

Abstract
The development and mechanistic investigation of a highly stereoselective methodology for preparing α-linked-urea neo-glycoconjugates and pseudo-oligosaccharides is described. This two-step procedure begins with the selective nickel-catalyzed conversion of glycosyl trichloroacetimidates to the corresponding α-trichloroacetamides. The α-selective nature of the conversion is controlled with a cationic nickel(II) catalyst, Ni(dppe)(OTf)2. Mechanistic studies have identified the coordination of the nickel catalyst with the equatorial C2-ether functionality of the α-glycosyl trichloroacetimidate to be paramount for achieving an α-stereoselective transformation. A cross-over experiment has indicated that the reaction does not proceed in an exclusively-intramolecular fashion. The second step in this sequence is the direct conversion of α-glycosyl trichloroacetamide products into the corresponding α-urea glycosides by reacting them with a wide variety of amine nucleophiles in presence of cesium carbonate. Only α-urea-product formation is observed, as the reaction proceeds with complete retention of stereochemical integrity at the anomeric C-N bond.
Keywords: Carbohydrates, Glycosyl Trichloroacetimidates, Stereoselectivity, Trichloroacetamides, Urea Glycosides
Introduction
As understanding continues to expand exponentially surrounding the indispensable role of carbohydrates in the living world, the development of synthetic methods to efficiently accomplish these ubiquitous structures becomes increasingly relevant. Advancing these technologies will not only facilitate biological studies, but will also provide access to modified structures that have both interesting and unique properties of their own. For carbohydrates, chemical and enzymatic degradation at the glycosidic linkage has been identified as a weak-point in the structural robustness of glycosylated compounds. As a result, having access to modified carbohydrate structures with enhanced resistance to enzymatic and chemical hydrolysis is highly desirable. The glycosyl urea, for example, has gained considerable attention in the development of neo-glycoconjugates,[1] pseudo-oligosacharides,[2] and N-linked glycopeptide mimetics,[3] to replace native O- and N-glycosidic linkages.These efforts aim to improve stability under physiological conditions while maintaining properties of the natural targets, having direct application in the design and development of anti-diabetic agents[4] and aminoglycoside antibiotics.[5]
In nature, the glycosyl urea is found as an integral part of both cinodine[6] and coumamidine[7] antibiotics. These compounds are of interest because they exhibit broad-spectrum activity against Gram-negative cell lines and have unusual structural features, including an α-urea-linkage at the anomeric center and a unique mode of action. These polycationic compounds do not inhibit bacterial protein synthesis, as do the aminoglycosides, but rather bind directly to bacterial DNA[8] and its organizational enzyme, DNA gyrase B.[9-11] The partial and/or total syntheses of cinodine and coumamidine antibiotics have never been reported potentially due to lack of technology for the general and efficient preparation of the α-urea glycosidic linkage.[12]
The stereoselective synthesis of glycosyl urea is by no means trivial.[13] Approaches can often be complicated with numerous steps and be forced to rely on neighboring group participation to facilitate a selective transformation.[13,14] In addition, there is a propensity for many donor types to undergo anomerization, which removes them from consideration in many stereoselective urea syntheses.[15] Although there are a handful of methods available for attaining the β-urea glycoside,[16] a general and selective preparation of α-urea glycoside is still lacking.[12.17] In Ichikawa's approach, α-glycosyl isocyanate 2 (Scheme 1a) is generated in situ from glycosyl azide 1 and subsequently trapped with amine to provide α-glycosyl urea 3.[17] Although this approach is able to retain α-stereochemical integrity at the anomeric C-N bond during the urea-forming step, moderate selectivity in starting material 2 (4:1 α/β-mixture) and additional step requirements to achieve it from α-glycosyl azide limit synthetic utility. Bernardi has reported a new method for the synthesis of α-glycosyl urea 6 using a modified Staudinger reduction (Scheme 1b). Bernardi's approach reduces the step requirement by directly converting α-azide 4 into α-iminophosphorane 5, then transforming it to urea 6 by reacting with isocyanate.[12] Although Bernardi's approach achieves the α-urea 6 (Scheme 1b) in a single-pot, the limited reactivity of the iminophosphorane intermediate places restrictions on scope in the conversion. A third methodology for forming α-glycosyl ureas has been developed in our lab employing a palladium-catalyzed stereoselective rearrangement of glycal trichloroacetimidate 7 to the 2,3-unsaturated trichloroacetamide product 8 (Scheme 1c).[18] After functionalizing glycal 8 to pyranoside with catalytic OsO4, the resulting trichloroacetamide is converted to α-glycosyl urea 9 with amine nucleophile in the presence of Cs2CO3. While this process is highly α-selective, the substrate scope is limited due to the 1,2-syn-diol-forming nature of the dihydroxylation reaction used to functionalize the 2,3-unsaturated glycal 8.
Scheme 1.

Methods for Selective Formation of α-Glycosyl Ureas
We report herein a new and efficient procedure for constructing α-urea glycoside, which has the potential to overcome the current limitations for the synthesis of this motif and can be applicable to a variety of carbohydrate substrates (Scheme 1d).[19] Based on our recent efforts using transition-metal catalysis in glycosylation reactions,[20] we envisioned that in the absence of an external nucleophile, a transition-metal catalyst would be able to promote the ionization and subsequent rearrangement of α-glycosyl trichloroacetimidate 10 to the corresponding α-trichloroacetamide 11 (Scheme 1d).[19] The trichloroacetamide intermediates 11 can be directly converted to the c α-glycosyl urea products 12 by reaction with a wide range of amines.[18] This two-step method of attaining α-urea linkage retains stereochemical integrity at the anomeric center; is highly atom-economical with chloroform being the only part of the original leaving group that is absent from the final urea structure; is tolerant to a variety of substrate types and protecting groups. This report will focus on the scope of this selective conversion, as well as the mechanistic investigation of the stereoselective transition-metal-catalyzed process.
Results and Discussion
A. Conversion of Glycosyl Trichloroacetimidates
Building on our previous successes using cationic transition-metal catalysis for the activation of glycosyl trichloroacetimidate donors in highly-selective glycosylation strategies,[20] we began our search for an appropriate catalyst to facilitate the transformation of α-glycosyl trichloroacetimidate 13 to glycosyl trichloroacetamide 14 with 5 mol% of the readily available Pd(CH3CN)4(BF4)2 catalyst (Table 1, entry 1). This system was found to be ill-suited for the conversion, showing no detectable product 14 after 5 h at room temperature. We continued our efforts by switching to a presumably more reactive Pd(PhCN)2(OTf)2 catalyst (entry 2), generated in situ from Pd(PhCN)2Cl2 and AgOTf, and discovered that with use of 5 mol% catalyst loading, the conversion proceeded smoothly to provide the desired trichloroacetamide 14 in 86% yield as a 10:1 mixture of α- and β-anomers (entry 2) within 1 h. The progress of this rearrangement was monitored by FT-IR spectroscopy, with completion of the reaction being noted after disappearance of the C=N stretching band of trichloroacetimidate 13 at 1670 cm-1.[21] A reduction in catalyst loading to 2 mol% (entry 3) maintained the yield and α-selectivity in the conversion. Switching from a cationic palladium to a cationic nickel species (entry 4), Ni(PhCN)4(OTf)2, provided a similar yield and anomeric selectivity with a notable improvement in rate (less than 30 min).
Table 1.
Screening Catalysts and Conventional Lewis Acids.[a]

