

Abstract
Retinitis pigmentosa (RP) is the leading cause of inherited blindness in the developed world, affecting approximately 1 in 3,000 individuals. While there is currently no cure for RP, the genetic pathology has been well established. In this study we developed a novel mouse model of RP (huRhoP347S) expressing a pathogenic human rhodopsin gene with a Pro347Ser mutation on a rhodopsin knockout background. These mice undergo severe retinal degeneration at one month of age. In contrast to prior studies, this model was administered a gene therapy treatment at 19 days post natal. We evaluated several self-complementary adeno-associated virus serotypes for photoreceptor tropism including scAAV2/2, scAAV2/5, scAAV2/6.2 and scAAV2/9, and found that scAAV2/9 transduced photoreceptors with greater efficiency and expression than other vectors. We engineered a scAAV2/9 vector to contain a microRNA sequence specifically targeting the human rhodopsin gene and demonstrated its ability to silence rhodopsin by 60.2 ± 8.2% in vitro. In addition, we constructed a scAAV2/9 vector to contain a replacement “codon-modified” rhodopsin transgene (RhoR2) that was resistant to degradation by the microRNA. We found that delivery of the RhoR2 by scAAV2/9 is capable of restoring vision to rhodopsin knockout mice, and rescuing our novel transgenic huRhoP347S mouse model of dominant RP. Average a-wave responses of RhoR2-injected eyes were 1.8-fold higher than those of control-injected eyes. We found that delivery of the microRNA and replacement rhodopsin in a 1:2 ratio produced an average ERG a-wave response of 17.4 ± 2.9 μV compared to 6.5 ± 2.8 μV for eyes injected with negative control virus.
Keywords: microRNA, P347S, gene therapy, RNAi, rhodopsin (Rho), retinitis pigmentosa (RP), AAV, AAV2/9
Introduction
Retinitis Pigmentosa (RP) is a group of retinal degenerative diseases that are characterized primarily by the loss of rod photoreceptor cells. RP is the most common cause of genetic blindness and affects approximately 1 in 3,000 individuals in the United States1. Patients with RP experience night blindness, a progressive loss of peripheral visual fields and eventually loss of central vision as cone-cell viability is compromised by rod-cell death2. There is currently no cure available for RP. Recent results in phase I clinical trials have validated the safety and efficacy of vector-based gene augmentation therapies for type 2 Leber’s congenital amaurosis (LCA)3, which is responsible for approximately 6% of LCA cases3,4.
In contrast to recessive diseases such as LCA, gene therapy strategies for retinal disorders such as rhodopsin-linked autosomal dominant retinitis pigmentosa (adRP) have included methods of gene suppression, such as RNA interference (RNAi)-mediated decay of mutant mRNA5. While earlier studies of gene suppression employing RNAi in animal models of adRP yielded mixed outcomes, later studies indicate significant promise of these strategies for the treatment of this subset of RP. While a 68% reduction in the mutant rhodopsin mRNA of P23H transgenic rats did not result in measurable anatomical or functional rescue6, a similar study involving suppression of the P23H-containing mRNA in transgenic mice resulted in a 33% increase in thickness of the retina7. Since then, RNAi-mediated reduction in photoreceptor loss has been observed in the RP10 murine model of adRP8, and has shown both anatomical and functional rescue of transgenic mice expressing a human rhodopsin containing the P347S mutation, up to 10 weeks post-injection of an AAV expressing a short hairpin RNA (shRNA)9.
In addition to gene suppression, our group and others have been exploring a mutation-independent approach involving both gene suppression and replacement7,9–18. Such an approach has been shown to provide both functional and anatomical rescue up to 20 weeks post-administration of treatment19; however, mice in this study were given a subretinal injection of the shRNA- and replacement rhodopsin- expressing AAVs at five days post-natal. Such early administration does not accurately reflect the circumstances observed in the clinic in which patients typically present symptoms for years before proper diagnosis and have lost the majority of rod photoreceptors before an interventional treatment can be considered20,21.
We have previously published an shRNA, RHOi2, which can induce RNAi-mediated decay of human rhodopsin in vitro in a mutation-independent manner13. In addition, we have engineered a RHOi2-resistant human rhodopsin (RhoR2) to replace the suppressed rhodopsin, using the degeneracy of the genetic code. While gene silencing strategies for RP have traditionally focused on RNA interference by shRNAs, recent evidence suggests that microRNA-based hairpins may offer a safer and more effective alternative22–26. Efficacy of a microRNA targeting peripherin-2 has been shown after delivery to the subretinal space of wild-type mice in which 50% loss of photoreceptors is observed within 5 weeks of injection22.
Mice carrying a mutant (P347S) human rhodopsin transgene on a wild-type background manifest a dominant form of RP, with progression of disease consistent with levels of transgene expression27. The Pro347Ser (P347S) mutation in the human rhodopsin gene is of particular interest since six different mutations affecting this residue have been identified among patients with RP, and mutations at this amino acid position in the human rhodopsin gene have been documented to cause a more severe form of RP than that found in patients with RP due to other mutations28,29. In addition, patients with autosomal dominant RP caused by rhodopsin mutations express both a wild-type and a mutant allele, and retinal degeneration is usually slow.
In this study, we incorporated the RHOi2 sequence into a microRNA (miRhoi2) and tested the ability of miRhoi2 to suppress the expression of the P347S mutant rhodopsin in vivo, as well as replacement of the mutant rhodopsin with RhoR2. To do this, we generated a novel mouse model of RP (huRhoP347S) by breeding mice carrying a mutant (P347S) human rhodopsin transgene27 onto a murine rhodopsin null background30. We chose a rhodopsin-deficient background because mice heterozygous for rhodopsin have almost normal retinal development and function30–33. In addition, we tested the ability of RhoR2 to rescue the rhodopsin null mice.
Recombinant AAV2/9 vectors expressing either miRhoi2 (AAV2/9miRhoi2) or RhoR2 (AAV2/9RhoR2) were generated. For both huRhoP347S and rhodopsin null mice, AAVs were injected at post-natal day 19. Retinal function was measured by electroretinography (ERG). Significant rescue of vision in both the huRhoP347S and rhodopsin null mouse models was observed after administration of the AAV2/9RhoR2. However, the level of rescue observed in huRhoP347S was not significantly augmented when AAV2/9RhoR2 was delivered in conjunction with the miRhoi2-expressing AAV2/9. These results illustrate the significant potential yet technical challenges for gene therapy of dominant retinal disorders.
Results
Novel model of Retinitis Pigmentosa
A novel mouse model of RP was generated by breeding a rhodopsin null (Rho−) mouse30 with a mouse transgenic27 for the human rhodopsin gene containing a P347S mutation. Offspring were genotyped and those that carry the P347S human rhodopsin and are homozygous null for murine rhodopsin are henceforth indicated as huRhoP347S. At four weeks of age, presence of rhodopsin was investigated in the retina of huRhoP347S mice by immunocytochemistry using an antibody (Rho4D2) that recognizes the N-terminus of both human and mouse rhodopsin. As expected, rhodopsin was observed in the photoreceptor outer segments (OS) of normal (Rho+) mice, and was not observed in the retina of rhodopsin null (Rho−) mice (Fig 1A). In huRhoP347S mice, rhodopsin was observed in the outer nuclear layer (ONL) of the photoreceptors (Fig 1A). That mislocalization of rhodopsin in huRhoP347S mice was due in part to a loss of OS was confirmed by scanning electron microscopy (Fig 1B). A comparison of normal (Rho+) and huRhoP347S mouse retina confirmed the presence of shortened OS at four weeks of age in huRhoP347S mice relative to Rho+ mice (Fig 1B). To corroborate the phenotypic findings in the huRhoP347S mouse, a functional analysis (electroretinography, ERG) was performed at four weeks of age. A representative ERG is shown for each genotype in Fig 1C. ERG is the most widely used and accepted test for diagnosing patients with RP. It measures the electrical responses of various cells in the retina, including the photoreceptors, inner retinal (bipolar and amacrine) cells, and the ganglion cells. A normal ERG will contain an a-wave (initial negative deflection, measured from 1–2 in Fig 1C (Rho+)), followed by a b-wave (positive deflection, measured from 2–3 in Fig 1C (Rho+)). The leading edge of the a-wave is produced by the photoreceptors, while the remainder of the signal is produced by a mixture of photoreceptors, bipolar, amacrine, and Muller cells. The ERG can be used to measure the functionality of the rod and cone photoreceptors and can be used to detect changes in rod function early in the disease process. The mouse ERG has been well-characterized and great efforts towards its standardization have been made (Jeffrey et al 2011, Weymouth et al 2008).
Figure 1. Novel Model of RP.
