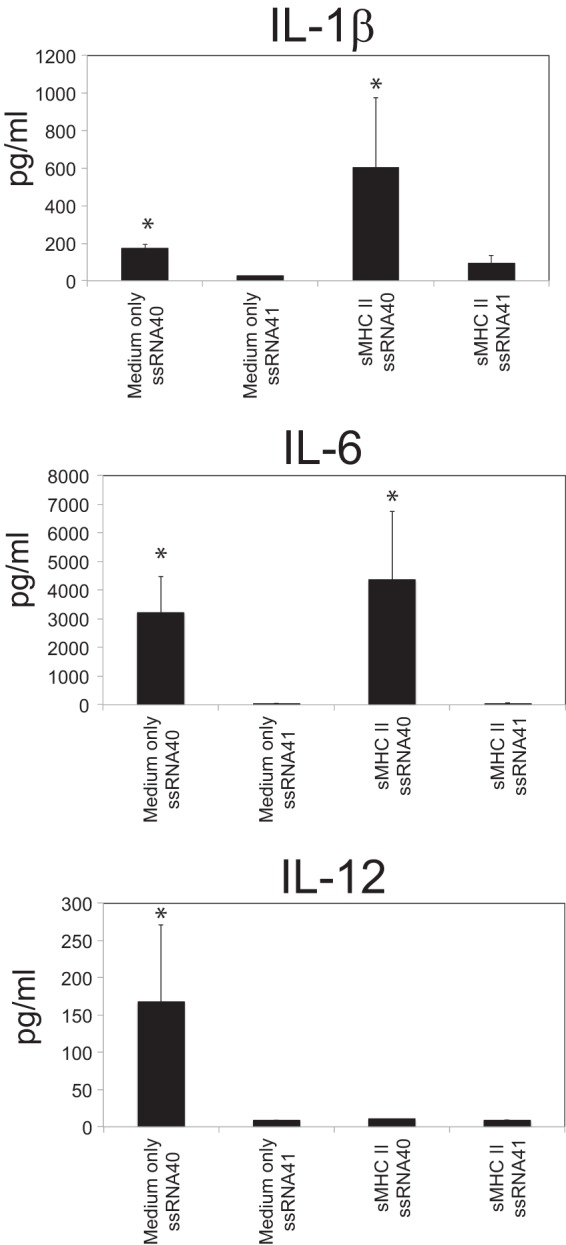
Functional TLR8 upregulation following CD4−-mediated macrophage differentiation. Fresh blood monocytes were purified by negative selection and differentiated for 2 days in the presence of medium only or sMHC-II and then separately stimulated with either the human TLR8-specific HIV-derived ligand ssRNA40 or control ssRNA41 for 24 h. IL-1β, IL-6, and IL-12p70 production was measured by cytokine bead array. The data represent the mean levels of expression of these cytokines from 3 separate experiments, each with separate donor cells. The error bars represent the standard deviations of the mean.
CD4 ligation by MHC-II on human monocytes results in modulation of gene expression and cytokine production.
In order to gain insight into pathways that may be relevant during macrophage differentiation following activation through CD4, we performed microarray analysis of monocytes activated by sMHC-II treatment. A number of factors known to be involved in macrophage differentiation and function were observed to be modulated 24 h following ligation (Fig. 4A). To assess potential transcriptional control mechanisms of the genes that were modulated, we performed differential expression analysis of the microarray data using TELiS (35). We found a significant overrepresentation in the incidence of the nuclear factor κB (NF-κB) (P = 5.15 × 10−5), nuclear factor 1 (P = 0.013), and Ikaros 3 (P = 0.0412) TFBMs in the genes that were modulated following activation with sMHC-II. The data suggest that these transcription factors, which are known to function during monocyte/macrophage differentiation (43–45), are highly involved in the signaling and regulation of monocyte differentiation following CD4 ligation by MHC-II.
FIG 4.

CD4 ligation by MHC-II modulates gene and cytokine expression. (A) Gene expression modulation following ligation of CD4 on negatively selected fresh blood monocytes was assessed 24 h following treatment of cells with medium only (−) and cells treated with sMHC-II (+) in 3 separate experiments with cells from 3 different donors. Genes that were significantly modulated are indicated on the right in order of levels of overall up- or downregulation. (B) The modulation of secreted cytokine expression was determined by comparison of multiple cytokines in supernatants 48 h following the treatment of cells with medium only or with sMHC-II. The values represent the average relative fold changes in cells treated with sMHC-II from cells treated with medium alone (set at a relative value of 1) (n = 3). The error bars represent the standard deviations of the mean.
To confirm the microarray results, we measured cytokine release following CD4 ligation by sMHC-II on freshly purified CD14+ cells. Multiple factors that were upregulated in the microarray and implicated in monocyte/macrophage development were modulated following CD4 ligation (Fig. 4B). In particular, expression of CXCL1 and CXCL5 was found to be consistently upregulated by both microarray and cytokine array in our analysis. Of note, we did not see a significant upregulation or downregulation of the HIV coreceptor CCR5 or CXCR4 within the time frame of this experiment. However, it is possible that these chemokine receptors may be modulated through secondary effects via the subsequent binding of the cytokines produced following the initial stimulation. Altogether, these results indicate that CD4 expressed by human monocytes has direct functional effects, modulating gene and cytokine expression following ligation by MHC-II.
CD4 expressed on human monocytes induces calcium flux and signal transduction through the ERK/MAPK/NF-κB pathway following ligation by MHC-II.
CD4 activation in T cells occurs primarily through the signaling adapter Lck. Since human monocytes lack expression of Lck (12), we investigated the direct intracellular effects of CD4-mediated signaling on primary human monocytes. Treatment with either sMHC-II, a cross-linking CD4 monoclonal antibody, or the CD4 ligand cytokine IL-16 (as a positive control that has been previously shown to induce calcium flux though CD4 ligation and monocyte migration [46, 47]) resulted in prolonged Ca2+ flux (Fig. 4A). This indicated the induction of secondary-messenger signaling by the CD4 molecule following its ligation on primary human monocytes. This prolonged Ca2+ flux was consistently observed in these experiments and may represent sustained triggering of this molecular response, similar to that observed with Ca2+ flux induced through CD4 signaling in the THP-1 cell line (48). Since MAPKs, which include the ERK family and the p38 MAPK family, are central in the induction of many monocyte/macrophage differentiation and activation pathways (49, 50), we investigated the phosphorylation of these molecules following CD4 ligation by MHC-II. We observed the phosphorylation of ERK1/2 and of the p38 MAPK molecule over time in cells treated with sMHC-II versus cells treated with medium alone (Fig. 5B). In addition, we observed increased phosphorylation of NF-κB following CD4 ligation by MHC-II, correlating with the activation of NF-κB-associated TFBMs that we had observed in our earlier TELiS-associated microarray data (see above). We did not, however, observe increased phosphorylation of STAT3, a central transcription factor associated with the expression of immune response genes in monocytes/macrophages and one that often antagonizes NF-κB-mediated gene activation (51, 52). Altogether, these results indicate that CD4 expressed by monocytes is functional and that its ligation can lead to the activation of MAPK-dependent signaling pathways.