| ||||
|---|---|---|---|---|
| Entry | Catalyst | Loading (mol%) | Time (h) | Yield (%)/[b] (α:β)[b] |
| 1 | Pd(CH3CN)4(BF4)2 | 5 | 5 | NR |
| 2 | Pd(PhCN)2(OTf)2 | 5 | 1 | 86 (10:1) |
| 3 | Pd(PhCN)2(OTf)2 | 2 | 1 | 85 (10:1) |
| 4 | Ni(PhCN)4(OTf)2 | 2 | 0.25 | 84 (11:1) |
| 5 | Ni(4-F-PhCN)4(OTf)2 | 2 | 0.25 | 88 (10:1) |
| 6 | Ni(4-MeO-PhCN)4(OTf)2 | 2 | 0.25 | 90 (10:1) |
| 7 | Ni(dppe)(OTf)2 | 2 | 1 | 85 (30:1) |
| 8 | Ni(dppe)Cl2 | 2 | 6 | NR |
| 9 | Ni(dppp)(OTf)2 | 2 | 1.75 | 70 (8:1) |
| 10 | Ni(dppb)(OTf)2 | 2 | 1.25 | 49 (9:1) |
| 11 | Ni(PPh3)(OTf)2 | 2 | 1.25 | 61 (12:1) |
| 12 | Ni(OTf)2 | 2 | 0.25 | 57 (19:1) |
| 13 | AgOTf | 6 | 14 | 72 (5:1) |
| 14 | TMSOTf | 4 | 2 | 72 (8:1) |
| 15 | BF3OEt2 | 6 | 6 | 65 (4:1) |
The conversions were performed at 0.2 M in CH2Cl2 with 2 −5 mol% of Pd/Ni catalysts or Lewis acids.
Isolated Yield.
1H NMR ratio.
We continued our optimization studies by varying the electronic properties of the ligands on the nickel catalyst (Table 1, entries 5–7) to observe the effects on yield and selectivity. We found that an optimal balance occurred with Ni(dppe)(OTf)2 (entry 7), providing 14 in 85% yield with α/β = 30:1.[22] We next probed the effect of varying the bite angle of the bisphosphine on the ligand-metal complex (entries 9–11). To this end, the reaction was investigated with 1,3-bis(diphenylphosphino)propane ligand (Ni(dppp)(OTf)2, entry 9) and 1,4-bis(diphenylphosphino)butane ligand (Ni(dppb)(OTf)2, entry 10), that have a larger bite angle than do 1,2-bis(diphenylphosphino) ethane ligand (Ni(dppe)(OTf)2, entry 7), led to reduced yield (47– 70%) and α-selectivity (α/β = 8:1–9:1).[23,24] We hypothesize that this is probably due to bite angle effects that impart both steric and electronic influences on metal catalyst.[24] Further switching to the monodentate triphenylphoshine ligand, (Ni(PPh3)2(OTf)2, entry 11) provided diminished yield and α- selectivity (61%, 12:1 α/β). To determine the necessity of the phosphine ligand, the conversion was carried out in absence of one (Ni(OTf)2, entry 12), which increased the rate and selectivity (19:1 α/β-ratio) of the reaction at the expense of yield (57%). In addition, a significant amount of decomposition was observed with use of Ni(OTf)2, suggesting the importance of dppe ligand to help modulate the nickel species and control the reaction. To evaluate if the catalytic activity observed in this series (Table 1) had arisen from the presence of residual AgOTf, a control experiment was conducted (entry 13) to determine if it would be able to facilitate a reaction on its own. The transformation required 14 h and diminished yield (72%) and anomeric selectivity (α/β = 5:1) were observed. Use of neutral nickel(II) catalyst, Ni(dppe)Cl2 (entry 8), resulted in no reaction.To determine if Lewis acid behavior alone was responsible for reactivity with the nickel(II) species, a control experiment was conducted with BF3OEt2 (entry 15), a commonly-employed activating agent in glycosylation with glycosyl imidates.[25] Trichloroacetamide 14 was obtained in 65% yield with α/β = 4:1. Faster reaction time was accomplished with TMSOTf (entry 14),[26] though yield (72%) and selectivity (8:1 α/β) were not on par with the results obtained using the nickel(II) catalyst (85%, 30:1 α/β, entry 7).
Next, we turned our attention to evaluating the effect of solvent in the transformation (Table 2). Increasing the polarity of the solvent (CH2Cl2 → THF, entry 1 to entry 2) decreased α-selectivity (α:β = 19:1) and yield (49%). We also observed that the prolonged reaction time (1 h vs. 3 h, entries 1 and 2) led to substantial loss in the form of hydrolysis. Similar results were obtained using MTBE (entry 3), though the reaction took less time to reach completion. Use of less polar toluene as solvent (entry 4) provided excellent α-selectivity in the transformation (20:1 α/β), though the yield of 14 was reduced (75% yield). Thus, 2 mol % of Ni(dppe)(OTf)2 in CH2Cl2 was chosen as the optimal balance between yield, α-anomeric selectivity, and reaction rate.
Table 2.
Screening of Solvents.[a]

