

Abstract
Fluorescence microscopy is an essential technique for the basic sciences, especially biomedical research. Since the invention of laser scanning confocal microscopy in 1980s, that enabled imaging both fixed and living biological tissue with three-dimensional precision, high-resolution fluorescence imaging has revolutionized biological research. Confocal microscopy, by its very nature, has one fundamental limitation. Due to the confocal pinhole, deep tissue fluorescence imaging is not practical. In contrast (no pun intended), two-photon fluorescence microscopy allows, in principle, the collection of all emitted photons from fluorophores in the imaged voxel, dramatically extending our ability to see deep into living tissue. Since the development of transgenic mice with genetically encoded fluorescent protein in neocortical cells in 2000, two-photon imaging has enabled the dynamics of individual synapses to be followed for up to two years. Since the initial landmark contributions to this field in 2002, the technique has been used to understand how neuronal structure are changed by experience, learning and memory and various diseases. Here we provide a basic summary of the crucial elements that are required for such studies, and discuss many applications of longitudinal two-photon fluorescence microscopy that have appeared since 2002.
Keywords: two-photon excitation, transgenic fluorescence, chronic surgery, cranial window, spine stability
In 2002, two laboratories published independently the first examples of long-term, repetitive two-photon fluorescent imaging of neurons in living mice(Grutzendler et al., 2002; Trachtenberg et al., 2002). The images and technique were breathtaking. The fate of many individual synapses had been followed over many days and weeks for the first time. Indeed, these were the experiments of which Cajal might have dreamt! In fact, four groups independently pioneered in vivo longitudinal two-photon fluorescence imaging. The first successfully reimaged amyloid plaques through the thinned skull by applying an exogenous dye through a small craniotomy(Christie et al., 2001). This study was followed by spine reimaging in the neocortex(Grutzendler et al., 2002; Trachtenberg et al., 2002) and olfactory bulb(Mizrahi and Katz, 2003) using transgenically fluorescence mice. Two of these studies established the chronic cranial window implantation as a viable technique for longitudinal reimaging of fluorescent structures in the living mammalian brain(Mizrahi and Katz, 2003; Trachtenberg et al., 2002).
The principle of two-photon microscopy for imaging biological specimens was first described in 1990(Denk et al., 1990). As a high-resolution, fluorescence imaging method it appeared very similar to laser-scanning confocal microscopy(Amos and White, 2003; Conchello and Lichtman, 2005) in that an image frame was made by raster scanning a sample with a laser beam focused through a lens of high numerical aperture. Fluorescence emission was detected using photomultiplier tubes and digitized by computer-controlled software. The key point for this new technique was that non-linear excitation with femtosecond pulsed laser enabled localization of chromophore excitation in the axial dimension. In other words, above and below a small excitation volume there was insufficient flux density of infrared photons to generate any chromophores with an excited singlet state. A high flux density was required because the photon that is absorbed first gives rise to what is called a “virtual excited state”, which has a lifetime of less than 10−15 s, the second photon must “catch” such molecules before decay back to the ground state. The most obvious advantage of this new excitation method, one that is perhaps somewhat over emphasized in the literature, was that photobleaching of chromophores outside the excitation volume is minimized. Whilst this is indeed true, a second advantage for fluorescence imaging was that two-photon excitation permitted the removal of the confocal pinhole(Denk and Svoboda, 1997). Perhaps the importance of this was not immediately apparent, but subsequent experiments showed very clearly that for imaging samples at depths beyond 10-20 microns there could be a dramatic improvement in image quality(Centonze and White, 1998). The reason for this is that biological tissue scatters emitted photons and such non-ballistic photons are blocked by the confocal pinhole(Denk and Svoboda, 1997). The effects of such scattering have been quantified and discussed in several studies(Helmchen and Denk, 2005; Oheim et al., 2001; Theer and Denk, 2006; Theer et al., 2003). The contribution of “out of focus excitation” is also of fundamental importance for deep tissue two-photon imaging. For depths less than one scattering length, this is not crucial, but at much greater depths the contribution of fluorescence excitation from the superficial portion of the brain becomes increasingly dominant part of the total signal(Theer and Denk, 2006).
The first reports using two-photon microscopy were from groups who had invented this laser-scanning method(Svoboda et al., 1997; Yuste and Denk, 1995). With the development of two-photon microscopes based on a modified commercial confocal microscope(Majewska et al., 2000), the technique started to become practical for neuroscience research. With the commercial development of one-box lasers that allowed simple computer-controlled mode-locking and wavelength tuning, two-photon microscopy became facile. Nevertheless, lasers remain irreducibly expensive, so it is fair to ask, “Why bother with longitudinal two-photon fluorescence imaging, especially as it seems technically challenging?” The attraction of longitudinal imaging lies in the fact that biological processes are dynamic, thus, cellular transitions are best captured by time-lapse imaging(Misgeld and Kerschensteiner, 2006). In the 19th century, think how Muybridge's “snaps” of motion completely revised our view of horses' motion. Traditional histologically based comparisons must be made between different animals, thus inter-individual variability is inherent to the measurement. A consequence of this is that changes smaller than the interanimal variability could be hidden. Perhaps even more important, if homeostatic processes are operative then these would render out important step-wise effects invisible to ex vivo imaging. For example, simultaneous addition and elimination of dendritic structures can only be observed using longitudinal studies of the same cells in situ. Following the choice between two competing pathways or the order of two successive steps in a mechanism are also much better suited to time-lapse imaging(Misgeld and Kerschensteiner, 2006).
Recently several reviews of the application of this powerful technique have appeared(Bhatt et al., 2009; Fu and Zuo, 2011; Holtmaat and Svoboda, 2009), as well as a number of detailed step-by-step protocols on their skull thinning and cranial window implantation techniques(Holtmaat et al., 2009; Yang et al., 2009b) and video how-to publications(Cao et al., 2013; Kelly and Majewska, 2010; Mostany and Portera-Cailliau, 2008; Shih et al., 2012b). These are superb sources for every detail involved in long-term two-photon imaging in living mice. In particular, the pooled experience of six laboratories involved in writing the cranial window implantation method is a unique resource(Holtmaat et al., 2009). Here we offer a basic guide to summarize these resources, one which reflects our own experience of using this method to study disease progression in a new mouse model of familial Alzheimer's disease(Crowe and Ellis-Davies, 2013a). This guide covers cover both surgical and non-surgical parameters of an optimized two-photon imaging system. The second part of our review integrates and discusses many of the important discoveries that have been made over the past 11 years using this technique around these guiding principles.
Materials and methods
Experiments were performed according to institutional IACUC guidelines.
In vivo two-photon imaging
Two-photon imaging was performed with a Prairie Ultima microscope and PrarieView software (Prairie Technologies, Middleton, WI, USA). Since 2004 a key feature of this software is that it allows subtle computer-controlled modulation of laser power with depth. This feature is an essential for the capture of images shown in herein. It has been copied by all major commercial imaging software and was introduced into scanimage(Pologruto et al., 2003) in December 2010 (version 3.7). Most images were taken with 20x water immersion objective (Olympus XLUMPLFL20XW, 0.95 NA or XLUMPLFLN20XW, 1.0 NA; Zeiss W Plan-APOCHROMAT, 1.0 NA). Some images were taken with a 60x water immersion objective (Olympus LUMFLN60XW, 1.1 NA) as designated. A mode-locked Ti:sapphire laser (Chameleon Ultra II or Vision II; Coherent, Santa Clara, CA, USA) was used to generate two-photon excitation, with power at the back aperture in the range of 10-50mW, depending on depth. Photomultiplier tubes (H7422-40, Hamamatsu Corporation, Bridgewater, NJ, USA) were used to detect the emitted light from the cells. We used a pixel dwell time of 4 μs, with a frame size of 512x512 pixels. The animal remained under anesthesia for the duration of the imaging session, using 20% ketamine/xylazine supplement of the initial dose (0.1 mg/g), as needed. Imaging sessions lasted approximately 1.5 hours per session.
Transgenic mice
The mouse models used in this publication were as follows: H-line YFP mouse line (B6.Cg-Tg(Thy1-YFPH)2Jrs/J), M-line GFP mouse line (B6.Cg-Tg(Thy1-EGFP)Mrs./J), GFAP-GFP mouse line (FVB/N-Tg(GFAPGFP)14Mes/J), Mito-CFP mouse line (B6.Cg-Tg(Thy1-CFP/COX8A)S2Lich/J), X98 mouse line (Tg(Gad1-EGFP)98Agmo/J), X94 mouse line (Tg(Gad1-EGFP)94Agmo/J), GIN mouse line (FVB-Tg(GadGFP)45704Swn/J), and the 5xFAD mouse line (B6.Cg-Tg(APPSwFlLon,PSEN1*M146L*L286V)6799Vas/Mmjax).
Surgical methods
Chronic cranial window implantation
Mice were anesthetized with an intraperitoneal (I.P.) injection of 0.1 mg/g xylazine/ketamine mixture, and supplemented with 20% of initial dose, as needed. The level of anesthesia was checked using the paw pinch test. Once fully anesthetized, all hair on the scalp was removed using a razor blade and the scalp was cleaned with alternating swabs of 70% ethanol and betadine. A midline incision in the skin was made from the base of the skull to the base of the nose. The periosteum was cleaned from the skull with a sterile cotton swab and then the animal was placed in a stereotaxic apparatus (Narishige International USA, Inc., East Meadow, NY, USA). Areas were marked in the designated stereotactic coordinate for the somatosensory cortex (−2 bregma, +2 midline). The surrounding area of skull was covered with a thin layer of Vetbond (3M, St. Paul, MN, USA). The craniotomy was performed using a burr drill bit with a microdrill. The skull was gently removed and a sterile 5 mm glass coverslip was placed over the exposed area and sealed with cyanoacrylate glue. The remaining exposed area was covered with dental cement, with a small well around the coverslip. Skin was glued and/or sutured around the window area.
Skull Thinning
Surgery was performed as described above to the point of the removal of the periosteum. The skull was swabbed with a sterile cotton swab and then a custom head plate was fixed to the skull with cyanoacrylate glue. A small burr-drill was used to thin an area of the skull approximately 1 mm in diameter. The skull was thinned to approximately 20 μm thick, which was checked manually with the microscope software.
Amyloid beta plaque labeling
Methoxy-XO4(Klunk et al., 2002) was used to label dense-core amyloid-beta deposits in the brain by I.P. injection of a heterogeneous solution in 10% DMSO/saline at a dosage of 12 mg/kg approximately 15-18 hours before each imaging session. Imaging wavelengths of either 780 nm or 840 nm were used.
Image Processing
Image adjustments to optimize contrast were performed in ImageJ. Z-stacks were constructed using the ZStackProject function in ImageJ. Volume reconstructions were performed using OsiriX (v3.6, www.osirix-viewer.com).
Basic guidelines for repetitive two-photon imaging
Use mice with transgenic expression of high levels of fluorescent protein
The “H-line” and “M-line” mouse lines were described in 2000(Feng et al., 2000), and it is no exaggeration to say their introduction was revolutionary for longitudinal, repetitive two-photon imaging (a full list of the transgenic mice described here, with Jackson Labs stock numbers and references is provided in the Supplemental Information). Since 1995(Helmchen et al., 1999; Svoboda et al., 1997; Yuste and Denk, 1995), the developers of the two-photon imaging technique had tried other methods of cell labeling to enable study of the details of dendritic structures, but comparison of two-photon images before(Lendvai et al., 2000) and after(Trachtenberg et al., 2002) the introduction of these two mouse lines reveal the fundamental importance of using intensely bright transgenic fluorescent mice for time-lapse imaging. Viral vector delivery of GFP into the brain can often result in dimly fluorescent structures or inconsistent labeling, making this technique unreliable for repetitive in vivo imaging(Dittgen et al., 2004). In contrast, bright transgenic fluorescence in the M-line and H-line mice is extremely stable and therefore allows one to see cells in a reliable way for very long periods of time.
However, not all transgenic mice produce enough fluorescent protein for clear in vivo imaging. We have imaged H-line and M-line mice, as well as several other commercially available transgenic mice that express fluorescent proteins in the neocortex. Two early reports labeling subpopulations of cortical interneurons looked very promising in vitro(Ma et al., 2006; Oliva et al., 2000). The ability to pick out selective categories of interneurons is a very attractive possibility for providing a means to study neuronal function in vivo and in vitro. However, we have found that only one subset of these interneuron mice, the “GIN” line(Oliva et al., 2000), are bright enough for in vivo imaging (but note (Lee et al., 2008) says otherwise). The other two lines of GFP labeled interneuron lines, the “X94” and “X98” mice(Ma et al., 2006), yielded no clear signals above background fluorescence (Figure 1). In our discussion section we compare “traditional” transgenic methods with the more recently developed cre/lox systems, in particular the ROSA26 mice that have been developed in the past few years(Madisen et al., 2010).