FIG 5.
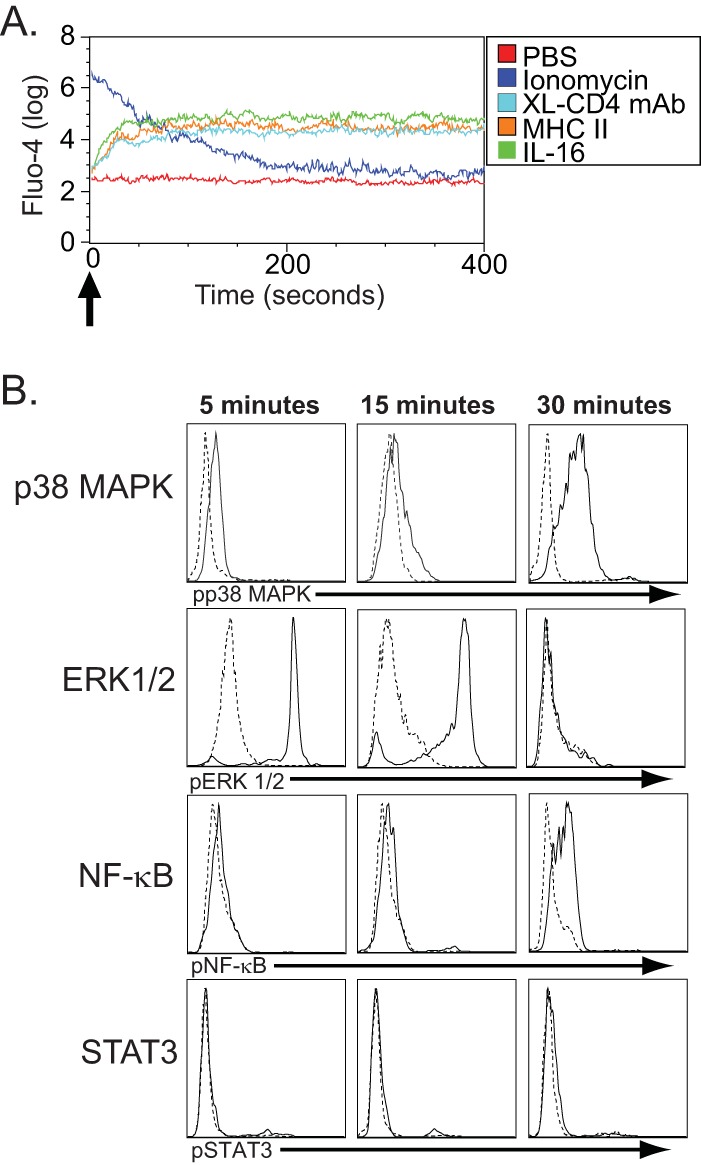
CD4-mediated signal transduction in negatively selected fresh human monocytes. (A) Ca2+ flux following CD4 ligation on human monocytes. Freshly isolated human monocytes were labeled with Fluo-4 and then treated (indicated by the arrow) with either PBS alone (negative control), ionomycin (positive control), cross-linked CD4-specific monoclonal antibodies (XL-CD4 MAb), sMHC-II, or IL-16. They were immediately measured for Ca2+ flux by flow cytometry and analyzed for fluorescence of Fluo-4 (y axis) for the indicated time (x axis) following stimulation. (B) Phosphorylated p38 MAP kinase (pp38MAPK), ERK1/2 (pERK1/2), NF-κB (pNF-κB), and STAT3 (pSTAT3) were assessed in cells treated with medium only (dashed lines) and cells treated with sMHC-II (solid lines) 5, 15, and 30 min following stimulation. The histogram depicts the relative fluorescence intensities, as determined by flow cytometry, for cells staining for pp38MAPK, pERK1/2, pNF-κB, and pSTAT3 and is representative of 3 separate experiments.
CD4-mediated differentiation of human monocytes involves Src family and MAPK pathways.
Given that CD4 ligation leads to Ca2+ flux and ERK1/2 and p38 phosphorylation, we investigated whether these signaling pathways were involved in mediating the differentiation of monocytes into macrophages. We utilized small-molecule signal transduction inhibitors to investigate the mechanism by which activation through CD4 triggers macrophage differentiation. Monocytes were pretreated either with medium alone or with the mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1 (MEK1) inhibitor PD98059 (53), the MEK1/MEK2 inhibitor U0126 (54), the Src family kinase inhibitor PP2, the protein kinase C inhibitor BIM (55), or the protein kinase A inhibitor H89 (56). The cells were then treated with medium alone or with sMHC-II, and markers of phenotypic macrophage differentiation were assessed. Inhibition of MEK1/MEK2 by PD98059/U1026 treatment, which directly inhibits the phosphorylation and activation of ERK1/2 (57), significantly blocked CD4-mediated differentiation into macrophages (Fig. 6). This directly correlates with the ability of CD4 to trigger phosphorylation of ERK1/2 and p38 MAPK (Fig. 5), further implicating this signaling pathway in CD4-mediated macrophage differentiation. In addition, we found that inhibition of Src family kinases by PP2 treatment significantly inhibited the phenotypic differentiation of monocytes into macrophages following CD4–MHC-II interaction (Fig. 6). In this analysis, differentiation of cells was assessed by expression of either CD209, CD163, or TLR8 following treatment to simplify the analysis. The differentiation index represents the relative fold changes of cells expressing any of the differentiation markers CD209, CD163, or TLR8 (calculated together), comparing sMHC-II-treated cells with cells that were treated with medium only. In contrast, treatment with BIM or H89 did not significantly inhibit the differentiation of monocytes following CD4 ligation. Together, these data suggest that CD4 activation leads to activation of Src family kinase(s), ERK1/2, and MAPK-dependent pathways, resulting in the differentiation of monocytes into macrophages.
FIG 6.