| |||
|---|---|---|---|
| Entry | Solvent | Time (h) | Yield (%)/[b] (α:β)[c] |
| 1 | CH2Cl2 | 1 | 85 (30:1) |
| 2 | THF | 3 | 49 (19:1) |
| 3 | MTBE | 1.5 | 50 (16:1) |
| 4 | Dioxane | 5 | 67 (15:1) |
| 5 | Toluene | 2 | 75 (20:1) |
| 6 | Trifluorotoluene | 1.25 | 67 (13:1) |
The rearrangements were performed at 0.2 M in CH2Cl2 with 2 mol% of Ni(dppe)(OTf)2.
Isolated Yield.
1H NMR ratio.
With the reaction conditions identified, we set out to establish the substrate scope of the transformation (Table 3). While the nickel catalyst was tolerant of the benzyl protecting group at C2 (Tables 1 and 2), we also found that incorporating allyl and silyl ethers at C2 of imidates 15 and 16 provided trichloroacetamides 20 and 21 (entries 1 and 2) in 90 – 91% with high α-selectivity (α/β = 10:1 – 20:1). It is interesting to note that although the C2-silyl group provided trichloroacetamide 21 in higher α-selectivity than both the C2-allyl and benzyl group, the conversion was much more sluggish (10 h vs. 1 h), presumably due to steric encumbrance of the bulky triisopropylsilyl (TIPS) group. Next, we investigated xylose 17 (entry 3) and quinovose 18 (entry 4) that lack C6-hydroxyl group.[27] These studies produced high yield (87–91%) and excellent α-selectivity (α/β = 15:1 – ≥ 25:1) in generating trichloroacetamide 22 and 23 (entries 3 and 4). A galactose 19 (entry 5) with an additional trichloroacetimidate group at the C6-position was also evaluated to determine what effect this might have on reactivity. While this reaction proceeded sluggishly (12 h), it was able to provide 72% yield of the exclusive α-trichloroacetamide 24. Interestingly, the C6-trichloroacetimidate group in 19 did not survive during the transformation, resulting in an unmasked hydroxyl group for further functionalization as a potential acceptor in the glycosylation reaction.
Table 3.
Nickel-Catalyzed Conversion of Monosaccharide Acetimidates.[a]

| ||||
|---|---|---|---|---|
| Entry | Acetimidates | Time (h) | Products (h) | Yield (%)/[b] (α:β)[c] |
| 1 |

|
1 |

|
91 (10:1) |
| 2 |

|
10 |

|
90 (20:1) |
| 3 |

|
0.5 |

|
87 (15:1) |
| 4 |

|
0.5 |

|
91 (α only) |
| 5 |

|
12 |

|
72 (α only) |
The rearrangements were performed at 0.2 M in CH2G2 with 2 mol% of Ni(dppe)(OTf)2.
Isolated Yield.
1H NMR ratio.
To investigate the robustness of our cationic nickel-catalyzed rearrangement methodology, a wide variety of disaccharide and trisaccharide trichloroacetimidates 25–29 (Table 4) were tested. These substrates provided oligosaccharide trichloroacetamides 30–34 in high yield (67 – 85%) with more anomeric homogeneity (nearly exclusive α-stereoselectivity) than was observed with their monosaccharide counterparts 20–24 (Table 3). The first study utilized the per-O-benzylated mannose-α-(1,6)-glucose imidate 25 (entry 1) provided disaccharide trichloroacetamide 30 in 83% yield with 33:1 α-selectivity. To determine how the transformation would proceed with differentially protected carbohydrate residues, glucose-α-(1,6)-glucose substrate 26 (entry 2) was examined and produced 84% of the α-disaccharide acetamide 31 as the sole product. Similar performance was achieved with the 1,4-linked disaccharide substrate 28 (entry 4) as well as with the N-acetyl glucosamine-containing-disaccharide 27 (entry 3). The reaction were complete in 1 h, providing disaccharide trichloroacetamides 32 and 33 in 85% and 91% yield, respectively, as the exclusive α-isomers (entries 3 and 4). A trisaccharide trichloroacetimidate 29 (entry 5) was also explored. Although it took longer for the reaction to reach completion (4 h), α-trichloroacetamide 34 (entry 5) was isolated in 67% yield as the only product.
Table 4.
Nickel-Catalyzed Conversion of Oligosaccharide Acetimidates.[a]

| |||
|---|---|---|---|
| Entry | Acetimidates | Products | Yield (%)/[e] (α:β)[f] |
| 1 |

|

|
83[b] (33:1) |
| 2 |

|

|
84[b] (α only) |
| 3 |

|

|
85[c] (α only) |
| 4 |

|

|
91[c] (α only) |
| 5 |

|

|
67[d] (α only) |
The rearrangements were performed at 0.2 M in CH2Cl2 with 2 mol% of Ni(dppe)(OTf)2.
The reaction was complete within 2 h.
The reaction was complete within 1 h.
The reaction was complete within 4 h.
Isolated Yield.
1H NMR ratio.
While other methodologies available lack compatibility with C2-aminosugars,[12,17] we proceeded to investigate our own level of compatibility in this regard. We initially examined the C2 N-acetyl glucosamine substrate 35 (Scheme 2a); only the decomposition of starting material was observed. We then evaluated substrate 36 (Scheme 2b) containing the C2-azido group, which also led to decomposition. Upon switching to the N-benzylidene protected 2-deoxy glucosamine acetimidates 37 and 38 (Scheme 2c),[20d,e] we were able to efficiently achieve an α-selective transformation (71 – 79%, α/β = 11:1 – 20:1). The Ni(4-F-PhCN)4(OTf)2 catalyst was found to be more reactive that the Ni(dppe)(OTf)2 catalyst. While the para-methoxy-N-benzylidene protected imidate 37 performed quite sluggishly in the conversion compared to the para-fluoro-N-benzylidene derivative 38 (20 h vs. 3 h), it can be rationalized that competing addition from the β-face takes place with prolonged conversion time and is likely to account for the diminished α-selectivity. The 1,2-cis-2-amino urea linkage found in 39 and 40 (Scheme 2c) also suggests the feasibility of applying this method in the stereoselective synthesis of cinodine and coumamidine antibiotics,[6,7] where the structural arrangement is prevalent.
Scheme 2.

Transformation of C2-Amino Trichloroacetimidate Substrates
B. Mechanistic Studies of Nickel-Catalyzed Transformation of Trichloroacetimidates
Once satisfied with the robustness and versatility of our nickel-catalyzed methodology, we became interested in elucidating a possible mechanism for the selective transformation of trichloroacetimidate substrates. We hypothesize that the nickel catalyst would first coordinate the imidate nitrogen and the C2- ether oxygen of substrate 41 to form complex 43 (Figure 1). Subsequent ionization of the trichloroacetimidate leaving group followed by delivery of the trichloroacetamide group to the anomeric center from the α-face of the oxocarbenium intermediate 44 provides the corresponding complex 45 (Pathway A). Dissociation of the cationic nickel(II) catalyst will provide the desired α-glycosyl trichloroacetamide 42. Alternatively, 44 can dissociate to generate tight ion pair 46 (Pathway B),[28] which then recombines in a stereoelectronically favored mode to form α-trichloroacetamide 42α as the major product.
Figure 1.

Proposed Mechanism of Nickel-Catalyzed Conversion of Imidate
We hypothesized that a coordination event between the nickel catalyst and the C2-oxygen of an equatorial ether group was necessary for achieving the high α-selectivity observed in the trichloroacetamide product 42 (Figure 1). Thus, our first goal was to determine if the α-orientation of the trichloroacetimidate group of substrate 41 outlined in Figure 1 is important for coordination and subsequent ionization. Accordingly, β-trichloroacetimidate 47 (Scheme 3) was attempted in the presence of 5 mol % of Ni(dppe)(OTf)2.The rearrangement was able to proceed at 25 °C and reached completion after 12 h.[29] The yield of the desired rearrangement product 14 was greatly diminished (50%, 20:1 α/β) compared to the result obtained with α-trichloroacetimidate 13 (85%, 30:1 α/β, Table 1, entry 7) and was accompanied by a significant amount of starting material decomposition.
Scheme 3.