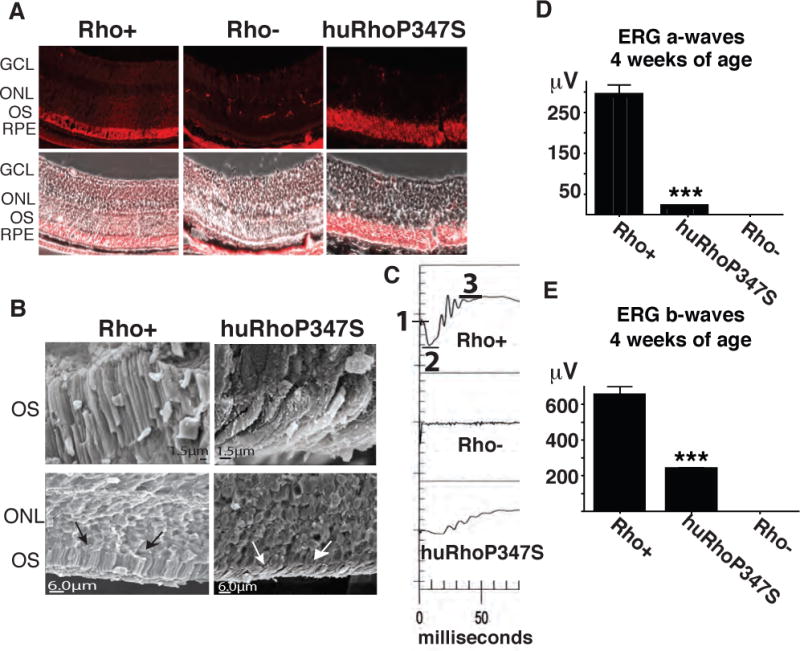
A) Rhodopsin localizes to photoreceptor outer segments (OS) in normal (Rho+) and to the ONL in huRhoP347S mice, but is not observed in rhodopsin null mice (Rho−) (magnification = 40X). Note: In Rho− retina, non-specific stain is observed in muscle tissue behind RPE. B) Scanning electron microscopy of normal (Rho+) and huRhoP347S mouse photoreceptor OS reveal shortened OS (white arrows) in huRhoP347S mice relative to those of Rho+ mice (black arrows). C) The huRhoP347S mice have decreased ERG a-waves and b-waves relative to mice with normal mouse rhodopsin (Rho+). Rhodopsin null (Rho−) mice do not exhibit a-waves or b-waves. Top panel illustrates ERG a-wave determination from baseline (1) to trough (2), and b-waves from trough (2) to crest (3). All recordings shown at 4 weeks of age. D) The huRhoP347S mice have a 15-fold reduction (***n=24, p<0.0001) in a-wave amplitudes relative to normal rhodopsin mice (Rho+) at 4 weeks of age. Rhodopsin null (Rho−) mice have no a-waves. E) The huRhoP347S mice have a 3-fold reduction (***n=24, p<0.0001) in b-wave amplitudes relative to normal rhodopsin mice (Rho+) at 4 weeks of age. Rhodopsin null (Rho−) mice have no b-waves. GCL, ganglion cell layer; ONL, outer nuclear layer; OS, outer segments; RPE, retinal pigment epithelium.
While rhodopsin null mice (Rho−) exhibited no a-waves or b-waves (Fig 1C, middle panel), huRhoP347S mice had reduced a-waves and b-waves (Fig 1C, bottom panel) relative to those observed with normal C57Bl6/J (Rho+) mice (Fig 1C, top panel). The huRhoP347S mice exhibited a significant 15-fold reduction in a-waves (18.8 ± 1.6μV, p<0.0001) relative to Rho+ mice (291.1 ± 26.0 μV; Fig 1D). A significant 3-fold reduction was also observed in the b-waves of huRhoP347S mice (232.5 ± 12.7μV, p<0.0001) relative to those of wild-type (Rho+) mice (646.3 ± 53.6μV; Fig 1E).
Degeneration of huRhoP347S Retina
To examine the degeneration of the huRhoP347S mouse retina, sections through the retina were taken at 3 time-points over a 24-week time course and stained with DAPI (nuclear stain) to distinguish the cell bodies of the inner and outer neurons (inner nuclear layer, INL/outer nuclear layer, ONL). By 24 weeks of age, the ONL of the huRhoP347S retina has completely degenerated (Fig 2A). While an ONL is observed in the huRhoP347S mouse retina at 12 weeks of age, an ERG analysis shows complete loss of a- and b-waves at this time point (Fig 2B). 14-micron cryosections transecting the retina were stained with DAPI (Fig. 2C) and ONL thickness measured at distances up to 2500μm from the optic nerve head. To quantify the degeneration of the retina, ONL thickness was measured at 4, 12 and 24 weeks in Rho+, huRhoP347S, and Rho− mice (Fig. 2D, 2E, 2F). Values were additionally plotted as a ratio of ONL to INL thickness to normalize for any variability in orientation while cryosectioning (Supplemental Fig A). At four weeks of age, the ONL of huRhoP347S and Rho− mice had begun to degenerate compared to Rho+ mice (Fig 2D). The average ONL thickness was 55.8 ± 6.9μm for Rho+ mice, compared to 35.3 ± 4.1μm for huRhoP347S and 38.2 ± 4.9μm for Rho− mice. At 12 weeks of age, while the Rho− mice had an almost completely degenerated ONL, the huRhoP347S mice had approximately 10% of their original ONL remaining (Fig 2E). The average ONL thickness at 12 weeks was 43.0 ± 7.4μm for Rho+ mice, compared to 7.4 ± 1.5μm for huRhoP347S and 3.5 ± 1.0μm for Rho− mice. At 24 weeks of age, the ONL of huRhoP347S mice was completely degenerated compared to the ONL of Rho+ (45.7 ± 6.1μm) and Rho− (6.7 ± 1.9μm) mice.
Figure 2. Retinal Degeneration of huRhoP347S Mouse.
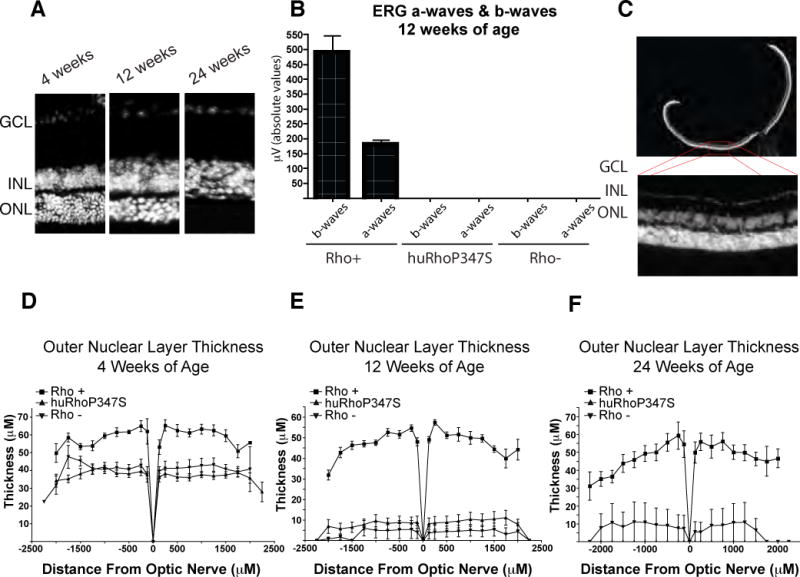
A) DAPI staining of transverse sections of retina of huRhoP347S mouse at 4, 12 and 24 weeks of age showing progressive loss of ONL. B) Average ERG response of Rho+, huRhoP347S, and Rho− mice at 12 weeks of age shows loss of a- and b-waves at this time point in huRhoP347S mice. C) DAPI staining of Rho+ retinal 14μm cryosection. D) ONL thickness of Rho+, huRhoP347S and Rho− mice at 4 weeks of age. E) ONL thickness of Rho+, huRhoP347S and Rho− mice at 12 weeks of age. F) ONL thickness of Rho+, huRhoP347S and Rho− mice at 24 weeks of age. Note*: all Rho− mice had completely degenerated ONL by 24 weeks, except one mouse, which had a single row of ONL remaining. GCL, ganglion cell layer; INL/ONL, inner/outer nuclear layer.
A microRNA (miRhoi2) targeting the human rhodopsin gene and codon-modified rhodopsin (RhoR2)
We have previously described a short hairpin RNA (shRNA), RHOi213, which targets the coding sequence for amino acids 73 to 79 in the first exon of the human rhodopsin gene and has previously been shown to silence human rhodopsin in vitro13. The miRhoi2 microRNA was developed by insertion of that portion of the RHOi2 sequence homologous to codons 73–79 into the miR-155 structural sequence (Fig 3A) to allow proper processing of the pre-miRNA sequence. The miRhoi2 microRNA and a negative control microRNA (miNEG) were each cloned into pCAGEN (kind gift of C. Cepko), a plasmid containing a chicken β-actin promoter/CMV enhancer and a thymidine kinase (TK) pA signal from Herpes Simplex Virus.
