

Abstract
Background
Maternal smoking during pregnancy (MSDP) is associated with early and long-term neurobehavioral deficits; however mechanisms remain unknown. We tested the hypothesis that MSDP programs the hypothalamic pituitary adrenocortical (HPA) axis of the offspring leading to adverse outcomes. In an intensive, prospective study, we investigated associations between MSDP and infant cortisol stress response and explored whether alterations in cortisol response were mediated by epigenetic modulation of the placental glucocorticoid receptor gene (NR3C1).
Methods
Participants were 100 healthy mother-infant pairs (53% MSDP-exposed; 42% female) from a low income, racially/ethnically diverse sample (55% minorities). MSDP was assessed by timeline followback interview verified by saliva and meconium cotinine. Infant cortisol responses to a neurobehavioral exam were assessed 7 times over the first postnatal month. Methylation of placental NR3C1 promoter exon 1F was assessed using bisulfite pyrosequencing in a subsample (n=45).
Results
MSDP-exposed infants showed significantly and persistently attenuated basal and reactive cortisol levels over the first postnatal month vs. unexposed infants. Exploratory analyses revealed that MSDP was associated with altered methylation of the placental NR3C1 promoter; degree of methylation of the placental NR3C1 was associated with infant basal and reactive cortisol over the first postnatal month and mediated effects of MSDP on infant basal cortisol.
Conclusions
Results provide initial support for our hypothesis that MSDP programs offspring HPA (dys) regulation. Epigenetic regulation of placental GR may serve as a novel underlying mechanism. Results may have implications for delineating pathways to adverse outcomes from MSDP.
Keywords: Smoking, tobacco, pregnancy, infant, cortisol, stress, epigenetic, fetal, programming, methylation, glucocorticoid receptor (GR), placenta, NR3C1
INTRODUCTION
Maternal smoking during pregnancy (MSDP) is one of the most common prenatal substance exposures (Hamilton, 2012; Tong et al., 2013) and a recalcitrant public health problem. Offspring exposed to MSDP are at increased risk for costly and adverse medical outcomes, including low birth weight, preterm delivery, andincreased length of neonatal intensive care unit stays (Shah and Bracken, 2000; Dietz et al., 2010; Adams et al., 2011). Exposure to MSDP has also been implicated in long-term child and adolescent behavioral problems, with the strongest and most consistent links to offspring disruptive behaviors/conduct disorder, attention deficits/ADHD, and smoking/nicotine dependence (Buka et al., 2003; Langley et al., 2005; Wakschlag et al., 2006). Previous work has also revealed effects of MSDP on behavioral dysregulation in the neonatal period, including increased irritability and motor activity, and attention and self-regulation deficits (Law et al., 2003; Stroud et al., 2009; Yolton et al., 2009). However, mechanisms underlying effects of MSDP on early and long-term neurobehavioral deficits remain unclear.
Numerous preclinical and human studies have highlighted the fundamental importance of the fetal environment in “programming” physiologic systems and structures leading to adult health and disease (Gluckman et al., 2008; Xiong and Zhang, 2012). Specifically, dysregulation of offspring hypothalamic pituitary adrenocortical (HPA) axis has been proposed as a final common pathway underlying prenatal programming of adult disease (Meaney et al., 2007; Cottrell and Seckl, 2009). In the present study, we test the hypothesis that exposure to MSDP will be associated with persistent dysregulation, or programming, of the offspring HPA axis over the neonatal period. The plausibility of our hypothesis is supported by preclinical studies showing increased HPA stress response in prenatal nicotine-exposed offspring, and human studies demonstrating increased cord blood cortisol in MSDP-exposed offspring (Poland et al., 1994a; Poland et al., 1994b; Poland et al., 1996; McDonald et al., 2006; Varvarigou et al., 2006; Varvarigou et al., 2009). Further and most relevant, Ramsay et al. (1996) found increased baseline and attenuated response to inoculation at two months in MSDP and low-level alcohol-exposed infants, while Schuetze et al. (2008) showed increased peak cortisol response to stressful behavioral tasks in MSDP-exposed infants at seven months. However, little is known regarding the influence of MSDP on offspring cortisol stress response in the early neonatal period (first postnatal month), despite the importance of this information for identifying the earliest biological pathways to later neurobehavioral deficits and parsing persistent effects of MSDP from acute effects of nicotine or a withdrawal process. Thus, the primary goal of the present study was to investigate the influence of MSDP on the evolution of the cortisol stress response over the first postnatal month.
The enduring nature of offspring HPA dysregulation following perturbations in the prenatal environment has pointed to the plausibility of epigenetic factors as mechanisms underlying prenatal programming (Meaney et al., 2007; Wadhwa et al., 2009). In particular, the hippocampal glucocorticoid receptor (GR) has been proposed as a key candidate for epigenetic modification of offspring HPA regulation following prenatal environmental perturbations (Meaney et al., 2007; Cottrell and Seckl, 2009). In concert with brain mineralocorticoid receptors (MR), GR regulates HPA and behavioral stress response, including initial reactivity and recovery, and memory storage and retrieval (Oitzl et al., 2010). Animal models have revealed persistent alterations in hippocampal GR and HPA hyper-reactivity following exposure to pre and early postnatal stress and in relation to postnatal maternal care/parenting mediated by differential methylation of the promoter region of the hippocampal GR gene (NR3C1, exon 17) (Meaney, 2001; Weaver et al., 2004; Szyf et al., 2007). Novel human studies further highlighted the importance of site-specific methylation of the human analog of the NR3C1 promoter (exon 1F; homologous to rat exon 17) in mediating enduring influences of prenatal and early life adversity on offspring HPA regulation (Oberlander et al., 2008; Tyrka et al., 2012).
The placenta resides at the maternal-fetal interface and serves as a vital source of fetal nutrition and immune-endocrine communication between mother and fetus. Emerging studies highlight the critical and active role of the placenta in supporting fetal brain development through adaptive responses to the maternal environment and protection from environmental insults (Maccani and Marsit, 2009; Bonnin and Levitt, 2011; Zeltser and Leibel, 2011). Epigenetic alterations in the placenta may both mimic and uniquely influence fetal brain and neuroendocrine development. Thus, the secondary goal of this study was to explore links between MSDP and site-specific methylation of the placental NR3C1 promoter (exon 1F), and between NR3C1 promoter methylation and evolution of the cortisol stress response over the first postnatal month. In sum, the present study is a short-term, intensive, prospective study designed to test a prenatal programming hypothesis: a) MSDP is associated with neonatal HPA (dys) regulation, and b) alterations in epigenetic regulation of placental NR3C1 are associated with MSDP exposure and with neonatal HPA (dys) regulation.
METHODS AND MATERIALS
Participants
The Behavior and Mood in Babies and Mothers (BAM BAM) study was an intensive, short-term, prospective study of MSDP and infant neurobehavior and stress response over the first postnatal month. Study eligibility was determined through maternal-report and medical record review. Maternal exclusion criteria included age <18 or >40, illicit drug use besides marijuana (meconium confirmed), current/prior involvement with child protective services prior to birth, serious medical conditions (e.g., pre-eclampsia, severe obesity). Infants were healthy singletons born >36 weeks gestational age (GA). Infants with congenital anomalies or serious medical complications were excluded. Participants provided written informed consent, and followed procedures reviewed and approved by Institutional Review Boards at Women and Infants’ Hospital of Rhode Island and Lifespan Hospitals.