Rearrangement of β-Glycosyl Trichloroacetimidates
The result of the control experiment in Scheme 3 suggests that a) β-glycosyl trichloroacetimidate 47 is much less reactive than α-trichloroacetimidate 13,[30] and b) the ion-pair mechanism (Figure 2, Pathway B) is likely to be one of the major operative pathways in this transformation. To further validate our hypothesis that coordination of the nickel catalyst to both the equatorial C2-ether and α-C1-trichloroacetimidate is pivotal in the rearrangement, control experiment was performed with 2-deoxy glucosyl imidate 48 (Scheme 4a), though the endeavor would only lead to decomposition. On the other hand, β-2-deoxy-2-benzylidenamino glycosyl imidate 49 (Scheme 4b) resulted in no conversion, which is in stark contrast with β-glucose substrate 47 (Scheme 3).
Figure 2.

HRMS Signals (M+NH4) of Products from Crossover Experiment
Scheme 4.

Substrates Lacking Equatorial C2-Ether Group
Another control experiment was conducted with tetrabenzylated mannose substrate 50 (Scheme 4c), where we hypothesized that the expanse between the leaving group and of the axial C2-ether group would be prohibitively large for a simultaneous coordination with the nickel catalyst to occur.[31] Confirming our prediction, the conversion proceeded with a complete lack of stereoselectivity, providing a 1:1 mixture of α- and β- isomers 51 and 52 in 86% yield (Scheme 4c). The next substrate examined was the 4,6-O-benzylidene mannosyl trichloroacetimidate 53 (Scheme 4d), the donor used by Crich[32] and Schmidt[33] in β-mannosylation. In contrast with tetrabenzylated mannosyl imidate 50, a β-selective conversion was achieved with substrate 53, providing acetamide 54 (Scheme 4d) in 74% yield with excellent β-selectivity (α:β = 1:18).[34] The discrepancy in stereoselectivity between 50 and 53 (Schemes 4c–d) could be accounted for by the twist boat (B2,5) conformation of the oxocarbenium ion[35a] generated from the reaction of 4,6-O-benzylidene mannose 53 with Ni(dppe)(OTf)2 catalyst. We hypothesize that this perturbed ring structure of the cation intermediate brings the C2-ether functional group in close-enough proximity to the trichloroacetimidate leaving group for joint coordination with the nickel catalyst on the β-face. The β-selective formation of 54 (Scheme 4d) fits our hypothesis nicely that the coordination of the nickel catalyst with the C2-ether group of trichloroacetimidate substrate (Pathway A, Figure 1) is crucial for achieving a highly stereoselective transformation.
Having established the critical function of the ether functional group at the C2-position of trichloroacetimidate substrate, we set out to determine if the reaction proceeded intramolecularly (i.e. 1,3-rearrangement), or if an intermolecular transformation was possible. A crossover-labeling experiment (Scheme 5) was then conducted using an equimolar mixture of both unlabeled per-O-benzylated glucosyl trichloroacetimidate 13 and fully-labeled substrate 55 (containing deuterium labeled benzyl ether groups and oxygen-18 incorporated into the trichloroacetimidate group) under standard reaction conditions. If an intramolecular reaction was the operational pathway, it is expected to find only the two trichloroacetamide products 14 and 56 (Scheme 5) in the reaction mixture.
Scheme 5.

Crossover-Labeling Experiments
Interestingly, it was determined that in addition to the expected products 14 and 56, two partially-labeled cross-over acetamide products 57 and 58 (Scheme 5) were also observed in the reaction. HRMS signals corresponding to intramolecularly-rearranged products 14 and 56 and crossover products 57 and 58 with partial labeling are shown in Figure 2. Interested in understanding if nickel-coordinated delivery of the trichloroacetamide to the anomeric carbon was taking place in the transformation, a control experiment with tetrabenzylated glucosyl imidate 13 and an equivalent of external trichloroacetamide 59 was performed (Scheme 6). Our logic in this endeavor was that if we found the transformation to proceed with 30:1 α/β-selectivity, (as was observed in the absence of trichloroacetamide as shown Table 1), we could assume that delivery of the trichloroacetamide group to the anomeric center is facilitated by complexation with nickel catalyst. What we found instead, was that the α-selectivity obtained from this reaction had been greatly diminished (5.4:1 α/β, Scheme 6); indicating that a change in mechanism had taken place during the course of the transformation. It is postulated that competition between the α-facial selective re-delivery by nickel and an SN2-like displacement of the leaving group (generating the β-trichloroacetamide) is likely to operate, simultaneously; as a result, flooding the conversion with additional trichloroacetamide shifts this balance towards the SN2-type displacement.
Scheme 6.

Rearrangement in the Presence of External Trichloroacetamide
Overall, the above experiments allow a clearer definition of the nickel-catalyzed transformation of glycosyl trichloroacetimidates to the corresponding α-trichloroacetamides. First, it appears that the equatorial C2-ether and benzylideneamino functional groups of the glycosyl trichloroacetimidates are paramount for achieving α-selective transformation. Second, while both α- and β-isomers of the glycosyl C2-ether trichloroacetimidates can transform into the corresponding trichloroacetamides with excellent levels of α-selectivity, only the α-isomer of C2-benzylideneamino acetimidate substrates undergo rearrangement. Third, it is clear from a crossover experiment that the transformation does not proceed in an exclusively-intramolecular fashion, and that there is competition between the re-delivery of the trichloroacetamide group by nickel catalyst (Pathway A, Figure 1) and an intermolecular ion-pair (Pathway B, Figure 1).
C. Transformation of Glycosyl Trichloroacetamides into α-Urea Glycosides
Having established the versatility of our nickel-catalyzed transformation and gaining insight into the mechanistic nature of the conversion, we turned our attention to the final step in constructing α-urea glycosides: the conversion of the resulting trichloroacetamide into glycosyl urea. Our group has previously illustrated the efficacy of treating α-mannosyl trichloroacetamides with a variety of amine nucleophiles in the presence of cesium carbonate for generating α-urea mannosides.[18] As such, we set out to determine if the procedure would be amenable to additional types of glycosyl trichloroacetamides.
A model reaction was conducted (Scheme 7) using per-O-benzylated trichloroacetamide 14 and benzyl amine (60) in the presence of cesium carbonate in DMF, resulting in the formation of 80% yield of α-urea glycoside 61. The ambient temperature condition avoids deprotection of the trichloroacetamide group, which occurs at 100 °C.[36] This new approach requires only one step for direct transformation of α-glycosyl trichloroacetamide to the α-urea glycoside with the retention of the stereochemistry at the anomeric carbon. A control study was conducted to determine if the formation of urea would occur without Cs2CO3 (Scheme 7). In this experiment, there was no indication that the reaction was occurring after 72 h, the starting material 14 was recovered. This result suggests that formation of α-urea glycoside is unlikely to occur via direct nucleophilic acyl substitution reaction.[37]
Scheme 7.