Figure 3. A microRNA (miRhoi2) targeting the human rhodopsin gene and codon-modified rhodopsin (RhoR2).
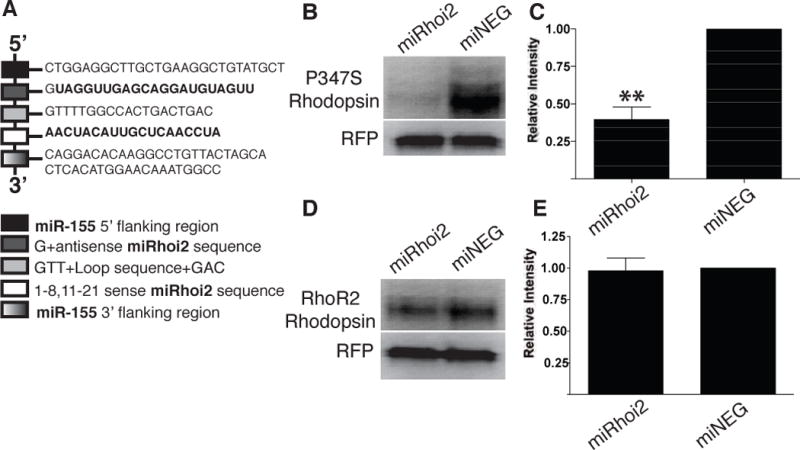
A) An artificial microRNA (miRhoi2) was engineered to target the 3′ end of the first exon in the human rhodopsin gene, specifically targeting codons 73 to 79. B) A representative western blot of lysate from human embryonic retinoblasts co-transfected with plasmids expressing either miRhoi2 or miNEG and a plasmid expressing both P347S rhodopsin and RFP. C) Quantitation of rhodopsin signal observed on western blots of lysate from HERs co-transfected with plasmids expressing either miRhoi2 or miNEG and a plasmid expressing both P347S rhodopsin and RFP. Rhodopsin levels were decreased by approximately 60% in the miRhoi2-transfected cells relative to those transfected with miNEG (**n=4, p<0.001). D) A representative western blot of human embryonic retinoblasts co-transfected with a plasmid co-expressing codon-modified rhodopsin (RhoR2) and RFP and a plasmid expressing either miRhoi2 or miNEG. E) Quantitation of rhodopsin signal observed on western blots of HERs co-transfected with a plasmid co-expressing codon-modified rhodopsin (RhoR2) and RFP and a plasmid expressing either miRhoi2 or miNEG. Rhodopsin levels were insignificantly changed in the presence of miRhoi2 relative to miNEG (n=4).
In addition, we modified a plasmid co-expressing human rhodopsin and red fluorescent protein (RFP)13 to contain a P347S mutation in the rhodopsin cDNA. Gene silencing of the P347S-containing rhodopsin mRNA by miRhoi2 was subsequently assayed in vitro. The P347S rhodopsin-expressing plasmid was co-transfected with a plasmid containing either the miRhoi2 or miNEG into human embryonic retinoblasts (HERs)35 and assayed for rhodopsin protein 72 hours post transfection (Fig 3B). The level of rhodopsin (normalized to RFP) expressed in the miRhoi2-transfected HERs, was reduced by 60.2± 8.2% relative to HER cells transfected with the miNEG control plasmid (Fig 3C).
A codon-modified rhodopsin (RhoR2) was generated as previously described by our group13. Using the degeneracy of the genetic code a rhodopsin transgene was engineered to encode normal rhodopsin protein, while specifically altering 10 of the 21 nucleotides that encode amino acids 73–79 of human rhodopsin to confer resistance to the miRhoi2 microRNA used in this study. A plasmid co-expressing RhoR2 and RFP was transiently transfected with plasmids expressing either miRhoi2 or miNEG into HERs. Rhodopsin protein levels were assayed 72 hours post transient transfection by Western blot analysis (Fig 3D). The expression of the replacement rhodopsin (normalized to RFP) in the miRhoi2-transfected HERs was not significantly altered from that produced by cells transfected with miNEG (Fig 3E), confirming resistance of the RhoR2 to miRhoi2.
Adeno-associated virus (AAV) serotype 2/9 infects photoreceptors
In order to identify the optimal vector for delivery of miRhoi2 and RhoR2 to photoreceptors in vivo, we injected several self-complementary AAV serotypes expressing GFP into the subretinal space of C57Bl6/J mice and vector tropism was determined by analysis of GFP expression in transverse sections of retina 3 weeks post-injection (Fig 4A–D). AAV2/2 preferentially infected the RPE, and a small number of photoreceptors. AAV2/5 infected the photoreceptors and RPE. AAV2/6.2 infected only the RPE, and AAV2/9 showed the strongest tropism for photoreceptor cells of all the serotypes tested. In addition to strong infectivity of the photoreceptors, AAV2/9 was capable of infecting a large portion of the retina 3 weeks post injection as observed by retinal whole mount and sectioning (Fig 4E). Based on these observations, we selected AAV2/9 as the optimal vector for gene delivery to the photoreceptors. AAV2/9 vectors expressing each of miRhoi2 (AAV2/9miRhoi2), miNEG (AAV2/9miNEG), and RhoR2 (AAV2/9RhoR2) were generated (Fig 4F). The AAV2/9miRHOi2 and AAV2/9miNEG vectors also express GFP, the miRhoi2 and miNEG sequences being embedded in the 3′ untranslated region of the GFP mRNA (see methods). Silver stain confirmed presence of adeno-associated virus genes VP1, VP2, VP3 in AAV2/9miRhoi2, AAV2/9miNEG and AAV2/9RhoR2 (Supplemental Fig B). Electron microscopy-negative staining was used to determine ratio of empty to packaged viral particles (Supplemental Fig B).
Figure 4. Adeno-associated virus (AAV) serotype 2/9 infects photoreceptors.
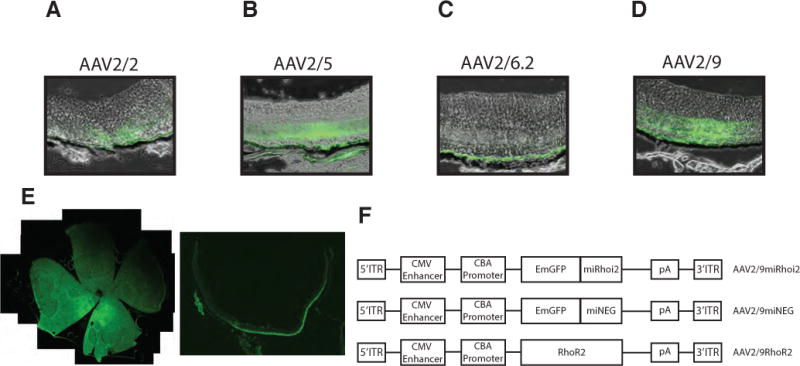
A–D) Several AAV serotypes were injected into the retina of Rho+ mice and vector tropism was determined by GFP localization 3 weeks post injection. AAV2/2 infected the RPE and some photoreceptors. AAV2/5 infected both photoreceptors and RPE. AAV2/6.2 infected the RPE exclusively. AAV2/9 showed the strongest photoreceptor tropism of all the serotypes tested. E) Whole mount and 14μm cryosection of AAV2/9 injected Rho+ retina. F) AAV constructs for AAV2/9miRhoi2, AAV2/9miNEG, and AAV2/9RhoR2. miRhoi2 and miNEG are situated in the 3′ untranslated region of GFP. ITR, inverted terminal repeat; CBA, chicken- β Actin; CMV, cytomegalovirus; pA, polyadenylation signal.
Rescue of Rho− mice by replacement rhodopsin (RhoR2)
Rhodopsin null mice (Rho−) were administered subretinal injections of a negative control virus (AAV2/9miNEG) into one eye and AAV2/9RhoR2 spiked 1:10 with AAV2/9GFP (GFP only-expressing virus) into the contralateral eye at 19 days post natal (P19). GFP expression was confirmed by fluorescence microscopy of retinal sections (data not shown). At two weeks post-injection, Rho− eyes injected with AAV2/9RhoR2 exhibited measurable ERG responses (Fig 5A), whereas eyes of Rho− mice injected with AAV2/9miNEG did not exhibit ERG responses (Fig 5B). Eyes of Rho− mice injected with AAV2/9RhoR2 exhibited average a-wave responses of 17.4 ± 3.2μV (Fig 5C), and average b-wave responses of 131.2 ± 11.1μV (Fig 5D). Rho− mice injected with AAV2/9RhoR2 were monitored 2, 4, 6, and 8 weeks post injection by ERG. The level of rescue was consistent for the first 4 weeks post injection, but then declined to approximately one-third the maximal response by 8 weeks post-injection (the latest time point examined, Fig 5E).