Fig. 1.
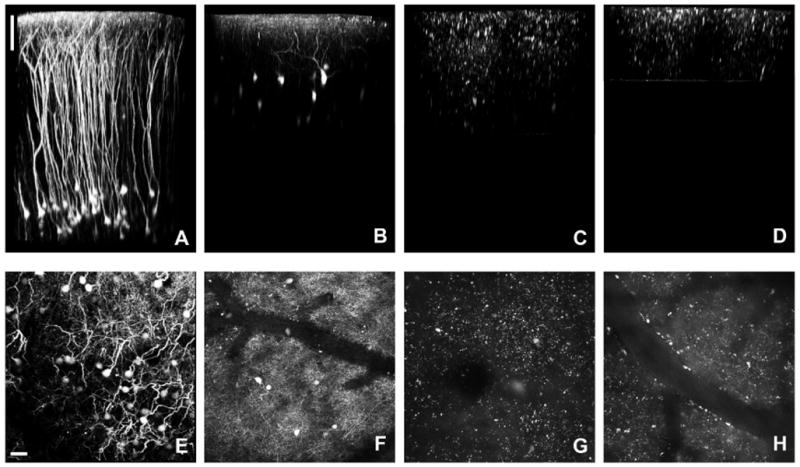
Comparison of transgenic fluorescent mouse lines. (A) “H-Line” mouse (B6.Cg-Tg(Thy1-YFPH)2Jrs/J) volume reconstruction. (B) “GIN” mouse (FVB/N-Tg(GadGFP)45704Swn/J) volume reconstruction. (C) “X94” mouse volume (Tg(Gad1-EGFP)94Agmo/J) reconstruction. No discernible neuronal structures can be seen. (D) “X98” mouse (Tg(Gad1-EGFP)98Agmo/J) volume reconstruction. Similar to the X94 mouse (C), no neuronal structures can be determined. (E) “H-line” mouse maximum projection (∼600μm in Z). (F) “GIN” mouse maximum projection (∼250μm in Z). (G) “X94” mouse maximum projection (∼300μm in Z). (H) “X98” mouse maximum projection (∼150μm in Z). Scale bar represents 100μm (A-D) and 30μm (E-H).
Use mice with sparse expression of fluorescent protein
If all pyramidal neurons are brightly labeled with a single fluorescent protein, then clear imaging of individual neurons and neurites is almost impossible in vivo. The importance of bright yet sparse labeling can be illustrated by images of “mitoCFP” mice, in which CFP is targeted to neurons by the Thy1 promoter, and targeted to the mitochondria by subunit 8A of the cytochrome c protein. Most L5 and some L2/3 neurons are brightly labeled. In these mice, in vivo imaging reveals some fine patterns against a general “haze” of fluorescence. Even though cell bodies in L5 can be imaged, too many cells are brightly labeled to allow clear imaging (Figure 2). While the signal is well above background fluorescence (c.f. Figure 1C,D), the required contrast is lost within the collected signal to distinguish individual structures.
Fig. 2.
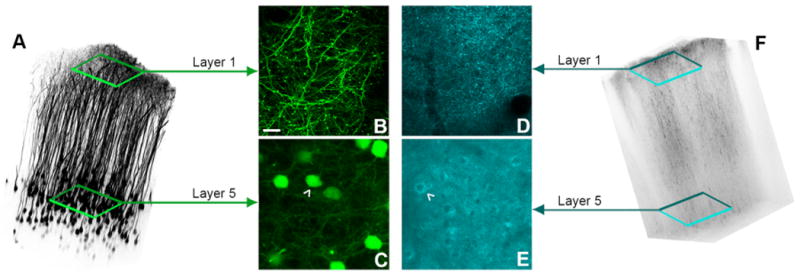
Comparison of a sparse fluorescent labeling to dense fluorescent labeling. (A) Volume reconstruction of a female “H-line” mouse. (B) A maximum projection (∼10μm) of Layer 1 dendrites and axons of neocortical L5 pyramidal neurons labeled with YFP under the Thy1 promoter. (C) A maximum projection of Layer 5 cell bodies of the pyramidal neurons. (D) A maximum projection (∼10μm) of Layer 1 dendrites and axons of L5 neocortical pyramidal neurons labeled with CFP under the Thy1 and COX8A promoters. (E) A maximum projection of Layer 5 cell bodies of the pyramidal neurons. (F) Volume reconstruction of a female B6/CB-Tg(Thy1-CFP/COX8A)S2Lich/J “Mito-CFP” female mouse. White arrowheads indicate a cell body. Scale bars represent 10 μm, and the field of view in the volume reconstructions was 500 x 500 μm.
In contrast to “mitoCFP” mice, M-line male mice(Feng et al., 2000; Trachtenberg et al., 2002) are the “gold standard” for bright and sparse transgenic labeling, with often only one cell in the field of view having high levels of GFP expression (Figure 3). This mouse line enables “easy” repetitive imaging. In contrast to male M-line mice, in both male and female H-line mice many more cells are labeled, but this level of labeling can also be very useful. For example, H-line mice have been used by a handful of groups to study neurodegenerative diseases using longitudinal, repetitive two-photon imaging. For such studies, often only a small percentage of neurons change their morphology, so the super sparse male M-line might not have enough labeled neurons to be useful. Within male and female M-line and H-line mice the number of cell bodies brightly labeled falls into three distinct categories, the sparsest being M-line males, then M-line females and H-line males being approximately equal, with H-line females being the densest (Figures 2A,3). Thus, it is important to consider these parameters of a study before deciding which mouse line would provide the most information.
Fig. 3.

Comparison of the density of fluorescent protein labeling in male H-line, male M-line and female M-line mice. (A) Three-dimensional reconstruction of pyramidal neurons in layer 5 in the sommatosensory cortex in a male H-line mouse. (B) Three-dimensional reconstruction of a single layer 5 pyramidal neuron in the sommatosensory cortex in a male M-line mouse. (C) Three-dimensional reconstruction of a volume from the entire sommatosensory cortex in a female M-line mouse. Transgenic expression of fluorescent protein in H-line and M-line mice varies considerably. H-line mice (A, and Figures 1A and 2A) have many layer 5 and some layer2/3 pyramidal cells brightly labeled. M-line mice (B,C) have the same population of layer 2/3 and 5 neurons labeled, as well as a significant number of layer 6 neurons. Neither line shows any labeling of layer 4 neurons. In both transgenic mouse lines, the females have many more cells per unit volume labeled with fluorescent protein. M-line male mice are especially useful for facile imaging of single cells, as they are so sparsely labeled. However, it is possible that a typical image frame (i.e. 500 x 500 microns) has no cells bodies brightly labeled. This is not the case with M-line females or H-line mice.
Surgical considerations for chronic window implantation
We have found that taking one, acute image is straightforward; it is repetitive imaging that presents the fundamental problem of reproducibility. The most carefully performed surgical implantation of a cranial window does not permit clear imaging of neuronal processes. This is because in brain beneath the window always goes cloudy within 1-3 days. This cloudiness is generally due to inflammation and sometimes the brain does not recover from the immune response(Holtmaat et al., 2009; Lee et al., 2008; Yang et al., 2009b). Even those “skilled in the art” state that the success rate varies tremendously (between 30-80%). The simple solution to this problem is to inject a dose of dexamethazone just before surgery(Holtmaat et al., 2009). Dexamethazone is steroid that acts as an anti-inflammatory and immunosuppressant. We typically only give one injection at the time of surgery and have seen a marked increase in our success rate. Since implantation of a cranial window is a major surgery, we normally wait at least two weeks before starting repetitive imaging. This allows time for recovery from any major inflammation to the brain. During this period the brain can be slightly cloudy, preventing clear imaging, but the dexamethazone injection normally prevents irreversible damage. We would note that this recovery period could sometimes take as long as 4-6 weeks. The “cloudiness” that disappears is very different than the milky white opaque substance that appears in an infected cranial window. Such mice must be discarded. In contrast, the thinned skull protocol does not really require drug administration, as imaging is always performed immediately after the less invasive surgery(Yang et al., 2010).
Parameters for repetitive deep tissue imaging
Protocols and reviews of the cranial window and thinned skull approaches to longitudinal 2P imaging have detailed the advantages and disadvantages of the two methods(Holtmaat et al., 2009; Yang et al., 2009b). Images of cellular structures in L1 taken through a cranial window or a thinned skull are essentially equivalent (Figure 4A,B). Note a protocol combining many of the advantages of both methods was described in 2010(Drew et al., 2010). However, imaging of structures in deeper cortical layers such as L5 cell bodies and L5 spines can only be accomplished with the cranial window method (Figure 4C). With the thinned skull technique it is possible in some instances to see L5 cell bodies, but no further detail can be obtained (Figure 4D). Therefore, in order to study changes in detailed structures 250 μm below the pia mater, a cranial window is required. For example, single synapses in L6 and axons in the white matter in M-line mice can be repetitively imaged through a chronically implanted window (Figure 5).
Fig. 4.
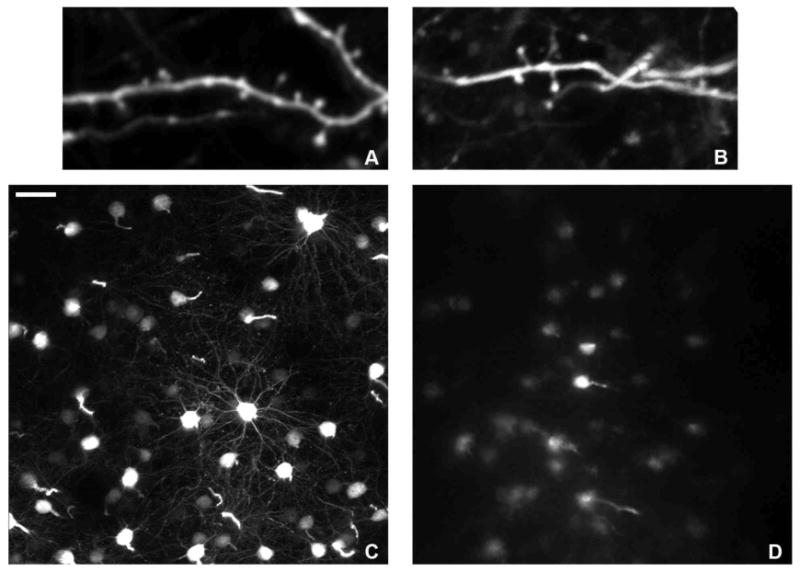
Comparison of images from a chronic window and thinned skull surgeries. High magnification of a dendritic segments in Layer 1 of the neocortex, in which individual spine heads can clearly be seen with both window implantation (A) and thinned skull (B). (C) Cell bodies in Layer 5 of the neocortex through a chronic cranial window. Basal dendrites can easily be seen projecting from the cell body. (D) Cell bodies in Layer 5 through a thinned skull. With this technique, cell bodies at this depth are “fuzzy” at best, without identifiable basal dendrites in the images. Scale bar represents 30 μm for (C,D).
Fig. 5.
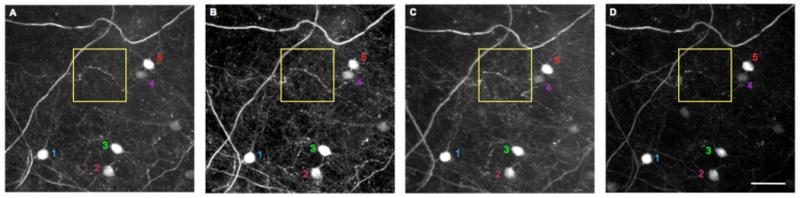
Repetitive deep tissue imaging through a cranial window.
A window was implanted into a female M-line mouse for imaging at depths up to 900 microns (full image stack in Fig. 3C). The images are maximum projections of images taken at approximately 770-830 microns below the pia mater. (A) Imaging session one at 105 days of age. (B) The second imaging session at 132 days of age. (C) Imaging session three at 211 days of age. (D) The final imaging session occurred at 323 days of age. In each image a few myelinated axons are apparent. Five re-imaged layer 6 neurons are numbered. The yellow box surrounds the same set of boutons imaged in each session. Note that by the final session the image quality had degraded slightly, so some fine detail was lost. Scale bar: 50 microns.