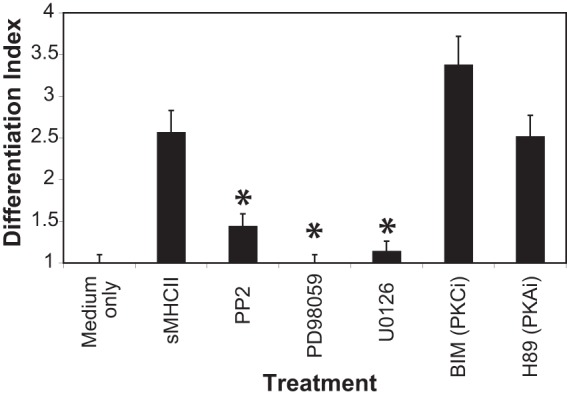
Inhibition of CD4-mediated signal transduction inhibits MHC-II-directed macrophage differentiation. Blood monocytes purified by negative selection were pretreated with medium only, PP2 (SRC family kinase inhibitor), PD98059 (MEK/MAPK 1 inhibitor), U0126 (MEK1/MEK2/MAPK inhibitor), bisindolylmaleimide (protein kinase C inhibitor [PKCi]), and H89 (protein kinase A inhibitor [PKAi]) and then subjected to stimulation with medium only or with sMHC-II. The differentiation index represents the fold changes in the percentages of cells expressing any of the differentiation markers CD209, CD163, or TLR8 following sMHC-II ligation compared to cells treated with medium only. The error bars represent the standard deviations of the mean (n = 3). Statistically significant differences from untreated cells stimulated with sMHC-II (P ≤ 0.05; t test) are indicated by asterisks.
MHC-II on HUVECs can ligate CD4 on monocytes and trigger differentiation.
A natural ligand for CD4 is MHC-II, which is expressed on stimulated endothelial cells, including HUVECs, following treatment with IFN-γ (58–61). We therefore investigated whether MHC-II on the surfaces of activated endothelial cells can ligate CD4 and induce human macrophage differentiation. HUVECs were either pretreated with IFN-γ or left untreated and washed, and then human monocytes were added to the culture. We found that IFN-γ treatment induced the expression of MHC-II (HLA-DR) on HUVECs, and this increased expression correlated with the increased maintenance of CD14 expression on monocytes following exposure to the endothelial cells (Fig. 7). The maintenance of CD14-expressing cells was inhibited in cultures that were pretreated with blocking CD4-specific MAbs, indicating that the CD4 molecule expressed by monocytes contributed to the maintenance of the cells (Fig. 7A, summarized in B). Interestingly, HUVEC cultures pretreated with IFN-γ support robust CD14 expression. However, following activation through CD4–MHC-II interaction, the ratio of CD14+ CD16+ to CD14+ CD16− cells is lower than that for untreated HUVECs (Fig. 7A). Overall maintenance of CD14+ cells, however, is higher in HUVEC cultures preactivated with IFN-γ (Fig. 7B). Further, we examined the development of cells expressing any of the differentiation markers CD209, CD163, or TLR8 (calculated together as the differentiation index) and found that the differentiation of these cells was inhibited in cultures that were pretreated with blocking CD4-specific MAbs. This indicates that the CD4 molecule expressed by monocytes contributed to the differentiation of these cells (Fig. 7C). Thus, the interaction of CD4 expressed on monocytes with MHC-II on endothelial cells results in maintenance of CD14+ cells, as well as facilitating the differentiation of these cells into macrophages.
FIG 7.
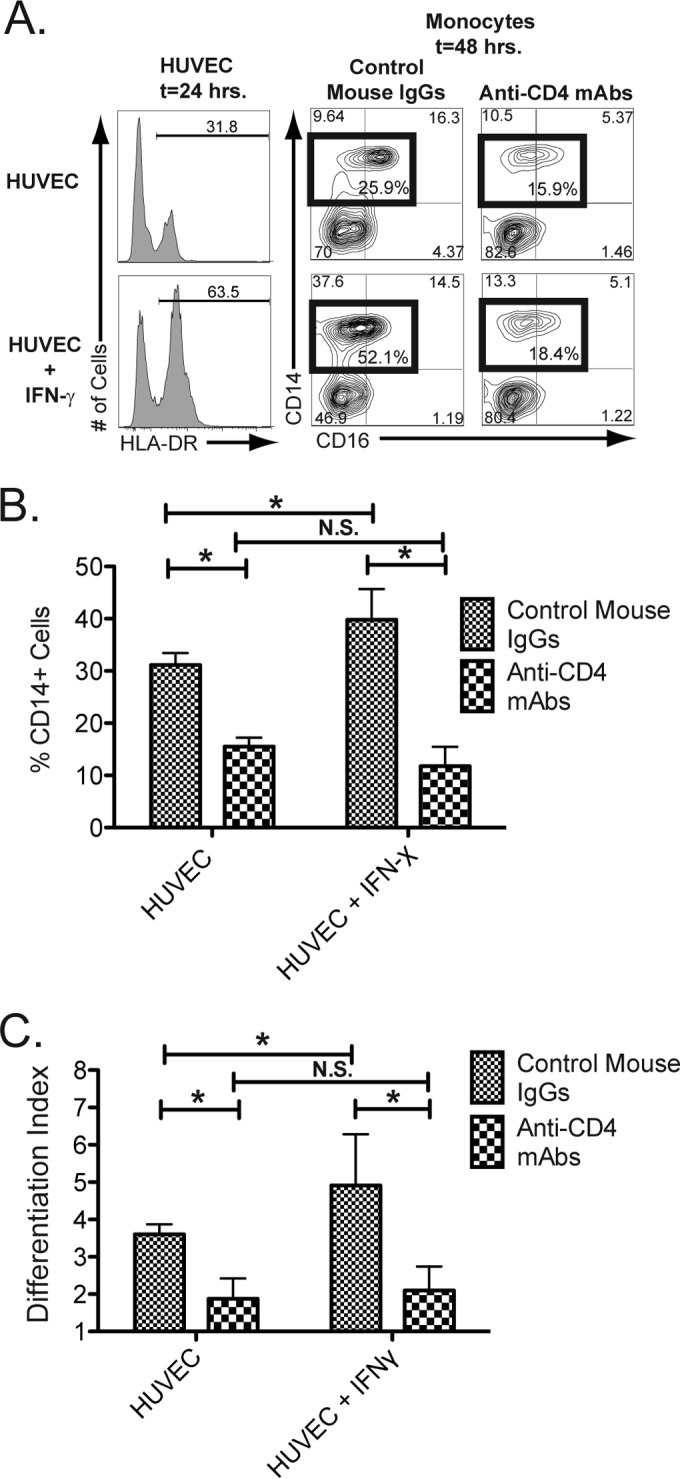
Endothelial cell activation of MHC-II results in CD4-mediated macrophage differentiation. HUVEC monolayers were stimulated either with medium only (top row) or with medium containing IFN-γ (bottom row) for 24 h. The histograms in the left column display the levels of MHC-II (HLA-DR) expression following 24 h of stimulation. Freshly purified human blood monocytes, isolated through negative selection, were labeled with CFSE and then pretreated either with mouse IgG or with anti-CD4 blocking antibodies for 30 min. The HUVECs were then washed, and the pretreated monocytes were placed in the cultures. (A) Macrophage differentiation was assessed in cultures 48 h following the initiation of the HUVEC-monocyte cultures by flow cytometry and gating on CFSE+ cells (to distinguish added monocytes from HUVECs) for expression of CD14 and CD16 in cells pretreated with mouse IgGs (middle column) and with cells pretreated with CD4-blocking monoclonal antibodies (right column). Total mature CD14+ cells, including CD14+ and CD14+ CD16+ cells, are highlighted by the box gate, and the percentage of cells within each box gate is provided within the gate. The additional numbers denote the percentages of cells within each gate. The data are from one representative experiment out of five. (B) Mean percentages (n = 5) of CFSE+ cells expressing CD14. The asterisks indicate statistically significant differences between the indicated groups (P < 0.05; t test; N.S., not significant). (C) Macrophage differentiation induced by CD4-HUVEC ligation. CD14+ cells were treated with mouse control IgG or with anti-CD4 blocking MAbs and then with HUVECs or HUVECS pretreated with IFN-γ. The cells were then assessed by flow cytometry, and the fold changes in the percentages of cells expressing any of the differentiation markers CD209, CD163, or TLR8 following sMHC-II ligation compared to cells treated with medium only were calculated as the differentiation index (n = 3). The error bars represent the standard deviations of the mean. The asterisks indicate statistically significant differences between the indicated groups (P < 0.05; t test; N.S., not significant).