Figure 5. Rescue of Rho− Mice with RhoR2.
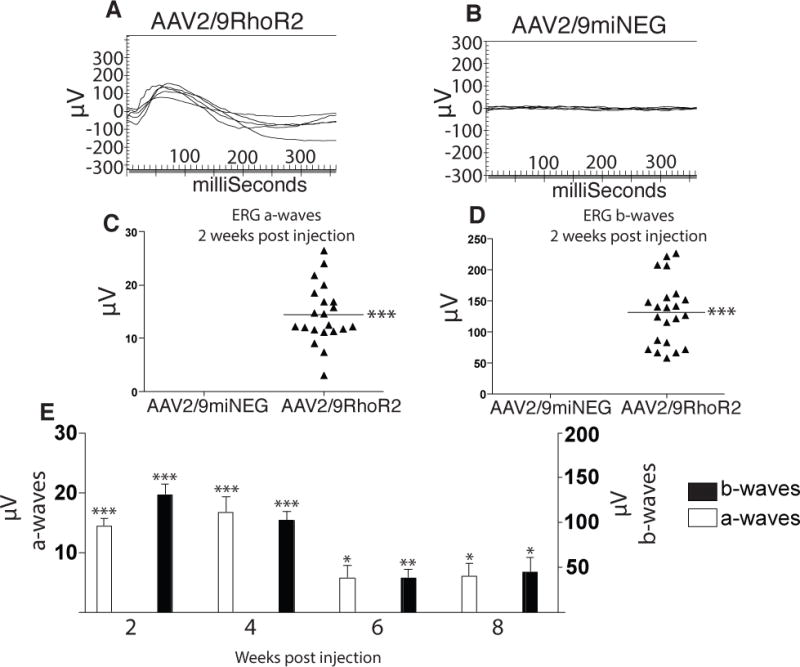
A) ERG tracings of Rho− mice illustrating responses evoked two weeks post injection of AAV2/9RhoR2. B) Rho− mice injected with AAV2/9miNEG did not yield ERG responses two weeks post injection. C) Average a-wave responses of AAV2/9RhoR2-injected eyes two weeks post injection was 17.42 ± 3.2μV (***n=21, p<0.0001). D) Average b-wave responses of AAV2/9RhoR2-injected eyes two weeks post injection was 131.2 ± 11.12μV (***n=21, p<0.0001). E) Average a-wave and b-wave responses of AAV2/9RhoR2- and AAV2/9miNEG-injected eyes were calculated two weeks post injection (***n=21, p<0.0001 (both waves)), 4 weeks post injection (***n=9, p≤0.0002 (both waves)), 6 weeks post injection (*,**n=9, p<0.05 (a-wave), p<0.009 (b-wave)), and 8 weeks post injection (*n=9, p<0.05).
Rescue of huRhoP347S mice by RhoR2
After confirming that the codon-modified replacement rhodopsin (RhoR2) was able to restore visual function in a rhodopsin null mouse, we investigated the effect of AAV2/9RhoR2 as a gene augmentation therapy in huRhoP347S mice. The huRhoP347S mice were administered contralateral subretinal injections of AAV2/9miNEG or AAV2/9RhoR2 spiked 1/10 with AAV2/9GFP at P19. ERGs were recorded two weeks post injection. The huRhoP347S mice injected with either AAV2/9RhoR2 (Fig 6A) or AAV2/9miNEG (Fig 6B) exhibited ERG responses at two weeks post injection. Eyes injected with AAV2/9RhoR2 exhibited a significant increase in average a-wave amplitudes (15.4 ± 1.5μV) relative to those injected with the control virus (8.5 ± 2.4μV; Fig 6C). The average b-wave amplitudes of AAV2/9RhoR2-injected eyes was 124.5 ± 12.1μV compared to that of 104.1 ± 21.7μV observed for AAV2/9miNEG-injected eyes at two weeks post injection (Fig 6D). The huRhoP347S mice injected with AAV2/9RhoR2 were monitored at 2 and 8 weeks post injection by ERG (Fig 6E). We observed significant (n=8, p<0.05) rescue of the a-wave only by ERG two weeks post injection in AAV2/9RhoR2-injected eyes relative to those eyes injected with the control virus. At 8 weeks post injection, the a-wave amplitudes of AAV2/9RhoR2-injected eyes were also significantly (n=8, p<0.05) increased relative to the eyes injected with the control virus.
Figure 6. Rescue of huRhoP347S Mice by RhoR2.
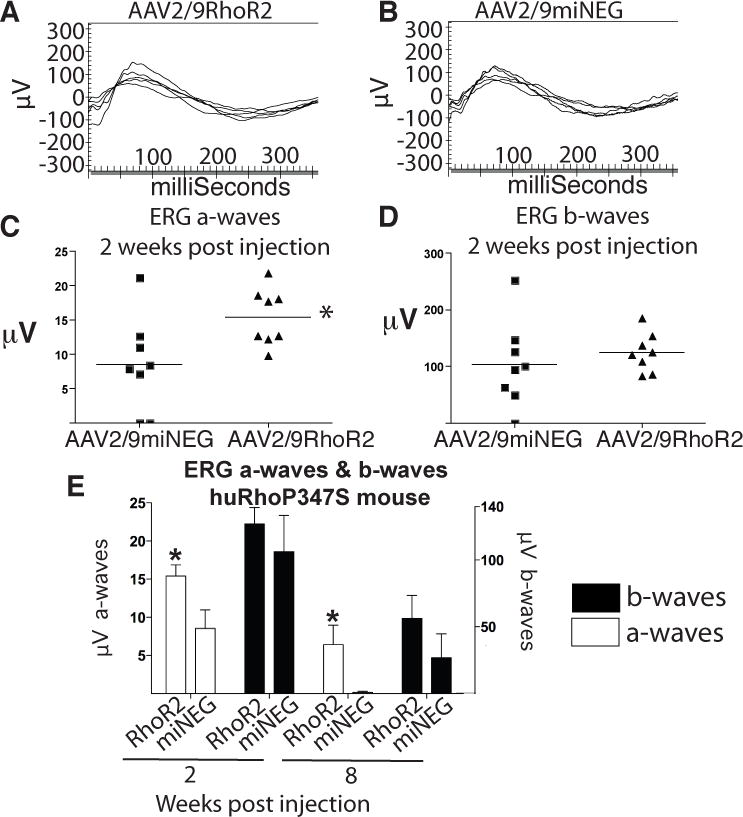
A) ERG tracings illustrating electrical response of retina of huRhoP347S mice evoked two weeks post injection of AAV2/9RhoR2. B) ERG tracings illustrating electrical response of huRhoP347S mice evoked two weeks post injection of AAV2/9miNEG. C) Average a-wave responses of AAV2/9RhoR2 injected eyes two weeks post injection was 15.40 ± 1.46μV compared to 8.53 ± 2.43μV for control injected eyes (n=8). D) Average b-wave responses of huRhoP347S mice injected with AAV2/9RhoR2 two weeks post injection was 124.5 ± 12.05μV compared to 104.1 ± 21.74μV for control injected eyes two weeks post injection (n=8). E) Average a-wave and b-wave ERG responses were calculated at two weeks post injection (*n=8, p<0.05) and 8 weeks (*n=8, p<0.05) post injection.
Comparison of miRhoi2/RhoR2 combination and RhoR2 in rescue of huRhoP347S mice
To ascertain the combinatorial effects of silencing the human pathogenic P347S rhodopsin using miRhoi2 and replacement with a miRhoi2-resistant rhodopsin (RhoR2), AAV2/9miRhoi2 was co-injected with AAV2/9RhoR2 into the subretinal space of huRhoP347S mice. The contralateral eye was injected with AAV2/9miNEG. Silencing of the human P347S rhodopsin by the miRhoi2 microRNA in vivo was confirmed by injecting AAV2/9miRhoi2 only into huRhoP347S mice at P19 and quantitation of rhodopsin mRNA levels by quantitative real-time PCR. We observed a significant (p<0.005) decrease in rhodopsin mRNA levels in huRhoP347S eyes injected with AAV2/9miRhoi2 relative to those injected with AAV2/9miNEG (Fig 7A). Eyes of huRhoP347S mice injected with AAV2/9miRhoi2 and AAV2/9RhoR2 in a ratio of 1:1 displayed a significant (p<0.05) increase in ERG a-wave response (11.8 ± 1.0μV) at two weeks post injection relative to those injected with AAV2/9miNEG (6.0 ± 2.8μV; Fig 7B). The huRhoP347S eyes injected with AAV2/9miRhoi2 and AAV2/9RhoR2 in a ratio of 1:1 displayed an ERG b-wave response of 134.7 ± 22.7μV at two weeks post injection compared to 87.8 ± 19.7μV for huRhoP347S eyes injected with AAV2/9miNEG (Fig 7C).