Conversion of Trichloroacetamide to Glycosyl Urea
Satisfied with the above result, we proceeded to examine a number of monosaccharide α-trichloroacetamides with secondary amines (Table 5). In the coupling of pyrrolidine (62, entry 1) with quinovose acetamide substrate 23, 88% yield of the desired α-urea glycoside 66 was attained. Similar yield (94%) for the urea glycoside product 67[38] (entry 2) was achieved in the reaction of xylose trichloroacetamide 22 with piperidine (63). In addition, N-piperidylpiperazine (64) was successfully coupled to acetamide 14 to afford the α-urea glycoside 69 (entry 3) in 93% yield. In all case, the α-urea products were formed exclusively as α-isomers without any observable epimerization at the anomeric C-N bond.
Table 5.
Conversion of Trichloroacetamides to α-Urea Glycosides with Secondary Amines.[a]

| ||||
|---|---|---|---|---|
| Entry | Amines | Acetamides | Ureas | Yield (%)[b] |
| 1 |

|
23 |

|
88 |
| 2 |

|
22 |

|
94 |
| 3 |

|
14 |

|
93 |
All urea-forming reactions were performed with 2-3 equiv. amine and 3-5 equiv. Cs2CO3 at 25 °C in DMF (0.2 M).
Isolated Yield.
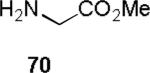
Although stereoselective methods for attaining β-urea-linked glycopeptides have been developed,[1b,39, 40] a direct and selective formation of α-urea-linked glycopeptides remains elusive. To test the scope and limitations of our method, a number of amino acid nucleophiles 70–72 (Table 6) were evaluated. The methyl esters of glycine 70 (entry 1) was efficiently coupled to α-acetamide 14, providing α-urea glycopeptide 74 in 85% yield. Phenylalanine 71 (entry 2) and derivatized proline 72 (entry 3) also reacted well to afford α-glycoconjugates 76 and 77, respectively, in 80 – 90% yield.[41] These results validate the efficacy of our methodology for generating high-yielding α-urea-linked glycopeptides in only two steps from trichloroacetimidates. In contrast, methods used for synthesizing α-urea-linked glycopeptides require five steps and employ the C(2)-acetyl group to control the formation of the β-anomeric selectivity.[1b,28]
Table 6.
Transformation of Trichloroacetamide 14 to α-Urea Glycosides with Amino Acid and Carbohydrate Amine Nucleophiles.[a]

| |||
|---|---|---|---|
| Entry | Amines | Ureas | Yield (%)[b] |
| 1 |
|

|
85 |
| 2 |

|

|
80 |
| 3 |

|

|
90 |
| 4 |

|

|
66 |
| 5 |

|

|
81 |
All urea-forming reactions were performed with 2-3 equiv. amine and 3-5 equiv. Cs2CO3 at 25 °C in DMF (0.2 M).
Isolated Yield.
We also demonstrated the versatility of our two-step approach by applying it in the synthesis of unsymmetrical α-urea-linked oligosaccharides (Table 6, entries 4 and 5). Although there are several efficient methodologies for the stereoselective synthesis of symmetrical urea-linked oligosaccharides,[16a,41] approaches for unsymmetrical preparation of this urea-linked motif remain under-developed.[3a] This warrants the development of efficient and stereoselective strategies for forming unsymmetrical urea-linked oligosaccharides. Accordingly, primary carbohydrate amines 79 and 80 (Table 6, entries 4 and 5) were explored with α-glycosyl trichloroacetamide 14. The coupling process proceeded smoothly to provide α-urea linked disaccharides 82 and 83, respectively, in 66 – 81% yield with complete retention of the stereochemical integrity at the anomeric center. Attempts to couple disaccharide trichloroacetamides 30 – 33 with either 79 or 80 resulted in poor conversions under similar conditions.
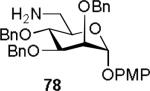
The operational simple of our two-step procedure was also highlighted in the coupling of number of amine nucleophiles with the 4,6-benzylidene mannosyl β-trichloroacetamide 54 (Table 7). Accordingly, treatment of substrate 54 with N-methyl piperazine (65) provided the corresponding β-glycosyl urea 68 (entry 1) in 75% yield with no observable epimerization at the anomeric C-N bond. Encouraged by this result, the reaction of phenylalanine 73 (entry 2) and carbohydrate amine 78 (entry 3) with 54 was also attempted, and the corresponding α-urea glycosides 75[41] and 81 were isolated in good yield (76 – 80%).
Table 7.
Stereoselective Synthesis of β-Urea-Linked Glycosides.[a]