CD4 ligation by MHC-II enhances HIV infection of monocytes/macrophages.
The majority of circulating monocytes in the peripheral blood are not susceptible to HIV infection. However, the virus has been identified in certain populations of cells, particularly the more mature monocytes and macrophages, which have increased permissiveness for HIV infection in vivo and in vitro (17–19, 62–65). We were interested to determine if ligation of CD4 expressed on blood monocytes through a proinflammatory interaction with MHC-II, which drives monocyte maturation into macrophages, would increase the susceptibility of blood-derived monocytes to HIV infection. Purified blood monocytes were either treated with medium alone as a control or with sMHC-II to drive differentiation. The cells were then treated with cell-free infectious HIV-1, and productive infection was assessed. sMHC-II-differentiated cells become infected and express HIV-1 proteins at higher levels than cells treated with medium alone (Fig. 8A). In fact, the more mature macrophages, CD209hi, had the greatest levels of virus-expressing cells. Ligation of CD4 with sMHC increased the ability of the resultant macrophages to become infected with and express HIV approximately 5-fold over treatment of cells with medium alone (Fig. 8B). This indicates that CD4–MHC-II ligation induces macrophage differentiation into cells that have enhanced ability to bind to and become infected with HIV. To asses a potential mechanism that allows greater infection of CD4–MHC-II-differentiated cells, purified blood monocytes were treated with medium alone or with sMHC-II and then with cell-free infectious HIV-1, and virus attachment was assessed. We found that treatment with sMHC-II triggered macrophage differentiation, and the resultant cells had an increased ability to bind and retain virus following exposure (Fig. 8C). We examined whether this was due to binding of HIV by increased expression of CD209. We found that HIV binding was significantly reduced by pretreating cells with a mouse anti-human blocking CD209 antibody versus pretreating cells with mouse IgG (control), indicating that the increased levels of HIV binding following MHC-II-induced differentiation were largely due to increased expression of CD209. In sum, these results suggest that circulating monocytes can be triggered to become more susceptible to infection with HIV in HIV-infected individuals at sites of inflammation and that the CD4–MHC-II interaction is a potential mechanism that can contribute to greater numbers of HIV-infected macrophages and can allow greater sequestration of virus through binding of newly induced CD209.
FIG 8.
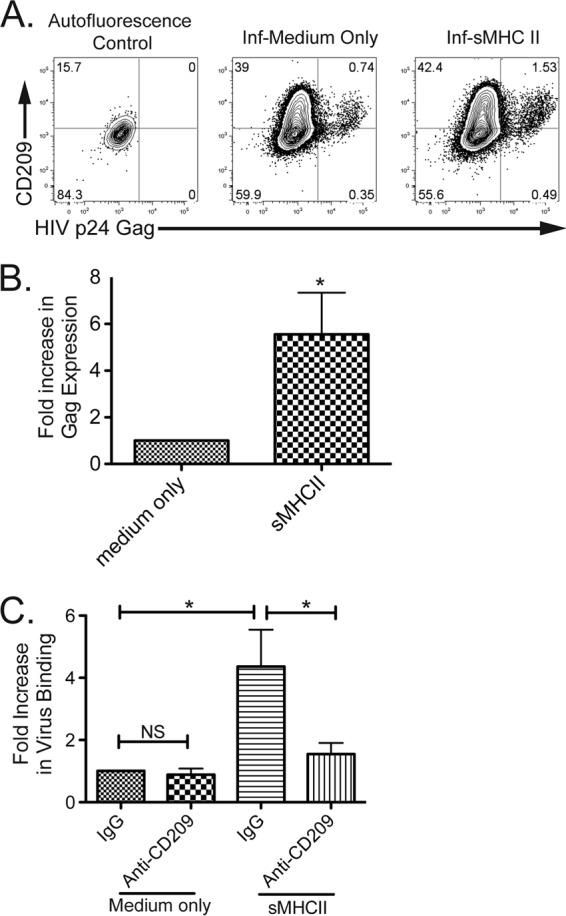
Macrophage differentiation triggered by CD4 enhances HIV infection. (A) Infection and expression of HIV following differentiation. Intracellular expression of HIV p24 gag (x axis) and extracellular expression of CD209 (y axis) were determined by flow cytometry 3 days after treatment with medium alone (left; autofluorescence control) or with HIV (middle and right). The data are representative of six separate experiments. (B) Increased expression of HIV following differentiation of cells by sMHC-II. The expression of HIV in differentiated cells was assessed by flow cytometry 3 days following treatment/infection (n = 6 experiments). The asterisk indicates that treatment was statistically significantly (t test following logarithmic transformation; P ≤ 0.05) different from treatment with medium only. The range of viral expression was 0.22% to 1.7% in cells treated with medium only and 0.68% to 15.7% in sMHC-II-treated cells; the variation was donor dependent. (C) sMHC-II differentiation increases HIV attachment, and pretreatment of cells with anti-CD209 blocks virus attachment. Purified, negatively selected blood monocytes pretreated either with mouse IgG (control) or with anti-CD209 blocking antibody, as indicated, were then treated with medium alone or sMHC-II for 2 days. The cells were then exposed to cell-free infectious HIV-1 and subsequently washed and assessed for HIV binding by ELISA of cell lysates. The values represent the fold increase in virus binding compared to IgG and medium-only control (left bar). The graph depicts the means of 3 total experiments. The asterisks indicate statistically significant differences (t test; P ≤ 0.05) under the compared conditions.