Fig 7. Comparison of miRhoi2/RhoR2 combination and RhoR2 in rescue of huRhoP347S mice.
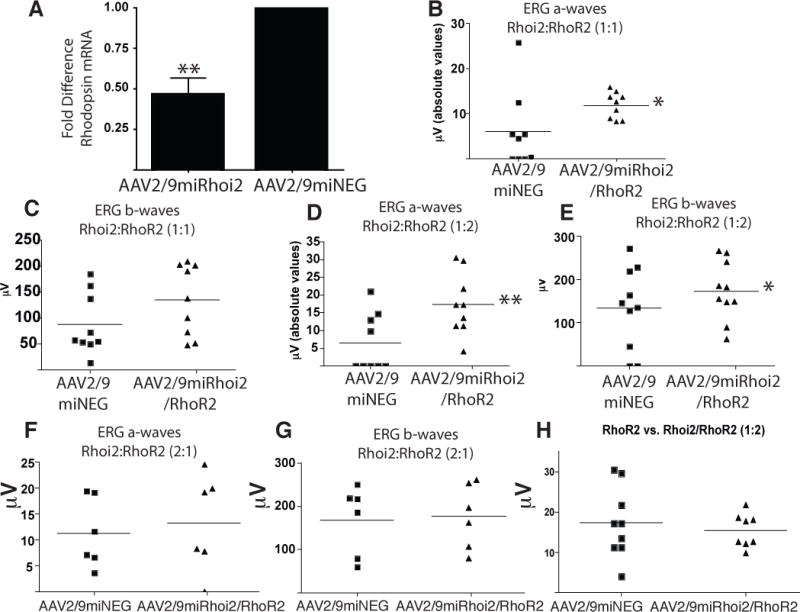
A) Quantitative real-time PCR of rhodopsin expression in AAV2/9miRhoi2 and AAV2/9miNEG injected huRhoP347S eyes (**n=6, p<0.005). B) ERG a-waves recorded at two weeks post injection in huRhoP347S mice injected in the subretinal space with either AAV2/9miNEG or in a 1:1 ratio shows a significant increase when eyes are administered both AAV2/9miRhoi2 and AAV2/9RhoR2 relative to injection with AAV2/9miNEG (*n= 9, p<0.05). C) ERG b-waves recorded at two weeks post injection in huRhoP347S mice injected with either AAV2/9miNEG or AAV2/9miRhoi2 and AAV2/9RhoR2 in a 1:1 ratio showing no significant difference between the two groups (n=9). D) ERG a-waves recorded at two weeks post injection in huRhoP347S mice injected with AAV2/9miRhoi2 and AAV2/9RhoR2 in a 1:2 ratio were significantly increased relative to those injected with AAV2/9miNEG (**n=10, p<0.005). Note: one of the data points in each of the conditions, AAV2/9miNEG and AAV2/9miRhoi2 combined with AAV2/9RhoR2, had such low amplitudes as to not be visible on the graph. E) ERG b-waves recorded at two weeks post injection in huRhoP347S mice injected with either AAV2/9miNEG or AAV2/9miRhoi2 and AAV2/9RhoR2 in a 1:2 ratio shows a significant increase in those injected with miRhoi2/RhoR2 relative to control-injected (*n=10, p<0.05). F) ERG a-waves recorded at two weeks post injection in huRhoP347S mice injected with either AAV2/9miNEG or AAV2/9miRhoi2 and AAV2/9RhoR2 in a 2:1 ratio shows no significant difference between the two groups (n=6). G) ERG b-waves recorded at two weeks post injection in huRhoP347S mice injected with either AAV2/9miNEG or AAV2/9miRhoi2 and AAV2/9RhoR2 in a 2:1 ratio shows no significant difference between the groups (n=6). H) No difference was observed in ERG a-waves recorded at two weeks post injection in huRhoP347S mice injected with either AAV2/9RhoR2 (data from Fig 6C) or AAV2/9miRhoi2 and AAV2/9RhoR2 (data from Fig 7D) in a 1:2 ratio.
We then altered the ratio of the codon-modified rhodopsin (AAV2/9RhoR2) to rhodopsin-targeting microRNA (AAV2/9miRhoi2) to determine the effect on visual rescue. The huRhoP347S eyes injected with AAV2/9miRhoi2 and AAV2/9RhoR2 in a ratio of 1:2 displayed a significant (p<0.005) increase in ERG a-wave response (17.4 ± 2.9μV) at two weeks post injection compared with huRhoP347S eyes injected with AAV2/9miNEG (6.5 ± 2.8μV; Fig 7D). The huRhoP347S eyes injected with AAV2/9miRhoi2 and AAV2/9RhoR2 in a ratio of 1:2 also displayed a significant increase (p<0.05) in ERG b-wave response (173.5 ± 21.7μV) at two weeks post injection compared to huRhoP347S eyes injected with AAV2/9miNEG (133.7 ± 29.8μV; Fig 7E). The huRhoP347S eyes injected with AAV2/9miRhoi2 and AAV2/9RhoR2 in a ratio of 2:1 displayed an ERG a-wave response of 13.3 ± 3.8μV two weeks post injection compared to 11.3 ± 2.7μV for huRhoP347S eyes injected with AAV2/9miNEG (Fig 7F). The huRhoP347S eyes injected with AAV2/9miRhoi2 and AAV2/9RhoR2 in a ratio of 2:1 showed no increase in ERG b-wave response (177.0 ± 30.6μV) at two weeks post injection relative to huRhoP347S eyes injected with AAV2/9miNEG (168.7 ± 32.5μV; Fig 7G).
When the ERG a-wave response of huRhoP347S eyes injected with AAV2/9RhoR2 alone (Fig 6C) was compared with those of huRhoP347S eyes injected with AAV2/9miRhoi2 and AAV2/9RhoR2 in a ratio of 1:2 (Fig 7D), no significant difference was observed (Fig 7H).
Discussion
In this study we generated a novel mouse model of RP by breeding a rhodopsin knockout (Rho−) mouse with a mouse containing one copy of the human rhodopsin gene containing a P347S mutation (huRhoP347S). We considered that generation of a mouse transgenic for a mutant human rhodopsin gene on a mouse rhodopsin deficient background was necessary to test a combination of mutation-independent gene silencing and replacement with a resistant rhodopsin; previous studies have shown that mice heterozygous for rhodopsin have almost normal retinal development and function30–33.
The huRhoP347S mice present with a gradual loss of ERG responses and outer nuclear layer thickness over the course of 1 to 3 months. While rhodopsin was observed in the photoreceptors of huRhoP347S mice, it was confined to the ONL (Fig 1A). This could be due to a reduction in the length of the outer segments and/or inefficient trafficking of rhodopsin; the P347S mutation occurs within the C-terminus of rhodopsin, a region implicated in regulating the trafficking of the protein to the OS by binding to ADP-ribosylation factor 4 (ARF4)36. However, that the P347S rhodopsin is functional is evidenced by measurable ERG responses in the huRhoP347S mice.
The presence of outer segments and measurable ERGs at one month of age encouraged us to administer gene therapy at P19, an age at which the retina is no longer undergoing development, eyes are opened, and mice are ready to be weaned from their mother. Therapeutic intervention at this age more accurately reflects the potential clinical situation than has been the case in other studies of gene silencing using mouse models of dominant RP which have been performed within 5 days postnatal. In addition, this model does not require a significantly long period of time to demonstrate degeneration – making the impact of therapeutic intervention more rapidly determinable.