| |||
|---|---|---|---|
| Entry | Amines | Ureas | Yield (%)/[b] |
| 1 |

|
|
75 |
| 2 |

|
|
88 |
| 3 |
|

|
76 |
All urea-forming reactions were performed with 2-3 equiv. amine and 3-5 equiv. Cs2CO3 at 25 °C in DMF (0.2 M).
Isolated Yield.
Conclusion
In summary, we have developed an atom-economical and efficient approach to the synthesis of α-urea-linked glycoside via nickel-catalyzed α-selective transformation of trichloroacetimidate to the corresponding trichloroacetamide and subsequent, one-step transformation to α-glycosyl urea with amine in the presence of cesium carbonate. The nickel-catalyzed conversion of glycosyl trichloroacetimidate to the α-trichloroacetamide intermediate has been found to be rapid and highly selective and to proceed in the presence of Ni(dppe)(OTf)2. This method is tolerant of a variety of sugar types and protecting groups and can applied to a range of monosaccharide, disaccharide, and trisaccharide substrates.
Control experiments with 2-deoxy glucose and mannose substrates have highlighted the importance of the C2-ether functionality in directing the α-selective transformation.
Crossover-labeling study has identified the conversion is not proceeding in strictly intramolecular fashion. There is competition between the re-delivery of the acetamide by cationic nickel(II) catalyst (Pathway A, Figure 1) and an intermolecular-ion-pair (Pathway B, Figure 1).
While both the α- and β-isomers of trichloroacetimidates containing C2-ether group undergo conversion, only the α-isomer of C2-benzylideneamino substrates can transform into the corresponding trichloroacetamide products.
The conversion of trichloroacetamide into α-urea glycoside has been achieved by reacting with amine in the presence of Cs2CO3. The procedure has been successfully applied to secondary amines, amino acids, and aminosugars to provide α-ureas in good yields with no epimerization of the anomeric C-N bond.
This atomic-economical two-step method is also applicable to the synthesis of β-urea glycosides from the coupling of 4,6-benzylidene mannosyl β-trichloroacetamide with amine nucleophiles.
Insights gained from our studies will help future advances in the selective formation of α-urea-linked pseudo-oligosaccharides and neo-glycoconjugates with the potential for increased stability over their O-linked and N-linked glycoside counterparts.
Experimental Section
Standard Procedure for Preparation of Trichloroacetamide
A 10 mL oven-dried Schlenk flask was charged with α-glycosyl trichloroacetimidate (0.2 mmol, 1 equiv.) and CH2Cl2 (1.5 mL). A preformed solution of Ni(dppe)OTf2, which was generated in situ from a reaction of Ni(dppe)Cl2 (0.004 mmol, 2 mol%) and AgOTf ( 0.008 mmol, 4 mol%) in CH2Cl2 (0.5 mL) for 15 min, was added. The resulting mixture was then monitored by FT-IR spectroscopy (C=N stretch at 1670 cm−1 → C=O stretch at 1726 cm−1) at room temperature. The mixture was loaded directly onto silica and purified by silica gel flash column chromatography to provide the corresponding trichloroacetamide as a viscous oil.
Standard Procedure for Preparation of α-Urea Glucoside
A 10 mL oven-dried Schlenk was charged with trichloroacetamide (0.1 mmol, 1 equiv.). and amine nucleophile (0.3 mmol, 3 equiv.) and toluene (1 mL) before concentrating in vacuo for azeotropic removal of water. This process was repeated three times (3×) before adding anhydrous cesium carbonate (0.3 – 0.5 mmol, 3 – 5 equiv). The resulting mixture was flushed with argon, and anhydrous DMF was (2 mL) was then added. The mixture was allowed to stir at room temperature for 10 h. The reaction mixture was poured into a saturated aqueous solution of NaHCO3 (25 mL) and extracted with ethyl acetate (5 × 25 mL). The combined organic extracts were dried over MgSO4, filtered, and then concentrated in vacuo. The crude product was purified by silica gel flash chromatography to provide α-urea glycoside.
Supplementary Material
Acknowledgements
We are grateful for the financial support from National Science Foundation (NSF-CHEM 1106082) and National Institutes of Health (R01 GM098285). We thank Dr. Fei Yu for preparing and spectroscopically analyzing trichloroacetamide 54. A portion of this work was performed at Montana State University.
References
- 1.Ichikawa Y, Nishiyama T, Isobe M. J. Org. Chem. 2001;66:4200–4205. doi: 10.1021/jo0100751. [DOI] [PubMed] [Google Scholar]
- 2.a Prosperi D, Ronchi S, Lay L, Rencurosi A, Russo G. Eur. J. Org. Chem. 2004:395–405. [Google Scholar]; b Prosperi D, Ronchi S, Panza L, Rencurosi A, Russo G. Synlett. 2004:1529–1532. [Google Scholar]
- 3.a Ichikawa Y, Nishiyama T, Isobe M. Synlett. 2000;9:1253–1256. [Google Scholar]; b Ichikawa Y, Ohara F, Kotsuki H, Nakano K. Org. Lett. 2006;8:5009–5012. doi: 10.1021/ol0616788. [DOI] [PubMed] [Google Scholar]
- 4.a Tewari N, Tiwari VK, Mishra RC, Tripathi RP, Srivastava AK, Ahmad R, Srivastava R, Srivastava BS. Bioorg. Med. Chem. 2003;11:2911–2922. doi: 10.1016/s0968-0896(03)00214-1. [DOI] [PubMed] [Google Scholar]; b Paulsen H, Todt K. Adv. Carbohydr. Chem. 1968;23:115–232. doi: 10.1016/s0096-5332(08)60169-1. [DOI] [PubMed] [Google Scholar]; c Truscheit E, Frommer W, Junge B, Mueller L, Schmidt DD, Wingender W. Angew. Chem. Int. Ed. 1981;20:744–761. [Google Scholar]; d Inouye S, Tsuruoka T, Ito T, Niida T. Tetrahedron. 1968;24:2125–2144. doi: 10.1016/0040-4020(68)88115-3. [DOI] [PubMed] [Google Scholar]; e Anzeveno PB, Creemer LJ, Daniel JK, King CHR, Liu PS. J. Org. Chem. 1989;54:2539–2542. [Google Scholar]
- 5.a Umezewa S, Tsuchiya T. In: Aminoglycosides Antibiotics. Umezewa H, Hooper RI, editors. Springer; Berlin: 1982. pp. 37–110. [Google Scholar]; b Kirst HA. In: Bruger's Medicinal Chemistry and Drug Discovery. Wolff ME, editor. Wiley; New York: 1996. pp. 463–525. [Google Scholar]