DISCUSSION
Peripheral blood monocytes are a heterogeneous population of cells that essentially “patrol” the blood vessels for signals that initiate their effector or differentiation potential (1–3). Many soluble triggers, including cytokines and PAMPs, have been identified that influence the ability of a monocyte to differentiate into a macrophage or dendritic cell (6, 66). We have identified a novel differentiation pathway mediated by the activation of CD4 expressed on peripheral monocytes. Macrophages derived from CD4-mediated differentiation expressed the C-type lectin CD209, the scavenger receptor CD163, the Fc receptor CD16, and/or the pattern recognition receptors CD14 and TLR8 and were efficient at phagocytosis. Therefore, given that MHC-II is expressed by activated B cells, DC, thymic epithelial cells, and activated T cells at sites of tissue inflammation or damage (29, 67), our data suggest that monocytes, which express CD4, could encounter MHC-II at sites of blood vessel egress or at sites of antigen reactivity, leading to their differentiation into a macrophages. These data also demonstrate a novel function for the CD4 molecule on human peripheral monocytes.
Following ligation of CD4 on peripheral monocytes, we observed increased protein expression of the proinflammatory cytokines leptin and IGF-1, as well as the chemokines CXCL1, CXCL5, CXCL6, CCL26, CCL1, CCL9, and CCL13, and decreased expression of IL-4, IL-15, and IFN-γ. In inflammatory diseases, such as atherosclerosis, macrophages are found in abundance at atherosclerotic lesions and are believed to significantly contribute to the development of disease (68). Based on our observations, the interaction of CD4 on circulating monocytes with MHC-II expressed by activated endothelium could be a potential mechanism of monocyte activation and subsequent plaque formation or recruitment to tissues at the site of inflammation (50, 68). The modulated expression of many of these proinflammatory cytokines following CD4–MHC-II interaction could serve to promote other types of inflammatory responses and subsequent macrophage development.
The increase in expression of pattern recognition receptors such as intracellular TLR8 suggests that CD4–MHC-II stimulation of monocytes potentiates these cells for activation by secondary signals. We demonstrated that activation of CD4-differentiated cells with ssRNA40 resulted in increased secretion of IL-1β and IL-6, important cytokines for driving Th17 differentiation. In addition, CD4 activation leads to highly phagocytic cells, as demonstrated by the ability of these macrophages to phagocytose inert particles, E. coli, and Dil-oxLDL. Our data indicate that the increase in phagocytosis is likely to occur through the upregulation of scavenger receptors, including CD36 (69). Therefore, CD4 activation triggers monocytes to differentiate into macrophages that contribute to two key innate immune functions, cytokine secretion and phagocytosis.
An important aspect of CD4 is its ability to signal, which in T cells occurs via Lck. In contrast, monocytes lack expression of Lck, and thus, the mechanism of CD4 signaling in these cells is unique (70). Importantly, we have identified a Src and MAPK family-dependent pathway in human monocytes following CD4-mediated activation and differentiation. Our results indicate that CD4-mediated intracellular signal transduction occurs through Src family kinase(s) and MEK1/2 to result in cellular differentiation, as inhibition of these pathways blocks this effect. MEK1/2 has been demonstrated to catalyze the phosphorylation of ERK1 and ERK2, which leads to subsequent phosphorylation and activation of p38 MAPK (49, 57). We observed increased phosphorylation of ERK1/2 and p38 following ligation of CD4 expressed by monocytes with MHC-II, which correlates with the further activation of this pathway in CD4-mediated macrophage differentiation. We also observed increased phosphorylation of NF-κB and increased expression of genes containing NF-κB TFBMs. The activation of NF-κB and the lack of STAT3 transcription factor activation suggest a possible antagonism or skewing resulting from CD4-mediated differentiation (52). Activation of monocytes via CD4 also triggers a robust Ca2+ flux, which is consistent with results from studies of the monocytic THP-1 cell line (48). Studies have also identified a putative association between CD4 and the Src family kinase Hck, which is expressed in monocytes but not T cells (9). Further, another CD4 ligand, IL-16, can activate stress-activated protein kinase (SAPK) and MAPK pathways in CD4+ macrophage lines (71). However, IL-16 may have other cellular receptors than the CD4 molecule on monocytes (72).
The increased ability of monocytes to bind HIV through CD209 and to subsequently become infected following CD4-triggered differentiation has potential implications in HIV disease. HIV infection is associated with increased global immune activation and increases in cardiovascular disease (73, 74). HIV infection results in dramatic upregulation of a variety of factors that enable vascular inflammation, which would consequently upregulate MHC-II expression by the exposed vasculature. CD4 ligation could trigger macrophage differentiation at sites of inflammation, upregulate CD209 expression, and contribute to plaque formation. Our data suggest that this would enable virus trapping, which could permit further transfer of virus to neighboring cells, as well as facilitate direct infection of these newly differentiated macrophages, further contributing to viral reservoirs and HIV persistence. Increased monocyte activation is also observed in HIV-infected individuals, and the association with increased cardiovascular disease is unclear (73). However, increased vascular inflammation coupled with increased monocyte activation through CD4 is a potential mechanism.
In sum, we have identified a unique role for the CD4 molecule on human monocytes: mediating the differentiation of monocytes into macrophages. This significantly differs from its well-defined role in CD4+ T helper cells as a cofactor in T cell receptor-mediated cellular activation (75, 76). Our results provide insight into a nontraditional role of the CD4 molecule when expressed by an innate immune cell versus an adaptive immune cell. These studies set the stage for further examination of an in vivo role of CD4 expression on monocytes and the consequences of this expression in inflammatory diseases and HIV infection. Since CD4 cell surface expression on macrophage lineage cells is species specific, our studies suggest that the events leading to activation of macrophages and innate immune responses may differ between humans and rodents. Careful extrapolation of these effects on human monocytes and the uniqueness of these cells can provide a basis for the understanding of the overall role of CD4 in human immunity. Finally, this understanding could be useful in the development of a therapeutic approach that targets this molecule in monocyte-related disorders and in HIV infection.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by NIH grants RO1 AI078806 (S.G.K.) and R01 AI103385 (J.A.Z.).
Footnotes
Published ahead of print 18 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00616-14.
