

Abstract
Directed migration of neurons is critical in the normal and pathological development of the brain and central nervous system. In C. elegans, the bilateral Q neuroblasts, QR on the right and QL on the left, migrate anteriorly and posteriorly, respectively. Initial protrusion and migration of the Q neuroblasts is autonomously controlled by the transmembrane proteins UNC-40/DCC, PTP-3/LAR, and MIG-21. As QL migrates posteriorly, it encounters and EGL-20/Wnt signal that induces MAB-5/Hox expression that drives QL descendant posterior migration. QR migrates anteriorly away from EGL-20/Wnt and does not activate MAB-5/Hox, resulting in anterior QR descendant migration. A forward genetic screen for new mutations affecting initial Q migrations identified alleles of cdh-4, which caused defects in both QL and QR directional migration similar to unc-40, ptp-3, and mig-21. Previous studies showed that in QL, PTP-3/LAR and MIG-21 act in a pathway in parallel to UNC-40/DCC to drive posterior QL migration. Here we show genetic evidence that CDH-4 acts in the PTP-3/MIG-21 pathway in parallel to UNC-40/DCC to direct posterior QL migration. In QR, the PTP-3/MIG-21 and UNC-40/DCC pathways mutually inhibit each other, allowing anterior QR migration. We report here that CDH-4 acts in both the PTP-3/MIG-21 and UNC-40/DCC pathways in mutual inhibition in QR, and that CDH-4 acts cell-non-autonomously. Interaction of CDH-4 with UNC-40/DCC in QR but not QL represents an inherent left-right asymmetry in the Q cells, the nature of which is not understood. We conclude that CDH-4 might act as a permissive signal for each Q neuroblast to respond differently to anterior-posterior guidance information based upon inherent left-right asymmetries in the Q neuroblasts.
Introduction
Directed neuronal migration is of fundamental importance in nervous system development. The Q neuroblasts of Caenorhabditis elegans are an established and useful model for understanding the molecular mechanisms of directed neuronal migration. The Q neuroblasts are born embryonically in the posterior lateral region of the worm on the right (QR) and left side (QL), and are the sister cells of the V5 hypodermal seam cells (Chalfie and Sulston, 1981; Sulston and Horvitz, 1977). QR and its descendants migrate anteriorly, and QL and descendants migrate posteriorly. Q migration can be divided into two steps, the first of which involves the protrusion and migration of the Q cells, QR to the anterior over the V4 hypodermal seam cell and QL to the posterior over V5 (Chapman et al., 2008; Honigberg and Kenyon, 2000). The Q cells then undergo their first division. The second stage of migration is regulated by EGL-20/Wnt signaling and the Hox molecule MAB-5 (Chalfie et al., 1983; Eisenmann, 2005; Harris et al., 1996; Herman, 2003; Kenyon, 1986; Korswagen et al., 2000; Salser and Kenyon, 1992; Whangbo and Kenyon, 1999). As QL migrates to the posterior, it encounters an EGL-20/Wnt signal from posterior cells, which through canonical Wnt signaling, activates expression of MAB-5/Hox in QL. QR migrates anteriorly away from the EGL-20/Wnt signal and does not activate MAB-5/Hox. MAB-5/Hox is a determinant for further posterior migration of QL descendants. QR continues anterior migration because it does not express MAB-5/Hox.
Initial Q neuroblast protrusion and migration resembles neuroblast migration in the developing vertebrate central nervous system, which extend leading processes followed by nuclear translocation in a saltatory fashion (reviewed in (Solecki et al., 2006)). At 1–2.5 h after hatching to L1 larvae, QR extends a protrusion anteriorly over V4, and QL posteriorly over V5. At 3–3.5 h post-hatching, the cell bodies follow the protrusions and migrate over the respective seam cells. At 4–4.5 h post hatching, the Q cells divide.
Clues about the molecules that control initial Q neuroblast directed protrusion and migration were first provided by (Honigberg and Kenyon, 2000), who showed that the Immunoglobulin-superfamily receptor UNC-40, similar to vertebrate Deleted in Colorectal Cancer, DCC, was required for directed protrusion and migration. Subsequent work delineated a group of transmembrane molecules that interact genetically in regulating Q directional migration, including UNC-40/DCC, the LAR receptor protein tyrosine phosphatase PTP-3, and the small thrombospondin type I-repeat containing protein MIG-21 (Honigberg and Kenyon, 2000; Middelkoop et al., 2012; Sundararajan and Lundquist, 2012). Mutations in all three genes cause misdirected QL and QR migrations. In QL, UNC-40/DCC acts redundantly in parallel to MIG-21 and PTP-3 in posterior QL migration (Middelkoop et al., 2012; Sundararajan and Lundquist, 2012). These molecules interact distinctly in QR, as genetic analysis indicates that UNC-40 and PTP-3/MIG-21 mutually inhibit each other’s roles in posterior migration, allowing anterior migration of QR (Sundararajan and Lundquist, 2012). Finally, cell autonomy experiments indicate that UNC-40/DCC, PTP-3/LAR, and MIG-21 act autonomously in the Q cells (Sundararajan and Lundquist, 2012), possibly as receptors for extracellular guidance information. Other molecules have been identified that act in cytoplasmic signaling involving Q cell migrations, including the DPY-19 C-mannosyltransferase that glycosylates thrombospondin repeat proteins including MIG-21 (Buettner et al., 2013; Honigberg and Kenyon, 2000), the MIG-15 NIK-family kinase (Chapman et al., 2008), the Rac GTPases CED-10 and MIG-2, and the GTP exchange factors UNC-73/Trio and PIX-1/PIX (Dyer et al., 2010). These molecules might act downstream of receptor signals to regulate cellular and or cytoskeletal polarity in initial Q migrations.
To identify genes that interact with unc-40, ptp-3 and mig-21 in in QR and QL migration, we performed a forward genetic screen for mutations that disrupted both QR and QL directional migration. We isolated three novel mutations in cdh-4 gene, which encodes a cadherin repeat-containing transmembrane protein most similar to the Fat family of cadherins (Ackley, 2013; Najarro et al., 2012; Schmitz et al., 2008). In C. elegans, CDH-4 has been implicated in axon development and cell migration, including migration of Q cell descendants (Najarro et al., 2012; Schmitz et al., 2008). In Drosophila, Fat is thought to coordinate cellular asymmetry generated by the core planar cell polarity pathway components with tissue and organismal polarity in a non-autonomous fashion, and to be distributed asymmetrically across imaginal disc primordia in response to Wnt signaling (Cho and Irvine, 2004; Loveless and Hardin, 2012; Ma et al., 2003; Matakatsu and Blair, 2004; Rawls et al., 2002; Strutt and Strutt, 2002; Tanoue and Takeichi, 2004; Thomas and Strutt, 2012; Yang et al., 2002).
In C. elegans, initial Q cell migration is independent of EGL-20/Wnt and MAB-5/Hox (Chapman et al., 2008), although the initial migration can influence the expression of MAB-5 in the Q descendants (Middelkoop et al., 2012). Furthermore, CDH-4 acts independently of MAB-5/Hox in Q descendant migration (Schmitz et al., 2008). Consistent with Wnt-independence of initial Q migrations and of CDH-4 function, we found that the triple Wnt mutant egl-20 cwn-2; cwn-1 displayed severe Q descendant migration defects but no initial directional migration defects. To determine if CDH-4 acts with the previously-defined signaling system of UNC-40/DCC, PTP-3/LAR and MIG-21 in directing early Q neuroblast migration, we analyzed double and triple mutants. In QL migration, CDH-4 acted genetically in the PTP-3/LAR pathway in parallel to UNC-40/DCC in posterior migration. In QR migration, CDH-4 acted genetically in both the PTP-3/LAR and UNC-40/DCC pathways in mutual inhibition of posterior migration. Mosaic analysis indicated that CDH-4 acts cell-non-autonomously to control QR and QL migration, possibly providing extracellular information to the Q cells about migration. Our results suggest a differential genetic interaction of CDH-4 with UNC-40/DCC in QL versus QR, acting in parallel in QL and acting with both PTP-3/LAR and UNC-40/DCC in QR in mutual inhibition. The nature of this inherent left-right asymmetry in the Q cells is unknown, but QL and QR display other left-right asymmetries, such as sensitivity to the EGL-20/Wnt signal and to MAB-5/Hox (Middelkoop et al., 2012; Whangbo and Kenyon, 1999). Possibly, an as-yet unidentified molecule that is expressed in QR but not QL mediates CDH-4 interaction with UNC-40/DCC in QR but not QL, thus establishing the mutual inhibition mechanism between PTP-3/LAR and UNC-40/DCC that allows anterior migration of QR.