One hundred forty-eight pregnant women aged 18–40 were originally enrolled in the study. Of these, 6 were excluded for regular opiate or cocaine use/positive meconium, 2 for current or prior involvement with child protective services, 5 for serious maternal medical conditions (3 for pre-eclampsia, 1 for severe obesity (BMI=61), 1 for blood clotting disorder),, and 6 for delivery <36 weeks. Ten participants who did not have cortisol response data were excluded from analyses. In order to investigate effects of continuous MSDP exposure, 19 participants who quit smoking prior to 28 weeks gestation (biochemically verified) were also excluded from these analyses.
The final analytic sample (n=100) included 53 smokers and 47 biochemically-verified controls. Participants were assigned to the smoking or control group based on maternal report of cigarette use during at least 2 trimesters of pregnancy and/or positive cotinine bioassay of maternal saliva (≥10 ng/mL) or meconium nicotine markers (≥10 ng/g). A subset of participants (n=45), balanced across smokers (n=22) and controls (n=23) were included in the placenta methylation substudy.
Procedures
Maternal Interviews
Mothers were interviewed prospectively over second and third trimesters of pregnancy and at delivery (M=3 interviews (range 2–4) between 24–42 weeks gestation). At each interview, mothers completed the calendar/anchor-based Timeline Follow Back (TLFB) interview regarding smoking, drug, and alcohol use over pregnancy and three months prior to conception (Robinson et al., 2014). Mothers also completed a socioeconomic status (SES) interview from which education, occupation, income, and Hollingshead four-factor index of SES were extracted (Gottfried, 1985). Mothers also reported on their health and pregnancy history, pre-pregnancy weight, and symptoms of depression (Hamilton, 1960). Maternal caffeine consumption and hours of environmental tobacco smoke (ETS) exposure over pregnancy were also assessed using detailed interviews covering each trimester. Maternal saliva for cotinine verification of smoking status was collected at each interview. Maternal weight was measured in late third trimester (M=35 weeks, SD=1).
Delivery/Birth
Information regarding maternal and infant health and medical conditions was extracted by medical chart review. Mothers and study staff collected diapers containing meconium for 3 days post-birth for verification of MSDP and other drug use. Placental tissue was collected from the n=45 subsample (M=1.2 hours after delivery) for extraction of DNA for NR3C1 methylation analysis.
Infant Stress Response Assessment over the First Postnatal Month
Cortisol stress response was elicited using the NICU Network Neurobehavioral Scale (NNNS) (Lester et al., 2004; Tronick and Lester, 2013) administered by certified examiners blind to MSDP status. The NNNS is a comprehensive evaluation of neurobehavioral performance including observation, neurologic and behavioral components, and exposure to auditory, visual, social and non-social stimuli (Lester et al., 2004; Tronick and Lester, 2013). The NNNS lasts approximately 30 minutes and involves mild stress as the infant is observed and handled during periods of sleep, wake, crying, and non-crying states. The NNNS was administered up to 7 times (Med=7, M=6) over the first postnatal month at days 0 (M=8 hours), 1, 2, 3–4, 5, 12, and 32. One hundred percent of day 0 and 1 NNNS were administered in the hospital, 54% of day 2–4 NNNS were administered in the hospital; 100% of day 5–32 NNNS were conducted at homes. Four saliva samples were collected for cortisol during and after the NNNS (baseline, end of NNNS, 20 and 40 minutes post-NNNS) using the sorbette sampling device (Salimetrics LLC, State College, Pennsylvania). Saliva samples were also assayed for cotinine to determine infant exposure to nicotine via ETS/breast milk.
Bioassays
Saliva cotinine is a reliable biomarker for nicotine levels (Jarvis et al., 1987). Maternal and infant saliva samples were frozen until analysis by Salimetrics LLC using highly-sensitive enzyme immunoassay (HS EIA). Intra and inter-assay coefficients of variation were 6.4% and 6.6% respectively.
Meconium analysis reflects cumulative substance use over primarily the third trimester (Ostrea, 1999; Gray et al., 2009b). Diapers were collected for 3 days after birth with meconium samples from multiple soiled diapers combined until the appearance of milk stool. Samples were frozen at −80° C until analysis. Meconium was analyzed for nicotine markers (nicotine, cotinine, trans-3′-hydroxycotinine), cannabinoid markers (Δ9-tetrahydrocannabinol (THC), 11-nor-9-carboxy-THC, cannabinol, 11 hydroxy THC, di-OH-THC) opiates, cocaine, and amphetamines via EMIT screens, tandem liquid chromatography mass spectrometry or gas chromatography mass spectroscopy confirmation (Moore et al., 1998; Gray et al., 2009b). Samples from all participants in the final sample were negative for cocaine, opiates, and amphetamines. Based on precedent in the field and sensitivity of the assays, samples with nicotine markers ≥10 ng/g or cannabinoid markers ≥40 ng/g were considered positive for nicotine and cannabinoids, respectively (Gray et al., 2009a).
Saliva cortisol provides a reliable, non-invasive estimate of free (unbound) cortisol (Gunnar et al., 1985). Following collection, infant saliva samples were frozen until analysis by expanded range HS-EIA by Salimetrics. Intra and inter-assay coefficients of variation were 4.6% and 6%, respectively.
Placental NR3C1 promoter methylation
Placental tissue free from maternal decidua was excised and placed immediately in RNAlater solution (Life Technologies, Grand Island, NY) then stored at 4° C. At least 72 hours later, placenta samples were removed from RNAlater, blotted dry, snap-frozen in liquid nitrogen, pulverized to homogeneity, and stored at −80° C until analysis. Placental genomic DNA was extracted using the QIAmp DNA Mini kit (Qiagen, Inc.), and assessed for quantity and quality using a ND-1000 Spectophotometer (Nanodrop, DE). DNA samples were sodium bisulfite modified using the EZ DNA Methylation Kit (Zymo Research, CA). Extent of methylation at the NR3C1 promoter region was examined with quantitative pyrosequencing (Dupont et al., 2004) following methods of Oberlander et al. (2008) using the PyroMark MD Pyrosequencing System. The region analyzed contains 13 CpGs and encompasses exon 1F (human homologue of rat exon 17). Reactions were performed in triplicate; averaged, and SDs for individual sites calculated. Any sample with SD>3% was re-analyzed. Sodium bisulfite-modified, fully-methylated referent positive control and fully-unmethylated (whole genome amplified) negative control DNA (Qiagen, Valencia, CA) was examined with each batch.
Potential Covariates
A number of potential maternal, perinatal, and neonatal covariates were tested for associations with cortisol outcomes (baseline and area under the curve (AUC)) in multivariate models. Maternal demographic information: age, race/ethnicity (% Non-Hispanic White), Hollingshead SES (low SES ≥ 4), weight gain over pregnancy (pre-pregnancy to 35±1 weeks); and pregnancy history: gravida, parity, were assessed by maternal report. Maternal medical conditions, e.g., gestational hypertension, gestational diabetes and medications: steroids, antidepressants were determined by maternal report and medical chart review at delivery. Maternal depression was assessed by structured interview using the 21-item Hamilton Depression Rating Scale (Hamilton, 1960). Maternal alcohol use was assessed through TLFB interview (Robinson et al., 2014); maternal cannabis use was assessed by TLFB interview and/or meconium. Maternal caffeine use and environmental tobacco smoke (ETS) exposure were assessed by structured interview; infant ETS exposure was assessed by infant saliva cotinine levels. Neonatal characteristics: sex, delivery mode, GA at birth, small for GA (SGA; birth weight <10th percentile for GA), Apgar were assessed by medical chart review.