Deep tissue imaging requires optimized excitation and detection
The most important advantage of 2P imaging compared to tradition confocal microscopy is that detectors can be positioned on a non-descanned light path(Denk and Svoboda, 1997; Mainen et al., 1999). As a result of this maneuver the confocal pinhole is removed for 2P imaging, enabling the detection of non-ballistic photons. Furthermore, some microscopes objectives offer a more favorable configuration for deep tissue imaging as they permit collection of more scattered photons(Oheim et al., 2001). Low magnification lenses having a large sized effective back aperture (EBA>15 mm) are to be preferred for deep tissue imaging as they can collect more photons because of their larger field of view and transmit more photons because of their larger back aperture. However, for imaging fine details in L1, lenses with a small EBA (6 mm) give the best results. The quality of such images depends upon two factors: the lens numerical aperture (NA) and the degree to which the 2P beam fills/overfills the lens. Traditional laser scanning microscopes overfill a lens with a small EBA, enabling diffraction-limited imaging. Similarly, when we over-filled the Olympus 60x 1.1 NA lens, it gave the best images of spines in L1 (Fig. 6A). In contrast, the Olympus 20x 1.0 NA lens yielded slightly poorer images of L1 (Fig. 6B). Subdiffraction-limited fluorescent beads (diameter 0.1 μm) revealed that on our system the 60x lens had a point-spread function of 0.43 μm ± 0.08 μm, while the 20x lens was slightly lower at 0.61 μm ± 0.07 μm, the latter is significantly worse than ideal (0.46 μm). When a superficial layer 2/3 dendrite was imaged, both lenses produced comparable results (data not shown), but when a dendritic segment was imaged at ∼400 μm below the pia, the 20x lens showed superior results compared to the 60x lens, as predicted by Oheim et al., (2001). There have been relatively few reports of imaging below layer 2/3. To our knowledge, our recent work using longitudinal imaging of a bigenic H-line/familial Alzheimer's disease mouse is the first example of repetitive imaging of cellular details in layer 5(Crowe and Ellis-Davies, 2013a). Such deep-tissue imaging could only be accomplished through a cranial window as the remaining bone from skull thinning prevents such details from being imaged (Figure 4). Secondly, use of microscope objectives with large EBA and field of view is vital for deep tissue imaging. Since the scattering length is about 200 microns at 800 nm(Helmchen and Denk, 2005) and 430 microns at 1280 nm(Kobat et al., 2009). Thus, the collection of photons emitted from cells below 500 microns is improved dramatically with such lenses(Oheim et al., 2001).
Fig. 6.
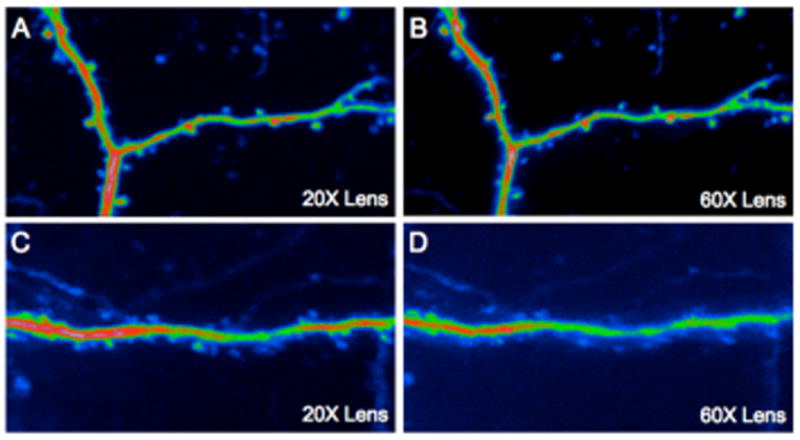
Comparison of image quality and two-photon efficiency using lenses with small and large back apertures. Dendrites in superficial and deep neocortical layers in a male M-line mouse were imaged through a chronically implanted cranial window. (A) A layer 1 dendritic segment imaged with a 20x objective with a 15 mm aperture (3 mW used). (B) The same dendritic segment imaged with a 60x objective with a small aperture (4 mW used). The aperture of the former is 15 mm and the latter 6 mm, and with numerical apertures of 1.0 and 1.1, the relative two-photon excitation efficiency is 1.9 in favor of the 60x lens. However (A,B) show that even though the 60x produces a slightly crisper image, it is not as bright indicating that even at superficial depths photon scattering is important. (C) A layer 5 dendritic segment imaged with the 20x objective using 61 mW of power at the front aperture. (D) The same layer 5 dendritic segment imaged with the 60x objective with 72 mW of power at the front aperture. These data show an even more pronounced effect of photon scattering on image quality and two-photon efficiency than the images from layer 1. Note also the general background signal in (D) is significantly higher than (C), even though the dendrite is dimmer. This may be due to the higher power used with the 1.1 NA producing greater excitation at the sample surface (Theer and Denk, 2006).
A second advantage of non-linear fluorescence imaging is that photobleaching out of the focal volume is minimized using two-photon excitation. This property does not fundamentally limit imaging depth like non-descanned detection noted above. However, from a practical point of view it may be very important, since less photobleaching could dramatically extend the number of imaging time points available.
Finally, laser alignment for two-photon microscopy is crucial consideration for day-to-day imaging. In contrast to traditional confocal microscopes, two-photon lasers cannot be delivered into the microscope scan head via a fiber optic cable. This is because glass increases the pulse-width of the output from a Ti:sapphire laser significantly, and two-photon excitation is inversely proportional to pulse-width. Therefore, two-photon beams are projected from the laser exit to the microscope via a series of mirrors. Unfortunately, wavelength tuning and the daily off-on-off duty cycle of the two-photon laser causes the beam to point to slightly different positions. These differences in the way light is delivered to confocal and two-photon scan heads means that former remain aligned from day to day in a much more reliable way. A misaligned 2-photon beam will cause distortions in the image, so we routinely check the alignment before each imaging session.
The effect of misalignment is illustrated in Figure 7. A simple lateral displacement of the beam in one dimension required significant increases in laser energy to produce the same signal. We routinely align the laser for each imaging session. Our microscopes allow positioning of fluorescent targets on the entrance to the scan head and at the back aperture of the objective. We also have two pinholes on the light path between the laser and microscope in order to keep track of the beam before it enters the microscope. However, most commercial microscopes are not open systems like this, and alignment is normally performed adjusting only one or perhaps two mirrors on the light table during imaging a standard sample, such as a fluorescent circular bead, until optimal signals are detected. For Figure 7, we deliberately slightly misaligned the beam on one axis, such that the amount of energy passing through a 20x lens was slightly diminished. In the case of imaging a dendrite in layer 2/3 the energy exiting the lens was reduced by 17%, but the misalignment caused a dramatic reduction in signal and image quality that really surprised us (Figure 7B)! Thus, even though daily alignment is not absolutely required to obtain signals on a two-photon microscope, the short time required for such procedures is more than rewarded with optimized two-photon images.
Fig. 7.
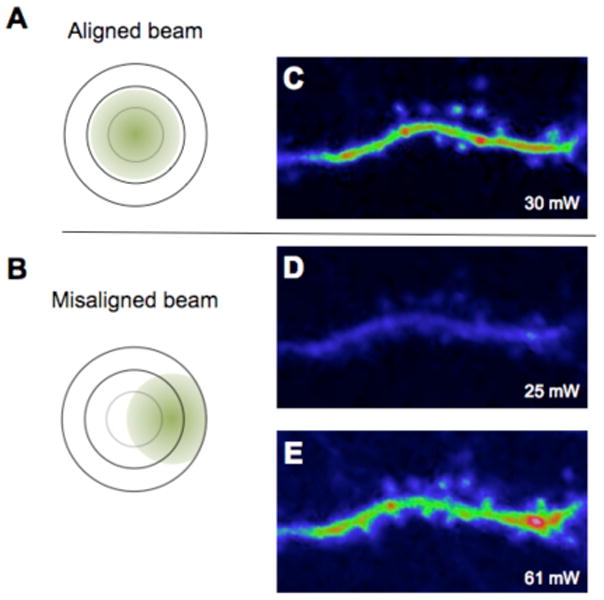
Slight misalignment of the laser beam reduces the two-photon effect in the sample and degrades the image quality. Two-photon images of a basal dendritic segment on layer 2/3 neuron in a male M-line mouse. A slight misalignment of the beam leaving the scan head, illustrated in (A) and (B), caused a 17% loss in energy when measured at the exit of the objective. (A) A well-aligned imaging beam produced crisp images of spines (C). (B) A misaligned beam results in image degradation (D) and more energy is required to produce a similar level of fluorescence (E) as with the aligned laser beam (C).
Applications of repetitive two-photon imaging
The fundamental rule of all imaging is that one must be able to detect a signal against background noise (Figure 1). The power of trangenically fluorescent mice such as M-and H-line is readily apparent in this context as labeling is so bright that relatively low amounts of laser energy can be used (Figure 7), such that individual synapses can be clearly seen against an almost black background. This can be seen most strikingly in male M-line mice, as often only one neuron is brightly labeled in the field of view, allowing the entire cell to be re-imaged (Figure 8). Over the past 10 years many laboratories have used this approach to study a range of basic questions concerning neuronal stability that may be grouped into several categories, including (a) what is the nature of the homeostatic stability of dendrites and spines; (b) how does experience change spines; (c) how does learning change spines; (d) how does pathophysiology change spines; and (e) how does drug treatment change spines. Furthermore, the development of genetically encoded calcium indicators (GECI) has started to enable a set of functional questions to be addressed, including: how does learning and experience become functionally encoded in synaptic activity and how does pathophysiology change neuronal calcium signaling? Such functional imaging does not yet use transgenic animals, but replies on other labeling methods. Thus, relatively few longitudinal imaging studies have been reported, and so we will only discuss these briefly.
Fig. 8.
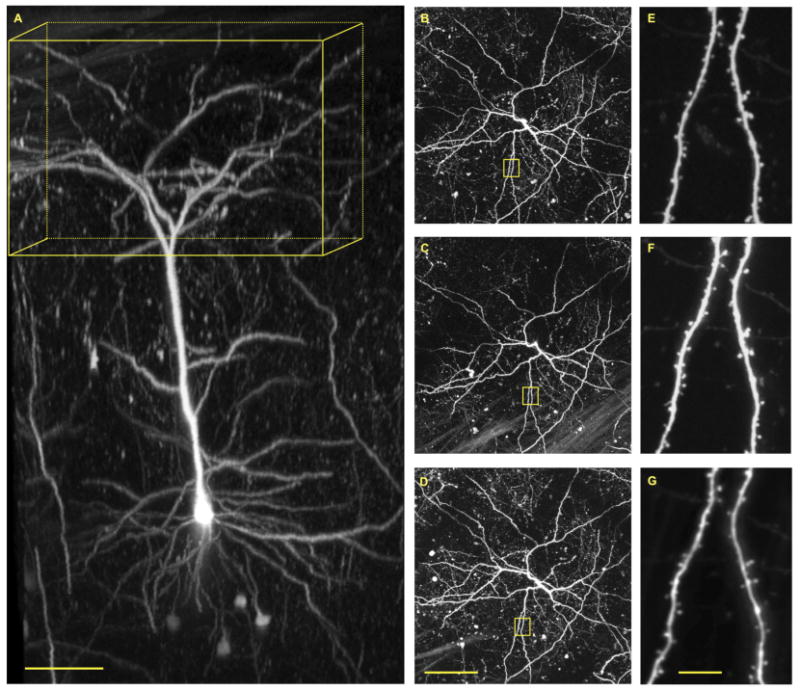
Repetitive imaging through a cranial window of M-line male mouse. (A) Three-dimensional reconstruction of a single layer 5 pyramidal neuron in the sommatosensory cortex. Yellow box has dimensions of 400 x 400 x 200 microns (x/y/z). (B) Maximum intensity projections of the boxed volume in A. (C) Maximum intensity projection of the boxed volume in A taken 7 days after the first image. (D) Maximum intensity projection of the boxed volume in A taken 21 days after the first image. (E-G) Magnified images of dendritic tree in yellow squares in B-D. Cellular labeling in M-line male mice with eGFP is so sparse (see Figure 3) and intense that it allows facile two-photon fluorescence imaging of single neurons with great clarity. Note, basal dendrites are visible at a depth of 500-600 microns below the pia. Repetitive images of the entire neuron revealed the overall structure of the dendritic tree is very stable (B-D). High-resolution imaging revealed most spine heads are also stable over a 21-day period (E-G). The window was implanted when the mouse was 121 days old, and imaging commenced at 62 days later. Scale bars: 100 microns (A,D) 100 microns; G 10 microns.