The goal of this study was to investigate the efficiency of gene silencing using a microRNA in combination with gene replacement in an adRP mouse model. To do this we designed a microRNA (miRhoi2) sequence that specifically and robustly silenced the pathogenic human rhodopsin gene, and a replacement rhodopsin that was codon-modified to be resistant to this microRNA while still encoding a functional human rhodopsin protein (RhoR2). We first confirmed the functionality of RhoR2 through delivery into the subretinal space of rhodopsin null (Rho−) mice using AAV2/9. When administered as late as P19, the replacement rhodopsin was able to provide rescue of visual function as measured by ERG to Rho− mice. The observed functional rescue reached a maximum at 4 weeks post subretinal injection and then declined to a lower level of rescue for the remaining 4 weeks of the experimental time course. This may be due to loss of expression from the specific AAV serotype used (see below), or due to insufficient photoreceptor transduction. Administration of rhodopsin replacement via AAV2/5 at a much earlier timepoint, P0, has been shown to provide functional rescue of rhodopsin null mice out to 12 weeks17. Others have reported longer-term (21 weeks) rescue of a photoreceptor-specific deficiency with AAV2/837; however, in this case untreated mice express low levels of the implicated protein (Aipl1). Consistent with this, we did observe functional rescue of huRhoP347S, which expresses a mutant but functional rhodopsin, out to 8 weeks post-injection when augmented with AAV2/9RhoR2 at P19.
Given that the P347S allele used in this study is a dominant mutation on a normal mouse rhodopsin background27, we expected the P347S allele to have a dominant negative effect on RhoR2 and limit the extent of rescue observed. The magnitude of rescue therefore would be expected to be greater when replacement rhodopsin is co-delivered with the rhodopsin-targeting microRNA (miRhoi2). Several ratios of AAV2/9miRhoi2 to AAV2/9RhoR2 were investigated in this study to test the effect of gene suppression and replacement. The most efficient rescue was observed when a ratio of 1 miRhoi2 virus particle per 2 RhoR2 virus particles was administered. However, the degree of visual rescue observed at two weeks post-injection under this paradigm was not significantly greater than that achieved through administration of AAV2/9RhoR2 alone. The results from the 2:1 ratio of AAV2/9miRhoi2:AAV2/9RhoR2 suggest that there is a critical threshold of replacement rhodopsin required for physiological rescue since the 1:2 and 1:1 ratios (AAV2/9miRhoi2:AAV2/9RhoR2) provided significantly increased visual function relative to control-injected. This is not unexpected considering that silencing of P347S rhodopsin would not be expected to restore visual function to huRhoP347S mice without replacement with functional rhodopsin.
The threshold of replacement rhodopsin required to permit functional rescue of the huRhoP347S mouse, prevented increasing the amount of miRhoi2-containing virus to have a significant additional effect on rescue. While we validated the efficacy of miRhoi2 to suppress expression of P347S rhodopsin in vivo, one possibility as to why we did not observe an enhanced rescue when co-delivered with the RhoR2-containing virus in vivo may be due to the two-virus approach in which the miRhoi2 and RhoR2 were delivered in separate viral vectors. This permits the possibility that certain photoreceptors were transduced with only one of the two viruses, in which case photoreceptors receiving only AAV2/9miRhoi2 would exhibit a decrease in visual function and photoreceptors receiving only AAV2/9RhoR2 would not exhibit visual rescue beyond that observed in the AAV2/9RhoR2-treated eyes. We chose the two-vector approach to ascertain the combinatorial effects of microRNA suppression and gene replacement; however, one could imagine a single vector approach in which miRhoi2 would be incorporated into the 3′ untranslated region of the replacement rhodopsin, instead of GFP as was done in this study, to generate an AAV co-expressing RhoR2 and miRhoi2.
A number of gene silencing studies have been performed by other groups on mice transgenic for either P23H or P347S rhodopsin. Interestingly, rats carrying a transgene for P23H on a normal rat rhodopsin background exhibited no rescue when administered an AAV-expressing a shRNA targeting the mutant rhodopsin mRNA, despite significant silencing of the mutant transgene expression6. In another study, anatomical rescue only was observed in a mouse carrying a P23H rhodopsin and heterozygous for the endogenous wild-type rhodopsin gene7; in this case mice were administered both a shRNA-expressing AAV, as well as an AAV expressing the shRNA-resistant rhodopsin at P10. Rescue of a P347S transgenic mouse has been observed out to 20 weeks post-injection following gene suppression and replacement administered at P5, but unlike our model the mouse used in this study was heterozygous for normal mouse rhodopsin19. In addition, while all of the above studies utilized AAV2/5 vectors, we observed more efficient transduction of photoreceptors in vivo using self complementary AAV2/9 vectors relative to AAV2/56,7,19,35,38. The high level of photoreceptor transduction observed with AAV2/9 in our study is consistent with that observed by Lei and colleagues39 when GFP expression was assayed within weeks of injection. However, a study of AAV2/9 retinal transduction in which GFP expression was assessed six months after injection showed no evidence of photoreceptor transduction at this later time point40; this could potentially explain the short-term rescue observed in our study.
The results of this study have implications for potential treatment designs for dominant RP as well as other genetic disorders that are candidates for RNAi-based therapies.
Methods
Transgenic Mice
The huRhoP347S mice were generated by breeding mice carrying a mutant (Pro347Ser) human rhodopsin transgene27 onto a rhodopsin null background30. The P347S allele is expressed at a ratio of 0.25:1 relative to endogenous opsin mRNA. Mice were genotyped by polymerase chain reaction using primers specific for the neomycin resistance gene NE04 (5′ CGGG AGCG GCGA TACC GTAA AGC 3′) and NE07 (5′ GAAG CGGG AAGG GACT GGCT GCTA 3′), the mouse rhodopsin gene using the primers W11 (5′ GTGC CTGT GATC ACAG CACT T 3′) and W75 (5′ AGAC TCAC ATGG GGAG GAAT TC 3′)34, and the human rhodopsin gene using the primers K296EF2 (5′ TGAC CATC CCAG CGTT CTTT G 3′) and K296ER2 (5′ CATT TCTC ACAC ACTC CCCA TTG 3′). Genotyping was performed by Transnetyx, Inc. (www.transnetyx.com).
MicroRNA Design
We have previously described a shRNA, RHOi213, specifically targeting the human rhodopsin gene that has been shown to silence rhodopsin in vitro. The microRNA based on this sequence, miRhoi2, was developed by insertion of that portion of the short hairpin RNA homologous to codons 73–79 of human rhodopsin into the miR-155 sequence of pcDNA6.2GW/EmGFP-miR using the BLOCK-iT Pol II miR RNAi Expression Vector Kits (Invitrogen, Carlsbad, CA) and oligonucleotides mRHOi2F (5′ TGCT GTAG GTTG AGCA GGAT GTAG TTGT TTTG GCCA CTGA CTGA CAAC TACA TTGC TCAA CCTA 3′) and mRHOi2R (5′ CCTG TAGG TTGA GCAA TGTA GTTG TCAG TCAG TGGC CAAA ACAA CTAC ATCC TGCT CAAC CTAC 3′).
As a negative control we utilized a sequence that can form a pre-miRNA hairpin structure, but is not predicted to target any known vertebrate gene (Invitrogen, Carlsbad, CA). The sequence of the miNEG control microRNA is 5′ GAAA TGTA CTGC GCGT GGAG ACGT TTTG GCCA CTGA CTGA CGTC TCCA CGCA GTAC ATTT 3′.
Codon-modified Rhodopsin
The codon-modified rhodopsin (RhoR2) has previously been described by our group13. Briefly, the degeneracy of the genetic code was used to construct a rhodopsin transgene that would code for a normal rhodopsin protein via a codon-modified RNA. 10 of the 21 nucleotides encoding amino acids 73–79 of human rhodopsin, which had been altered to confer resistance to the shRNA, RHOi2, would also confer resistance to the miRhoi2 microRNA used in this study. Specifically, nucleotide 793 of the rhodopsin cDNA was changed from T to C, 796 was changed from T to C, 799 and 800 were changed from T to C, 802 was changed from A to G, 803 was changed from T to C, 805 was changed from G to C, 808 and 809 were changed from T to C, and 811 was changed from G to A.
P347S Rhodopsin
A previously published13 dual CMV IE (immediate early) promoter plasmid expressing both red fluorescent protein (RFP) and the human rhodopsin cDNA (pCMVRhoRFP) was modified by site-directed PCR to contain the P347S mutation. The nucleotides CCG (Pro) were modified using site-directed mutagenesis to TCG (Ser) using the primers P347SF (5′ ACCA CACA GAAG GCAG AGAA GGAG 3′) and P347SR (5′ GGTG GCCT CGGC CTAA GACC TGCC TAGG ATCC GGGG 3′). This design incorporated a novel BamHI restriction site into the PCR product, which was then cloned into the pCMVRhoRFP plasmid by digestion with BstEII and BamHI.
Western Blot Analysis
Western blots were performed using standard procedures. In brief, 12.5% polyacrylamide criterion zymogram precast gels were purchased from BioRad (Hercules, CA), and protein signals were detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Waltham, MA). Protein signals were quantified using Universal Hood II (Bio Rad, Hercules, CA), and software from Quantity One (version 4.5.0). Mouse monoclonal antibody Rho 1D4 was used against the C-terminus and Rho4D2 against the N-terminus of rhodopsin at a concentration of 1:500 (both antibodies courtesy Prof R. Molday, University of British Columbia). All western blots were analyzed by averaging the pixel intensity relative to control on four different exposures. Representative blots are shown.