Statistical analysis
Group comparisons (smokers vs. controls) for demographics, pregnancy history, and infant characteristics utilized t-, chi-square, Mann-Whitney and Fisher’s tests, as appropriate. A trapezoidal rule was applied to the four cortisol measures taken at each infant exam to produce an integrated cortisol response measure (area under the curve; AUC (Pruessner et al., 2003)). Both baseline and AUC cortisol were transformed to the logarithmic scale for statistical analyses.
Primary, full-sample analyses (n=100)
Effects of MSDP on infant cortisol response (baseline and AUC) over the first postnatal month were investigated using longitudinal regression modeling with MSDP as a between-subjects factor and baby age (modeled in hours) as a within-subjects factor. The MSDP by baby age interaction was also tested. Cortisol measures were analyzed jointly over time via generalized least squares in Splus 8.2 (TIBCO Spotfire, 2010), assuming heteroscedastic variances by MSDP group and a continuous AR(1) correlation structure. AUC analyses were corrected for differences in baseline cortisol via covariate adjustment. Although infant age was modeled in hours and baseline and AUC cortisol transformed to the logarithmic scale for statistical analyses, results in Figures 1a and 1b are presented by day of life and with raw mean cortisol values for ease of visual display. Potential covariates (above) were evaluated for associations with cortisol outcomes in models that included time since feeding (time-varying covariate) and infant age at NNNS, as well as baseline cortisol for AUC-specific analyses. Those showing significant association with cortisol outcome (p<.05) were included in multivariate models: GA at birth and maternal regular caffeine use for baseline cortisol; GA and regular alcohol use for cortisol AUC. Offspring gender was not associated with basal or AUC cortisol.
Figure 1.

Figure 1a. Evolution of the Cortisol Stress Response over the First Month in Exposed and Unexposed Infants
Note. Cortisol stress response to the NICU Network Neurobehavioral Scale (NNNS) was assessed up to 7 times over the first postnatal month at days 0 (M=8 hours), 1, 2, 3–4, 5, 12, and 32. Four saliva samples were collected for cortisol during and after the NNNS (baseline, end of NNNS, 20 and 40 minutes following the NNNS). Although age was modeled in hours and baseline and area under the curve (AUC) cortisol in the logarithmic scale for statistical analyses, results are presented by day of life and with raw mean cortisol values for ease of visual display.
Figure 1b. Magnified view of Cortisol Stress Response at Days 3–4 and Day 32 in Exposed and Unexposed Infants.
Secondary, subsample epigenetic analyses (n=45)
Mann-Whitney tests were utilized to determine effects of MSDP on methylation of the NR3C1 promoter across all CpG sites. Associations between NR3C1 and baseline and AUC cortisol over time were investigated utilizing longitudinal regression modeling with and without adjustment for MSDP. Alpha was set to .10 in longitudinal regression models due to the decreased sample size. CpG3 and CpG4 sites were the focus of analyses based on findings of Oberlander et al. (2008) and preclinical studies highlighting CpG3 and CpG4 as key NGFI-A binding sites and homologous to regions differentially methylated by maternal care (Weaver et al., 2004). A causal steps approach (Baron and Kenny, 1986) was utilized to explore NR3C1 methylation as a mediator of the MSDP/cortisol link, requiring significant associations between: a) predictor (MSDP) and outcome (cortisol), b) predictor and mediator (NR3C1 methylation), c) mediator and outcome adjusted for the predictor, and d) attenuation of the predictor-outcome association when the putative mediator is included in the model.
RESULTS
Sample Characteristics
The final analytic sample comprised of 100 mothers (mean age 25, SD=5) and their healthy infant offspring (singleton births; 42% female; ages 0–43 days. The sample was primarily low socioeconomic status (57% had a high school education or less, 59% was unemployed, and 46% had an annual income <$20,000), and racially/ethnically diverse (55% minorities: 20% African American, 23% Hispanic, and 12% Multiracial/Other; 45% Non-Hispanic White). Seventy-seven percent of pregnancies were unplanned; 72% of mothers were unmarried. The 53 smokers reported smoking an average of 9 (SD=6) cigarettes per day across pregnancy, with averages of 12 (SD=7), 8 (SD=6), and 7 (SD=5) cigarettes per day during first, second, and third trimesters, respectively. Mean saliva cotinine levels over maternal perinatal interviews ranged from 77–131 ng/ml (SD’s =66–130). Average gravida was 3 (SD=2); average parity was 1 (SD=1). Infants ranged in age from 0–43 days over the course of the study; 42% were female. Average gestational age was 39.5 weeks (SD=1.3); average birth weight was 3333 grams (SD=454). Average Apgar score at 5 minutes was 9. Descriptive statistics for the overall sample and stratified by smoking group are presented in Table 1. A greater percentage of mothers who smoked were low SES (58% vs. 24%, p<.01). Pregnant smokers were also more likely to use marijuana (13% vs. 0%, p<.05) and caffeine (42% vs. 4%, p<.01) and reported more depressive symptoms (M (SD)’s=6 (6) vs. 2 (3), p<.01). Smokers were also exposed to greater ETS (M (SD)’s=25 (24) vs. 1 (3) hours per week, p<.001). Smokers and controls did not differ significantly in age, race/ethnicity, parity, or weight gain. MSDP-exposed infants were less likely to be breast-fed (40% vs. 72%, p<.01) and showed increased saliva cotinine levels (M (SD)’s=14 (23) vs. 1 (2) ng/ml, p<.01). Exposed and unexposed infants did not differ significantly by gender, GA at birth, SGA, or delivery mode.
Table 1.
Maternal and infant characteristics by smoking group and full sample
| Smokers (n=53) Mean (SD)/ % |
Controls (n=47) Mean (SD)/ % |
Total (n=100) Mean (SD)/ % |
|
|---|---|---|---|
| Maternal Characteristics | |||
| Maternal age (years) | 25 (4) | 25 (5) | 25 (5) |
| Race/Ethnicity (% Non-Hispanic White) | 49% | 41% | 45% |
| Low SES1 | 58% | 24% | 42%** |
| Parity | 1 (2) | 1 (1) | 1 (1) |
| Weight gain (pounds)2 | 28 (17) | 32 (16) | 30 (16) |
| Alcohol Use (>1 drink/week) | 6% | 0% | 3% |
| Marijuana Use (>1 joint/week or meconium)3 | 13% | 0% | 7%* |
| ETS Exposure (hours per week)4 | 25 (24) | 1 (3) | 14 (21)*** |
| Caffeine Use (>200 mg/day caffeine)5 | 42% | 4% | 24%** |
| Gestational Medical Condition6 | 21% | 6% | 14%* |
| Maternal Depressive Symptoms7 | 6 (6) | 2 (3) | 4 (5)** |
|
| |||
| Infant Characteristics | |||
| Sex (% male) | 53% | 64% | 58% |
| Delivery Mode (% vaginal delivery) | 77% | 77% | 77% |
| Gestational age at birth (weeks) | 39 (1) | 40 (1) | 39 (1) |
| Small for gestational age8 | 6% | 2% | 4% |
| Apgar score, 5 minutes | 9 (.28) | 9 (.36) | 9 (.31) |
| Any breast-feeding | 40% | 79% | 58%** |
| ETS Exposure: saliva cotinine (ng/ml)9 | 14.2 (23.4) | .98 (2) | 7.9(3)** |
NOTE:
p<.05;
p<.01,
p<.001.
Based on a score of 4 or 5 on the Hollingshead Index.
Weight gain in pounds between pre-pregnancy and 35±1 weeks.