Dendritic stability
From the first two longitudinal studies onwards(Grutzendler et al., 2002; Trachtenberg et al., 2002), [reviews: (Bhatt et al., 2009; Holtmaat and Svoboda, 2009)], it is widely agreed that the basic structure of pyramidal neurons (the overall branching of the dendritic tree and relative disposition of cells) in adult mice does not change (Figures 8, 9, and supplemental video 1). Furthermore, spine density is stable over many months. The 2002 studies did disagree on the rate of homeostatic turn over of spines. Svoboda and co-workers reported that in mice of 6-11 weeks of age about 20% spines disappeared between daily imaging sessions and only 60% of spines survived more than 8 days. Of the latter group a further 15% disappeared over the next 20 days(Trachtenberg et al., 2002). In contrast to this instability, Gan and co-workers reported(Grutzendler et al., 2002) that spines were strikingly stable. In adult mice (age at least 4 months) 96% of spines were present for at least a month corresponding to a halflife of 13 months. In 2005 Svoboda and co-workers reported that in adult mice (age 6 months) about 73% of spines survived more than 1 month(Holtmaat et al., 2005). Obviously these differences are significant, and therefore important. In an attempt to understand this disparity, in 2007 Gan and co-workers addressed the effects of the two different surgical techniques used by groups (Figure 4). Comparing the effects of cranial windows implanted in the barrel cortex of H-line male mice with imaging through a thinned skull revealed that the former induced significant spine turn over compared to the latter (35% vs. 6% over 1 month)(Xu et al., 2007). Finally, Svoboda and colleagues compared thinned skull and window implantation survival fractions in their 2009 protocol(Holtmaat et al., 2009). They reported that 80% of spines survived 1 month under thinned skull and 60% under a cranial window (see supplemental figure 2 in Holtmaat, et al., 2009). Subsequently the two camps have attempted to reconcile these differences, but the numbers still seem to have a large gap.
Fig. 9.
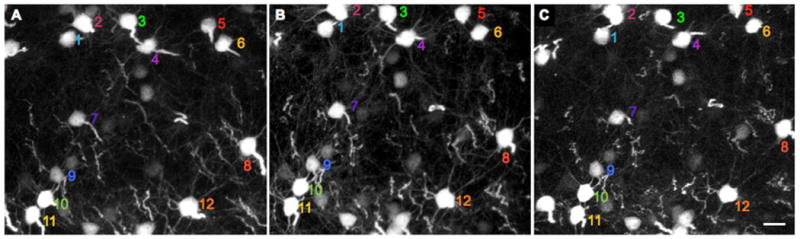
Repetitive imaging through a cranial window of layer 5 neurons in H-line male mouse taken over 100 days. (A-C) Maximum intensity projections of volume in layer 5 (125 x 110 x 85 microns, x/y/z) taken at 45, 63 and 108 days after window implantation. H-line male mice have many more layer 5 pyramidal neurons than their M-line counter parts (see Fig. 3), this allows many more cells bodies to be imaged repetitively in layer 5 in H-line transgenic mice. The supplemental video shows animated three-dimensional reconstructions of the imaged neurons. Scale bar: 20 microns.
In parallel to the pioneering studies of structural stability of pyramidal cells, Mizrahi and Katz used G-line mice to image dendritic trees of mitral and tufted cells in the olfactory bulb(Mizrahi and Katz, 2003). They found that under natural conditions cell dendrites remained highly stable. In contrast, the fourth group to independently use longitudinally imaging reported that the dendritic structure of interneurons is highly plastic, and this plasticity is further modified by experience(Lee et al., 2006). For this work Nedivi and co-workers used S-line mice, which have moderately dense GFP labeling of pyramidal and non-pyramidal cells. This result was the first example of large-scale structural plasticity of dendritic arbors in the adult cortex. Recently, Trachtenberg and co-workers studied the effect of conditional Pten deletion on dendritic arborization of layer 2/3 pyramidal neurons(Chow et al., 2009). Longitudinal imaging of adult M-line male mice revealed growth of new regions of existing dendritic arbors of at least 25% over a period of 1 month.
All the initial studies of H-, M-, S- and G-line mice took advantage of the very high levels of fluorescent protein expression in these transgenic mice, as this produced neurons that were intensely fluorescent with modest amounts of energy from Ti:sapphire lasers used for two-photon microscopy. Certainly male M-line mice exhibit extremely sparse labeling, so that it is feasible to find only one neuron in the field of view (Figure 8). This makes re-imaging dendrites and spines quite easy (Figure 8) compared to all other transgenic fluorescent mice. If this mouse line was used for thinned skull imaging, detecting any dendrites in the 0.2 mm square area available would be very problematic. For this reason, Gan and co-workers used H-line male mice (Figure 3A). Thus, the choice of the density of neuronal labeling is probably determined by the requirements envisioned in a particular study. Furthermore, it must be noted that the sparseness of M-line and H-line might imply that fluorescence labeling might not be truly random within layer 5 neurons, and this may explain some of the divergent conclusions published on spine stability by various groups(Holtmaat and Svoboda, 2009).
Experience changes spines and dendrites
In their 2002 report, Svoboda and co-workers described the effects of chessboard whisker removal on spine turnover: spine turner over was enhanced about 50%. A subsequent, more detailed study confirmed these initial findings(Holtmaat et al., 2006). Using a different manipulation of the barrel cortex, Gan and co-workers removed all the whiskers from one pad, and found that in adult (> 4 month) mice this had no effect over 2 weeks(Zuo et al., 2005). But when whiskers were removed for 2 months, they found that fewer spines were eliminated than in controls, while the same number of new spines appeared in both groups. The latter was mimicked by chronic blockade of NMDA-receptors, suggesting they may be involved in the effects of sensory depravation.
Long-term changes in the visual cortex have also been studied using longitudinal imaging of fluorescent mice. Hubener and co-workers used local retinal lesions induced by photoablation to probe how dendrites and spines in the visual cortex respond to such lesions(Keck et al., 2008). Dendritic branches remained stable, but >90% of spines disappeared and were “replaced” over a two month period subsequent to the lesion. The total spine density did not change during this period. Monocular deprivation (MD) is a classic manipulation used to understand the visual cortex. Hubener and co-workers found that the number of spines in the superficial layers of neocortical neurons in the binocular region were selectively enhanced by MD in adult M-line mice(Hofer et al., 2009). A second MD experience only affected these new spines (selective enlargement), but did not induce a further batch of new spines, suggesting certain branches may become “saturated” by experience but that individual spines may be “re-activated”.
Whisker trimming and MD of rodents are widely used and are powerful means to manipulate well-defined circuits in the CNS. How do other learning modalities change spines stability, number or kind? Several studies have appeared recently that report a specific enhancement or change in dendritic spines that correlated very strongly with an experience. First, in 2009, two laboratories independently reported experience-driven spine enrichment using H-line male mice. Zuo and co-workers using longitudinal imaging to observe rapid spinogenesis in mice that were training to grasp and successfully eat food(Xu et al., 2009). Furthermore, old spines were preferentially eliminated by further training, and new spines subsisted for months after training. Similar findings were reported by Gan and co-workers using a rotor-rod learning task(Yang et al., 2009a). Perhaps an even more remarkable longitudinal imaging study was published in 2010 in which lentivirus was used to label neurons in juvenile songbirds. Obviously there are no transgenic GFP songbirds, but their unique tutoring paradigm, involving a well-defined circuit made them attractive target for two-photon fluorescence imaging. Mooney and co-workers were able to show that spine dynamics decreased with age and that song learning at P60 was correlated with rapid dendritic stabilization of mushroom spines(Roberts et al., 2010). They also observed that a high degree of spine turnover before tutoring correlated with an increased ability for song imitation. Finally, Gan and co-workers showed that fear conditioning of male H-line mice specifically increased the rate of spine elimination in the frontal association cortex, and extinction increased the rate of spine formation. Strikingly, the spine associated with extinction formed within a distance of 2 μm from spines eliminated after fear conditioning. Finally, Gan and co-workers found that reconditioning preferentially induced elimination of extinction-associated spines. They concluded that “within vastly complex neuronal networks, fear conditioning, extinction and reconditioning lead to opposing changes at the level of individual synapses. These findings also suggest that fear memory traces are partially erased after extinction”.(Lai et al., 2012).
Imaging cells and axons
Neurons in the olfactory bulb and hippocampus are well known to undergo adult neurogenesis. The former is physically accessible to two-photon microscopy and so several elegant longitudinal imaging studies of neuronal stability have been reported. For example, the fine detail of how experience (exposure to odor) stabilizes newly formed synapses by as much as 50% (imaging was perform 8 times over 30 days through a cranial window)(Livneh and Mizrahi, 2012). In contrast to such fine-scaled imaging, the same laboratory imaged the rate of appearance and disappearance of cells (cell bodies of dopaminergic interneurons, cf. cells imaged in H-line mice, Figure 9 and supplemental video) in the olfactory bulb over a 9-month period and found that there was a net increase of about 13% (Adam and Mizrahi, 2011).
Dendritic stability and pathophysiology
With a baseline set for dendritic stability from the initial reports of longitudinal two-photon imaging, application of the technique to the study of how neurons change with time in mouse models of disease was inevitable. Several areas have been studied, including Alzheimer's disease, fragile X syndrome, stroke, cancer, and prion disease.
Gan and co-workers again pioneered this field, by creating a fluorescent familial Alzheimer's disease (FAD) mouse model. Crossing the H-line YFP mouse with the PSAPP mouse gave a bigenic mouse that allowed longitudinal imaging of dendritic rupture and axonal damage(Tsai et al., 2004). Specifically, of 198 neurites imaged twice during a 1-2 week period 7.6% were eliminated, and of the 30 imaged twice over a 4-5 week period 37% were eliminated when the neurites were within 15 microns of an amyloid plaque. These observations gave strong support to the idea that amyloid plaque deposits found in AD are highly toxic towards neurons and therefore may be an important therapeutic target for drug treatment. In 2008, Hyman and co-workers reported longitudinal imaging of another bigenic mouse (cross of an unspecified YFP mouse with the APPswe/PS1d9) in which the appearance of plaques was very rapid (over 1 day), but no equivalent neurite fragmentation to that reported by Gan was seen(Meyer-Luehmann et al., 2008). Interestingly, Hyman reported that once plaques were deposited they did not change in size, but rapidly attracted microglia to their periphery. In 2010, Herms and co-workers published a remarkable study in which they reported the complete disappearance of neuron in a mouse model of FAD(Fuhrmann et al., 2010). They created a trigenic mouse by crossing H-line, 3xTgAD and GFP-microglia transgenic mice. Three-color two-photon imaging allowed them to visualize neurons (yellow), microglia (green) and vasculature (red dye injected in the tail vein). In 6-month old mice, weekly imaging sessions revealed the loss of entire layer 2/3 cells. Such loss was pre-figured by the accumulation of microglia around the dying cell, and prevented by deletion of the fractalkine receptor from microglia. This result is even more striking as it happens at pre-plaque stage of the 3xTgAD model. In a follow up study the Herms groups described the gradual loss of spines in old (age 13-20 months) 3xtgAD mice(Bittner et al., 2010). The Herms group has also used longitudinal imaging of H-line mice to reveal that drug treatment with a gamma-secretase inhibitor (LY 450139) for 4 days caused significant reduction in spine density within 2-3 weeks after treatment(Bittner et al., 2009). This effect was abrogated by deletion of the amyloid precursor protein. Finally, we have recently report longitudinal imaging of another bigenic FAD mouse (5xFAD/H-line). In this mouse model of AD, we observed the first signs of structural dystrophy were with axons mainly located in layer 5, whilst spines on dendrites were stable at this time (4 month old female mice)(Crowe and Ellis-Davies, 2013a). Ex vivo imaging confirmed this result and revealed that by 6 months of age there was a significant reduction of spine density on basal dendrites in layer 5, but not apical dendrites in layer 1(Crowe and Ellis-Davies, 2013b). We also found that over time plaques increased in size by significant amounts (3.2-fold over 3 weeks). Two other similar reports have appeared recently(Burgold et al., 2010; Hefendehl et al., 2011). Figure 10 shows representative longitudinal two-photon fluorescence imaging of increasing dystrophy and plaque deposition in a male 5xFAD/H-line mouse.