Results
cdh-4 mutations were identified in a screen for neuronal migration defects
Our earlier work showed that the transmembrane molecules UNC-40/DCC, PTP-3/LAR and MIG-21 act cell-autonomously in a genetic pathway directing anterior-posterior Q neuroblast migrations (Sundararajan and Lundquist, 2012). To identify new genes that could function with unc-40, ptp-3 and mig-21, we performed an ethyl methansulfonate forward genetic screen for new mutations affecting Q migrations (see Materials and Methods), using the positions of Q descendant neurons AQR and PQR. AQR and PQR are descendants of QR and QL, respectively, and make the longest anterior and posterior migrations of Q descendants (Sulston and Horvitz, 1977; White et al., 1986). AQR migrates anteriorly to a position near the anterior deirid just behind the posterior pharyngeal bulb, and PQR migrates into the phasmid ganglion posterior to the anus. Directional migrations of AQR and PQR are disrupted when earlier Q migrations are defective, and defects in AQR and PQR migrations serve as a proxy for earlier defects in Q neuroblast migrations (Chapman et al., 2008). We focused on new mutations that affected both AQR and PQR directional migration, as do unc-40, ptp-3, and mig-21. The new mutations lq56, lq83, and lq97 were identified (Table 1). lq56 and lq83 were mapped to linkage group III by single nucleotide polymorphism (snp) mapping against the CB4856 Hawaiian background (Davis et al., 2005). The strains were then subject to whole genome sequencing (see Methods) to identify potential mutations. lq97 was mapped to LGIII and sequenced using the CloudMap strategy (see Materials and Methods) (Minevich et al., 2012). Each of the three strains carried a novel premature stop codon in the cdh-4 gene on LGIII (Figure 1A). The lesions were confirmed by polymerase chain reaction of the genomic region and Sanger sequencing.
Table 1.
The Fat-like cadherin CDH-4 controls AQR and PQR migration
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||||||
| Genotype | AQR position (%) | PQR position (%) | ||||||||||
|
|
|
|||||||||||
| 1 | 2 | 3 | 4 | 5 | N | 1 | 2 | 3 | 4 | 5 | N | |
|
|
|
|||||||||||
| wild-type | 100 | 0 | 0 | 0 | 0 | 100 | 0 | 0 | 0 | 0 | 100 | 100 |
| cdh-4(lq56) | 39 | 3 | 2 | 7 | 49 | 100 | 28 | 3 | 4 | 4 | 60 | 100 |
| cdh-4(lq56); Ex[cdh-4(+)] | 90* | 0 | 1 | 0 | 9 | 100 | 2* | 0 | 1 | 1 | 96 | 100 |
| cdh-4(lq83) | 48 | 2 | 0 | 3 | 47 | 100 | 23 | 0 | 0 | 2 | 75 | 100 |
| cdh-4(lq97) | 80** | 3 | 1 | 0 | 16 | 205 | 19 | 2 | 1 | 1 | 77 | 205 |
| cdh-4(lq97); smg-1(RNAi) | 82 | 3 | 1 | 0 | 14 | 200 | 13 | 1 | 1 | 1 | 84*** | 200 |
| cdh-4(rh310) | 50 | 1 | 1 | 2 | 46 | 100 | 19 | 0 | 0 | 2 | 79 | 100 |
| cdh-4(ok1323) | 45 | 0 | 2 | 1 | 52 | 100 | 24 | 0 | 1 | 0 | 75 | 100 |
| cdh-4(hd40) | 42 | 1 | 1 | 2 | 54 | 100 | 20 | 0 | 0 | 1 | 80 | 100 |
| cdh-4(RNAi) | 100 | 0 | 0 | 0 | 0 | 100 | 0 | 0 | 0 | 0 | 100 | 100 |
| unc-40(n324) | 96 | 1 | 0 | 0 | 3 | 76 | 32 | 3 | 8 | 6 | 51 | 76 |
|
unc-40(n324); cdh-4(RNAi) egl-20(n585) |
99 | 0 | 0 | 1 | 0 | 100 | 76 | 4 | 2 | 5 | 13**** | 100 |
| cwn-2(ok895); cwn-1(ok546) | 39 | 49 | 10 | 2 | 0 | 100 | 3 | 16 | 19 | 25 | 37 | 100 |
p < 0.0001 compared to other cdh-4(lq56).
p < 0.001 compared to other cdh-4 alleles lq56, lq83, rh310, ok1323, and hd40.
p = 0.0416 compared to cdh-4(lq97) alone.
p < 0.0001 compared to unc-40(n324) alone.
Figure 1. CDH-4 is a Fat-like cadherin.

A) A diagram representing the genomic region of the cdh-4A isoform. Exons are indicated by boxes, and introns by lines. Positions of the lesions are marked and the corresponding nucleotide changes are noted. lq56 was a T to A transversion resulting in a leucine to stop (TTG to TAG) at LGII position 4525658 in Wormbase WS240. lq83 was a C to T transition resulting in an arginine to stop (CGA to TGA) at position 4521154. lq97 was C to T transition resulting in an arginine to stop (CGA to TGA) at position 4535721. B) A diagram of the 4292 residue CDH-4A polypeptide. The position of the stop codons introduced by lq56, lq83, and lq97 are indicated.
CDH-4 is a member of the Cadherin superfamily most similar to Fat, a transmembrane molecule with multiple extracellular cadherin repeats, EGF-like repeats, and a laminin G domain (Figure 1B) (Schmitz et al., 2008). lq83 was in the second exon, lq56 was in the eighth exon that codes for the sixth cadherin repeat, and lq97 was in penultimate exon eighteen, introducing a premature stop thirty codons upstream of the region coding for the transmembrane domain (Figure 1A, B). lq56 and lq83 failed to complement each other and the previously identified cdh-4(hd40) mutation for AQR and PQR migration (data not shown), and a fosmid transgene harboring a wild-type cdh-4(+) gene rescued AQR and PQR migration defects of lq56 animals (Table 1).
Previous studies showed that CDH-4 is required for migration of the Q descendants AVM/PVM and SDQL/R (Schmitz et al., 2008). We found that AQR often migrated posteriorly and PQR often migrated anteriorly in cdh-4 mutants, including in the previously-characterized alleles rh310, ok1323, and hd40 (Schmitz et al., 2008), and the new alleles lq56, lq83, and lq97 (Table 1). The AQR migration defects of lq97 were significantly less severe than lq56, lq83, rh310, ok1323, and hd40 (Table 1), indicating that the lq97 allele might retain some cdh-4 function. That the lq97 premature stop is in the second-to-the-last exon, it is possible that some lq97 transcripts escape nonsense mediated mRNA decay (Popp and Maquat, 2013) and produce a truncated CDH-4 protein with a signal sequence but no transmembrane domain or cytoplasmic region. This putative truncated protein would be expected to be secreted, and might retain some function, resulting in the less-severe lq97 phenotype. Double mutants of lq97 with smg-1(e1228) (Grimson et al., 2004) and smg-2(e2008) (Johns et al., 2007) mRNA surveillance mutants were lethal, suggesting that in the absence of nonsense-mediated decay, a truncated CDH-4 is produced that results in lethality. smg-1 feeding RNAi slightly but significantly suppressed PQR migration defects of lq97 (Table 1). Together, these results suggest that cdh-4(lq97) might produce a truncated, secreted version of CDH-4 that is functional.