- 6.a Tresner HD, Korshalla JH, Fantini AA, Korshalla JD, Kirby JP, Goodman JJ, Kele RA, Shay AJ, Borders DB. J. Antibiot. 1978;31:394–397. doi: 10.7164/antibiotics.31.394. [DOI] [PubMed] [Google Scholar]; b Martin JH, Kuntsmann MP, Barbatschi F, Hertz M, Ellestad GA, Dann M, Redin GS, Dornbush AC, Kuck NA. J. Antibiot. 1978;31:398–404. doi: 10.7164/antibiotics.31.398. [DOI] [PubMed] [Google Scholar]; c Ellestad GA, Cosulich DB, Broschard RW, Martin JH, Kuntsmann MP, Morton GO, Lancaster JE, Fulmor W, Lovell FM. J. Am. Chem. Soc. 1978;100:2515–2524. [Google Scholar]
- 7.Fernandes P, Swanson R, Hardy D, Hanson C, McDaniel D, Beyer J, Chen R, Antibiot J. 1989;42:538–541. doi: 10.7164/antibiotics.42.538. [DOI] [PubMed] [Google Scholar]
- 8.Greenstein M, Speth J, Maiese W. Antimicrob. Agents Ch. 1981;20:425–432. doi: 10.1128/aac.20.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Osburne M, Maiese w., Greenstein M. Antimicrob. Agents Ch. 1990;34:1450–1452. doi: 10.1128/aac.34.7.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.A simplified structure, glycocinnasperimicin D, that contains β-linked anomeric urea bond, also bind directly to bacterial DNA and its DNA gyrase, see: Dobashi K, Nagaoka K, Watanabe Y, Nishida M, Hamada M, Naganawa J, Takita T, Takeuchi T, Umezawa H. J. Antibiot. 1985;38:1166–1170. doi: 10.7164/antibiotics.38.1166.
- 11.Only the synthesis of β-urea-linked glycocinnasperimicin D has been reported, see: Nishiyama T, Isobe M, Ichikawa Y, Angew Y. Chem. Int. Ed. 2005;44:4372–4375. doi: 10.1002/anie.200500892. Nishiyama T, Kusumoto Y, Okumura K, Hara K, Kusaba S, Hirato K, Kamiya Y, Isobe M, Nakano K, Kotsuki H, Ichikawa Y, Chem Y. Eur. J. 2010;16:600–610. doi: 10.1002/chem.200901745.
- 12.Bianchi A, Ferrario D, Bernandi A. Carbohyd. Res. 2006;341:1438–1446. doi: 10.1016/j.carres.2006.03.042. [DOI] [PubMed] [Google Scholar]
- 13.For recent review on the synthesis of carbohydrate ureas, see: McKay MJ, Nguyen HM. Carbohydr. Res. 2014;385:18–44. doi: 10.1016/j.carres.2013.08.007.
- 14.For recent review on the synthesis of carbohydrate ureas, see: Spanu P, Ulgheri F. Curr. Org. Chem. 2008;12:1071–1092.
- 15.Ogawa T, Nakabayashi S, Shibata S. Agric. Biol. Chem. 1983;47:281–285. [Google Scholar]
- 16.a Benn MH, Jones AS. J. Chem. Soc. 1960:3837–3841. [Google Scholar]; b García-Fernández JM, Ortiz-Mellet C, Díaz-Pérez VM, Fuentes J, Kovács J, Pintér I. Tetrahedron Lett. 1997;38:4161–4164. [Google Scholar]; c García-Moreno MI, Benito JM, Ortiz-Mellet C, García-Fernández JM. Tetrahedron: Asymmetry. 2000;11:1331–1341. [Google Scholar]; d Ichikawa Y, Matsukawa Y, Isobe M. Synlett. 2004:1019–1022. [Google Scholar]; e Ichikawa Y, Matsukawa Y, Isobe M. J. Am. Chem. Soc. 2006;128:3934–3938. doi: 10.1021/ja056253f. [DOI] [PubMed] [Google Scholar]; f Ichikawa Y, Matsukawa Y, Tamura M, Ohara F, Isobe M, Kotsuki H. Chem. Asian. J. 2006;1:717–723. doi: 10.1002/asia.200600190. [DOI] [PubMed] [Google Scholar]
- 17.a Ichikawa Y, Nishiyama T, Isobe M. Synlett. 2000:1253–1256. [Google Scholar]; b Ichikawa Y, Nishiyama T, Isobe M. J. Org. Chem. 2001;66:4200–4205. doi: 10.1021/jo0100751. [DOI] [PubMed] [Google Scholar]; c Ichikawa Y, Nishiyama T, Isobe M. M. Tetrahedron. 2004;60:2621–2627. [Google Scholar]
- 18.Mercer GJ, Yang J, McKay MJ, Nguyen HM. J. Am. Chem. Soc. 2008;130:11210–11218. doi: 10.1021/ja803378k. [DOI] [PubMed] [Google Scholar]
- 19.Park NH, Nguyen HM. Org. Lett. 2009;11:2433–2436. doi: 10.1021/ol900670a. [DOI] [PubMed] [Google Scholar]
- 20.a McConnell MS, Mensah EA, Nguyen HM. Carbohydr. Res. 2013;381:146–152. doi: 10.1016/j.carres.2013.09.006. [DOI] [PubMed] [Google Scholar]; b McConnell MS, Yu F, Nguyen HM. Chem. Comm. 2013;49:4313–4315. doi: 10.1039/c2cc35823a. [DOI] [PubMed] [Google Scholar]; c Yu F, Nguyen HM. J. Org. Chem. 2012;77:7730–7343. doi: 10.1021/jo301050q. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Mensah EA, Yu F, Nguyen HM. J. Am. Chem. Soc. 2010;132:14288–14302. doi: 10.1021/ja106682m. [DOI] [PubMed] [Google Scholar]; e Mensah EA, Nguyen HM. J. Am. Chem. Soc. 2009;131:8778–8780. doi: 10.1021/ja903123b. [DOI] [PubMed] [Google Scholar]; f McKay MJ, Naab BD, Mercer GJ, Nguyen HM. J. Org. Chem. 2009;74:4705–4711. doi: 10.1021/jo9002807. [DOI] [PubMed] [Google Scholar]; g Mensah EA, Azzarelli JA, Nguyen HM. J. Org. Chem. 2009;74:1650–1657. doi: 10.1021/jo802468p. [DOI] [PubMed] [Google Scholar]; h Yang J, Cooper-Vanosdell C, Mensah EA, Nguyen HM. J. Org. Chem. 2008;73:794–800. doi: 10.1021/jo702436p. [DOI] [PubMed] [Google Scholar]; i Schuff BP, Mercer GJ, Nguyen HM. Org. Lett. 2007;9:3173–3176. doi: 10.1021/ol071268z. [DOI] [PubMed] [Google Scholar]
- 21.Pretsch E, Simon W, Seibl J, Clerc T. In: Spectral Data for Structure Determination of Organic Compounds. Fresenius W, Huber JFK, Rechnitz GA, Simon W, West Th. S., editors. Springer-Verlag; Berlin Heidelberg: 1989. pp. 1150–1160. [Google Scholar]
- 22.In order to maintain high α-selectivity, it is crucial to use Ni(dppe)(OTf)2 immediately upon generating in situ from Ni(dppe)Cl2 and AgOTf. We also noted that extended pre-stirring Ni(dppe)(OTf)2 mixture significantly deteriorates the α-selectivity.
- 23.Considerable loss in the form of hydrolysis was observed in the reaction of trichloroacetimidate 13 with use of Ni(dppb)(OTf)2.
- 24.Freixa Z, van Leeuwen PWNM. Dalton Trans. 2003:1890–1901. [Google Scholar]
- 25.a Schmidt RR, Michel J. Angew. Chem. Int. Ed. 1980;19:731–732. [Google Scholar]; b Schmidt RR, Michel J. J. Carbohydr. Chem. 1985;4:141–169. [Google Scholar]; c Zimmermann P, Bommer R, Bär T, Schmidt RR. J. Carbohydr. Chem. 1988;7:435–452. [Google Scholar]