Fig. 10.
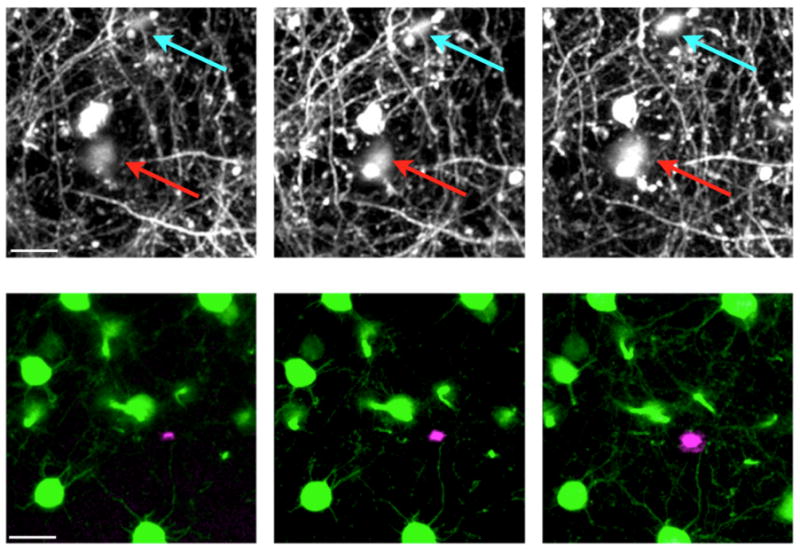
Longitudinal two-photon imaging of Alzheimer's disease pathology in the 5xFAD mouse model crossed with H-line YFP mice. (Top row) Presence of AD transgenes causes an increase in axonal dystrophy in the layer 1 of the somatosensory cortex (arrows). (Bottom row) Two-color, two-photon imaging of neurons and plaques shows the growth of an amyloid plaque (magenta) near pyramidal cell bodies (green) in layer 5 of the somatosensory cortex. Plaques were stained acutely with methoxy-XO4. Images were taken at weekly intervals. Scale bars: 20 microns.
The status of pre-plaque amyloid beta oligomers is of great current interest in the AD field, but the classic Congo Red-based dyes only bind to dense core plaque deposits. To address this technical issue, Grutzendler and co-workers injected synthetic amyloid beta-42 monomers labeled with a red dye into the neocortex of bigenic FAD/GFP-microglia mice. Within minutes of injection, in vivo two-photon imaging revealed that monomers bound to pre-existing plaques areas(Liu et al., 2010). Confocal analysis of ex vivo brain slices showed that microglia could also phagocytose injected monomers, but not dense core plaques. This process also occurred when the fractalkine receptor was deleted from microglia. In contradiction to this work, Jucker and co-workers reported that microglia could internalize particles stained with a dense core dye(Bolmont et al., 2008). The same dye (methoxy-XO4) and GFP-microglia mouse was used for both studies, however different FAD mice were used.
Use of in vivo two-photon imaging to study the effects on neurons of various stroke models in mice has been reported extensively by Murphy and co-workers, and more recently by Portera-Cailliau and co-workers. Several acute imaging studies by the Murphy group have shown that when ischemia was induced in H-line mice, a rapid loss of spines and/or beading on the dendritic structure was induced, following reperfusion there was recovery or restoration of the original structures(Brown et al., 2007; Li and Murphy, 2008; Murphy et al., 2008; Zhang et al., 2005). Even acute studies such as these demonstrate the power of in vivo two-photon imaging to reveal subtleties in neuronal dynamics that would not be apparent in classic histologically based methods. Longitudinal imaging of M-line mice revealed that after stroke, apical dendritic arbor remodeling increased significantly, particularly within the first two weeks after stroke. Interestingly, there was an apparent compensation in the length of arbors, those lost near the infarct were “replaced” by dendritic extensions away from the stroke volume. According to Murphy these results demonstrated that “fully mature cortical pyramidal neurons retain the capacity for extensive structural plasticity and remodel in a balanced and branch-specific manner”(Brown et al., 2010). In contrast to these results, Portera-Cailliau and co-workers found no evidence of compensatory dendritic remodeling in their stroke model(Mostany and Portera-Cailliau, 2011). Rather, they reported a slight shrinkage of the apical dendritic tips of 10 microns over 90 days of imaging male M-line mice. They used middle cerebral artery occlusion as their stroke model, whereas Murphy's group used photo-thrombic infarct with Rose Bengal. In a second study by Portera-Cailliau and coworkers they found that middle cerebral artery occlusion induced a temporary loss dendritic spines in the region local to the infarct. In fact, the number of spines eventually slightly increased in density in these regions. This lovely study combined longitudinal imaging of male M-line mice (15 times over 100 days!) with two-photon imaging of blood flow in the affected regions in order to correlate the amount of local occlusion with the degree of spine loss(Mostany et al., 2010). Note injection of dye-labeled dextran into rodent plasma can be used for repetitive imaging of the vascular network and blood flow(Shih et al., 2012a; Yoder and Kleinfeld, 2002). In the graphical abstract we illustrate how this allows blood vessels to be used as fiduciary markers for structural changes. Recently, Kleinfeld and co-workers have used short-term, longitudinal two-photon imaging of blood flow to access how optically targeted microinfarcts compromise cognitive tasks in adult rats. In a technical tour-de-force, they found that “microinfarcts were not innocuous, as has been often assumed from their small volume. Instead, the occlusion of only a single penetrating vessel led to cognitive dysfunction in a behavioral task”(Shih et al., 2013). Longitudinal two-photon fluorescent imaging has been applied to other areas of pathophysiology, for example prion disease(Fuhrmann et al., 2007), neuropathic pain(Kim and Nabekura, 2011), fragile-X syndrome(Nimchinsky et al., 2001; Pan et al., 2010), and cancer (Lam et al., 2010) have all used this method.
Non-transgenic labeling methods
The usefulness of the many Thy1.2 transgenically fluorescent mice introduced by Sanes and Litchman in 2000 is obvious. However, relatively few other transgenic mice have proved useful for longitudinal two-photon imaging, suggesting other means of producing reliably brightly fluorescent labeling of defined subsets of neurons is highly desirable. It should be noted that the extremely sparse labeling seen M-line mice raises the possibility that a select population of neurons may be labeled by GFP(Holtmaat and Svoboda, 2009). To our knowledge this idea has been tested at the physiological level. In the case of H-line mice, Shepherd and co-workers has shown that labeled neurons are part of different circuits than unlabeled neurons(Yu et al., 2008). How such differences might be influence the use of these mice in longitudinal imaging experiments is not clear.
Several alternative labeling strategies to Thy1 transgenics are feasible. Perhaps the most attractive approach is through the development of Rosa26 FP cre reporter mice. Zeng and co-workers generated several such mouse lines in 2010 by insertion of the CAG promoter behind two loxP sites to control the expression of eYFP, tdTomato and ZsGreen(Madisen et al., 2010). Crossing with more than 20 different transgenic mice with cell-selective expression of cre allowed the generation of many different fluorescent mice, of which several were of comparable brightness to H-line (see supplemental figure 7 of Madisen et al., 2010). These mice lines have been used for identification of interneurons in acute in vivo calcium imaging experiments, but we are not aware of any longitudinal imaging applications. Labeling of neurons by injection of lentivirus or AAV is another alternative that has been used in several recent studies(Dombeck et al., 2010; Harvey et al., 2012; Huber et al., 2012; O'Connor et al., 2010; Osakada et al., 2011). This approach is particularly useful for GECI, as until recently no transgenic mice with GECI have been reported. Indeed, it seems that since GECI are being a developed a fast pace (GCaMP5(Akerboom et al., 2012), GCaMP6, RCaMP(Ohkura et al., 2012), etc.), GECOs(Zhao et al., 2011), etc.), any transgenic mice might be “out of date” before they are widely available. However, by its very nature, viral injection is unlikely to create really sparse labeling equivalent to M-line male mice (Fig. 3B), dense bright labeling can be useful but is more challenging to quantify longitudinally. A more refined version of viral injection is the delivery of floxed AAV to the appropriate cre mouse line to provided selective labeling of a well-defined subset of neurons. Cell-specific labeling in the neocortex has been reported via knock-in of the CreERT2 gene into the Nex1 locus(Agarwal et al., 2012). The extent of GFP labeling could be controlled by the period of tamoxifen treatment. In utero electroporation is another method that has recently been used for the delivery of two colors of FP probes. Nedivi and co-workers found that spines and GABA-A receptors in layer 2/3 pyramidal cells in the visual cortex are quite plastic(Chen et al., 2012). Longitudinal imaging before and after MD revealed that such plasticity is highly clustered showing coordination of excitatory and inhibitory synapses during sensory experience. Using a similar approach, Levelt and co-workers found that GABA-A receptors on spines were more sensitive to MD than those on dentritic shafts(van Versendaal et al., 2012). Further, most spines that lost GABA-A puncta were persistent. These two elegant studies demonstrate the real potential of two-color longitudinal two-photon imaging.
Functional imaging
In 1997 four groups independently introduced molecular optical sensors using derivatives of GFP(Miesenbock and Rothman, 1997; Miyawaki et al., 1997; Romoser et al., 1997; Siegel and Isacoff, 1997), thus initiating a field that has come to be known as “optogenetics”(Miesenbock, 2009) (strictly this is a misnomer, as it is genetics that is used to deliver optical probes). Of the four types of sensors, GECI have subsequently attracted the most developmental effort, with hundreds of person-years being devoted to the improvement of their properties(Mank and Griesbeck, 2008). Whilst it seems impossible that GECI will match electrodes as faithful reporters of spiking, they have several distinctive properties that make them useful for in vivo imaging of cellular activity. The first proof-of-principle example of repetitive longitudinal in vivo imaging using TN-XXL appeared in 2008 (Mank et al., 2008), followed by GCaMP3 in 2009 (Tian et al., 2009) and YC3.6 in 2010 (Andermann et al., 2010). Recently, a handful of studies have been published using longitudinal imaging of GECI that moved beyond proof-of-principle reports to interesting biological problems(Huber et al., 2012; Kato et al., 2012; Margolis et al., 2012). Such reports suggest these GECI may start to rival small organic molecular calcium dyes for in vivo imaging (Grienberger and Konnerth, 2012), with the huge benefit of allowing longitudinal imaging of learning and behavior.
Concerns about window implantation for longitudinal deep tissue imaging
A major concern of window implantation is that it is a major surgery. Complete removal of even a small piece of piece of the skull inevitably presents a toxic challenge to the exposed brain(Xu et al., 2007). Our initial attempts at longitudinal imaging without post-operative injection of anti-inflammatory drugs met with almost complete failure (10 out of 11 windows became irreversible opaque). Following the suggestion of Holtmaat et al., 2009, we have found that dexamethazone can dramatically improve the success rate of surgery. It is important to note that after about 3-4 days virtually all implanted windows still become hazy for 2-3 weeks. One should not be discouraged by this event, and discard these mice, as the vast majority of windows will eventually clear. Thus, during our imaging of about 150 H-line mice, we found that more than 90% of windows remained clear for at least two months. Use of non-steroidal anti-inflammatory drugs such as carprofen is an alternative to dexamethazone(Adam and Mizrahi, 2011). And chronic administration of sulfamethoxazole and trimethoprim in drinking water until the first imaging session has been used in other studies(Lee et al., 2008). Interestingly, in the latter study, Nedivi and co-workers found that clear windows had no activated microglia or astrocytes 4 weeks after window implantation, whereas windows that did not allow imaging had significant reactive glia in layers 1-4. It should be noted that the thinned skull does not require anti-inflammatory drugs(Yang et al., 2009b). Bone re-growth naturally occurs for both skull thinning surgeries and chronic window implantation. In skull thinning, bone re-growth is common and requires a subsequent thinning surgery for each imaging session. For window implantation, bone has been reported to grow back into the craniotomy space, obscuring the imaging area (Holtmaat et al., 2009). However, we have personally never seen re-growth significant enough to interfere with imaging, even after seven months of repetitive sessions(Crowe and Ellis-Davies, 2013a).