CDH-4 is required for anterior-posterior Q neuroblast migrations
We showed previously that defects in Q descendent AQR and PQR migrations can be due to defects in initial QR and QL protrusions and migrations (Chapman et al., 2008; Dyer et al., 2010; Sundararajan and Lundquist, 2012). We next analyzed the early Q neuroblast migrations in the putative null cdh-4 (lq56) and cdh-4(lq83) using the scm::GFP::CAAX reporter to observe QR and QL migration as described previously (Chapman et al., 2008). In wild-type, at 1–2.5 h post hatching, QR and QL extend protrusions anteriorly and posteriorly over seam cells V4 and V5, respectively (Figure 2A, E). At 3–3.5 h post hatching, the cell bodies follow the protrusions and migrate over the seam cells. At 4–4.5 h post hatching, the Q cells divide on top of their respective seam cells to produce two daughter cells (Figure 2B, F) (Chapman et al., 2008; Dyer et al., 2010; Ou and Vale, 2009; Sundararajan and Lundquist, 2012). In cdh-4 mutants, we observed directional migration defects and failures to migrate of both QR and QL. Figure 2C shows a QR that has migrated posteriorly over V5 at 3–3.5h, and figure 2D shows a QR that has divided over V5 at 4–4.5h. Figure 2G shows a QL that protruded anteriorly over V4 at 1–2.5 h, and Figure 2H a QL that has divided between V4 and V5, but in contact with the anterior V4.
Figure 2. Q neuroblast migration defects in cdh-4 mutants.

Micrographs of animals with scm::myr::gfp expression in the Q cells and seam cells. Anterior is left, and dorsal is up. The tracing below each micrograph indicates the position of the Q cells and seam cells. (A–D) QR migration in wild-type and cdh-4. A) A QR of a wild-type L1 animal at 2–2.5h post-hatch protruded anteriorly over V4. B) A wild-type QR at 4–4.5 h post-hatch divided over V4. C) A cdh-4(lq56) mutant QR migrated posteriorly over V5 at 3–3.5 h post-hatch. D) A cdh-4(lq56) mutant QR divided over V5 at 4–4.5 h post-hatch. (E–H) QL migration in wild-type and cdh-4. E) A wild-type QL at 2–1.5 h post-hatch protruded posteriorly over V5. F) A wild-type QL at 4–4.5 h post-hatch divided over V5. G) A cdh-4(lq83) QL at 2–2.5 h post-hatch protruded anteriorly over V4. H) A cdh-4(lq56) mutant QL divided over V4 at 4–4.5 h post-hatch.
We quantified the extent of QR and QL migration defects by determining the cell position upon division at 4–4.5 h (Figures 3 and 4). Defects observed at this stage were similar in percentage to defects at the protrusion and migration stages (data not shown) (Sundararajan and Lundquist, 2012). In wild type, no migration defects were observed in QR and QL (Figure 3A and 4A). In putative null cdh-4(lq56 and lq83) mutants, 42% and 27% of QLs migrated anteriorly and divided over V4 (Figure 3A), and 72% and 73% of QRs migrated posteriorly and divided over V5 (Figure 4A). These results suggest that CDH-4 is required for directional migration of both QR to the anterior and QL to the posterior. QR posterior migration was greater than 50% in each mutant, suggesting that QR migration was not randomized. Rather, this result suggests that CDH-4 might alter QR’s response to A-P guidance information.
Figure 3. Quantification of QL migration in cdh-4 mutants.


Graphs represent the division stage of early QL neuroblast migrations at 4–4.5 hours post hatching. Genotype is the Y-axis, and the percentage of defective anterior QL migration and division is the X-axis. Migration was scored as defective if QL migration was reversed and it divided while in contact with the anterior V4 seam cell. Failure to migrate, resulting in division between the seam cells, was also observed in cdh-4 mutants but is not included in the graphs. unc-40(RNAi) and ptp-3(RNAi) represent the Pscm::unc-40(RNAi) and Pscm::ptp-3(RNAi) transgenes (Sundararajan et al., 2012), and cdh-4 RNAi by feeding is indicated (see Materials and Methods). The error bars represent two times the standard error of proportion, and the statistical difference between the genotypes were determined by Fisher’s Exact test. Twenty-five animals or more were scored for each genotype. A) cdh-4 double mutants with unc-40. B) cdh-4 double mutants with ptp-3 and mig-21.
Figure 4. Quantification of QR migration in cdh-4 mutants.


Graphs represent the division stage of early QR neuroblast migrations at 4–4.5 hours post hatching. Genotype is the Y-axis, and the percentage of defective posterior QR migration and division is the X-axis. Migration was scored as defective if QR migration was reversed and it divided while in contact with the posterior V5 seam cell. Failure to migrate, resulting in division between the seam cells, was also observed in cdh-4 mutants but is not included in the graphs. unc-40RNAi and ptp-3(RNAi) represent the Pscm::unc-40(RNAi) and Pscm::ptp-3(RNAi) transgenes (Sundararajan et al., 2012), and cdh-4 RNAi by feeding is indicated (see Materials and Methods). The error bars represent two times the standard error of proportion, and the statistical difference between the genotypes were determined by Fisher’s Exact test. Twenty-five animals or more were scored for each genotype. A) cdh-4 double mutants with unc-40. B) cdh-4 double mutants with ptp-3 and mig-21.
cdh-4(lq97) showed significantly reduced AQR migration defects compared to lq56 and lq83 (Table 1). Early QL and QR migrations were both significantly less severe in lq97 compared to lq56 and lq83 (Figure 3A and 4A), further indicating that lq97 might retain function, possibly via the production of a truncated, secreted form of CDH-4.
Fat-like cadherins have been implicated in acting in parallel to Wnt signaling in planar cell polarity, (Cho and Irvine, 2004; Loveless and Hardin, 2012; Matakatsu and Blair, 2004; Rawls et al., 2002; Strutt and Strutt, 2002; Tanoue and Takeichi, 2004; Yang et al., 2002). EGL-20/Wnt, which controls Q descendant migrations including PQR to the posterior, showed no early Q migration defects (Chapman et al., 2008). The five C. elegans Wnts act in a complex and redundant fashion in guiding Q descendants (Zinovyeva et al., 2008). The triple mutant cwn-1; egl-20 cwn-2 displayed extensive defects in the migrations of AQR and PQR (Table 1) but showed no apparent defects in early QL or QR migrations (n = 10; data not shown). While we cannot rule out a role of Wnt in early Q protrusion and migration, these results are consistent with a Wnt-independent role of CDH-4 in early Q protrusion and migration.
CDH-4 acts genetically in the PTP-3/LAR pathway, in parallel to UNC-40/DCC, in posterior QL migration
Previous studies revealed that UNC-40/DCC, PTP-3/LAR, and MIG-21 each stimulated posterior migration of QL and QR, and that distinct interactions between these molecules in QL versus QR governed posterior versus anterior migration (Sundararajan and Lundquist, 2012). In QL, UNC-40 acts redundantly with PTP-3 and MIG-21 to control posterior QL migration, and in QR, UNC-40 and PTP-3/MIG-21 mutually repress each other’s posterior migration function to allow anterior migration. We sought to determine how CDH-4 acts with these molecules in Q cell migration.
We first focused on interactions of cdh-4 with unc-40, ptp-3 and mig-21 in QL posterior migration. Double mutants of unc-40 with ptp-3 or mig-21 showed synergistic defects in QL posterior migration (Sundararajan and Lundquist, 2012). unc-40(n324) mutants alone displayed 4% QL migration defects wherein QL migrated anteriorly and divided over V4 (Fig 3A). unc-40; cdh-4 double mutants were lethal, so we reduced unc-40 function using expression of unc-40 RNAi from the scm promoter expressed in the seam cells and Q cells as described previously (Sundararajan and Lundquist, 2012). unc-40(RNAi) showed no defects alone, but cdh-4; unc-40(RNAi) double mutants displayed a significant increase in the percentage of anterior QL compared to cdh-4 alone (Figure 3A). This result indicates that CDH-4 acts in parallel to UNC-40/DCC in posterior QL migration, similar to PTP-3 and MIG-21 (Sundararajan and Lundquist, 2012).
We used ptp-3(RNAi) to reduce ptp-3 gene function, as double mutants of cdh-4 with the null ptp-3(mu245) allele were inviable. The previously-described scm::ptp-3(RNAi) transgene was used in these studies, which enhanced QL defects of unc-40 mutants (Sundararajan and Lundquist, 2012). We found that QL defects in cdh-4; ptp-3(RNAi) double mutants did not differ significantly from cdh-4 single mutants (Figure 3B). The double mutant of cdh-4(lq56) with the hypomorphic ptp-3(mu256) allele (Sundararajan and Lundquist, 2012) was also no more severe than cdh-4(lq56) alone. These data suggest that PTP-3 and CDH-4 might act in the same pathway in parallel to UNC-40 to direct posterior QL migration. Consistent with this idea, the triple mutant cdh-4(feeding RNAi); unc-40(RNAi); ptp-3(mu256) showed no enhancement relative to unc-40; ptp-3 and cdh-4; unc-40 double mutants (Figure 3A). While cdh-4 RNAi by feeding alone had no effect, it did significantly enhance PQR migration defects of unc-40(n324) (p < 0.0001) (Table 1), suggesting that cdh-4 gene function is reduced by feeding RNAi.