>1 joint per week or meconium positive for marijuana metabolites.
Hours of ETS exposure per week measured by structured interview.
Equivalent of 2 cups of coffee per day.
e.g., gestational hypertension, gestational diabetes.
Score on 21-item Hamilton Depression Rating Scale.
Birthweight <10th percentile for gestational age.
Environmental Tobacco Smoke (ETS) exposure measured by infant saliva cotinine (ng/ml).
Primary Analyses: Effects of MSDP on evolution of cortisol stress response over the first postnatal month
Baseline Cortisol
Effects of MSDP on trajectories of infant baseline cortisol are shown in Table 2 and Figures 1a and 1b. Baseline cortisol (modeled in the logarithmic scale) showed a linear decline with the logarithm of infant age (β=−.226, t=−8.020, p<.001), which translated to a 77% decline in baseline cortisol level over the first postnatal month. Further, after entry of relevant covariates (time since feeding, GA at birth, maternal caffeine), a significant effect of MSDP group was detected with exposed infants showing 23% attenuated baseline cortisol vs. unexposed infants (β=−.258, t=−3.031, p<.01) across the first postnatal month. No significant interaction emerged between exposure group and infant age (p=.787), suggesting patterns of group differences were not altered over time.
Table 2.
Effects of maternal smoking during pregnancy (MSDP) on infant baseline and Area Under the Curve (AUC) cortisol over the first month of life (N=100)
| Predictor variables | Baseline Cortisol1
|
AUC Cortisol1
|
||||
|---|---|---|---|---|---|---|
| B | SE | p-value | B | SE | p-value | |
| Intercept | .992 | .262 | <.001 | 3.879 | .284 | <.001 |
| Time since feeding (min)1 | −.192 | .047 | <.001 | .187 | .047 | <.001 |
| GA at birth (weeks)2 | −.064 | .032 | .049 | −.071 | .032 | .029 |
| Caffeine use3 | .225 | .097 | .021 | - | - | - |
| Alcohol use4 | - | - | - | .458 | .232 | .049 |
| Baseline cortisol (linear)1 | - | - | - | .674 | .067 | <.001 |
| Baseline cortisol (quadratic)1 | - | - | - | .178 | .031 | <.001 |
| Infant age (hours)1 | −.226 | .028 | <.001 | −.148 | .031 | <.001 |
| MSDP | −.258 | .085 | .003 | −.277 | .081 | <.001 |
Baseline and AUC cortisol, time since feeding, and infant age were natural log transformed.
Gestational age was centered at the mean.
0:≤200 mg/day; 1:>200 mg/day.
0:≤1 drink per week, 1:>1 drink per week.
Area under the curve (AUC)
Effects of MSDP on trajectories of infant cortisol AUC are also shown in Table 2 and Figures 1a and 1b. Similar to baseline cortisol, cortisol reactivity (modeled in the logarithmic scale) showed a linear decline with the logarithm of infant age (β=−.148, t=−4.822, p<.001), translating to a 62% decline in cortisol reactivity over the first month. Further, after entry of relevant covariates (time since feeding, GA at birth, maternal alcohol) and baseline cortisol, a significant effect of MSDP group was detected with exposed infants showing 28% attenuated cortisol reactivity vs. unexposed infants (β=−.277, t=−3.427, p<.001) across the first month. No significant interaction emerged between exposure group and infant age (p=.772), suggesting patterns of group differences were not altered over time.
Secondary Analyses: Role of NR3C1 promoter methylation in associations between MSDP and evolution of the infant cortisol stress response
MSDP and NR3C1 methylation
Figure 2 shows effects of MSDP on placental NR3C1 methylation across all CpG sites. MSDP was associated with attenuated methylation at CpG 3 and CpG 4 (Z’s=2.032 and 2.639; p’s<.05), equivalent to a 29% and 75% attenuation of methylation, respectively, in exposed vs. unexposed offspring.
Figure 2.

Methylation of the Placental NR3C1 Promoter in Exposed and Unexposed Infants Note. *p<.05; **p<.01.
A series of longitudinal regression models are presented in Table 3: a) effects of MSDP on infant baseline and AUC cortisol in the subsample (Model 1), b) effects of methylation of NR3C1 promoter CpG 3 and 4 on baseline and AUC cortisol (Model 2), and c) effects of methylation of NR3C1 promoters CpG 3 and 4 and of MSDP on baseline and AUC cortisol (Model 3).
Table 3.
Role of NR3C1 promoter methylation in associations between MSDP and evolution of infant baseline and AUC cortisol over the first month of life (N=45).
| Predictor variables | Model 11
|
Model 22
|
Model 33
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| β | SE | p | β | SE | p | β | SE | P | |
| Baseline Cortisol | |||||||||
| Intercept | .559 | .403 | .167 | .331 | .410 | .421 | .396 | .414 | .340 |
| Time since feeding (min)4 | −.164 | .068 | .017 | −.169 | .068 | .013 | −.161 | .068 | .019 |
| GA at birth (weeks)5 | −.012 | .051 | .808 | −.034 | .053 | .515 | −.039 | .053 | .463 |
| Caffeine use6 | .396 | .150 | .009 | .235 | .148 | .114 | .302 | .158 | <.001 |
| Infant age (hours)4 | −.155 | .044 | <.001 | −.160 | .044 | <.001 | −.158 | .044 | <.001 |
| MSDP | −.223 | .123 | .071 | - | - | - | −.155 | .132 | .241 |
| NR3C1 methylation CpG 3 | - | - | - | .105 | .046 | .024 | .089 | .048 | .064 |
| NR3C1 methylation CpG 4 | - | - | - | .008 | .022 | .731 | .002 | .023 | .933 |
|
| |||||||||
| AUC Cortisol | |||||||||
| Intercept | 4.104 | .379 | <.001 | 3.672 | .389 | <.001 | 3.951 | .392 | <.001 |
| Time since feeding (min)4 | .149 | .063 | .019 | .137 | .064 | .035 | .149 | .063 | .020 |
| GA at birth (weeks)5 | −.094 | .045 | .036 | −.078 | .046 | .090 | −.102 | .045 | .026 |
| Alcohol use7 | .470 | .347 | .178 | .351 | .353 | .321 | .494 | .349 | .158 |
| Baseline cortisol (linear)4 | .644 | .083 | <.001 | .656 | .085 | <.001 | .637 | .084 | <.001 |
| Baseline cortisol (quadratic)4 | .191 | .042 | <.001 | .205 | .044 | <.001 | .194 | .043 | <.001 |
| Infant age (hours)4 | −.143 | .042 | <.001 | −.143 | .043 | <.001 | −.147 | .042 | <.001 |
| MSDP | −.376 | .106 | <.001 | - | - | - | −.364 | .117 | .002 |
| NR3C1 methylation CpG 3 | - | - | - | .085 | .048 | .079 | .049 | .049 | .318 |
| NR3C1 methylation CpG 4 | - | - | - | .008 | .022 | .698 | −.010 | .022 | .655 |
Model with MSDP, but not NR3C1 methylation.
Model with NR3C1 methylation but not MSDP.
Model with NR3C1 methylation and MSDP.
Natural log transformed.
Centered at the mean.
0:≤200 mg/day, 1:>200 mg/day.
0:≤1 drink per week, 1:>1 drink per week.