Recent developments and future directions
It is readily apparent from our survey that longitudinal two-photon in vivo fluorescence imaging is a uniquely powerful technique for neurophysiology that has become well established in the biomedical research community. The ability to observe structural and/or functional changes in the same cells in the CNS over time allows one to test a different set of questions than traditional histologically-based methods of studying changes in cellular structure. However, no technique is perfect and several groups have made important recent contributions to improving the method. These pioneering studies fall broadly into two categories, namely, improvements to microscope hardware or to fluorescent proteins.
Hardware
A very active area of research for the improvement of hardware involves rapid scanning. Frame rates are normally of the order of 1 Hz, and for structural studies illustrated in our figures this is more than adequate. But for fast physiological processes such as calcium or membrane potential imaging this rate is not optimal. Since there is an inherent problem to standard galvanometer scanning, namely that of the inertia of the mirrors, several alternative approaches have been advanced. Using a resonant galvanometer for x scanning increasing the natural frame rate to 30 Hz. Initially developed for confocal imaging(Bacskai et al., 1995; Callamaras and Parker, 1999), it has been applied to two-photon microscopy(Nguyen et al., 2001). An alternative means of fast scanning is use of acousto-optical deflectors (AODs). Again this technology was first deployed for confocal imaging(Draaijer and Houpt, 1998), and subsequently adapted to two-photon imaging to increase the x-scan rate(Ng et al., 2002). More recently, two AODs have been applied to x and y dimensions (Grewe et al., 2010; Katona et al., 2010; Salome et al., 2006) and three to z/y/z rapid scanning (Kirkby et al., 2010; Reddy and Saggau, 2005). Other methods for fast z scanning have also been developed, involving mirrors(Botcherby et al., 2013), electronically focused lenses(Grewe et al., 2011), or piezo motors.(Gobel et al., 2007). Increasing the rate of raster scanning does not come without cost to image quality for two-photon microscopy, as the signal-to-noise ratio decreases significantly at video-rate, partly due to the fact that only 7-8 pulses per pixel from the mode-locked laser are feasible. A potential solution to this problem could be provided by passive pulse splitting(Ji et al., 2008). Arguably a more practical solution is to image only the voxels that contain useful information by directing the laser beam appropriately using regular mirrors(Gobel and Helmchen, 2007; Gobel et al., 2007). However, meaningful image recovery from selective illumination is not trivial, as normal software such as imageJ (or its commercial equivalent) does not allow reconstruction from random scanning. Another area of development has been increasing signal strength, either by improving the efficiency of photon collection using additional detectors(Engelbrecht et al., 2009) or larger apertures(Ducros et al., 2011; Oheim et al., 2001) or by use of adaptive optics(Ji et al., 2011). Unfortunately most of these useful hardware developments have not had any commercial deployment. Furthermore, the exact details in the original publications are not complete to the level of a protocol so as to allow one to apply them in a facile way to one's own microscope.
Novel fluorescent proteins and transgenic mice
Attempts to develop transgenic mice with RFP have failed, probably because fluorophores derived from the red coral FP DsRed were used. It seems that these proteins are not well tolerated transgenically. Recent development of RFP from evolution of jellyfish GFP might offer a solution to this problem(Chudakov et al., 2010). We believe that transgenic mice in which neocortical interneurons(Ascoli et al., 2008) labeled with RFP would be very useful, as these would complement H-line and M-line mice. FP-based indicators continue to be developed, including the first GE glutamate indicator that is effective for two-photon imaging in living mice.(Marvin et al., 2013). Whilst there are many GE indicators for protein activity that have been used for two-photon imaging in culture(Patterson and Yasuda, 2011), it is not clear how effective these are in vivo. For obvious reasons, GE membrane potential indicators have attracted a good deal of attention(Knopfel, 2012), but compared to GE calcium indicators these probes do not seem ready for longitudinal two-photon imaging in vivo. Since intracellular calcium concentration is a very poor surrogate for neuronal firing, this might be regarded as the single most important area for GE indicator development. It must be noted that the immediate distribution by the pioneers of these new GE proteins through Addgene and the Penn vector core is to be applauded
Software
Finally, we would like to highlight the pressing need for new software for automatic reconstruction of fluorescence imaging(Svoboda, 2011). Currently all spine counting from repetitive imaging must be done manually. This is tremendously tedious and time consuming. Moore's law implies that we will not lack computational power for automatic reconstruction and analysis, but the software gap is huge. The recent Diadem challenge from HHMI only served to highlight this(Baker and Ascoli, 2011). Such software should be open-source, and compatible across multiple OS platforms.
Supplementary Material
The video shows three slides of layer 5 pyramidal neurons from a male H-line mouse for which the cell bodies in layer 5 are shown in Figure 9. The first slide shows the sequence of 135 x/y planes gathered at 45 days after window implantation, with maximal intensity side projects in x/z and y/z. Each subsequent imaging time point gathered the same number of image frames. The second slide shows 3D reconstructions of the imaged volume at t = 45d and 63d. The final slide shows maximum intensity projections of the cell bodies in layer 5, with 12 cells numbered as in Figure 9.
Acknowledgments
In developing this guide we benefitted from a day's tutoring in Wenbiao Gan's laboratory, by seeing a working draft of an early version of the cranial window implant protocol kindly provided by Karl Svoboda in June 2008 and by discussion with several colleagues experienced in different forms of rodent survival surgery. The Ellis-Davies laboratory is supported by the National Institutes of Health (USA) grants GM053395 and NS069720. The bigenic mouse model (Fig 10) was initially developed by support from the PA CURE Fund. The two-photon microscopes used for most of our studies were supported by GM53395, the McKnight Endowment Fund and the Human Frontier Science Program (Program Grant RGP0071/2002-C). No institutional startup funds supported this equipment until 2010.
Footnotes
References
- Adam Y, Mizrahi A. Long-term imaging reveals dynamic changes in the neuronal composition of the glomerular layer. J Neurosci. 2011;31(22):7967–7973. doi: 10.1523/JNEUROSCI.0782-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal A, Dibaj P, Kassmann CM, Goebbels S, Nave KA, Schwab MH. In vivo imaging and noninvasive ablation of pyramidal neurons in adult NEX-CreERT2 mice. Cereb Cortex. 2012;22(7):1473–1486. doi: 10.1093/cercor/bhr214. [DOI] [PubMed] [Google Scholar]
- Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderon NC, Esposti F, Borghuis BG, Sun XR, Gordus A, Orger MB, Portugues R, Engert F, Macklin JJ, Filosa A, Aggarwal A, Kerr RA, Takagi R, Kracun S, Shigetomi E, Khakh BS, Baier H, Lagnado L, Wang SS, Bargmann CI, Kimmel BE, Jayaraman V, Svoboda K, Kim DS, Schreiter ER, Looger LL. Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci. 2012;32(40):13819–13840. doi: 10.1523/JNEUROSCI.2601-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amos WB, White JG. How the confocal laser scanning microscope entered biological research. Biol Cell. 2003;95(6):335–342. doi: 10.1016/s0248-4900(03)00078-9. [DOI] [PubMed] [Google Scholar]
- Andermann ML, Kerlin AM, Reid RC. Chronic cellular imaging of mouse visual cortex during operant behavior and passive viewing. Front Cell Neurosci. 2010;4:3. doi: 10.3389/fncel.2010.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascoli GA, Alonso-Nanclares L, Anderson SA, Barrionuevo G, Benavides-Piccione R, Burkhalter A, Buzsaki G, Cauli B, Defelipe J, Fairen A, Feldmeyer D, Fishell G, Fregnac Y, Freund TF, Gardner D, Gardner EP, Goldberg JH, Helmstaedter M, Hestrin S, Karube F, Kisvarday ZF, Lambolez B, Lewis DA, Marin O, Markram H, Munoz A, Packer A, Petersen CC, Rockland KS, Rossier J, Rudy B, Somogyi P, Staiger JF, Tamas G, Thomson AM, Toledo-Rodriguez M, Wang Y, West DC, Yuste R. Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nature reviews. 2008;9(7):557–568. doi: 10.1038/nrn2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacskai BJ, Wallen P, Lev-Ram V, Grillner S, Tsien RY. Activity-related calcium dynamics in lamprey motoneurons as revealed by video-rate confocal microscopy. Neuron. 1995;14(1):19–28. doi: 10.1016/0896-6273(95)90237-6. [DOI] [PubMed] [Google Scholar]
- Baker JL, Ascoli GA. Tracking the source of quantitative knowledge in neuroscience: a neuroinformatics role for computational models. Neuroinformatics. 2011;9(1):1–2. doi: 10.1007/s12021-011-9100-7. [DOI] [PubMed] [Google Scholar]
- Bhatt DH, Zhang S, Gan WB. Dendritic spine dynamics. Annu Rev Physiol. 2009;71:261–282. doi: 10.1146/annurev.physiol.010908.163140. [DOI] [PubMed] [Google Scholar]
- Bittner T, Fuhrmann M, Burgold S, Jung CK, Volbracht C, Steiner H, Mitteregger G, Kretzschmar HA, Haass C, Herms J. Gamma-secretase inhibition reduces spine density in vivo via an amyloid precursor protein-dependent pathway. J Neurosci. 2009;29(33):10405–10409. doi: 10.1523/JNEUROSCI.2288-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner T, Fuhrmann M, Burgold S, Ochs SM, Hoffmann N, Mitteregger G, Kretzschmar H, LaFerla FM, Herms J. Multiple events lead to dendritic spine loss in triple transgenic Alzheimer's disease mice. PLoS One. 2010;5(11):e15477. doi: 10.1371/journal.pone.0015477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28(16):4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botcherby EJ, Smith CW, Kohl MM, Debarre D, Booth MJ, Juskaitis R, Paulsen O, Wilson T. Aberration-free three-dimensional multiphoton imaging of neuronal activity at kHz rates. Proc Natl Acad Sci U S A. 2013;109(8):2919–2924. doi: 10.1073/pnas.1111662109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Boyd JD, Murphy TH. Longitudinal in vivo imaging reveals balanced and branch-specific remodeling of mature cortical pyramidal dendritic arbors after stroke. J Cereb Blood Flow Metab. 2010;30(4):783–791. doi: 10.1038/jcbfm.2009.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Li P, Boyd JD, Delaney KR, Murphy TH. Extensive turnover of dendritic spines and vascular remodeling in cortical tissues recovering from stroke. J Neurosci. 2007;27(15):4101–4109. doi: 10.1523/JNEUROSCI.4295-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgold S, Bittner T, Dorostkar MM, Kieser D, Fuhrmann M, Mitteregger G, Kretzschmar H, Schmidt B, Herms J. In vivo multiphoton imaging reveals gradual growth of newborn amyloid plaques over weeks. Acta Neuropathol. 2010;121(3):327–335. doi: 10.1007/s00401-010-0787-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callamaras N, Parker I. Construction of a confocal microscope for real-time x-y and x-z imaging. Cell Calcium. 1999;26(6):271–279. doi: 10.1054/ceca.1999.0085. [DOI] [PubMed] [Google Scholar]
- Cao VY, Ye Y, Mastwal SS, Lovinger DM, Costa RM, Wang KH. In vivo two-photon imaging of experience-dependent molecular changes in cortical neurons. J Vis Exp. 2013;71 doi: 10.3791/50148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze VE, White JG. Multiphoton excitation provides optical sections from deeper within scattering specimens than confocal imaging. Biophys J. 1998;75(4):2015–2024. doi: 10.1016/S0006-3495(98)77643-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JL, Villa KL, Cha JW, So PT, Kubota Y, Nedivi E. Clustered dynamics of inhibitory synapses and dendritic spines in the adult neocortex. Neuron. 2012;74(2):361–373. doi: 10.1016/j.neuron.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow DK, Groszer M, Pribadi M, Machniki M, Carmichael ST, Liu X, Trachtenberg JT. Laminar and compartmental regulation of dendritic growth in mature cortex. Nat Neurosci. 2009;12(2):116–118. doi: 10.1038/nn.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie RH, Bacskai BJ, Zipfel WR, Williams RM, Kajdasz ST, Webb WW, Hyman BT. Growth arrest of individual senile plaques in a model of Alzheimer's disease observed by in vivo multiphoton microscopy. J Neurosci. 2001;21(3):858–864. doi: 10.1523/JNEUROSCI.21-03-00858.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 2010;90(3):1103–1163. doi: 10.1152/physrev.00038.2009. [DOI] [PubMed] [Google Scholar]