The small transmembrane thrombospondin repeat containing protein MIG-21 acts in the PTP-3 pathway in parallel to UNC-40 in posterior QL migration (Middelkoop et al., 2012; Sundararajan and Lundquist, 2012). Due to close proximity on LGIII and similarity in Q cell phenotype, we were unable to construct mig-21; cdh-4 double mutants. cdh-4 RNAi by feeding did not modify the mig-21(u787) QL phenotype (Figure 3B). The consistent lack of enhancement between cdh-4 and ptp-3/mig-21 support the idea that CDH-4 acts in the PTP-3/MIG-21 pathway, in parallel to UNC-40, in posterior QL migration. While our double and triple mutant analysis using RNAi was consistent with CDH-4 acting in the PTP-3 pathway in parallel to UNC-40, it is also possible that RNAi did not effectively reduce gene function in ptp-3 double mutants and that CDH-4 defines a third pathway in parallel to both UNC-40 and PTP-3.
CDH-4 acts genetically in both the UNC-40/DCC and PTP-3/LAR pathways in QR anterior migration
unc-40, ptp-3 and mig-21 mutations each displayed QRs that migrated to the posterior over V5 (Sundararajan and Lundquist, 2012). In unc-40; ptp-3 and unc-40; mig-21 double mutants, this aberrant posterior migration was reduced, suggesting a mutual inhibition between UNC-40 and PTP-3/MIG-21 in QR that allows anterior migration (Sundararajan and Lundquist, 2012). In other words, in the absence of PTP-3, UNC-40 is free to drive posterior migration, and vice versa.
cdh-4 mutants each displayed over 70% of QRs migrating posteriorly and dividing over V5, significantly more severe than unc-40, ptp-3, and mig-21 mutants (Figure 4A). unc-40(RNAi) significantly reduced posterior QR migration of cdh-4(lq56 and lq83) (Figure 4A), as it did with ptp-3 (Sundararajan and Lundquist, 2012). Double mutant combinations with ptp-3 and cdh-4 showed reduced posterior QR compared to cdh-4 alone, but only one was significant at the p < 0.05 level (the ptp-3(RNAi); cdh-4(lq83) double) (Figure 4B). cdh-4(RNAi) did not significantly modify the QR defects of mig-21(u787) (Figure 4B). CDH-4 was clearly involved in inhibition of UNC-40 in QR, as unc-40(RNAi) suppressed posterior QR migration in cdh-4 mutants. CDH-4 might also act in inhibition of PTP-3, as ptp-3; cdh-4 doubles showed consistent reductions in posterior QR, only one of which was significant at p < 0.05. These data are consistent with CDH-4 acting in both the PTP-3/LAR and UNC-40/DCC pathways to facilitate a mutual inhibition between them to allow anterior QR migration. The triple unc-40(RNAi); ptp-3(mu256); cdh-4(feeding RNAi) showed reduced posterior QR, but was not significantly different compared to unc-40(RNAi) ptp-3(mu256) alone. It is possible that posterior QR defects in unc-40(RNAi) ptp-3(mu256) are already too low to detect any significant enhancement. However, this result is consistent with CDH-4 acting in both the UNC-40/DCC and PTP-3/LAR pathways in QR. This model accounts for why the QR defects of cdh-4 mutants are significantly stronger than unc-40 and ptp-3, as in cdh-4 mutants, both UNC-40 and PTP-3 would be free to drive posterior migration. cdh-4; unc-40 and cdh-4; ptp-3 double mutants have significantly more posterior QR than the unc-40; ptp-3 double mutant, suggesting that CDH-4 is not the only molecule that acts with UNC-40 and PTP-3 in mutual inhibition.
cdh-4::gfp is expressed in Q cells and surrounding cells
A recombineered version of a fosmid clone containing the full-length cdh-4 genomic locus and surrounding region with gfp fused in frame at the cdh-4 3′ end was obtained from the TransgeneOme project (Sarov et al., 2006). This transgene is predicted to drive expression of CDH-4::GFP under its endogenous promoter and regulatory regions. Expression of cdh-4::gfp was observed in many cells at the time of Q cell polarization and migration in early L1 larvae (1–4.5h post hatching), including hypodermal seam cells and P cells (Figure 5). cdh-4::gfp expression was also observed in the Q cells, as noted by overlapping expression with egl-17::mCherry, expression of which is limited to the Q cells in the posterior of early L1 animals (Branda and Stern, 2000; Cordes et al., 2006; Sundararajan and Lundquist, 2012). Thus cdh-4::gfp is expressed in the Q cells but also in neighboring hypodermal cells. CDH-4::GFP accumulated throughout the cell bodies and margins, incuding the nucleus. This accumulation could reflect endogenous localization of CDH-4, or could be an artifact of transgenic expression, possibly overexpression. However, cdh-4::gfp rescued the Q migration defects of cdh-4 mutants, indicating that some functional CDH-4::GFP is expressed from the transgene.
Figure 5. Expression of cdh-4::gfp in the Q cells and neighboring cells.
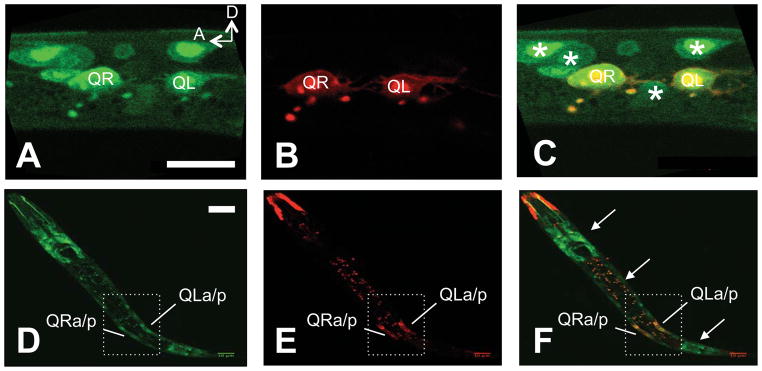
Images are micrographs of L1 animals with transgenic expression of the cdh-4::gfp recombineered fosmid (green) and the Pegl-17::mCherry transgene expressed in the Q neuroblasts (red). The scale bar in A represents 103m for AC, and in C for C–E. A) cdh-4::gfp expression in the Q cells an L1 animal 3–3.5 h post-hatch. Neighboring cells expressing cdh-4::gfp can also be seen (asterisks in C). B) The same animal in A showing Pegl-17::mCherry expression in the Q cells. C) A merge of images in A and B. (C–E) A lower magnification image of expression in an L1 animal at 4–4.5 h post-hatch with the same parameters as A–C. cdh-4::gfp is expressed along the length of the animal (arrows in F), including in the Q cells, which have divided.
CDH-4 acts non-cell autonomously in Q migrations
To test the functional requirement of cdh-4 in the Q cells, we performed a mosaic analysis using the cdh-4::gfp fosmid transgene and a strategy used previously to demonstrate autonomy of function of mig-15/NIK kinase in the Q cells (Chapman et al., 2008) (see Materials and Methods). A stably-integrated gcy-32::cfp transgene was used to score the positions of AQR and PQR, and an unstable extrachromosomal array containing rescuing cdh-4::gfp and gcy-32::yfp was used to generate genetic mosaics of cdh-4(+) activity in the cdh-4(lq56) mutant (Figure 6A). The cdh-4::gfp extrachromosomal array rescued AQR and PQR defects in cdh-4(lq56) mutants, indicating that it supplies cdh-4(+) activity (Figure 7). We next screened for mosaic animals in which the cdh-4::gfp extrachromosomal array was specifically lost in either AQR or PQR but retained in the other and in the URX neurons using gcy-32::cfp expression (Figure 6). The positions of AQR or PQR that lost the array were then determined by gcy-32::yfp. In such mosaic animals, AQR and PQR migration defects were not significantly different from non-mosiac, rescued animals (Figure 7). These results indicate that cdh-4 is not required in AQR and PQR for their proper migration. A similar strategy with mig-15/NIK showed opposite results, that loss of the rescuing array in AQR and PQR led to migration defects, indicating autonomy of function (Chapman et al., 2008). These results suggest that CDH-4 acts non-autonomously in directing AQR and PQR migrations. These results do not indicate which tissues require cdh-4(+) for AQR and PQR migration, but cdh-4::gfp is expressed broadly, including in hypodermal cells neighboring the Q cells (Figure 5).