NR3C1 methylation and baseline cortisol
As shown in Model 1 of Table 3, MSDP was associated with 20% attenuated baseline cortisol over the first postnatal month after entry of relevant covariates (time since feeding, GA, caffeine; β=−.223, t=−1.815, p=.071). In Model 2, NR3C1 CpG 3 methylation was significantly associated with attenuated baseline cortisol over the first postnatal month after entry of relevant covariates (β=.105, t=2.273, p=.024), with each one-percentile drop in methylation associated with a 10% decrease in baseline cortisol. Finally, as shown in Figure 3, when both NR3C1 CpG 3 and MSDP were included in Model 3, the MSDP coefficient was deflated (β=−.155, t=−1.175, p=.241) while the NR3C1 CpG 3 coefficient (β=.089, t=1.861, p=.064) approached statistical significance. Results were consistent with 14.4% attenuation in baseline cortisol as a direct effect of MDSP and 4.9% attenuation in baseline cortisol via the NR3C1 CpG 3 pathway (change in methylation equivalent to mean difference between exposure groups). Thus, NR3C1 CpG3 methylation mediated approximately 25% of the effects of MSDP on baseline cortisol. NR3C1 CpG 4 methylation did not add significantly to Models 2 or 3 for baseline cortisol.
Figure 3.
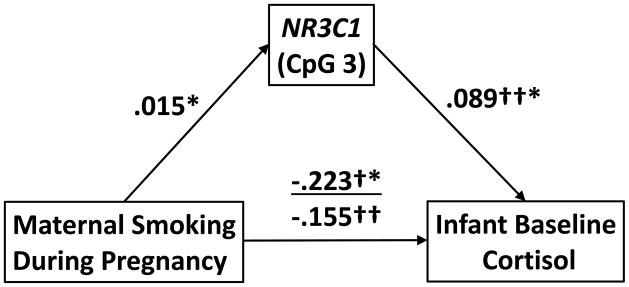
Placental NR3C1 Promoter Methylation Partially Mediates Associations between maternal smoking during pregnancy (MSDP) and Evolution of the Infant Baseline Cortisol. Note. *p<.05; †Model without NR3C1 CpG3; ††Model with NR3C1 CpG3. Path coefficients from MSDP and NR3C1 CpG 3 to baseline/AUC cortisol were derived from longitudinal regression models; coefficient for the MSDP to NR3C1 CpG 3 path corresponds to differences in average methylation levels between smoking groups.
NR3C1 methylation and AUC cortisol
As shown in Model 1 of Table 3, MSDP was associated with 31.3% decrease in AUC cortisol over the first postnatal month after entry of relevant covariates (time since feeding, GA, alcohol; β=−.376, t=−3.540, p<.001). Furthermore, in Model 2, each one-percentile drop in NR3C1 CpG 3 methylation was associated with a 8.1% decrease in infant cortisol reactivity over the first month after entry of relevant covariates and baseline cortisol (β=.085, t=1.764, p=.079). Finally, when both NR3C1 CpG 3 and MSDP were included in Model 3, effects of MSDP remained significant (β=−.364, t=−3.108, p=.002), but effects of NR3C1 CpG 3 methylation were deflated (β=.049, t=1.001, p=.318), suggesting no evidence for mediation of MSDP effects on AUC cortisol. NR3C1 CpG 4 methylation did not add significantly to Models 2 or 3 for cortisol AUC.
In sum, exploratory methylation analyses suggest associations between methylation of the placental NR3C1 promoter and attenuated infant baseline cortisol and cortisol reactivity, and further suggest that methylation of the NR3C1 promoter may serve as a mediator of effects of MSDP on baseline cortisol.
DISCUSSION
The present study reveals persistent associations between MSDP and infant basal and reactive cortisol response over the first postnatal month. Our data provide suggestive evidence in humans for prenatal programming of offspring stress response by MSDP. These findings point to offspring HPA dysregulation as one potential common pathway leading to long-term adverse outcomes from MSDP. Our results also indicate large maturational declines in both baseline and stress-responsive cortisol over the first postnatal month. Finally, the present study reveals preliminary evidence for associations between site-specific methylation (exon 1F) of the NR3C1 promoter in the placenta and infant basal and reactive cortisol response over the first postnatal month. Exploratory analyses suggest that methylation of the placental NR3C1 promoter mediates effects of MSDP on infant basal cortisol.
One key strength of the study is the detailed, prospective phenotypic characterization of mothers and infants in our sample. MSDP was measured prospectively with two types of biochemical verification, (non) use of illicit drugs was also biochemically confirmed, key covariates were measured by detailed interview and/or medical chart review, infant baseline and stress responsive cortisol were measured seven times over the first postnatal month, including 4 saliva samples at each time point, and placental samples for methylation analyses were collected and preserved immediately following delivery. Additional strengths include the low-income, racially/ethnically diverse sample including the control group, and consistent tobacco exposure in our smoking group (at least 2 trimesters of pregnancy, average ½ pack per day smoking level).
Exposure to MSDP was associated with a twenty-three percent attenuation of baseline cortisol and a twenty-eight percent attenuation of cortisol stress response over the first month. Effects of MSDP on offspring baseline and reactive cortisol remained consistent over the first postnatal month; we found no significant interactions between MSDP and infant maturation in influencing infant cortisol. Given the short half-life of nicotine (approximately 2 hours), this finding is consistent with persistent neurotoxic effects of MSDP rather than acute effects of nicotine/tobacco or a withdrawal process. Results are supportive of the hypothesis that MSDP “programs” the fetal HPA axis, resulting in persistent dysregulation of offspring HPA axis/stress response over the neonatal period. Results complement prior findings of attenuated cortisol response to inoculation at two months (Ramsay et al., 1996), but contrast with findings of increased cortisol response to behavioral stressors at seven months (Schuetze et al., 2008) in MSDP-exposed infants. Reversal in direction of MSDP effects between one to two months (present study and Ramsay et al., 1996) and seven months (Schuetze et al., 2008) may be related to normative maturational shifts in adaptive stress response patterns over the first year. Specifically, prior normative studies have shown a more robust response in the first four months, but decreasing cortisol stress response over the remainder of the first year (Tarullo and Gunnar, 2006; Gunnar et al., 2009). Future longitudinal studies of MSDP are needed to further elucidate effects of MSDP on trajectories of cortisol response over the first year.
Our study also highlighted maturational influences on trajectories of baseline and reactive cortisol over the first month. Collapsing across MSDP groups, we found a 77 percent decrease in baseline cortisol and a 62 percent decrease in stress responsive cortisol over the first postnatal month. Prior primarily cross-sectional studies of cortisol regulation in human infants have highlighted decreasing cortisol response to stress over the first year despite equivalent behavioral responses to stress. It has been proposed that this period of stress response quiescence is analogous to the stress hyporesponsive period in rodents, which is believed to protect the brain from the deleterious effects of steroid exposure (Tarullo and Gunnar, 2006; Gunnar et al., 2009). However, the trajectory of infant cortisol stress response over the immediate neonatal period was previously unclear. Our results suggest continuing decrease in baseline and stress responsive cortisol over the first month. Given the profound importance of early life experience in influencing risk for adult psychiatric disease, future intensive longitudinal studies of stress response trajectories are needed to determine key inflection points and potential points of vulnerability for more enduring influences of environmental adversity and interventions.