REFERENCES
- 1.Auffray C, Sieweke MH, Geissmann F. 2009. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu. Rev. Immunol. 27:669–692. 10.1146/annurev.immunol.021908.132557 [DOI] [PubMed] [Google Scholar]
- 2.Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F. 2007. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317:666–670. 10.1126/science.1142883 [DOI] [PubMed] [Google Scholar]
- 3.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. 2010. Development of monocytes, macrophages, and dendritic cells. Science 327:656–661. 10.1126/science.1178331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martinez FO, Helming L, Gordon S. 2009. Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 27:451–483. 10.1146/annurev.immunol.021908.132532 [DOI] [PubMed] [Google Scholar]
- 5.Montoya D, Cruz D, Teles RM, Lee DJ, Ochoa MT, Krutzik SR, Chun R, Schenk M, Zhang X, Ferguson BG, Burdick AE, Sarno EN, Rea TH, Hewison M, Adams JS, Cheng G, Modlin RL. 2009. Divergence of macrophage phagocytic and antimicrobial programs in leprosy. Cell Host Microbe 6:343–353. 10.1016/j.chom.2009.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krutzik SR, Tan B, Li H, Ochoa MT, Liu PT, Sharfstein SE, Graeber TG, Sieling PA, Liu YJ, Rea TH, Bloom BR, Modlin RL. 2005. TLR activation triggers the rapid differentiation of monocytes into macrophages and dendritic cells. Nat. Med. 11:653–660. 10.1038/nm1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krutzik SR, Hewison M, Liu PT, Robles JA, Stenger S, Adams JS, Modlin RL. 2008. IL-15 links TLR2/1-induced macrophage differentiation to the vitamin D-dependent antimicrobial pathway. J. Immunol. 181:7115–7120. 10.4049/jimmunol.181.10.7115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crocker PR, Jefferies WA, Clark SJ, Chung LP, Gordon S. 1987. Species heterogeneity in macrophage expression of the CD4 antigen. J. Exp. Med. 166:613–618. 10.1084/jem.166.2.613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lynch GW, Turville S, Carter B, Sloane AJ, Chan A, Muljadi N, Li S, Low L, Armati P, Raison R, Zoellner H, Williamson P, Cunningham A, Church WB. 2006. Marked differences in the structures and protein associations of lymphocyte and monocyte CD4: resolution of a novel CD4 isoform. Immunol. Cell Biol. 84:154–165. 10.1111/j.1440-1711.2005.01403.x [DOI] [PubMed] [Google Scholar]
- 10.Maddon PJ, Littman DR, Godfrey M, Maddon DE, Chess L, Axel R. 1985. The isolation and nucleotide sequence of a cDNA encoding the T cell surface protein T4: a new member of the immunoglobulin gene family. Cell 42:93–104. 10.1016/S0092-8674(85)80105-7 [DOI] [PubMed] [Google Scholar]
- 11.Ravichandran KS, Collins TL, Burakoff SJ. 1996. CD4 and signal transduction. Curr. Top. Microbiol. Immunol. 205:47–62 [DOI] [PubMed] [Google Scholar]
- 12.Pelchen-Matthews A, da Silva RP, Bijlmakers MJ, Signoret N, Gordon S, Marsh M. 1998. Lack of p56lck expression correlates with CD4 endocytosis in primary lymphoid and myeloid cells. Eur. J. Immunol. 28:3639–3647. 10.1002/(SICI)1521-4141(199811)28:11<3639::AID-IMMU3639>3.0.CO;2-Q [DOI] [PubMed] [Google Scholar]
- 13.Stevenson M. 2003. HIV-1 pathogenesis. Nat. Med. 9:853–860. 10.1038/nm0703-853 [DOI] [PubMed] [Google Scholar]
- 14.Perno CF, Svicher V, Schols D, Pollicita M, Balzarini J, Aquaro S. 2006. Therapeutic strategies towards HIV-1 infection in macrophages. Antiviral Res. 71:293–300. 10.1016/j.antiviral.2006.05.015 [DOI] [PubMed] [Google Scholar]
- 15.Ancuta P, Kunstman KJ, Autissier P, Zaman T, Stone D, Wolinsky SM, Gabuzda D. 2006. CD16+ monocytes exposed to HIV promote highly efficient viral replication upon differentiation into macrophages and interaction with T cells. Virology 344:267–276. 10.1016/j.virol.2005.10.027 [DOI] [PubMed] [Google Scholar]
- 16.Crowe SM, Westhorpe CLV, Mukhamedova N, Jaworowski A, Sviridov D, Bukrinsky M. 2010. The macrophage: the intersection between HIV infection and atherosclerosis. J. Leukoc. Biol. 87:589–598. 10.1189/jlb.0809580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellery P, Tippett E, Chiu Y, Paukovics G, Cameron P, Solomon A, Lewin S, Gorry P, Jaworowski A, Greene W. 2007. The CD16+ monocyte subset is more permissive to infection and preferentially harbors HIV-1 in vivo. J. Immunol. 178:6581–6589. 10.4049/jimmunol.178.10.6581 [DOI] [PubMed] [Google Scholar]
- 18.Jaworowski A, Kamwendo D, Ellery P, Sonza S, Mwapasa V, Tadesse E, Molyneux M, Rogerson S, Meshnick S, Crowe S. 2007. CD16+ monocyte subset preferentially harbors HIV-1 and is expanded in pregnant Malawian women with Plasmodium falciparum malaria and HIV-1 infection. J. Infect. Dis. 196:38–42. 10.1086/518443 [DOI] [PubMed] [Google Scholar]
- 19.Ziegler-Heitbrock L. 2007. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J. Leukoc. Biol. 81:584–592. 10.1189/jlb.0806510 [DOI] [PubMed] [Google Scholar]
- 20.Zhu T, Muthui D, Holte S, Nickle D, Feng F, Brodie S, Hwangbo Y, Mullins JI, Corey L. 2002. Evidence for human immunodeficiency virus type 1 replication in vivo in CD14(+) monocytes and its potential role as a source of virus in patients on highly active antiretroviral therapy. J. Virol. 76:707–716. 10.1128/JVI.76.2.707-716.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McElrath MJ, Pruett JE, Cohn ZA. 1989. Mononuclear phagocytes of blood and bone marrow: comparative roles as viral reservoirs in human immunodeficiency virus type 1 infections. Proc. Natl. Acad. Sci. U. S. A. 86:675–679. 10.1073/pnas.86.2.675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Llewellyn N, Zioni R, Zhu H, Andrus T, Xu Y, Corey L, Zhu T. 2006. Continued evolution of HIV-1 circulating in blood monocytes with antiretroviral therapy: genetic analysis of HIV-1 in monocytes and CD4+ T cells of patients with discontinued therapy. J. Leukoc. Biol. 80:1118–1126. 10.1189/jlb.0306144 [DOI] [PubMed] [Google Scholar]
- 23.Lewin SR, Kirihara J, Sonza S, Irving L, Mills J, Crowe SM. 1998. HIV-1 DNA and mRNA concentrations are similar in peripheral blood monocytes and alveolar macrophages in HIV-1-infected individuals. AIDS 12:719–727. 10.1097/00002030-199807000-00008 [DOI] [PubMed] [Google Scholar]
- 24.Lambotte O, Taoufik Y, de Goer MG, Wallon C, Goujard C, Delfraissy JF. 2000. Detection of infectious HIV in circulating monocytes from patients on prolonged highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 23:114–119. 10.1097/00126334-200002010-00002 [DOI] [PubMed] [Google Scholar]