Figure 6. Mosaic analysis of cdh-4 function.

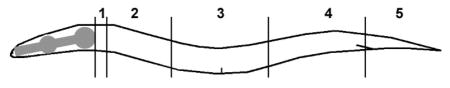
A) A simplified representation of the C. elegans cell lineage showing the origins of all Pgcy-32::gfp-expressing cells (AQR, PQR, URX L/R). Not all cell divisions are shown. B) A diagram of an animal subdivided into five segments along its body is shown. The wild-type positions of AQR (QR descendent) and PQR (QL descendent) are indicated in the diagram in positions 1 and 5 respectively. C–D) Micrographs of an early L2 larval animal with stably-integrated Pgcy-32::cfp expression to mark the positions of AQR and PQR, as well as an unstable extrachromosomal array with rescuing cdh-4::gfp and Pgcy-32::yfp to identify genetic mosaics of this array in AQR and PQR. C) Pgcy-32::cfp expression shows that AQR is in its normal position 1, and PQR is in its normal position 5. D) Pgcy-32::yfp expression shows that the rescuing cdh-4::gfp array has been lost in AQR, but retained in PQR and the URX neurons. Despite loss of the array in AQR, it has migrated normally as judged by Pgcy-32::cfp expression in C.
Figure 7. Quantification of AQR and PQR migration in cdh-4 genetic mosaics.

Graphs plot the percentage of AQR or PQR migration defects (Y-axis) in different genotypes (X-axis). Error bars represent 2x SEP, and at least 100 animals of each genotype were scored. Statistical significance was determined by Fisher’s exact test. A) PQR migration defects in animals that lost the rescuing cdh-4::gfp array in PQR (PQR loss) but retained the array in AQR and URX L/R. Animals with and without the array in PQR showed reduced defects compared to cdh-4(lq56) alone that were not significantly different from one another. B) AQR migration defects in animals that lost the array in AQR (AQR loss) but retained it in PQR and URX L/R. Animals with and without the array in AQR showed reduced defects compared to cdh-4(lq56) alone that were not significantly different from one another.
Materials and Methods
C. elegans genetics and microscopy
Experiments were performed at 20° C using standard C. elegans techniques (Sulston and Brenner, 1974). The following mutations and transgenics were used: LGI unc-40(n324), smg-1(e1228), smg-2(e2008); LGII ptp-3(mu256), cwn-1(ok546); LGIII mig-21(u787), cdh-4(lq56), cdh-4(lq83), cdh-4(lq97), cdh-4(rh310), cdh-4(ok1323), cdh-4(hd40); LGIV egl-20(n585), cwn-2(ok895), lqIs80[Pscm::gfp::caax]; LGV lqIs58[Pgcy-32::cfp]. Chromosomal locations for the following transgenes were not assigned: lqIs146[Pscm::unc-40(RNAi)], lqIs166[Pscm::ptp-3(RNAi)], lqIs249[Pegl-17::mCherry]. Extrachromosomal arrays used were lhEx397[unc-119; cdh-4(+)::gfp fosmid], and lqEx755[Pgcy-32::yfp; cdh-4::gfp fosmid]. The extrachromosomal arrays were built using standard gonadal microinjection techniques and were integrated into the genome using UV-TMP techniques (Mello and Fire, 1995). cdh-4 and smg-1 feeding RNAi was conducted using the standard RNAi by feeding protocol with bacterial stocks form the “Ahringer” library (Source Bioscience, UK) (Kamath and Ahringer, 2003). The cdh-4::gfp fosmid clone 77167333417072 A06 was obtained from the TransgeneOme project (Sarov et al., 2012).
Micrographs were obtained using a Leica DM550 compound microscope with filters for GFP, YFP, CFP, and mCherry and a Qimaging Retiga camera. The micrographs in Figure 5 were obtained using a Nikon laser scanning confocal microscope.
Imaging and scoring Q neuroblast defects
Previously described larval synchronization techniques were used (Chapman et al., 2008; Sundararajan and Lundquist, 2012). Adult and larval animals were washed from plates using M9 buffer. Eggs remained adhered to the agar surface of the plates. Newly-hatched L1 larvae were collected every 30 minutes by washing the plates with M9 and placing the larvae onto fresh NGM plates. These larvae were then visualized at 2, 3 and 4 hours post hatching using a the Pscm::gfp::caax transgene lqIs80. All the wild-type larvae visualized at 2 hours post synchronization (2–2.5 h timepoint) displayed defined anterior QR protrusions over the seam cell V4 and defined posterior QL protrusions over the seam cell V5. At 3 hours post synchronization (the 3–3.5 h timepoint), QR had migrated anteriorly on V4 and QL posteriorly on V5. At 4 hours post synchronization (4–4.5 h timepoint) QR had divided on V4 and QL on V5. Defects in mutant backgrounds were scored for direction of protrusion, migration and division. QR that protruded, migrated and divided posteriorly on V5 were scored as defective. QL that protruded, migrated and divided anteriorly on V4 were scored as defective. Cells also failed to migrate in cdh-4 and other mutants, but only directional migration defects were scored and included in Figures 3 and 4. At least 25 cells were scored for all stages, but only the 4–4.5 h division stage is included in Figures 3 and 4, as the position of division correlates with defects in protrusion and migration (Sundararajan and Lundquist, 2012). Statistical differences between genotypes were determined by Fisher’s exact test.
Scoring and analysis of AQR and PQR defects in Table 1 and Figure 7
Previously described quantification methods were used (Chapman et al., 2008; Sundararajan and Lundquist, 2012). The gcy-32::cfp transgene lqIs58 was used to visualize the positions of AQR and PQR in L4 larval animals, well after AQR and PQR undergo their migrations in L1 larvae. The final positions of AQR and PQR were binned into five positions along the body of the worm. Position 1 is the wild-type position of AQR in the anterior deirid ganglion just posterior to the pharyngeal bulb. Position 2 represents the region posterior to the pharyngeal bulb and just anterior to the vulva. Position 3 represents the region around the vulva. Position 4 represents the region posterior to the vulva and anterior to posterior deirid, where the Q cells are born and begin their migrations. Position 5 is the wild-type position of PQR, behind the anus in the phasmid ganglion. To best represent the defects in direction of migration, only the percentage of PQR in position 1 and the percentage of AQR in position 5 were included in Figure 7, but Table 1 shows each position. At least 100 worms were scored for each mutant background and all comparisons were done using Fisher’s exact test.
Mosaic analysis
The cdh-4(+)::gfp fosmid transgene lqEx755 rescued the defects in AQR and PQR migration observed in a cdh-4(lq56) background (Table 1 and Figure 7). A Pgcy-32::yfp construct was included with the cdh-4(+) extra chromosomal array to visualize the presence and absence of cdh-4(+) in AQR, PQR, and URX L/R. During mitotic divisions, extrachromosomal arrays are spontaneously lost from some daughter cells, resulting in genetically mosaic animals. Animals with losses in AQR and PQR were identified by lack of Pgcy-32::yfp expression in these cells (Figure 6). The strain also carried the stably-integrated Pgcy-32::cfp transgene lqIs58 to mark the positions of AQR in all animals, including those that lost the rescuing cdh-4(+) array in AQR and PQR. Animals that lost the rescuing cdh-4(+) array in AQR, but retained it in PQR and the URX neurons were scored for AQR position; and those that lost the rescuing cdh-4(+) array in PQR, but retained it in AQR and the URX neurons were scored for PQR position (Figure 7).