Secondary analyses revealed that MSDP was associated with 29%–75% attenuated methylation within two CpG sites (3 and 4) of the placental NR3C1 promoter--sites previously shown to be differentially methylated in studies of prenatal adversity and homologous to key sites from preclinical models (Weaver et al., 2004; Oberlander et al., 2008). Results highlight effects of MSDP on epigenetic regulation of placental glucocorticoid passage and complement a small number of prior studies demonstrating effects of MSDP on global and site-specific methylation of the placental epigenome (Suter et al., 2010; Suter and Aagaard, 2012; Maccani et al., 2013). Results also extend studies of prenatal depression and adversity, in which maternal stress/depression was associated with altered methylation of NR3C1 in cord or peripheral blood, to prenatal substance exposure and a focus on placental NR3C1 methylation (Mulligan et al 2012; Oberlander et al 2008; Radtke et al 2011). In contrast to associations between prenatal adversity/depression and increasedNR3C1 methylation in prior studies (Oberlander et al., 2008; Conradt et al., 2013), here, MSDP was associated with attenuated NR3C1 methylation. Differences in direction of effects between studies may be due to our focus on placental NR3C1, where increased methylation leading to decreased plasticity of GR gene expression might be a compensatory mechanism to protect the fetus from glucocorticoid overload, whereas increased methylation and increased GR plasticity might be more adaptive in offspring tissue (Bromer et al., 2013). Additional studies of MSDP and placental NR3C1 methylation are needed to replicate results reported here.
For each one-percentile drop in placental NR3C1 promoter methylation, we found a 10% decrease in baseline cortisol and an 8% decrease in cortisol reactivity over the first postnatal month. Results corroborate findings by Oberlander et al. (2008) showing positive associations between methylation of cord blood NR3C1 and infant cortisol reactivity at 3 months in relation to exposure to maternal depression. Our study highlights the importance of epigenetic regulation of placentalNR3C1 in programming of offspring HPA (dys) regulation. In addition, results suggest that small absolute differences/variability in NR3C1 methylation due to maternal characteristics have meaningful functional consequences in their effects on offspring cortisol (Oberlander et al., 2008; Conradt et al., 2013). Mechanistic pathways underlying links between placental NR3C1 methylation and altered cortisol stress response have not been fully elucidated. Our group has shown inverse associations between NR3C1 methylation and GR expression in human placenta and placental cell lines (Filiberto et al., 2011; Bromer et al., 2013). Thus, it could be speculated that attenuated methylation would lead to increased placental GR expression leading to increased sensitivity to glucocorticoid overload in the placenta. We could further speculate increased fetal exposure to glucocorticoids and altered plasticity of fetal hippocampal GR expression would result in altered set point of the fetal/neonatal HPA axis (Xiong and Zhang, 2012). Future studies are needed to delineate components and direction of molecular pathways between placental NR3C1 methylation, placental and fetal GR expression, and infant cortisol response.
Finally, we found preliminary evidence that methylation of placental NR3C1 partially explains associations between MSDP and altered infant basal HPA regulation. Results highlight the importance of glucocorticoid regulation pathways in mediating adverse influences of MSDP on infant outcomes and highlight a potential role for placental NR3C1 in mediating long-term influences of MSDP. Although alterations in methylation of NR3C1 were significantly and positively associated with both basal and reactive cortisol over the first month, NR3C1 methylation emerged as a mediator only for links between MSDP and infant basal cortisol, not between MSDP and reactive cortisol. Despite their preliminary nature, results highlight the possibility of differing pathways linking MSDP to infant basal versus reactive cortisol. Future work is needed to elucidate the mechanistic role of glucocorticoid versus additional pathways in explaining links between MSDP and short and long-term adverse outcomes.
We acknowledge a number of limitations of our study that point to directions for future research. First, a key limitation of the study is that smokers and controls differed on a number of characteristics including socioeconomic status, gestational medical conditions, and other substance use. All confounders were tested statistically; however, it is possible that programming effects are not due solely to MSDP. In fact, we believe prenatal programming of offspring stress response via epigenetic modulation of placental NR3C1 represents an underlying pathway common to numerous perinatal substance and environmental exposures. Second, by design, our study selected for healthy infants (4% small for gestational age, 39 weeks average gestational age) in order to attribute alterations in offspring stress response to prenatal exposure rather than birth complications. However, given associations between small for gestational age (SGA) and diminished placental function and reserves, it is possible that the MSDP-exposed infants in the present study are those who have shown some degree of placental compensation for MSDP-related effects. If this is the case, we might speculate that effects of MSDP on placental epigenetic modifications would be more profound in samples including both SGA/preterm births. Future studies including both healthy and SGA/premature births are needed to investigate this question.
Third, epigenetic analyses were conducted on only a subsample of participants. Although epigenetic regulation of NR3C1 was associated with clinically significant decreases in baseline and stress-responsive cortisol, sample sizes were small and alphas were set to .10. Replication of these effects in a larger sample is needed. Relatedly, we did not investigate associations between NR3C1 methylation and GR gene expression in the present sample. Finally, the present study was not powered to investigate interactions with offspring gender. Given that offspring gender has been an important modifier of both MSDP and glucocorticoid programming effects (Owen et al., 2005; Cornelius and Day, 2009; Slotkin et al., 2010), future studies designed to investigate sex-specific programming effects of MSDP are needed.
Conclusions
Exposure to MSDP was associated with consistent attenuation of infant baseline and stress responsive cortisol over the first postnatal month. Exploratory analyses revealed that associations between MSDP and infant baseline cortisol were mediated by altered epigenetic regulation of the placental glucocorticoid receptor promoter (NR3C1). This study provides supporting evidence for prenatal programming of offspring HPA regulation by MSDP. Results have implications for delineating pathways to long-term adverse outcomes from MSDP.
Acknowledgments
ROLE OF THE FUNDING SOURCE
Preparation of this manuscript was supported by the National Institutes of Health (R01 DA019558 and R01 DA031188 to L.R.S.) and the Flight Attendant Medical Research Institute Clinical Innovator Award to L.R.S. The funding agencies had no further role in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
We are grateful to mothers and infants who contributed to this study, and to the Maternal-Infant Studies Laboratory staff for their assistance with data collection, and to Polly Gobin for her assistance with data management. We are also grateful to Cheryl Boyce and Nicolette Borek, Program Officers, for their support of this work and this field.
Footnotes
CONFLICT OF INTEREST
The authors have no biomedical financial interests or potential conflicts of interest.