Adeno-Associated Virus (AAV) Production
AAV vectors were produced by the UMass Vector Core facility. A pseudotyping strategy was used to produce AAV vectors packaged with AAV2, AAV5, AAV6.2 and AAV9 capsid proteins. Recombinant AAV genomes containing AAV2 inverted terminal repeats (ITRs) were packaged by triple transfection of 293 cells with cis-plasmid, adenovirus helper plasmid and a chimeric packaging construct in which the AAV2 replication gene is fused with capsid genes of novel AAV serotypes. All recombinant vectors were purified by standard CsCl2 sedimentation. Genome copy (GC) titers of AAV vectors were determined by TaqMan (Applied Biosystems) analysis using probes and primers targeting the SV40 poly(A) region. Ratios of packaged versus unpackaged virions were determined by transmission electron microscopy (data not shown). Self complementary AAV vectors containing the rhodopsin-targeting microRNA (AAV2/9miRhoi2), negative control microRNA (AAV2/9miNEG), and replacement rhodopsin (AAV2/9RhoR2) were generated using AAV9 capsid genes (Fig 4F). The microRNA-expressing viruses also express EmGFP, an emerald green fluorescent protein, which is co-expressed with the microRNA as part of the Invitrogen BLOCK-iT Pol II miR RNAi Expression Vector system.
Subretinal AAV Injections
All experiments involving animals were performed in accordance with the Statement for the Use of Animals in Ophthalmic and Vision Research, set out by the Association for Research in Vision and Ophthalmology. Mice were anesthetized by intraperitoneal injection of Xylazine (Xyla-Ject, Phoenix Pharmaceutical, Inc., St Joseph, MO, USA; 10 mg/ml)/Ketamine HCl (Ketaset, Fort Dodge Animal Health, Fort Dodge, IA, USA; 1 mg/ml). Subretinal injections containing 1μl of virus at a concentration of 5×109 virus particles per microliter were performed with a 32-G needle (Becton Dickinson, Franklin Lakes, NJ, USA) and a 5μl glass syringe (Hamilton, Reno, NV, USA) by a trans-scleral trans-choroidal approach. Animals were sacrificed by CO2 inhalation followed by cervical dislocation.
Quantitative Real-Time Polymerase Chain Reaction
Rhodopsin gene expression was measured by quantitative real time-PCR (qRT-PCR) using a commercially available primer/probe reagent. 1μl of AAV2/9miRhoi2, AAV2/9miNEG, or AAV2/9RhoR2 was subretinally injected into rhodopsin null or P347S mice 19 days post natal. Mice were sacrificed three weeks post injection, and sections of retina expressing GFP were isolated. Retinal tissue was then homogenized and total mRNA was extracted using QIAGEN total RNA kit. 1.5μg of RNA was treated with Turbo DNAse (Ambion, Foster City, CA) for 1hr at 37C, and DNAse inactivation reagent for 5 minutes prior to qRT-PCR reaction. 3.5μl of mRNA was used in a standard 1-step qRT-PCR protocol measuring human rhodopsin mRNA (Hs00173452_m1 Rho, Applied Biosystems Foster City, CA) and was normalized to mouse -actin (Applied Biosystems Foster City, CA). All experiments were performed in quadruplicate using Bio-Rad iQ5 Multicolor real time PCR detection system.
Electroretinography (ERG)
Animals were dark adapted overnight and all procedures were carried out under dim red light. To anesthetize the animals, ketamine and xylazine were injected intraperitoneally as described above. Pupils were dilated with 2.5% phenylephrine hydrochloride ophthalmic solution USP (Bausch & Lomb, Tampa, FL, USA) and 1% Tropicamide (Akorn, Inc., Lake Forest, IL, USA). Eyes were maintained in a proptosed position throughout the examination. Reference and ground electrodes were positioned subcutaneously, ~1 mm from the temporal canthus and anterior to the tail, respectively. ERG responses were recorded simultaneously from both eyes by means of gold wire electrodes, which were positioned to touch the central cornea of each eye. Corneal hydration was maintained after the examination by the application of phosphate buffered saline to the cornea. Scotopic electroretinograms were recorded at a flash intensity of −10 dB using contact lens electrodes and the UTAS system with BigShot ganzfeld (LKC Technologies, Inc., Gaithersburg, MD, USA). Ten flashes were averaged for each recording. ERG results were analyzed using standard statistical convention (paired Student’s t test) and represented as mean absolute values and standard error (SE).
Supplementary Material
Supplementary Figure A: Ratio of ONL to INL thickness of the retinas of Rho+, huRhoP347S and Rho− mice
ONL to INL thickness ratio of Rho+, huRhoP347S, and Rho− mice at 4 weeks of age (A), 12 weeks of age (B), and 24 weeks of age (C). Note*: all Rho− mice had completely degenerated ONL by 24 weeks, except one mouse, which had a single ONL row remaining.
Supplementary Figure B: A) Silver stain of VP1, VP2, VP3 for AAV2/9miRhoi2, AAV2/9miNEG, and AAV2/9RhoR2. B) Morphology and ultra structure of viral particles (electron microscopy-negative stain)
Acknowledgments
We would like to acknowledge Tiansen Li and Janis Lem for providing the founder mice used to generate the novel mouse model described in this investigation. We would also like to thank Robert Molday for providing the Rhodopsin 1D4 and 4D2 antibodies. This study was supported by the NIH/NEI, (EY013837), The Ellison Foundation and grants to the Department of Ophthalmology at Tufts University School of Medicine from the Lions Eye Foundation and Research To Prevent Blindness.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
Supplementary Information is available at Gene Therapy’s website.
References
- 1.Hartong D, Berson EL, Dryja TP. Retinitis Pigmentosa. The Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 2.Farrar GJ, Kenna PF, Humphries P. On the genetics of retinitis pigmentosa and on mutation-independent approaches to therapeutic intervention. EMBO J. 2002;21:857–864. doi: 10.1093/emboj/21.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maguire AM, High KA, Auricchio A, Wright JF, Pierce EA, Testa F, et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet. 2009;374:1597–1605. doi: 10.1016/S0140-6736(09)61836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27:391–419. doi: 10.1016/j.preteyeres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Baulcombe D. RNA silencing. Curr Biol. 2002;12:R82–84. doi: 10.1016/s0960-9822(02)00665-6. [DOI] [PubMed] [Google Scholar]
- 6.Tessitore A, Parisi F, Denti MA, Allocca M, Di Vicino U, Domenici L, et al. Preferential silencing of a common dominant rhodopsin mutation does not inhibit retinal degeneration in a transgenic model. Mol Ther. 2006;14:692–699. doi: 10.1016/j.ymthe.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 7.O’Reilly M, Palfi A, Chadderton N, Millington-Ward S, Ader M, Cronin T, et al. RNA interference-mediated suppression and replacement of human rhodopsin in vivo. Am J Hum Genet. 2007;81:127–135. doi: 10.1086/519025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tam LC, Kiang AS, Kennan A, Kenna PF, Chadderton N, Ader M, et al. Therapeutic benefit derived from RNAi-mediated ablation of IMPDH1 transcripts in a murine model of autosomal dominant retinitis pigmentosa (RP10) Hum Mol Genet. 2008;17:2084–2100. doi: 10.1093/hmg/ddn107. [DOI] [PubMed] [Google Scholar]
- 9.Chadderton N, Millington-Ward S, Palfi A, O’Reilly M, Tuohy G, Humphries MM, et al. Improved retinal function in a mouse model of dominant retinitis pigmentosa following AAV-delivered gene therapy. Mol Ther. 2009;17:593–599. doi: 10.1038/mt.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Millington-Ward S, O’Neill B, Tuohy G, Al-Jandal N, Kiang AS, Kenna PF, et al. Strategems in vitro for gene therapies directed to dominant mutations. Hum Mol Genet. 1997;6:1415–1426. doi: 10.1093/hmg/6.9.1415. [DOI] [PubMed] [Google Scholar]
- 11.Lewin AS, Drenser KA, Hauswirth WW, Nishikawa S, Yasumura D, Flannery JG, et al. Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nat Med. 1998;4:967–971. doi: 10.1038/nm0898-967. [DOI] [PubMed] [Google Scholar]