Discussion
Fat-like cadherins have been implicated extensively in cooperating with Wnt signaling and PCP core pathway components to generate tissue polarity (reviewed in (Thomas and Strutt, 2012)). Here we show that the Fat-like cadherin CDH-4 in C. elegans is required for anterior-posterior protrusion and migration of the Q neuroblasts. In cdh-4 mutants, QR often migrated posteriorly, and QL anteriorly. Greater than 50% of QR cells migrated in the wrong direction posteriorly in cdh-4 mutants, indicating that migration direction is not randomized in cdh-4 mutants, but rather that CDH-4 might change the direction of migration of Q cells in response to similar guidance information. Wnts control the anterior-posterior migration of Q cell descendants. EGL-20/Wnt activates expression of MAB-5/Hox in QL and descendants to cause posterior migration (Chapman et al., 2008; Eisenmann, 2005; Harris et al., 1996; Harterink et al., 2011; Herman, 2001; Kenyon, 1986; Maloof et al., 1999; Salser and Kenyon, 1992), and the five C. elegans Wnts display complex interactions in anterior-posterior migration of Q descendants (Zinovyeva et al., 2008). However, EGL-20/Wnt is not required for Q neuroblast initial migrations (Chapman et al., 2008), and despite having severe defects in AQR and PQR migration, the Wnt triple mutant egl-20 cwn-2; cwn-1 had no defects in initial Q migrations. These results are consistent with early Q cell migration and thus CDH-4 function in early Q migration being Wnt-independent.
We found that cdh-4; unc-40 and cdh-4; ptp-3 double mutants were inviable. cdh-4 mutants show some embryonic lethality coupled with variable failures in elongation and hypodermal organization (Schmitz et al., 2008). ptp-3 mutants when combined with vab-1/EphR display defects in embryonic morphogenesis (Harrington et al., 2002), and ptp-3; unc-40 double mutants are lethal (Sundararajan and Lundquist, 2012). Possibly, UNC-40/DCC, PTP-3/LAR, and CDH-4 all act in parallel to control aspects of embryonic morphogenesis and gastrulation.
CDH-4 acts in parallel to UNC-40, in the PTP-3/LAR pathway, in posterior QL migration
Previous work showed that UNC-40/DCC, PTP-3/LAR and MIG-21 are each required for posterior migration (Honigberg and Kenyon, 2000; Middelkoop et al., 2012; Sundararajan and Lundquist, 2012; Williams, 2003). In QL, MIG-21 and PTP-3 function in the same genetic pathway redundantly with UNC-40 to direct posterior migration (Sundararajan and Lundquist, 2012). In QL, cdh-4 mutations enhanced unc-40/DCC but not ptp-3/LAR or mig-21, suggesting that CDH-4 acts in the PTP-3/LAR pathway (Figure 3). CDH-4 acted non-autonomously (Figures 6 and 7), while UNC-40/DCC and PTP-3/LAR were required autonomously (Sundararajan and Lundquist, 2012). One model is that CDH-4 acts as an extracellular cue that directs posterior QL migration via PTP-3/LAR (Figure 8). This model implies that an unidentified, possibly redundant, cue would act through UNC-40/DCC. This cue is unlikely to be the canonical UNC-40/DCC ligand UNC-6/Netrin, as unc-6 mutants have no effect on anterior-posterior Q migrations (Honigberg and Kenyon, 2000). It is also possible that RNAi reduction of gene function was ineffective in cdh-4(RNAi); ptp-3 double mutants, and that CDH-4 defines a third pathway in parallel to both UNC-40 and PTP-3.
Figure 8. A model of CDH-4 genetic interaction with UNC-40/DCC, PTP-3/LAR, and MIG-21 that is consistent with data described in this work.

A) CDH-4 acts non-autonomously with PTP-3/LAR and MIG-21 in parallel to UNC-40/DCC in directing posterior QL migration. Grouping of molecules does not imply physical interaction in this model, rather action in the same genetic pathway. The non-autonomous cellular source of CDH-4 is unknown (?). B) CDH-4 acts in both the PTP-3/LAR and UNC-40/DCC pathways in QR in mutual inhibition, allowing anterior QR migration. The QR-specific factor that mediates UNC-40/DCC function with PTP-3/LAR in QR but not QL is unknown (?).
CDH-4 acts in both the UNC-40/DCC and PTP-3/LAR pathways in QR, and possibly facilitates mutual inhibition
Previous results indicate that UNC-40/DCC and PTP-3 interact differently in QR versus QL. In QR, UNC-40/DCC and PTP-3/LAR mutually inhibit each others’ roles in posterior migration, resulting in anterior migration (Sundararajan and Lundquist, 2012). cdh-4 mutants displayed greater than 70% posterior QR migration, a significantly higher defect than unc-40 and ptp-3 (Figure 4). unc-40(RNAi) reduced posterior QR migration in cdh-4 (Figure 4), suggesting that when CDH-4 is absent, UNC-40 drives posterior QR migration and that CDH-4 normally inhibits UNC-40. ptp-3 loss also reduced posterior QR migration in cdh-4 mutants. This effect was not as dramatic as with unc-40, but three different double mutant combinations showed reduced posterior QR, one of which was significant. Thus, CDH-4 might also normally inhibit PTP-3/LAR. The triple unc-40(RNAi); ptp-3(mu256); cdh-4(feeding RNAi) showed reduced posterior QR, but was not significantly different compared to unc-40(RNAi) ptp-3(mu256) alone. It is possible that posterior QR defects in unc-40(RNAi) ptp-3(mu256) are already too low to detect any significant enhancement. However, this result is consistent with CDH-4 acting in both the UNC-40/DCC and PTP-3/LAR pathways in QR, explaining why QR defects are so much more severe in cdh-4 mutants versus unc-40 and ptp-3 (i.e. both UNC-40 and PTP-3 drive posterior QR in the absence of CDH-4) (Figure 8).
Our results are consistent with a model in which CDH-4 facilitates the mutual inhibition of UNC-40/DCC and PTP/LAR in QR by interacting with both pathways in a non-autonomous manner (Figure 8). However, CDH-4 does not act in the UNC-40/DCC pathway in QL. This model requires that a QR-specific function, such as a co-receptor or a cytoplasmic molecule, mediates interaction of CDH-4 with the UNC-40 pathway in QR and not QL, and thus facilitates mutual inhibition in QR but not QL. QL and QR have inherent differences, such as response to EGL-20/Wnt and MAB-5/Hox. QL is more sensitive to both than QR (Middelkoop et al., 2012; Whangbo and Kenyon, 1999). The conserved transmembrane protein MIG-13 specifically affects QR descendant migrations (Sym et al., 1999; Wang et al., 2013). However, MIG-13 does not affect initial QR migration (Sym et al., 1999). Instead, MIG-13 expression is activated specifically in QR descendants by the Hox transcription factors LIN-39 and MAB-5 after initial QR migration (Wang et al., 2013). A molecule that mediates CDH-4/UNC-40 interaction in QR and not QL to mediate early migration has not yet been identified.
CDH-4 acts non-autonomously
UNC-40/DCC and PTP-3/LAR are required in the Q cells to guide their migrations (Sundararajan and Lundquist, 2012). In contrast, mosaic analysis of cdh-4 suggests that it acts non-autonomously to guide Q migrations. Loss of cdh-4(+) activity in either AQR or PQR had no effect on their migrations (Figures 6 and 7), suggesting that cdh-4 is required in other cells that retained the cdh-4(+) activity in the mosaic animals. Our experiments could not determine which cells these were, but cdh-4(+)::gfp is expressed in cells neighboring the Q cells, such as the hypodermal seam cells and the hypodermal P cells (Figure 5). In QL, our data support a model in which CDH-4 might be a cue that directs posterior QL migrations via PTP-3/LAR (Figure 8). In QR, our data suggest CDH-4 that facilitates mutual inhibition of UNC-40/DCC and PTP-3/LAR because of an inherent left-right asymmetry in QR versus QL (Figure 8). This inherent asymmetry would allow CDH-4 to interact with the UNC-40/DCC pathway in QR but not QL, thus inhibiting both UNC-40/DCC and PTP-3/LAR and allowing anterior migration. This is similar to the role of Fat in planar cell polarity, which acts non-autonomously to orient cells polarized by the core PCP pathway to the tissue or organismal axis (Yang et al., 2002; Zeidler et al., 1999).
Despite non-autonomous function in determining direction of migration in QR and QL, cdh-4::gfp is expressed in the Q cells. Possibly, CDH-4 has a role in the Q cells that does not involve direction of migration, such as cell adhesion. Indeed, cdh-4 mutants display defects in attachment of the pharynx to the hypodermis, suggestive of a defect in cell adhesion (Schmitz et al., 2008).