- 26.a Schmidt RR. Angew. Chem. Int. Ed. 1986;25:212–235. [Google Scholar]; b Schmidt RR, Behrendt M, Toepfer A. Synlett. 1990:694–696. [Google Scholar]; c Schaubach R, Hemberger J, Kinzy W. Liebigs. Ann. Chem. 1991:607–614. [Google Scholar]; d Paulsen H, Wilkens R, Reck F, Brockhausen I. Liebigs. Ann. Chem. 1992:1303–1313. [Google Scholar]; e Kulkarni SS, Hung S-C. Lett. Org. Chem. 2005;2:670–677. [Google Scholar]
- 27.a Pereda-Miranda R, Kaatz GW, Gibbons J. J. Nat. Prod. 2006;69:406–409. doi: 10.1021/np050227d. [DOI] [PubMed] [Google Scholar]; b Lee K-H, Morris-Natschke SL. Pure. Appl. Chem. 1999;71:1045–1051. [Google Scholar]; c Sun I-C, Kashiwada Y, Morris-Natschke SL, Lee K-H. Curr. Med. Chem. 2003;3:155–169. doi: 10.2174/1568026033392435. [DOI] [PubMed] [Google Scholar]
- 28.a Calter M, Hollis TK, Overman LE, Ziller J, Zipp GG. J. Org. Chem. 1997;62:1449–1456. [Google Scholar]; b Yougai S, Miwa T. J. Chem. Soc., Chem. Commun. 1983:68–69. [Google Scholar]
- 29.The conversion of β-glycosyl trichloroacetimidate containing the C(2)-benzoyl participatory group to the corresponding β-trichloroacetamide under TMSOTf conditions has been recently reported, see: Tanaka H, Iwata Y, Takahashi D, Adachi M, Takahashi T. J. Am. Chem. Soc. 2005;127:1630–1631. doi: 10.1021/ja0450298.
- 30.It is not clear why use of β-trichloroacetimidate 47 provided only 50% yield of α-glycosyl trichloroacetamide 14. We hypothesize that because β-trichloroacetimidate 47 is not very reactive in the transformation, there is likely competition between degradative pathways (ie. hydrolysis) and formation of the desired product 14.
- 31.The mannosyl oxocarbenium of substrate 50 is likely to exist in one of two half-chair conformations. While one computation study shows a small preference for the 4H3 conformer with the maximum number of alkoxy substituents placed equatorially and C2-alkoxy group placed axially,a another calculation and biological data suggest that the 3H4 conformer (alkoxyl group at C2 favors equatorial position and alkoxy functional groups at the C3, C4, and C6 adopt axial orientations) is favored.b,c An alternative explanation relies on considering the fact that both conformers are in rapid equilibrium, leading to a 1:1 mixture of α-and β-trichloroacetamide products 51 and 52.[d] Nukakda T, Berces A, Wang L, Zgierski MZ, Whitfield DM. Carbohydr. Res. 2005;340:841–852. doi: 10.1016/j.carres.2004.12.021. Amat L, Carbo-Dorca R. J. Chem. Inf. Comput. Sci. 2000;40:1188–1198. doi: 10.1021/ci0000272. Winkler DA, Holan G. J. Med. Chem. 1989;32:2084–2089. doi: 10.1021/jm00129a011. Winkler DA. J. Med. Chem. 1996;39:4332–4334. doi: 10.1021/jm960237z. Lucero CG, Woerpel KA. J. Org. Chem. 2006;71:2641–2647. doi: 10.1021/jo0522963.
- 32.a Crich D, Sun S. J. Org. Chem. 1996;61:4506–4507. doi: 10.1021/jo9606517. [DOI] [PubMed] [Google Scholar]; b Crich D, Sun S. J. Org. Chem. 1997;62:1198–1199. [Google Scholar]; c Crich D, Sun S. J. Am. Chem. Soc. 1997;119:11217–11223. [Google Scholar]; d Crich D, Sun S. J. Am. Chem. Soc. 1998;120:435–436. [Google Scholar]; e Crich D, Sun S. Tetrahedron. 1998;54:8321–8348. [Google Scholar]; f Crich D, Cai W, Dai Z. J. Org. Chem. 2000;65:1291–1297. doi: 10.1021/jo9910482. [DOI] [PubMed] [Google Scholar]; g Crich D, Smith M. J. Am. Chem. Soc. 2001;123:9015–9020. doi: 10.1021/ja0111481. [DOI] [PubMed] [Google Scholar]; h Crich D. J. Carbohydr. Chem. 2002;21:667–690. [Google Scholar]
- 33.Weingart R, Schmidt R. Tetrahedron Lett. 2000;41:8753–8758. [Google Scholar]
- 34.The 1JCH coupling between the anomeric carbon of 54 and its associated proton was measured, and 1JCH = 160 Hz. It is known that the magnitude of the 1JCH coupling wherein a value of around 160 Hz is diagnostic for the β-configuration and 170 Hz the α-mannoside, see: Boch K, Pedersen C. J. Chem. Soc., Perkin Trans. 2. 1974:293–297. Crich D, Sun S. Tetrahedron. 1998;54:8321–8348.
- 35.a Crich D, Chandrasekera NS. Angew. Chem. Int. Ed. 2004;43:5386–5389. doi: 10.1002/anie.200453688. [DOI] [PubMed] [Google Scholar]; b Huang M, Retailleau P, Bohe L, Crich D. J. Am. Chem. Soc. 2012;134:14746–14749. doi: 10.1021/ja307266n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Urabe D, Sugino K, Nishikawa T, Isobe M. Tetrahedron Lett. 2004;45:9405–9407. [Google Scholar]
- 37.Although the mechanism for urea formation from trichloroacetamides has not been completely clarified yet, it is speculated that trichloroacetamides are used as in situ isocyanate generating reagents which react with carboxamides or sulfonamides to afford acylureas or sulfonylureas, see: Antanassova IA, Petrov JS, Mollov NM. Synthesis. 1987:734–736.
- 38.The anomeric effect of the nitrogen atom in the urea functional groups was studied using 1H NMR analysis of both α- and β-xylopyranosyl ureas. It was determined that the anomeric effect of the urea functionality is not observed and the urea substituent prefers the equatorial orientation to minimize steric interactions. In addition, the xylopyranosyl urea 67 (Table 5, entry 2) may adapt a 1C4 conformation in which the urea substituent occupies an equatorial position, see: Ichikawa Y, Watanabe H, Kotshuki H, Nakano K. Eur. J. Org. Chem. 2010:6331–6337.
- 39.a Dwek RA. Chem. Rev. 1996;96:683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]; b Varki A. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.a Christiansen-Brams I, Meldal M, Bock K. J. Chem. Soc., Perkin Trans. 1993;1:1461–1471. [Google Scholar]; b van Ameijde J, Albada HB, J. Liskamp RM. J. Chem. Soc., Perkin Trans. 1994;1:1042–1049. [Google Scholar]
- 41.There was no observable epimerization at the α-position of the amino acid moiety of α-urea-linked glycopeptides 75 and 76.
- 42.Kovacs J, Pinter I, Messmer A. Carbohydr. Res. 1987;166:101–111. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.