- Conchello JA, Lichtman JW. Optical sectioning microscopy. Nature methods. 2005;2(12):920–931. doi: 10.1038/nmeth815. [DOI] [PubMed] [Google Scholar]
- Crowe SE, Ellis-Davies GC. In vivo characterization of a bigenic fluorescent mouse model of Alzheimer's disease with neurodegeneration. J Comp Neurol. 2013a;521(10):2181–2194. doi: 10.1002/cne.23306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe SE, Ellis-Davies GC. Spine pruning in 5xFAD mice starts on basal dendrites of layer 5 pyramidal neurons. Brain Struct Funct. 2013b doi: 10.1007/s00429-013-0518-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248(4951):73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Denk W, Svoboda K. Photon upmanship: why multiphoton imaging is more than a gimmick. Neuron. 1997;18(3):351–357. doi: 10.1016/s0896-6273(00)81237-4. [DOI] [PubMed] [Google Scholar]
- Dittgen T, Nimmerjahn A, Komai S, Licznerski P, Waters J, Margrie TW, Helmchen F, Denk W, Brecht M, Osten P. Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proc Natl Acad Sci U S A. 2004;101(52):18206–18211. doi: 10.1073/pnas.0407976101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombeck DA, Harvey CD, Tian L, Looger LL, Tank DW. Functional imaging of hippocampal place cells at cellular resolution during virtual navigation. Nat Neurosci. 2010;13(11):1433–1440. doi: 10.1038/nn.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draaijer A, Houpt PM. A standard video-rate confocal laser-scanning reflection and fluorescence microscope. Scanning. 1998;10:139–145. [Google Scholar]
- Drew PJ, Shih AY, Driscoll JD, Knutsen PM, Blinder P, Davalos D, Akassoglou K, Tsai PS, Kleinfeld D. Chronic optical access through a polished and reinforced thinned skull. Nature methods. 2010;7(12):981–984. doi: 10.1038/nmeth.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducros M, van 't Hoff M, Evrard A, Seebacher C, Schmidt EM, Charpak S, Oheim M. Efficient large core fiber-based detection for multi-channel two-photon fluorescence microscopy and spectral unmixing. J Neurosci Methods. 2011;198(2):172–180. doi: 10.1016/j.jneumeth.2011.03.015. [DOI] [PubMed] [Google Scholar]
- Engelbrecht CJ, Gobel W, Helmchen F. Enhanced fluorescence signal in nonlinear microscopy through supplementary fiber-optic light collection. Opt Express. 2009;17(8):6421–6435. doi: 10.1364/oe.17.006421. [DOI] [PubMed] [Google Scholar]
- Feng GP, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28(1):41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fu M, Zuo Y. Experience-dependent structural plasticity in the cortex. Trends Neurosci. 2011;34(4):177–187. doi: 10.1016/j.tins.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM, Kretzschmar H, Herms J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat Neurosci. 2010;13(4):411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann M, Mitteregger G, Kretzschmar H, Herms J. Dendritic pathology in prion disease starts at the synaptic spine. J Neurosci. 2007;27(23):6224–6233. doi: 10.1523/JNEUROSCI.5062-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobel W, Helmchen F. New angles on neuronal dendrites in vivo. J Neurophysiol. 2007;98(6):3770–3779. doi: 10.1152/jn.00850.2007. [DOI] [PubMed] [Google Scholar]
- Gobel W, Kampa BM, Helmchen F. Imaging cellular network dynamics in three dimensions using fast 3D laser scanning. Nature methods. 2007;4(1):73–79. doi: 10.1038/nmeth989. [DOI] [PubMed] [Google Scholar]
- Grewe BF, Langer D, Kasper H, Kampa BM, Helmchen F. High-speed in vivo calcium imaging reveals neuronal network activity with near-millisecond precision. Nature methods. 2010;7(5):399–405. doi: 10.1038/nmeth.1453. [DOI] [PubMed] [Google Scholar]
- Grewe BF, Voigt FF, van 't Hoff M, Helmchen F. Fast two-layer two-photon imaging of neuronal cell populations using an electrically tunable lens. Biomed Opt Express. 2011;2(7):2035–2046. doi: 10.1364/BOE.2.002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grienberger C, Konnerth A. Imaging calcium in neurons. Neuron. 2012;73(5):862–885. doi: 10.1016/j.neuron.2012.02.011. [DOI] [PubMed] [Google Scholar]
- Grutzendler J, Kasthuri N, Gan WB. Long-term dendritic spine stability in the adult cortex. Nature. 2002;420(6917):812–816. doi: 10.1038/nature01276. [DOI] [PubMed] [Google Scholar]
- Harvey CD, Coen P, Tank DW. Choice-specific sequences in parietal cortex during a virtual-navigation decision task. Nature. 2012;484(7392):62–68. doi: 10.1038/nature10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefendehl JK, Wegenast-Braun BM, Liebig C, Eicke D, Milford D, Calhoun ME, Kohsaka S, Eichner M, Jucker M. Long-term in vivo imaging of beta-amyloid plaque appearance and growth in a mouse model of cerebral beta-amyloidosis. J Neurosci. 2011;31(2):624–629. doi: 10.1523/JNEUROSCI.5147-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen F, Denk W. Deep tissue two-photon microscopy. Nat Methods. 2005;2(12):932–940. doi: 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Svoboda K, Denk W, Tank DW. In vivo dendritic calcium dynamics in deep-layer cortical pyramidal neurons. Nat Neurosci. 1999;2(11):989–996. doi: 10.1038/14788. [DOI] [PubMed] [Google Scholar]
- Hofer SB, Mrsic-Flogel TD, Bonhoeffer T, Hubener M. Experience leaves a lasting structural trace in cortical circuits. Nature. 2009;457(7227):313–317. doi: 10.1038/nature07487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Bonhoeffer T, Chow DK, Chuckowree J, De Paola V, Hofer SB, Hubener M, Keck T, Knott G, Lee WC, Mostany R, Mrsic-Flogel TD, Nedivi E, Portera-Cailliau C, Svoboda K, Trachtenberg JT, Wilbrecht L. Long-term, highresolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc. 2009;4(8):1128–1144. doi: 10.1038/nprot.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nature reviews. 2009;10(9):647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Wilbrecht L, Knott GW, Welker E, Svoboda K. Experience-dependent and cell-type-specific spine growth in the neocortex. Nature. 2006;441(7096):979–983. doi: 10.1038/nature04783. [DOI] [PubMed] [Google Scholar]
- Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K. Transient and persistent dendritic spines in the neocortex in vivo. Neuron. 2005;45(2):279–291. doi: 10.1016/j.neuron.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Huber D, Gutnisky DA, Peron S, O'Connor DH, Wiegert JS, Tian L, Oertner TG, Looger LL, Svoboda K. Multiple dynamic representations in the motor cortex during sensorimotor learning. Nature. 2012;484(7395):473–478. doi: 10.1038/nature11039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji N, Magee JC, Betzig E. High-speed, low-photodamage nonlinear imaging using passive pulse splitters. Nature methods. 2008;5(2):197–202. doi: 10.1038/nmeth.1175. [DOI] [PubMed] [Google Scholar]
- Ji N, Milkie DE, Betzig E. Adaptive optics via pupil segmentation for highresolution imaging in biological tissues. Nature methods. 2011;7(2):141–147. doi: 10.1038/nmeth.1411. [DOI] [PubMed] [Google Scholar]
- Kato HK, Chu MW, Isaacson JS, Komiyama T. Dynamic sensory representations in the olfactory bulb: modulation by wakefulness and experience. Neuron. 2012;76(5):962–975. doi: 10.1016/j.neuron.2012.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona G, Szalay G, Maak P, Kaszas A, Veress M, Hillier D, Chiovini B, Vizi ES, Roska B, Rozsa B. Fast two-photon in vivo imaging with three-dimensional random-access scanning in large tissue volumes. Nature methods. 2010;9(2):201–208. doi: 10.1038/nmeth.1851. [DOI] [PubMed] [Google Scholar]
- Keck T, Mrsic-Flogel TD, Vaz Afonso M, Eysel UT, Bonhoeffer T, Hubener M. Massive restructuring of neuronal circuits during functional reorganization of adult visual cortex. Nat Neurosci. 2008;11(10):1162–1167. doi: 10.1038/nn.2181. [DOI] [PubMed] [Google Scholar]
- Kelly EA, Majewska AK. Chronic imaging of mouse visual cortex using a thinned-skull preparation. J Vis Exp. 2010;44 doi: 10.3791/2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Nabekura J. Rapid synaptic remodeling in the adult somatosensory cortex following peripheral nerve injury and its association with neuropathic pain. J Neurosci. 2011;31(14):5477–5482. doi: 10.1523/JNEUROSCI.0328-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby PA, Srinivas Nadella KM, Silver RA. A compact Acousto-Optic Lens for 2D and 3D femtosecond based 2-photon microscopy. Opt Express. 2010;18(13):13721–13745. doi: 10.1364/OE.18.013720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP, Debnath ML, Holt DP, Wang Y, Hyman BT. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol. 2002;61(9):797–805. doi: 10.1093/jnen/61.9.797. [DOI] [PubMed] [Google Scholar]
- Knopfel T. Genetically encoded optical indicators for the analysis of neuronal circuits. Nature reviews. 2012;13(10):687–700. doi: 10.1038/nrn3293. [DOI] [PubMed] [Google Scholar]
- Kobat D, Durst ME, Nishimura N, Wong AW, Schaffer CB, Xu C. Deep tissue multiphoton microscopy using longer wavelength excitation. Opt Express. 2009;17(16):13354–13364. doi: 10.1364/oe.17.013354. [DOI] [PubMed] [Google Scholar]
- Lai CS, Franke TF, Gan WB. Opposite effects of fear conditioning and extinction on dendritic spine remodelling. Nature. 2012;483(7387):87–91. doi: 10.1038/nature10792. [DOI] [PubMed] [Google Scholar]
- Lam CK, Yoo T, Hiner B, Liu Z, Grutzendler J. Embolus extravasation is an alternative mechanism for cerebral microvascular recanalization. Nature. 2010;465(7297):478–482. doi: 10.1038/nature09001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WC, Chen JL, Huang H, Leslie JH, Amitai Y, So PT, Nedivi E. A dynamic zone defines interneuron remodeling in the adult neocortex. Proc Natl Acad Sci U S A. 2008;105(50):19968–19973. doi: 10.1073/pnas.0810149105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WC, Huang H, Feng G, Sanes JR, Brown EN, So PT, Nedivi E. Dynamic remodeling of dendritic arbors in GABAergic interneurons of adult visual cortex. PLoS biology. 2006;4(2):e29. doi: 10.1371/journal.pbio.0040029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendvai B, Stern EA, Chen B, Svoboda K. Experience-dependent plasticity of dendritic spines in the developing rat barrel cortex in vivo. Nature. 2000;404(6780):876–881. doi: 10.1038/35009107. [DOI] [PubMed] [Google Scholar]
- Li P, Murphy TH. Two-photon imaging during prolonged middle cerebral artery occlusion in mice reveals recovery of dendritic structure after reperfusion. J Neurosci. 2008;28(46):11970–11979. doi: 10.1523/JNEUROSCI.3724-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Condello C, Schain A, Harb R, Grutzendler J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-beta phagocytosis. J Neurosci. 2010;30(50):17091–17101. doi: 10.1523/JNEUROSCI.4403-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livneh Y, Mizrahi A. Experience-dependent plasticity of mature adult-born neurons. Nat Neurosci. 2012;15(1):26–28. doi: 10.1038/nn.2980. [DOI] [PubMed] [Google Scholar]
- Ma Y, Hu H, Berrebi AS, Mathers PH, Agmon A. Distinct subtypes of somatostatin-containing neocortical interneurons revealed in transgenic mice. J Neurosci. 2006;26(19):5069–5082. doi: 10.1523/JNEUROSCI.0661-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13(1):133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainen ZF, Maletic-Savatic M, Shi SH, Hayashi Y, Malinow R, Svoboda K. Two-photon imaging in living brain slices. Methods. 1999;18(2):231–239. doi: 10.1006/meth.1999.0776. 181. [DOI] [PubMed] [Google Scholar]
- Majewska A, Yiu G, Yuste R. A custom-made two-photon microscope and deconvolution system. Pflugers Arch. 2000;441(2-3):398–408. doi: 10.1007/s004240000435. [DOI] [PubMed] [Google Scholar]