- 25.Fulcher JA, Hwangbo Y, Zioni R, Nickle D, Lin X, Heath L, Mullins JI, Corey L, Zhu T. 2004. Compartmentalization of human immunodeficiency virus type 1 between blood monocytes and CD4+ T cells during infection. J. Virol. 78:7883–7893. 10.1128/JVI.78.15.7883-7893.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gendelman HE, Orenstein JM, Martin MA, Ferrua C, Mitra R, Phipps T, Wahl LA, Lane HC, Fauci AS, Burke DS, Skillman D, Meltzer MS. 1988. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J. Exp. Med. 167:1428–1441. 10.1084/jem.167.4.1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orenstein JM, Meltzer MS, Phipps T, Gendelman HE. 1988. Cytoplasmic assembly and accumulation of human immunodeficiency virus types 1 and 2 in recombinant human colony-stimulating factor-1-treated human monocytes: an ultrastructural study. J. Virol. 62:2578–2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garaci E, Aquaro S, Lapenta C, Amendola A, Spada M, Covaceuszach S, Perno CF, Belardelli F. 2003. Anti-nerve growth factor Ab abrogates macrophage-mediated HIV-1 infection and depletion of CD4+ T lymphocytes in hu-SCID mice. Proc. Natl. Acad. Sci. U. S. A. 100:8927–8932. 10.1073/pnas.1332627100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Handunnetthi L, Ramagopalan SV, Ebers GC, Knight JC. 2010. Regulation of major histocompatibility complex class II gene expression, genetic variation and disease. Genes Immun. 11:99–112. 10.1038/gene.2009.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thedrez A, de Lalla C, Allain S, Zaccagnino L, Sidobre S, Garavaglia C, Borsellino G, Dellabona P, Bonneville M, Scotet E, Casorati G. 2007. CD4 engagement by CD1d potentiates activation of CD4+ invariant NKT cells. Blood 110:251–258. 10.1182/blood-2007-01-066217 [DOI] [PubMed] [Google Scholar]
- 31.Milia E, Di Somma MM, Majolini MB, Ulivieri C, Somma F, Piccolella E, Telford JL, Baldari CT. 1997. Gene activating and proapoptotic potential are independent properties of different CD4 epitopes. Mol. Immunol. 34:287–296. 10.1016/S0161-5890(97)00050-3 [DOI] [PubMed] [Google Scholar]
- 32.Kitchen SG, Jones NR, LaForge S, Whitmire JK, Vu BA, Galic Z, Brooks DG, Brown SJ, Kitchen CM, Zack JA. 2004. CD4 on CD8+ T cells directly enhances effector function and is a target for HIV infection. Proc. Natl. Acad. Sci. U. S. A. 101:8727–8732. 10.1073/pnas.0401500101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitchen SG, LaForge S, Patel VP, Kitchen CM, Miceli MC, Zack JA. 2002. Activation of CD8 T cells induces expression of CD4, which functions as a chemotactic receptor. Blood 99:207–212. 10.1182/blood.V99.1.207 [DOI] [PubMed] [Google Scholar]
- 34.Brooks DG, Arlen PA, Gao L, Kitchen CM, Zack JA. 2003. Identification of T cell-signaling pathways that stimulate latent HIV in primary cells. Proc. Natl. Acad. Sci. U. S. A. 100:12955–12960. 10.1073/pnas.2233345100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cole SW, Yan W, Galic Z, Arevalo J, Zack JA. 2005. Expression-based monitoring of transcription factor activity: the TELiS database. Bioinformatics 21:803–810. 10.1093/bioinformatics/bti038 [DOI] [PubMed] [Google Scholar]
- 36.Kreutz M, Hennemann B, Ackermann U, Grage-Griebenow E, Krause SW, Andreesen R. 1999. Granulocyte–macrophage colony-stimulating factor modulates lipopolysaccharide (LPS)-binding and LPS-response of human macrophages: inverse regulation of tumour necrosis factor-α and interleukin-10. Immunology 98:491–496. 10.1046/j.1365-2567.1999.00904.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sallusto F, Lanzavecchia A. 1994. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 179:1109–1118. 10.1084/jem.179.4.1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clayton LK, Sieh M, Pious DA, Reinherz EL. 1989. Identification of human CD4 residues affecting class II MHC-versus HIV-1 gp120 binding. Nature 339:548–551. 10.1038/339548a0 [DOI] [PubMed] [Google Scholar]
- 39.Kato M, Neil TK, Fearnley DB, McLellan AD, Vuckovic S, Hart DN. 2000. Expression of multilectin receptors and comparative FITC-dextran uptake by human dendritic cells. Int. Immunol. 12:1511–1519. 10.1093/intimm/12.11.1511 [DOI] [PubMed] [Google Scholar]
- 40.Endemann G, Stanton LW, Madden KS, Bryant CM, White RT, Protter AA. 1993. CD36 is a receptor for oxidized low density lipoprotein. J. Biol. Chem. 268:11811–11816 [PubMed] [Google Scholar]
- 41.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. 2004. Species-specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science 303:1526–1529. 10.1126/science.1093620 [DOI] [PubMed] [Google Scholar]
- 42.Iwasaki A, Medzhitov R. 2004. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5:987–995. 10.1038/ni1112 [DOI] [PubMed] [Google Scholar]
- 43.Baker RG, Hayden MS, Ghosh S. 2011. NF-[kappa]B, inflammation, and metabolic disease. Cell Metab. 13:11–22. 10.1016/j.cmet.2010.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosa A, Ballarino M, Sorrentino A, Sthandier O, De Angelis FG, Marchioni M, Masella B, Guarini A, Fatica A, Peschle C, Bozzoni I. 2007. The interplay between the master transcription factor PU.1 and miR-424 regulates human monocyte/macrophage differentiation. Proc. Natl. Acad. Sci. U. S. A. 104:19849–19854. 10.1073/pnas.0706963104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakayama H, Ishimaru F, Katayama Y, Nakase K, Sezaki N, Takenaka K, Shinagawa K, Ikeda K, Niiya K, Harada M. 2000. Ikaros expression in human hematopoietic lineages. Exp. Hematol. 28:1232–1238. 10.1016/S0301-472X(00)00530-0 [DOI] [PubMed] [Google Scholar]
- 46.Cruikshank WW, Greenstein JL, Theodore AC, Center DM. 1991. Lymphocyte chemoattractant factor induces CD4-dependent intracytoplasmic signaling in lymphocytes. J. Immunol. 146:2928–2934 [PubMed] [Google Scholar]
- 47.Cruikshank WW, Berman JS, Theodore AC, Bernardo J, Center DM. 1987. Lymphokine activation of T4+ T lymphocytes and monocytes. J. Immunol. 138:3817–3823 [PubMed] [Google Scholar]
- 48.Graziani-Bowering G, Filion LG, Thibault P, Kozlowski M. 2002. CD4 is active as a signaling molecule on the human monocytic cell line Thp-1. Exp. Cell Res. 279:141–152. 10.1006/excr.2002.5581 [DOI] [PubMed] [Google Scholar]