CONTRIBUTORS
Dr. Stroud designed the study, obtained funding, drafted the manuscript, and contributed to analysis and interpretation of data. Dr. Papandonatos undertook the statistical analysis, contributed to interpretation of data, and conducted critical revision of the manuscript. Dr. Rodriguez conducted statistical analyses and assisted in drafting and conducting critical revision of the manuscript. Ms. McCallum conducted statistical analyses and data collection and edited the manuscript. Dr. Salisbury contributed to the study design and data collection and edited the manuscript. Dr. Phipps contributed to the study design and sample recruitment and edited the manuscript. Dr. Lester contributed to the study design and edited the manuscript. Dr. Huestis conducted the meconium assays and conducted critical revision of the manuscript. Dr. Niaura contributed to the study design and edited the manuscript. Dr. Padbury contributed to the study design and data collection, and edited the manuscript. Dr. Marsit contributed to the study design and literature review, oversaw the epigenetic analyses, and edited the manuscript. All authors have contributed to and approved the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams EK, Melvin CL, Raskind-Hood C, Joski PJ, Galactionova E. Infant Delivery Costs Related to Maternal Smoking: An Update. Nic Tobac Res. 2011;13:627–637. doi: 10.1093/ntr/ntr042. [DOI] [PubMed] [Google Scholar]
- Baron RM, Kenny DA. The moderator-mediator variable distinction in social psychological research: conceptual, strategic, and statistical considerations. J Pers Soc Psychol. 1986;51:1173–1182. doi: 10.1037//0022-3514.51.6.1173. [DOI] [PubMed] [Google Scholar]
- Bonnin A, Levitt P. Fetal, maternal, and placental sources of serotonin and new implications for developmental programming of the brain. Neuroscience. 2011;197:1–7. doi: 10.1016/j.neuroscience.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromer C, Marsit CJ, Armstrong DA, Padbury JF, Lester B. Genetic and epigenetic variation of the glucocorticoid receptor (NR3C1) in placenta and infant neurobehavior. Dev Psychobio. 2013;55:673–683. doi: 10.1002/dev.21061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buka SL, Shenassa ED, Niaura R. Elevated risk of tobacco dependence among offspring of mothers who smoked during pregnancy: A 30-year prospective study. Am J Psychiatry. 2003;160:1978–1984. doi: 10.1176/appi.ajp.160.11.1978. [DOI] [PubMed] [Google Scholar]
- Conradt E, Lester BM, Appleton AA, Armstrong DA, Marsit CJ. The roles of DNA methylation of NR3C1 and 11beta-HSD2 and exposure to maternal mood disorder in utero on newborn neurobehavior. Epigenetics. 2013;8:1321–1329. doi: 10.4161/epi.26634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelius MD, Day NL. Developmental consequences of prenatal tobacco exposure. Curr Opin Neurol. 2009;22:121–125. doi: 10.1097/WCO.0b013e328326f6dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell EC, Seckl JR. Prenatal stress, glucocorticoids and the programming of adult disease. Front Behav Neurosci. 2009;3:19. doi: 10.3389/neuro.08.019.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz PM, England LJ, Shapiro-Mendoza CK, Tong VT, Farr SL, Callaghan WM. Infant Morbidity and Mortality Attributable to Prenatal Smoking in the US. Am J Prev Med. 2010;39:45–52. doi: 10.1016/j.amepre.2010.03.009. [DOI] [PubMed] [Google Scholar]
- Dupont JM, Tost J, Jammes H, Gut IG. De novo quantitative bisulfite sequencing using the pyrosequencing technology. Anal Biochem. 2004;333:119–127. doi: 10.1016/j.ab.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Filiberto AC, Maccani MA, Koestler D, Wilhelm-Benartzi C, Avissar-Whiting M, Banister CE, Gagne LA, Marsit CJ. Birthweight is associated with DNA promoter methylation of the glucocorticoid receptor in human placenta. Epigenetics. 2011;6:566–572. doi: 10.4161/epi.6.5.15236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359:61–73. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottfried AW. Measures of socioeconomic status in child development research: Data and recommendations. Merrill Palmer Q. 1985;31:85–92. [Google Scholar]
- Gray TR, LaGasse LL, Smith LM, Derauf C, Grant P, Shah R, Arria AM, Della Grotta SA, Strauss A, Haning WF, Lester BM, Huestis MA. Identification of prenatal amphetamines exposure by maternal interview and meconium toxicology in the Infant Development, Environment and Lifestyle (IDEAL) study. Ther Drug Monit. 2009a;31:769–775. doi: 10.1097/FTD.0b013e3181bb438e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray TR, Shakleya DM, Huestis MA. A liquid chromatography tandem mass spectrometry method for the simultaneous quantification of 20 drugs of abuse and metabolites in human meconium. Anal Bioanal Chem. 2009b;393:1977–1990. doi: 10.1007/s00216-009-2680-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunnar MR, Malone S, Fisch RO. The psychobiology of stress and coping in the human neonate: Studies of adrenocortical activity in response to aversive stimulation. In: Field T, McCabe P, Schneiderman N, editors. Stress and Coping. Erlbaum; Hillsdale, NJ: 1985. [Google Scholar]
- Gunnar MR, Talge NM, Herrera A. Stressor paradigms in developmental studies: what does and does not work to produce mean increases in salivary cortisol. Psychoneuroendocrinology. 2009;34:953–967. doi: 10.1016/j.psyneuen.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton BE, Martin JA, Ventura SJ. Births: preliminary data for 2011. National Vital Statistics Reports. 2012;61:1–18. [PubMed] [Google Scholar]
- Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis MJ, Tunstall-Pedoe H, Feyerabend C, Vesey C, Saloojee Y. Comparison of tests used to distinguish smokers from nonsmokers. Am J Public Health. 1987;77:1435–1438. doi: 10.2105/ajph.77.11.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley K, Rice F, van den Bree MB, Thapar A. Maternal smoking during pregnancy as an environmental risk factor for attention deficit hyperactivity disorder behaviour. A review. Minerva Pediatr. 2005;57:359–371. [PubMed] [Google Scholar]
- Law KL, Stroud LR, LaGasse LL, Niaura R, Liu J, Lester BM. Smoking during pregnancy and newborn neurobehavior. Pediatrics. 2003;111:1318–1323. doi: 10.1542/peds.111.6.1318. [DOI] [PubMed] [Google Scholar]
- Lester BM, Tronick EZ, Brazelton TB. The Neonatal Intensive Care Unit Network Neurobehavioral Scale procedures. Pediatrics. 2004;113:641–667. [PubMed] [Google Scholar]
- Maccani JZ, Koestler DC, Houseman EA, Marsit CJ, Kelsey KT. Placental DNA methylation alterations associated with maternal tobacco smoking at the RUNX3 gene are also associated with gestational age. Epigenomics. 2013;5:619–630. doi: 10.2217/epi.13.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccani MA, Marsit CJ. Epigenetics in the placenta. Am J Reprod Immunol. 2009;62:78–89. doi: 10.1111/j.1600-0897.2009.00716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald SD, Walker M, Perkins SL, Beyene J, Murphy K, Gibb W, Ohlsson A. The effect of tobacco exposure on the fetal hypothalamic-pituitary-adrenal axis. Bjog. 2006;113:1289–1295. doi: 10.1111/j.1471-0528.2006.01089.x. [DOI] [PubMed] [Google Scholar]
- Meaney MJ. Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu Rev Neurosci. 2001;24:1161–1192. doi: 10.1146/annurev.neuro.24.1.1161. [DOI] [PubMed] [Google Scholar]
- Meaney MJ, Szyf M, Seckl JR. Epigenetic mechanisms of perinatal programming of hypothalamic-pituitary-adrenal function and health. Trends Mol Med. 2007;13:269–277. doi: 10.1016/j.molmed.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Moore C, Negrusz A, Lewis D. Determination of drugs of abuse in meconium. J Chromatogr B Biomed Sci Appl. 1998;713:137–146. doi: 10.1016/s0378-4347(97)00479-9. [DOI] [PubMed] [Google Scholar]