- 12.LaVail MM, Yasumura D, Matthes MT, Drenser KA, Flannery JG, Lewin AS, et al. Ribozyme rescue of photoreceptor cells in P23H transgenic rats: long-term survival and late-stage therapy. Proc Natl Acad Sci U S A. 2000;97:11488–11493. doi: 10.1073/pnas.210319397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cashman SM, Binkley EA, Kumar-Singh R. Towards mutation-independent silencing of genes involved in retinal degeneration by RNA interference. Gene Ther. 2005;12:1223–1228. doi: 10.1038/sj.gt.3302512. [DOI] [PubMed] [Google Scholar]
- 14.Kiang AS, Palfi A, Ader M, Kenna PF, Millington-Ward S, Clark G, et al. Toward a gene therapy for dominant disease: validation of an RNA interference-based mutation-independent approach. Mol Ther. 2005;12:555–561. doi: 10.1016/j.ymthe.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 15.Gorbatyuk M, Justilien V, Liu J, Hauswirth WW, Lewin AS. Preservation of photoreceptor morphology and function in P23H rats using an allele independent ribozyme. Exp Eye Res. 2007;84:44–52. doi: 10.1016/j.exer.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palfi A, Ader M, Kiang AS, Millington-Ward S, Clark G, O’Reilly M, et al. RNAi-based suppression and replacement of rds-peripherin in retinal organotypic culture. Hum Mutat. 2006;27:260–268. doi: 10.1002/humu.20287. [DOI] [PubMed] [Google Scholar]
- 17.Palfi A, Millington-Ward S, Chadderton N, O’Reilly M, Goldmann T, Humphries MM, et al. Adeno-associated virus-mediated rhodopsin replacement provides therapeutic benefit in mice with a targeted disruption of the rhodopsin gene. Hum Gene Ther; 21:311–323. doi: 10.1089/hum.2009.119. [DOI] [PubMed] [Google Scholar]
- 18.Millington-Ward S, Chadderton N, O’Reilly M, Palfi A, Goldmann T, Kilty C, et al. Suppression and Replacement Gene Therapy for Autosomal Dominant Disease in a Murine Model of Dominant Retinitis Pigmentosa. Mol Ther. doi: 10.1038/mt.2010.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Millington-Ward S, Chadderton N, O’Reilly M, Palfi A, Goldmann T, Kilty C, et al. Suppression and Replacement Gene Therapy for Autosomal Dominant Disease in a Murine Model of Dominant Retinitis Pigmentosa. Mol Ther. 2011;19:642–649. doi: 10.1038/mt.2010.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kalloniatis M, Fletcher EL. Retinitis pigmentosa: understanding the clinical presentation, mechanisms and treatment options. Clin Exp Optom. 2004;87:65–80. doi: 10.1111/j.1444-0938.2004.tb03152.x. [DOI] [PubMed] [Google Scholar]
- 21.Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1:40. doi: 10.1186/1750-1172-1-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Georgiadis A, Tschernutter M, Bainbridge JW, Robbie SJ, McIntosh J, Nathwani AC, et al. AAV-mediated knockdown of peripherin-2 in vivo using miRNA-based hairpins. Gene Ther. 17:486–493. doi: 10.1038/gt.2009.162. [DOI] [PubMed] [Google Scholar]
- 23.Kim VN. MicroRNA precursors in motion: exportin-5 mediates their nuclear export. Trends Cell Biol. 2004;14:156–159. doi: 10.1016/j.tcb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 24.McBride JL, Boudreau RL, Harper SQ, Staber PD, Monteys AM, Martins I, et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci U S A. 2008;105:5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shan Z, Lin Q, Deng C, Li X, Huang W, Tan H, et al. An efficient method to enhance gene silencing by using precursor microRNA designed small hairpin RNAs. Mol Biol Rep. 2009;36:1483–1489. doi: 10.1007/s11033-008-9339-8. [DOI] [PubMed] [Google Scholar]
- 26.Boudreau RL, Martins I, Davidson BL. Artificial microRNAs as siRNA shuttles: improved safety as compared to shRNAs in vitro and in vivo. Mol Ther. 2009;17:169–175. doi: 10.1038/mt.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li T, Snyder WK, Olsson JE, Dryja TP. Transgenic mice carrying the dominant rhodopsin mutation P347S: evidence for defective vectorial transport of rhodopsin to the outer segments. Proc Natl Acad Sci U S A. 1996;93:14176–14181. doi: 10.1073/pnas.93.24.14176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stephen P. Daiger LSSaJAR. Genetic and functional complexity of inherited retinal degeneration. Behavioral and Brain Sciences. 1995;18:501–521. [Google Scholar]
- 29.Berson EL, Rosner B, Sandberg MA, Dryja TP. Ocular findings in patients with autosomal dominant retinitis pigmentosa and a rhodopsin gene defect (Pro-23-His) Arch Ophthalmol. 1991;109:92–101. doi: 10.1001/archopht.1991.01080010094039. [DOI] [PubMed] [Google Scholar]
- 30.Lem J, Krasnoperova NV, Calvert PD, Kosaras B, Cameron DA, Nicolo M, et al. Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc Natl Acad Sci U S A. 1999;96:736–741. doi: 10.1073/pnas.96.2.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang Y, Fotiadis D, Maeda T, Maeda A, Modzelewska A, Filipek S, et al. Rhodopsin signaling and organization in heterozygote rhodopsin knockout mice. J Biol Chem. 2004;279:48189–48196. doi: 10.1074/jbc.M408362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calvert PD, Govardovskii VI, Krasnoperova N, Anderson RE, Lem J, Makino CL. Membrane protein diffusion sets the speed of rod phototransduction. Nature. 2001;411:90–94. doi: 10.1038/35075083. [DOI] [PubMed] [Google Scholar]
- 33.Humphries MM, Rancourt D, Farrar GJ, Kenna P, Hazel M, Bush RA, et al. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet. 1997;15:216–219. doi: 10.1038/ng0297-216. [DOI] [PubMed] [Google Scholar]
- 34.Frederick JM, Krasnoperova NV, Hoffmann K, Church-Kopish J, Ruther K, Howes K, et al. Mutant rhodopsin transgene expression on a null background. Invest Ophthalmol Vis Sci. 2001;42:826–833. [PubMed] [Google Scholar]
- 35.Fallaux FJ, Kranenburg O, Cramer SJ, Houweling A, Van Ormondt H, Hoeben RC, et al. Characterization of 911: a new helper cell line for the titration and propagation of early region 1-deleted adenoviral vectors. Hum Gene Ther. 1996;7:215–222. doi: 10.1089/hum.1996.7.2-215. [DOI] [PubMed] [Google Scholar]
- 36.Deretic D, Williams AH, Ransom N, Morel V, Hargrave PA, Arendt A. Rhodopsin C terminus, the site of mutations causing retinal disease, regulates trafficking by binding to ADP-ribosylation factor 4 (ARF4) Proc Natl Acad Sci U S A. 2005;102:3301–3306. doi: 10.1073/pnas.0500095102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tan MH, Smith AJ, Pawlyk B, Xu X, Liu X, Bainbridge JB, et al. Gene therapy for retinitis pigmentosa and Leber congenital amaurosis caused by defects in AIPL1: effective rescue of mouse models of partial and complete Aipl1 deficiency using AAV2/2 and AAV2/8 vectors. Hum Mol Genet. 2009;18:2099–2114. doi: 10.1093/hmg/ddp133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allocca M, Mussolino C, Garcia-Hoyos M, Sanges D, Iodice C, Petrillo M, et al. Novel adeno-associated virus serotypes efficiently transduce murine photoreceptors. J Virol. 2007;81:11372–11380. doi: 10.1128/JVI.01327-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lei B, Zhang K, Yue Y, Ghosh A, Duan D. Adeno-associated virus serotype-9 mediated retinal outer plexiform layer transduction is mainly through the photoreceptors. Adv Exp Med Biol. 2010;664:671–678. doi: 10.1007/978-1-4419-1399-9_77. [DOI] [PubMed] [Google Scholar]
- 40.Lebherz C, Maguire A, Tang W, Bennett J, Wilson JM. Novel AAV serotypes for improved ocular gene transfer. J Gene Med. 2008;10:375–382. doi: 10.1002/jgm.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure A: Ratio of ONL to INL thickness of the retinas of Rho+, huRhoP347S and Rho− mice
ONL to INL thickness ratio of Rho+, huRhoP347S, and Rho− mice at 4 weeks of age (A), 12 weeks of age (B), and 24 weeks of age (C). Note*: all Rho− mice had completely degenerated ONL by 24 weeks, except one mouse, which had a single ONL row remaining.
Supplementary Figure B: A) Silver stain of VP1, VP2, VP3 for AAV2/9miRhoi2, AAV2/9miNEG, and AAV2/9RhoR2. B) Morphology and ultra structure of viral particles (electron microscopy-negative stain)