A non-autonomous role of CDH-4 is supported by the lq97 mutation, which is in the second to the last exon but is still 894 bp upstream of the endogenous stop codon and is thus likely subject to nonsense-mediated mRNA decay. It is possible that a fraction of the mRNA escapes nonsense mediated decay and produces a CDH-4 molecule that lacks a transmembrane domain and is potentially secreted. AQR migration defects of cdh-4(lq97) were significantly less severe than other cdh-4 alleles (Table 1), and smg-1 RNAi slightly suppressed PQR migration defects, indicating that a potentially secreted form of CDH-4 in cdh-4(lq97) mutants retains some function. That this secreted version retains function in AQR migration supports a non-autonomous role.
CDH-4::GFP showed no apparent asymmetric distribution in the anterior-posterior axis, suggesting that it might act as a permissive cue for migration, rather than a directional cue. Possibly, CDH-4 modifies the response of the Q cells to the same anterior-posterior guidance information. That QR migration is not randomized in cdh-4 mutants but rather skewed toward the posterior is consistent with a permissive role of CDH-4 in distinguishing the responses of QR and QL to similar anterior-posterior guidance information. The Fat gene in humans has been associated with neuropsychiatric disorders such as bipolar disorder and schizophrenia (Abou Jamra et al., 2008; Jung and Jun, 2013; Light et al., 2007; Redies et al., 2012). Mechanisms described here might be relevant in vertebrate central nervous system development and neuropsychiatric disorders.
Highlights.
The Fat-like cadherin CDH-4 controls QR and QL neuroblast direction of protrusion and migration.
CDH-4 acts with the LAR receptor tyrosine phosphatase PTP-3.
CDH-4 acts in parallel to the Ig-family receptor UNC-40, related to DCC.
CDH-4 displays left-right asymmetry of function in QL (left) versus QR (right).
CDH-4 acts cell-non-autonomously in Q migrations.
Acknowledgments
We thank E. Struckhoff for technical assistance, the Lundquist and Ackley labs for discussion and critical feedback, and O. Hobert, A. Boyanov, M. Doitsidu, and G. Minevich for assistance with next generation sequencing and CloudMap analysis. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). This work was supported by NIH grants R01NS040945 and R21NS070417 to E.A.L. and NIH grant P20GM103418, the Kansas Infrastructure Network of Biomedical Research Excellence, on which M.L.N. was an Undergraduate Scholar. M.L.N. and S.S were University of Kansas Undergraduate Research Award Scholars. Some next generation sequencing was provided by the University of Kansas Genome Sequencing Core Laboratory, funded by the NIH infrastructure grant P20GM103638.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Abou Jamra R, Becker T, Georgi A, Feulner T, Schumacher J, Stromaier J, Schirmbeck F, Schulze TG, Propping P, Rietschel M, Nothen MM, Cichon S. Genetic variation of the FAT gene at 4q35 is associated with bipolar affective disorder. Mol Psychiatry. 2008;13:277–284. doi: 10.1038/sj.mp.4002111. [DOI] [PubMed] [Google Scholar]
- Ackley BD. Wnt-signaling and planar cell polarity genes regulate axon guidance along the anteroposterior axis in C. elegans. Dev Neurobiol. 2013 doi: 10.1002/dneu.22146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branda CS, Stern MJ. Mechanisms controlling sex myoblast migration in Caenorhabditis elegans hermaphrodites. Dev Biol. 2000;226:137–151. doi: 10.1006/dbio.2000.9853. [DOI] [PubMed] [Google Scholar]
- Buettner FF, Ashikov A, Tiemann B, Lehle L, Bakker H. C. elegans DPY-19 is a C-mannosyltransferase glycosylating thrombospondin repeats. Mol Cell. 2013;50:295–302. doi: 10.1016/j.molcel.2013.03.003. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Sulston J. Developmental genetics of the mechanosensory neurons of Caenorhabditis elegans. Dev Biol. 1981;82:358–370. doi: 10.1016/0012-1606(81)90459-0. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Thomson JN, Sulston JE. Induction of neuronal branching in Caenorhabditis elegans. Science. 1983;221:61–63. doi: 10.1126/science.6857263. [DOI] [PubMed] [Google Scholar]
- Chapman JO, Li H, Lundquist EA. The MIG-15 NIK kinase acts cell-autonomously in neuroblast polarization and migration in C. elegans. Dev Biol. 2008;324:245–257. doi: 10.1016/j.ydbio.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho E, Irvine KD. Action of fat, four-jointed, dachsous and dachs in distal-to-proximal wing signaling. Development. 2004;131:4489–4500. doi: 10.1242/dev.01315. [DOI] [PubMed] [Google Scholar]
- Cordes S, Frank CA, Garriga G. The C. elegans MELK ortholog PIG-1 regulates cell size asymmetry and daughter cell fate in asymmetric neuroblast divisions. Development. 2006;133:2747–2756. doi: 10.1242/dev.02447. [DOI] [PubMed] [Google Scholar]
- Davis MW, Hammarlund M, Harrach T, Hullett P, Olsen S, Jorgensen EM. Rapid single nucleotide polymorphism mapping in C. elegans. BMC genomics. 2005;6:118. doi: 10.1186/1471-2164-6-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer JO, Demarco RS, Lundquist EA. Distinct roles of Rac GTPases and the UNC-73/Trio and PIX-1 Rac GTP exchange factors in neuroblast protrusion and migration in C. elegans. Small GTPases. 2010;1:44–61. doi: 10.4161/sgtp.1.1.12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenmann DM. Wnt signaling. WormBook. 2005:1–17. doi: 10.1895/wormbook.1.7.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, O’Connor S, Newman CL, Anderson P. SMG-1 is a phosphatidylinositol kinase-related protein kinase required for nonsense-mediated mRNA Decay in Caenorhabditis elegans. Mol Cell Biol. 2004;24:7483–7490. doi: 10.1128/MCB.24.17.7483-7490.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington RJ, Gutch MJ, Hengartner MO, Tonks NK, Chisholm AD. The C. elegans LAR-like receptor tyrosine phosphatase PTP-3 and the VAB-1 Eph receptor tyrosine kinase have partly redundant functions in morphogenesis. Development. 2002;129:2141–2153. doi: 10.1242/dev.129.9.2141. [DOI] [PubMed] [Google Scholar]
- Harris J, Honigberg L, Robinson N, Kenyon C. Neuronal cell migration in C. elegans: regulation of Hox gene expression and cell position. Development. 1996;122:3117–3131. doi: 10.1242/dev.122.10.3117. [DOI] [PubMed] [Google Scholar]
- Harterink M, Kim DH, Middelkoop TC, Doan TD, van Oudenaarden A, Korswagen HC. Neuroblast migration along the anteroposterior axis of C. elegans is controlled by opposing gradients of Wnts and a secreted Frizzled-related protein. Development. 2011;138:2915–2924. doi: 10.1242/dev.064733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman M. C. elegans POP-1/TCF functions in a canonical Wnt pathway that controls cell migration and in a noncanonical Wnt pathway that controls cell polarity. Development. 2001;128:581–590. doi: 10.1242/dev.128.4.581. [DOI] [PubMed] [Google Scholar]
- Herman MA. Wnt signaling in C. elegans. In: Kühl M, editor. Wnt signaling in Development. Landes Biosciences; Georgetown, TX: 2003. pp. 187–212. [Google Scholar]
- Honigberg L, Kenyon C. Establishment of left/right asymmetry in neuroblast migration by UNC-40/DCC, UNC-73/Trio and DPY-19 proteins in C. elegans. Development. 2000;127:4655–4668. doi: 10.1242/dev.127.21.4655. [DOI] [PubMed] [Google Scholar]