- Mulligan CJ, D’Errico NC, Stees J, Hughes DA. Methylation changes at NR3C1 in newborns associate with maternal prenatal stress exposure and newborn birth weight. Epigenetics. 2012;7:853–857. doi: 10.4161/epi.21180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3:97–106. doi: 10.4161/epi.3.2.6034. [DOI] [PubMed] [Google Scholar]
- Oitzl MS, Champagne DL, van der Veen R, de Kloet ER. Brain development under stress: hypotheses of glucocorticoid actions revisited. Neuroscience and biobehavioral reviews. 2010;34:853–866. doi: 10.1016/j.neubiorev.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Ostrea EM., Jr Testing for exposure to illicit drugs and other agents in the neonate: a review of laboratory methods and the role of meconium analysis. Curr Probl Pediatr. 1999;29:37–56. [PubMed] [Google Scholar]
- Owen D, Andrews MH, Matthews SG. Maternal adversity, glucocorticoids and programming of neuroendocrine function and behaviour. Neurosci Biobehav Rev. 2005;29:209–226. doi: 10.1016/j.neubiorev.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Poland RE, Lutchmansingh P, Au D, Edelstein M, Lydecker S, Hsieh C, McCracken JT. Exposure to threshold doses of nicotine in utero: I. Neuroendocrine response to restraint stress in adult male offspring. Life Sciences. 1994a;55:1567–1575. doi: 10.1016/0024-3205(94)00318-1. [DOI] [PubMed] [Google Scholar]
- Poland RE, Lutchmansingh P, Au D, Hsieh C, Acosta S, Lydecker S, McCracken JT, Afrane S. Exposure to threshold doses of nicotine in utero: II. Neuroendocrine response to nicotine in adult male offspring. Brain Res Dev Brain Res. 1994b;83:278–284. doi: 10.1016/0165-3806(94)00143-x. [DOI] [PubMed] [Google Scholar]
- Poland RE, Lutchmansingh P, Au D, McGeoy S, Que M, Acosta S, McCracken JT. Exposure to threshold doses of nicotine in utero: III. Augmentation of the prolactin and ACTH response to 8-OH DPAT by desipramine treatment is compromised in adult male offspring. Neurotoxicology. 1996;17:351–358. [PubMed] [Google Scholar]
- Pruessner JC, Kirschbaum C, Meinlschmid G, Hellhammer DH. Two formulas for computation of the area under the curve represent measures of total hormone concentration versus time-dependent change. Psychoneuroendocrinology. 2003;28:916–931. doi: 10.1016/s0306-4530(02)00108-7. [DOI] [PubMed] [Google Scholar]
- Radtke KM, Ruf M, Gunter HM, Dohrmann K, Schauer M, Meyer A, Elbert T. Transgenerational impact of intimate partner violence on methylation in the promoter of the glucocorticoid receptor. Transl Psychiatry. 2011;1:e21. doi: 10.1038/tp.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay DS, Bendersky MI, Lewis M. Effect of prenatal alcohol and cigarette exposure on two- and six-month-old infants’ adrenocortical reactivity to stress. J Pediatr Psychol. 1996;21:833–840. doi: 10.1093/jpepsy/21.6.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SM, Sobell LC, Sobell MB, Leo GI. Reliability of the Timeline Followback for cocaine, cannabis, and cigarette use. Psychol Addict Behav. 2014;28:154–162. doi: 10.1037/a0030992. [DOI] [PubMed] [Google Scholar]
- Schuetze P, Lopez FA, Granger DA, Eiden RD. The association between prenatal exposure to cigarettes and cortisol reactivity and regulation in 7-month-old infants. Dev Psychobiol. 2008;50:819–834. doi: 10.1002/dev.20334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NR, Bracken MB. A systematic review and meta-analysis of prospective studies on the association between maternal cigarette smoking and preterm delivery. Am J of Obstet Gynecol. 2000;182:465–472. doi: 10.1016/s0002-9378(00)70240-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin TA, Ryde IT, Seidler FJ. Additive and synergistic effects of fetal nicotine and dexamethasone exposure on cholinergic synaptic function in adolescence and adulthood: Implications for the adverse consequences of maternal smoking and pharmacotherapy of preterm delivery. Brain Res Bull. 2010;81:552–560. doi: 10.1016/j.brainresbull.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Stroud LR, Paster RL, Papandonatos GD, Niaura R, Salisbury AL, Battle C, Lagasse LL, Lester B. Maternal smoking during pregnancy and newborn neurobehavior: effects at 10 to 27 days. J Pediatr. 2009;154:10–16. doi: 10.1016/j.jpeds.2008.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter M, Abramovici A, Showalter L, Hu M, Shope CD, Varner M, Aagaard-Tillery K. In utero tobacco exposure epigenetically modifies placental CYP1A1 expression. Metabolism. 2010;59:1481–1490. doi: 10.1016/j.metabol.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter MA, Aagaard K. What changes in DNA methylation take place in individuals exposed to maternal smoking in utero? Epigenomics. 2012;4:115–118. doi: 10.2217/epi.12.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szyf M, Weaver I, Meaney M. Maternal care, the epigenome and phenotypic differences in behavior. Reprod Toxicol. 2007;24:9–19. doi: 10.1016/j.reprotox.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Tarullo AR, Gunnar MR. Child maltreatment and the developing HPA axis. Horm Behav. 2006;50:632–639. doi: 10.1016/j.yhbeh.2006.06.010. [DOI] [PubMed] [Google Scholar]
- TIBCO Spotfire. SPLUS 8.2 for Solaris/Linux User’s Guide. 8.2. TIBCO Software, Inc; Seattle, WA: 2010. [Google Scholar]
- Tong VT, Dietz PM, Morrow B, D’Angelo DV, Farr SL, Rockhill KM, England LJ. Trends in smoking before, during, and after pregnancy—Pregnancy Risk Assessment Monitoring System, United States, 40 Sites, 2000–2010. Morbidity and Mortality Weekly Report. 2013;62 (SS-6):1–19. [PubMed] [Google Scholar]
- Tronick E, Lester BM. Grandchild of the NBAS: the NICU network neurobehavioral scale (NNNS): a review of the research using the NNNS. Journal of child and adolescent psychiatric nursing. 2013;26:193–203. doi: 10.1111/jcap.12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrka AR, Price LH, Marsit C, Walters OC, Carpenter LL. Childhood Adversity and Epigenetic Modulation of the Leukocyte Glucocorticoid Receptor: Preliminary Findings in Healthy Adults. Plos One. 2012;7:e30148. doi: 10.1371/journal.pone.0030148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varvarigou AA, Liatsis SG, Vassilakos P, Decavalas G, Beratis NG. Effect of maternal smoking on cord blood estriol, placental lactogen, chorionic gonadotropin, FSH, LH, and cortisol. J Perinat Med. 2009;37:364–369. doi: 10.1515/JPM.2009.028. [DOI] [PubMed] [Google Scholar]
- Varvarigou AA, Petsali M, Vassilakos P, Beratis NG. Increased cortisol concentrations in the cord blood of newborns whose mothers smoked during pregnancy. J Perinat Med. 2006;34:466–470. doi: 10.1515/JPM.2006.091. [DOI] [PubMed] [Google Scholar]
- Wadhwa PD, Buss C, Entringer S, Swanson JM. Developmental origins of health and disease: brief history of the approach and current focus on epigenetic mechanisms. Semin Reprod Med. 2009;27:358–368. doi: 10.1055/s-0029-1237424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakschlag LS, Leventhal BL, Pine DS, Pickett KE, Carter AS. Elucidating early mechanisms of developmental psychopathology: the case of prenatal smoking and disruptive behavior. Child Dev. 2006;77:893–906. doi: 10.1111/j.1467-8624.2006.00909.x. [DOI] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Xiong F, Zhang L. Role of the hypothalamic-pituitary-adrenal axis in developmental programming of health and disease. Front Neuroendocrinol. 2012 doi: 10.1016/j.yfrne.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yolton K, Khoury J, Xu Y, Succop P, Lanphear B, Bernert JT, Lester B. Low-level prenatal exposure to nicotine and infant neurobehavior. Neurotoxicol Teratol. 2009;31:356–363. doi: 10.1016/j.ntt.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeltser LM, Leibel RL. Roles of the placenta in fetal brain development. Proc Natl Acad Sci U S A. 2011;108:15667–15668. doi: 10.1073/pnas.1112239108. [DOI] [PMC free article] [PubMed] [Google Scholar]