- 49.Rao KM. 2001. MAP kinase activation in macrophages. J. Leukoc. Biol. 69:3–10 [PubMed] [Google Scholar]
- 50.Tiwari RL, Singh V, Barthwal MK. 2008. Macrophages: an elusive yet emerging therapeutic target of atherosclerosis. Med. Res. Rev. 28:483–544. 10.1002/med.20118 [DOI] [PubMed] [Google Scholar]
- 51.Grivennikov SI, Karin M. 2010. Dangerous liaisons: STAT3 and NF-[kappa]B collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 21:11–19. 10.1016/j.cytogfr.2009.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu H, Pardoll D, Jove R. 2009. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9:798–809. 10.1038/nrc2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. 1995. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. U. S. A. 92:7686–7689. 10.1073/pnas.92.17.7686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. 1998. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 273:18623–18632. 10.1074/jbc.273.29.18623 [DOI] [PubMed] [Google Scholar]
- 55.Hers I, Tavare JM, Denton RM. 1999. The protein kinase C inhibitors bisindolylmaleimide I (GF 109203x) and IX (Ro 31-8220) are potent inhibitors of glycogen synthase kinase-3 activity. FEBS Lett. 460:433–436. 10.1016/S0014-5793(99)01389-7 [DOI] [PubMed] [Google Scholar]
- 56.Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. 1990. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J. Biol. Chem. 265:5267–5272 [PubMed] [Google Scholar]
- 57.English JM, Cobb MH. 2002. Pharmacological inhibitors of MAPK pathways. Trends Pharmacol. Sci. 23:40–45. 10.1016/S0165-6147(00)01865-4 [DOI] [PubMed] [Google Scholar]
- 58.Alexander I, Edelman ER, Methe H. 2009. Function and mode of regulation of endothelial major histocompatibility complex class II. Cell Transplant. 18:255–259. 10.3727/096368909788534997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McDouall RM, Batten P, McCormack A, Yacoub MH, Rose ML. 1997. MHC-class II expression on human heart microvascular endothelial cells: exquisite sensitivity to interferon-gamma and natural killer cells. Transplantation 64:1175–1180. 10.1097/00007890-199710270-00016 [DOI] [PubMed] [Google Scholar]
- 60.Pober JS, Collins T, Gimbrone MA, Jr, Cotran RS, Gitlin JD, Fiers W, Clayberger C, Krensky AM, Burakoff SJ, Reiss CS. 1983. Lymphocytes recognize human vascular endothelial and dermal fibroblast Ia antigens induced by recombinant immune interferon. Nature 305:726–729. 10.1038/305726a0 [DOI] [PubMed] [Google Scholar]
- 61.Wedgwood JF, Hatam L, Bonagura VR. 1988. Effect of interferon-[gamma] and tumor necrosis factor on the expression of class I and class II major histocompatibility molecules by cultured human umbilical vein endothelial cells. Cell. Immunol. 111:1–9. 10.1016/0008-8749(88)90046-9 [DOI] [PubMed] [Google Scholar]
- 62.Crowe SM, Sonza S. 2000. HIV-1 can be recovered from a variety of cells including peripheral blood monocytes of patients receiving highly active antiretroviral therapy: a further obstacle to eradication. J. Leukoc. Biol. 68:345–350 [PubMed] [Google Scholar]
- 63.Coleman C, Wu L. 2009. HIV interactions with monocytes and dendritic cells: viral latency and reservoirs. Retrovirology 6:51. 10.1186/1742-4690-6-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Collman R, Hassan N, Walker R, Godfrey B, Cutilli J, Hastings J, Friedman H, Douglas S, Nathanson N. 1989. Infection of monocyte-derived macrophages with human immunodeficiency virus type 1 (HIV-1). Monocyte-tropic and lymphocyte-tropic strains of HIV-1 show distinctive patterns of replication in a panel of cell types. J. Exp. Med. 170:1149–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu T, Muthui D, Holte S, Nickle D, Feng F, Brodie S, Hwangbo Y, Mullins J, Corey L. 2002. Evidence for human immunodeficiency virus type 1 replication in vivo in CD14(+) monocytes and its potential role as a source of virus in patients on highly active antiretroviral therapy. J. Virol. 76:707–716. 10.1128/JVI.76.2.707-716.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Conti L, Cardone M, Varano B, Puddu P, Belardelli F, Gessani S. 2008. Role of the cytokine environment and cytokine receptor expression on the generation of functionally distinct dendritic cells from human monocytes. Eur. J. Immunol. 38:750–762. 10.1002/eji.200737395 [DOI] [PubMed] [Google Scholar]
- 67.Mach B, Steimle V, Martinez-Soria E, Reith W. 1996. Regulation of MHC-class II genes: lessons from a disease. Annu. Rev. Immunol. 14:301–331. 10.1146/annurev.immunol.14.1.301 [DOI] [PubMed] [Google Scholar]
- 68.Galkina E, Ley K. 2009. Immune and inflammatory mechanisms of atherosclerosis (*). Annu. Rev. Immunol. 27:165–197. 10.1146/annurev.immunol.021908.132620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Woollard KJ, Geissmann F. 2010. Monocytes in atherosclerosis: subsets and functions. Nat. Rev. Cardiol. 7:77–86. 10.1038/nrcardio.2009.228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Palacios EH, Weiss A. 2004. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 23:7990–8000. 10.1038/sj.onc.1208074 [DOI] [PubMed] [Google Scholar]
- 71.Krautwald S. 1998. IL-16 activates the SAPK signaling pathway in CD4+ macrophages. J. Immunol. 160:5874–5879 [PubMed] [Google Scholar]
- 72.Qi JC, Wang J, Mandadi S, Tanaka K, Roufogalis BD, Madigan MC, Lai K, Yan F, Chong BH, Stevens RL, Krilis SA. 2006. Human and mouse mast cells use the tetraspanin CD9 as an alternate interleukin-16 receptor. Blood 107:135–142. 10.1182/blood-2005-03-1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hsue PY, Deeks SG, Hunt PW. 2012. Immunologic basis of cardiovascular disease in HIV-infected adults. J. Infect. Dis. 205(Suppl. 3):S375–S382. 10.1093/infdis/jis200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hunt PW. 2012. HIV and inflammation: mechanisms and consequences. Curr. HIV/AIDS Rep. 9:139–147. 10.1007/s11904-012-0118-8 [DOI] [PubMed] [Google Scholar]
- 75.Li QJ, Dinner AR, Qi S, Irvine DJ, Huppa JB, Davis MM, Chakraborty AK. 2004. CD4 enhances T cell sensitivity to antigen by coordinating Lck accumulation at the immunological synapse. Nat. Immunol. 5:791–799. 10.1038/ni1095 [DOI] [PubMed] [Google Scholar]
- 76.Parham P. 2009. The immune system, 3rd ed. Garland Science, New York, NY [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.