- Mank M, Griesbeck O. Genetically encoded calcium indicators. Chem Rev. 2008;108(5):1550–1564. doi: 10.1021/cr078213v. [DOI] [PubMed] [Google Scholar]
- Mank M, Santos AF, Direnberger S, Mrsic-Flogel TD, Hofer SB, Stein V, Hendel T, Reiff DF, Levelt C, Borst A, Bonhoeffer T, Hubener M, Griesbeck O. A genetically encoded calcium indicator for chronic in vivo two-photon imaging. Nature methods. 2008;5(9):805–811. doi: 10.1038/nmeth.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis DJ, Lutcke H, Schulz K, Haiss F, Weber B, Kugler S, Hasan MT, Helmchen F. Reorganization of cortical population activity imaged throughout long-term sensory deprivation. Nat Neurosci. 2012;15(11):1539–1546. doi: 10.1038/nn.3240. [DOI] [PubMed] [Google Scholar]
- Marvin JS, Borghuis BG, Tian L, Cichon J, Harnett MT, Akerboom J, Gordus A, Renninger SL, Chen TW, Bargmann CI, Orger MB, Schreiter ER, Demb JB, Gan WB, Hires SA, Looger LL. An optimized fluorescent probe for visualizing glutamate neurotransmission. Nature methods. 2013;10(2):162–170. doi: 10.1038/nmeth.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008;451(7179):720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenbock G. The optogenetic catechism. Science. 2009;326(5951):395–399. doi: 10.1126/science.1174520. [DOI] [PubMed] [Google Scholar]
- Miesenbock G, Rothman JE. Patterns of synaptic activity in neural networks recorded by light emission from synaptolucins. Proc Natl Acad Sci U S A. 1997;94(7):3402–3407. doi: 10.1073/pnas.94.7.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M. In vivo imaging of the diseased nervous system. Nature reviews. 2006;7(6):449–463. doi: 10.1038/nrn1905. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, Tsien RY. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388(6645):882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- Mizrahi A, Katz LC. Dendritic stability in the adult olfactory bulb. Nat Neurosci. 2003;6(11):1201–1207. doi: 10.1038/nn1133. [DOI] [PubMed] [Google Scholar]
- Mostany R, Chowdhury TG, Johnston DG, Portonovo SA, Carmichael ST, Portera-Cailliau C. Local hemodynamics dictate long-term dendritic plasticity in peri-infarct cortex. J Neurosci. 2010;30(42):14116–14126. doi: 10.1523/JNEUROSCI.3908-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostany R, Portera-Cailliau C. A craniotomy surgery procedure for chronic brain imaging. J Vis Exp. 2008;12 doi: 10.3791/680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostany R, Portera-Cailliau C. Absence of large-scale dendritic plasticity of layer 5 pyramidal neurons in peri-infarct cortex. J Neurosci. 2011;31(5):1734–1738. doi: 10.1523/JNEUROSCI.4386-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Li P, Betts K, Liu R. Two-photon imaging of stroke onset in vivo reveals that NMDA-receptor independent ischemic depolarization is the major cause of rapid reversible damage to dendrites and spines. J Neurosci. 2008;28(7):1756–1772. doi: 10.1523/JNEUROSCI.5128-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng M, Roorda RD, Lima SQ, Zemelman BV, Morcillo P, Miesenbock G. Transmission of olfactory information between three populations of neurons in the antennal lobe of the fly. Neuron. 2002;36(3):463–474. doi: 10.1016/s0896-6273(02)00975-3. [DOI] [PubMed] [Google Scholar]
- Nguyen QT, Callamaras N, Hsieh C, Parker I. Construction of a two-photon microscope for video-rate Ca(2+) imaging. Cell Calcium. 2001;30(6):383–393. doi: 10.1054/ceca.2001.0246. [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci. 2001;21(14):5139–5146. doi: 10.1523/JNEUROSCI.21-14-05139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor DH, Peron SP, Huber D, Svoboda K. Neural activity in barrel cortex underlying vibrissa-based object localization in mice. Neuron. 2010;67(6):1048–1061. doi: 10.1016/j.neuron.2010.08.026. [DOI] [PubMed] [Google Scholar]
- Oheim M, Beaurepaire E, Chaigneau E, Mertz J, Charpak S. Two-photon microscopy in brain tissue: parameters influencing the imaging depth. J Neurosci Methods. 2001;111(1):29–37. doi: 10.1016/s0165-0270(01)00438-1. [DOI] [PubMed] [Google Scholar]
- Ohkura M, Sasaki T, Kobayashi C, Ikegaya Y, Nakai J. An improved genetically encoded red fluorescent Ca2+ indicator for detecting optically evoked action potentials. PLoS ONE. 2012;7(7):e39933. doi: 10.1371/journal.pone.0039933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva AA, Jr, Jiang M, Lam T, Smith KL, Swann JW. Novel hippocampal interneuronal subtypes identified using transgenic mice that express green fluorescent protein in GABAergic interneurons. J Neurosci. 2000;20(9):3354–3368. doi: 10.1523/JNEUROSCI.20-09-03354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osakada F, Mori T, Cetin AH, Marshel JH, Virgen B, Callaway EM. New rabies virus variants for monitoring and manipulating activity and gene expression in defined neural circuits. Neuron. 2011;71(4):617–631. doi: 10.1016/j.neuron.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan F, Aldridge GM, Greenough WT, Gan WB. Dendritic spine instability and insensitivity to modulation by sensory experience in a mouse model of fragile X syndrome. Proc Natl Acad Sci U S A. 2010;107(41):17768–17773. doi: 10.1073/pnas.1012496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson M, Yasuda R. Signalling pathways underlying structural plasticity of dendritic spines. Br J Pharmacol. 2011;163(8):1626–1638. doi: 10.1111/j.1476-5381.2011.01328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K. ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng Online. 2003;2:13. doi: 10.1186/1475-925X-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy GD, Saggau P. Fast three-dimensional laser scanning scheme using acousto-optic deflectors. J Biomed Opt. 2005;10(6):064038. doi: 10.1117/1.2141504. [DOI] [PubMed] [Google Scholar]
- Roberts TF, Tschida KA, Klein ME, Mooney R. Rapid spine stabilization and synaptic enhancement at the onset of behavioural learning. Nature. 2010;463(7283):948–952. doi: 10.1038/nature08759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romoser VA, Hinkle PM, Persechini A. Detection in living cells of Ca2+-dependent changes in the fluorescence emission of an indicator composed of two green fluorescent protein variants linked by a calmodulin-binding sequence. A new class of fluorescent indicators. J Biol Chem. 1997;272(20):13270–13274. doi: 10.1074/jbc.272.20.13270. [DOI] [PubMed] [Google Scholar]
- Salome R, Kremer Y, Dieudonne S, Leger JF, Krichevsky O, Wyart C, Chatenay D, Bourdieu L. Ultrafast random-access scanning in two-photon microscopy using acousto-optic deflectors. J Neurosci Methods. 2006;154(1-2):161–174. doi: 10.1016/j.jneumeth.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Shih AY, Blinder P, Tsai PS, Friedman B, Stanley G, Lyden PD, Kleinfeld D. The smallest stroke: occlusion of one penetrating vessel leads to infarction and a cognitive deficit. Nat Neurosci. 2013;16(1):55–63. doi: 10.1038/nn.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Driscoll JD, Drew PJ, Nishimura N, Schaffer CB, Kleinfeld D. Two-photon microscopy as a tool to study blood flow and neurovascular coupling in the rodent brain. J Cereb Blood Flow Metab. 2012a;32(7):1277–1309. doi: 10.1038/jcbfm.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Mateo C, Drew PJ, Tsai PS, Kleinfeld D. A polished and reinforced thinned-skull window for long-term imaging of the mouse brain. J Vis Exp. 2012b;61 doi: 10.3791/3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel MS, Isacoff EY. A genetically encoded optical probe of membrane voltage. Neuron. 1997;19(4):735–741. doi: 10.1016/s0896-6273(00)80955-1. [DOI] [PubMed] [Google Scholar]
- Svoboda K. The past, present, and future of single neuron reconstruction. Neuroinformatics. 2011;9(2-3):97–98. doi: 10.1007/s12021-011-9097-y. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Denk W, Kleinfeld D, Tank DW. In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature. 1997;385(6612):161–165. doi: 10.1038/385161a0. [DOI] [PubMed] [Google Scholar]
- Theer P, Denk W. On the fundamental imaging-depth limit in two-photon microscopy. J Opt Soc Am A Opt Image Sci Vis. 2006;23(12):3139–3149. doi: 10.1364/josaa.23.003139. [DOI] [PubMed] [Google Scholar]
- Theer P, Hasan MT, Denk W. Two-photon imaging to a depth of 1000 microm in living brains by use of a Ti:Al2O3 regenerative amplifier. Opt Lett. 2003;28(12):1022–1024. doi: 10.1364/ol.28.001022. [DOI] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, Bargmann CI, Jayaraman V, Svoboda K, Looger LL. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nature methods. 2009;6(12):875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng GP, Sanes JR, Welker E, Svoboda K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420(6917):788–794. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004;7(11):1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- van Versendaal D, Rajendran R, Saiepour MH, Klooster J, Smit-Rigter L, Sommeijer JP, De Zeeuw CI, Hofer SB, Heimel JA, Levelt CN. Elimination of inhibitory synapses is a major component of adult ocular dominance plasticity. Neuron. 2012;74(2):374–383. doi: 10.1016/j.neuron.2012.03.015. [DOI] [PubMed] [Google Scholar]
- Xu HT, Pan F, Yang G, Gan WB. Choice of cranial window type for in vivo imaging affects dendritic spine turnover in the cortex. Nat Neurosci. 2007;10(5):549–551. doi: 10.1038/nn1883. [DOI] [PubMed] [Google Scholar]
- Xu T, Yu X, Perlik AJ, Tobin WF, Zweig JA, Tennant K, Jones T, Zuo Y. Rapid formation and selective stabilization of synapses for enduring motor memories. Nature. 2009;462(7275):915–919. doi: 10.1038/nature08389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pan F, Gan WB. Stably maintained dendritic spines are associated with lifelong memories. Nature. 2009a;462(7275):920–924. doi: 10.1038/nature08577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pan F, Parkhurst CN, Grutzendler J, Gan W-B. Thinned-skull cranial window technique for long-term imaging of the cortex in live mice. Nature protocols. 2010;5(2):201–208. doi: 10.1038/nprot.2009.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pan F, Parkhurst CN, Grutzendler J, Gan WB. Thinned-skull cranial window technique for long-term imaging of the cortex in live mice. Nature protocols. 2009b;5(2):201–208. doi: 10.1038/nprot.2009.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder EJ, Kleinfeld D. Cortical imaging though the intact mouse skull using two-photon excitation laser scanning microscopy. Micro Res Tech. 2002;56:304–305. doi: 10.1002/jemt.10002. [DOI] [PubMed] [Google Scholar]
- Yu JN, Anderson CT, Kiritani T, Sheets PL, Wokosin DL, Wood L, Shepherd GMG. Local-circuit phenotypes of layer 5 neurons in motor-frontal cortex of YFP-H mice. Frontiers in neural circuits 2. 2008 doi: 10.3389/neuro.04.006.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Denk W. Dendritic spines as basic functional units of neuronal integration. Nature. 1995;375(6533):682–684. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]
- Zhang S, Boyd J, Delaney K, Murphy TH. Rapid reversible changes in dendritic spine structure in vivo gated by the degree of ischemia. J Neurosci. 2005;25(22):5333–5338. doi: 10.1523/JNEUROSCI.1085-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Araki S, Wu J, Teramoto T, Chang YF, Nakano M, Abdelfattah AS, Fujiwara M, Ishihara T, Nagai T, Campbell RE. An expanded palette of genetically encoded Ca indicators. Science. 2011;333(6051):1888–1891. doi: 10.1126/science.1208592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Yang G, Kwon E, Gan WB. Long-term sensory deprivation prevents dendritic spine loss in primary somatosensory cortex. Nature. 2005;436(7048):261–265. doi: 10.1038/nature03715. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The video shows three slides of layer 5 pyramidal neurons from a male H-line mouse for which the cell bodies in layer 5 are shown in Figure 9. The first slide shows the sequence of 135 x/y planes gathered at 45 days after window implantation, with maximal intensity side projects in x/z and y/z. Each subsequent imaging time point gathered the same number of image frames. The second slide shows 3D reconstructions of the imaged volume at t = 45d and 63d. The final slide shows maximum intensity projections of the cell bodies in layer 5, with 12 cells numbered as in Figure 9.