- Johns L, Grimson A, Kuchma SL, Newman CL, Anderson P. Caenorhabditis elegans SMG-2 selectively marks mRNAs containing premature translation termination codons. Mol Cell Biol. 2007;27:5630–5638. doi: 10.1128/MCB.00410-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung YE, Jun TY. Association between FAT Gene and Schizophrenia in the Korean Population. Clin Psychopharmacol Neurosci. 2013;11:67–71. doi: 10.9758/cpn.2013.11.2.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–321. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- Kenyon C. A gene involved in the development of the posterior body region of C. elegans. Cell. 1986;46:477–487. doi: 10.1016/0092-8674(86)90668-9. [DOI] [PubMed] [Google Scholar]
- Korswagen HC, Herman MA, Clevers HC. Distinct beta-catenins mediate adhesion and signalling functions in C. elegans. Nature. 2000;406:527–532. doi: 10.1038/35020099. [DOI] [PubMed] [Google Scholar]
- Light KJ, Miller AL, Doughty CJ, Joyce PR, Olds RJ, Kennedy MA. FAT and bipolar affective disorder. Mol Psychiatry. 2007;12:899–900. doi: 10.1038/sj.mp.4002040. [DOI] [PubMed] [Google Scholar]
- Loveless T, Hardin J. Cadherin complexity: recent insights into cadherin superfamily function in C. elegans. Curr Opin Cell Biol. 2012;24:695–701. doi: 10.1016/j.ceb.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Yang CH, McNeill H, Simon MA, Axelrod JD. Fidelity in planar cell polarity signalling. Nature. 2003;421:543–547. doi: 10.1038/nature01366. [DOI] [PubMed] [Google Scholar]
- Maloof JN, Whangbo J, Harris JM, Jongeward GD, Kenyon C. A Wnt signaling pathway controls hox gene expression and neuroblast migration in C. elegans. Development. 1999;126:37–49. doi: 10.1242/dev.126.1.37. [DOI] [PubMed] [Google Scholar]
- Matakatsu H, Blair SS. Interactions between Fat and Dachsous and the regulation of planar cell polarity in the Drosophila wing. Development. 2004;131:3785–3794. doi: 10.1242/dev.01254. [DOI] [PubMed] [Google Scholar]
- Mello C, Fire A. DNA transformation. Methods Cell Biol. 1995;48:451–482. [PubMed] [Google Scholar]
- Middelkoop TC, Williams L, Yang PT, Luchtenberg J, Betist MC, Ji N, van Oudenaarden A, Kenyon C, Korswagen HC. The thrombospondin repeat containing protein MIG-21 controls a left-right asymmetric Wnt signaling response in migrating C. elegans neuroblasts. Dev Biol. 2012;361:338–348. doi: 10.1016/j.ydbio.2011.10.029. [DOI] [PubMed] [Google Scholar]
- Minevich G, Park DS, Blankenberg D, Poole RJ, Hobert O. CloudMap: a cloud-based pipeline for analysis of mutant genome sequences. Genetics. 2012;192:1249–1269. doi: 10.1534/genetics.112.144204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najarro EH, Wong L, Zhen M, Carpio EP, Goncharov A, Garriga G, Lundquist EA, Jin Y, Ackley BD. Caenorhabditis elegans flamingo cadherin fmi-1 regulates GABAergic neuronal development. J Neurosci. 2012;32:4196–4211. doi: 10.1523/JNEUROSCI.3094-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou G, Vale RD. Molecular signatures of cell migration in C. elegans Q neuroblasts. J Cell Biol. 2009;185:77–85. doi: 10.1083/jcb.200812077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp MW, Maquat LE. Organizing principles of mammalian nonsense-mediated mRNA decay. Annual review of genetics. 2013;47:139–165. doi: 10.1146/annurev-genet-111212-133424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls AS, Guinto JB, Wolff T. The cadherins fat and dachsous regulate dorsal/ventral signaling in the Drosophila eye. Curr Biol. 2002;12:1021–1026. doi: 10.1016/s0960-9822(02)00893-x. [DOI] [PubMed] [Google Scholar]
- Redies C, Hertel N, Hubner CA. Cadherins and neuropsychiatric disorders. Brain Res. 2012;1470:130–144. doi: 10.1016/j.brainres.2012.06.020. [DOI] [PubMed] [Google Scholar]
- Salser SJ, Kenyon C. Activation of a C. elegans Antennapedia homologue in migrating cells controls their direction of migration. Nature. 1992;355:255–258. doi: 10.1038/355255a0. [DOI] [PubMed] [Google Scholar]
- Sarov M, Murray JI, Schanze K, Pozniakovski A, Niu W, Angermann K, Hasse S, Rupprecht M, Vinis E, Tinney M, Preston E, Zinke A, Enst S, Teichgraber T, Janette J, Reis K, Janosch S, Schloissnig S, Ejsmont RK, Slightam C, Xu X, Kim SK, Reinke V, Stewart AF, Snyder M, Waterston RH, Hyman AA. A genome-scale resource for in vivo tag-based protein function exploration in C. elegans. Cell. 2012;150:855–866. doi: 10.1016/j.cell.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarov M, Schneider S, Pozniakovski A, Roguev A, Ernst S, Zhang Y, Hyman AA, Stewart AF. A recombineering pipeline for functional genomics applied to Caenorhabditis elegans. Nat Methods. 2006;3:839–844. doi: 10.1038/nmeth933. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Wacker I, Hutter H. The Fat-like cadherin CDH-4 controls axon fasciculation, cell migration and hypodermis and pharynx development in Caenorhabditis elegans. Dev Biol. 2008;316:249–259. doi: 10.1016/j.ydbio.2008.01.024. [DOI] [PubMed] [Google Scholar]
- Solecki DJ, Govek EE, Tomoda T, Hatten ME. Neuronal polarity in CNS development. Genes Dev. 2006;20:2639–2647. doi: 10.1101/gad.1462506. [DOI] [PubMed] [Google Scholar]
- Strutt H, Strutt D. Nonautonomous planar polarity patterning in Drosophila: dishevelled-independent functions of frizzled. Dev Cell. 2002;3:851–863. doi: 10.1016/s1534-5807(02)00363-5. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Brenner S. The DNA of Caenorhabditis elegans. Genetics. 1974;77:95–104. doi: 10.1093/genetics/77.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- Sundararajan L, Lundquist EA. Transmembrane Proteins UNC-40/DCC, PTP-3/LAR, and MIG-21 Control Anterior-Posterior Neuroblast Migration with Left-Right Functional Asymmetry in Caenorhabditis elegans. Genetics. 2012;192:1373–1388. doi: 10.1534/genetics.112.145706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sym M, Robinson N, Kenyon C. MIG-13 positions migrating cells along the anteroposterior body axis of C. elegans. Cell. 1999;98:25–36. doi: 10.1016/S0092-8674(00)80603-0. [DOI] [PubMed] [Google Scholar]
- Tanoue T, Takeichi M. Mammalian Fat1 cadherin regulates actin dynamics and cell-cell contact. J Cell Biol. 2004;165:517–528. doi: 10.1083/jcb.200403006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Strutt D. The roles of the cadherins Fat and Dachsous in planar polarity specification in Drosophila. Dev Dyn. 2012;241:27–39. doi: 10.1002/dvdy.22736. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhou F, Lv S, Yi P, Zhu Z, Yang Y, Feng G, Li W, Ou G. Transmembrane protein MIG-13 links the Wnt signaling and Hox genes to the cell polarity in neuronal migration. Proc Natl Acad Sci U S A. 2013;110:11175–11180. doi: 10.1073/pnas.1301849110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whangbo J, Kenyon C. A Wnt signaling system that specifies two patterns of cell migration in C. elegans. Mol Cell. 1999;4:851–858. doi: 10.1016/s1097-2765(00)80394-9. [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos Trans R Soc Lond. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- Williams L. PhD thesis. University of California; San Francisco: 2003. A genetic analysis of the left-right asymmetric polarizations and migrations of the Q neuroblasts in C. elegans. [Google Scholar]
- Yang CH, Axelrod JD, Simon MA. Regulation of Frizzled by fat-like cadherins during planar polarity signaling in the Drosophila compound eye. Cell. 2002;108:675–688. doi: 10.1016/s0092-8674(02)00658-x. [DOI] [PubMed] [Google Scholar]
- Zeidler MP, Perrimon N, Strutt DI. The four-jointed gene is required in the Drosophila eye for ommatidial polarity specification. Curr Biol. 1999;9:1363–1372. doi: 10.1016/s0960-9822(00)80081-0. [DOI] [PubMed] [Google Scholar]
- Zinovyeva AY, Yamamoto Y, Sawa H, Forrester WC. Complex Network of Wnt Signaling Regulates Neuronal Migrations During Caenorhabditis elegans Development. Genetics. 2008;179:1357–1371. doi: 10.1534/genetics.108.090290. [DOI] [PMC free article] [PubMed] [Google Scholar]