

Abstract
Aminoglycosides (AGs) are highly potent antibacterial agents, which are known to exert their deleterious effects on bacterial cells by interfering with the translation process, leading to aberrant protein synthesis that usually results in cell death. Nearly 45 years ago, AGs were shown to induce read-through activity in prokaryotic systems by selectively encoding tRNA molecules at premature termination codon (PTC) positions; resulting in the generation of full length functional proteins. However, only in the last 20 years this ability has been demonstrated in eukaryotic systems, highlighting their potential as therapeutic agents to treat PTC induced genetic disorders. Despite the great potential, AGs use in these manners is quite restricted due to relatively high toxicity values observed upon their administration. Over the last few years several synthetic derivatives were developed to overcome some of the enhanced toxicity issues, while in parallel showed significantly improved PTC suppression activity in various in-vitro, ex-vivo and in-vivo models of a variety of different diseases models underling by PTC mutations. Although these derivatives hold great promise to serve as therapeutic candidates they also demonstrate the necessity to further understand the molecular mechanisms of which AGs confer their biological activity in eukaryotic cells for further rational drug design. Recent achievements in structural research shed light on AGs mechanism of action and opened a new avenue in the development of new and improved therapeutic derivatives. The following manuscript highlights these accomplishments and summarizes their contributions to the state of art rational drug design.
1. Aminoglycosides
Aminoglycosides (AGs) were first established as antibiotics in the 1940s with the discovery of streptomycin and are still widely used worldwide for the treatment of various infectious diseases 1, 2. Chemically AGs are cationic oligosaccharides composed of between two and five amino sugar rings. At physiological pH, the amino groups are nearly all protonated, giving AGs a net positive charge 3, 4. AGs can be categorized structurally into two groups based upon the identity of conserved aminocyclitol - 2-deoxystreptamine (2-DOS) ring around which they are built. The amino sugar moieties are distributed about the 2-DOS ring in two major substitution patterns: 4,5-disubtituted which include neomycin and paromomycin, and 4,6-disubsituted, which include gemtamicins and kanamycins. Members of the 4,6-disubstituted class are mostly used clinically (Fig. 1). The ribosome is thought to be the primary target of AG antibiotics. These antibacterial agents bind selectively to the bacterial ribosomal RNA (rRNA) and induce a conformational change that eventually leads to miscoding and bactericidal effects. AGs that contain 2-DOS ring target the same region of rRNA and interfere with the ribosomal process involved in decoding and processivity of proteins. In addition to the ribosome, AGs have been shown to bind numerous RNA constructs such as ribozymes 5, introns 6, 7, HIV-1 Tat-responsive element (TAR) 8 and HIV-1 Rev-responsive element (RRE) 9 and modulate their structure and function.
Figure 1.
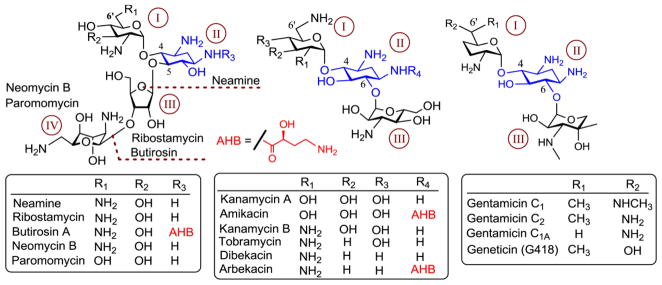
Representative aminoglycoside antibiotics with a 2-deoxystreptamine (2-DOS) ring. The 2-DOS ring and ring numbers are highlighted in blue and brown, respectively. The substitution by (S)-4-amino-2-hydroxybutanoyl (AHB) group is shown in red.
2. Aminoglycosides and genetic diseases
AGs antibacterial activity as miscoding agents has been extensively investigated over the last several decades. However, nearly 2 decades after the discovery of streptomycin, it was discovered that AGs are able to suppress premature termination codons (PTCs), restoring full-length protein production in Escherichia coli (E. coli) 10. PTCs are genetic code mutations usually occurring due to base pair insertions, deletions, or substitutions that result in the replacement of an amino-acid codon in DNA by one of the three universal stop codons (TAA, TGA or TAG). These mutations generally lead to the production of truncated, nonfunctional proteins (Fig. 2). In humans, PTCs have been linked to nearly 2,000 genetic disorders, such as cystic fibrosis (CF), Duchenne muscular dystrophy (DMD), ataxia-telangiectasia (AT), Hurler syndrome (HS), Rett syndrome, Usher syndrome (USH), Hemophilia A, Hemophilia B and Tay-Sachs 11, 12. For many of these diseases there is presently no effective treatment.
Figure 2.
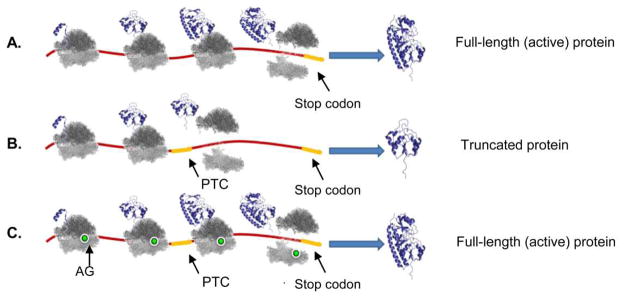
A comparison of the (A) normal translation process leading to functional protein production, (B) translation which is interrupted by a premature termination codon (PTC) leading to a truncated non-functional protein production, and (C) translation process which was restored by a read-through inducing compound such as an aminoglycoside. Terminal codons and PTCs are highlighted in yellow, aminoglycoside in green.
In the last several years, a new therapeutic strategy has suggested the use of PTC suppression in the treatment of PTC induced genetic disorders 13. This therapeutic strategy, also called “suppression therapy” or “translational readthrough”, utilizes small molecule pharmacological agents to selectively suppress translation termination at PTCs but not at normal stop codons to restore translation of full-length, functional proteins (for the recent reviews see Keeling et. al. 2012 14 and Bidou et. al. 2012 15). Recent scientific evidences have demonstrated the ability of some natural AGs, such as gentamicin, G418 and paromomycin to induce PTC read-through in various eukaryotic systems 16–19, via the selective insertion of a near cognate tRNA at the PTC position; restoring the production of full length functional proteins (Fig. 2C). These proof-of-principle studies have suggested the use of such AGs as a possible treatment for human genetic diseases caused by PTCs, and indeed, recent clinical studies performed in CF patients carrying one of the CFTR stop mutations indicated gentamicin's ability to improve patients' symptoms 20, 21.
These encouraging results were further exploited for the establishment of several clinical trials in DMD patients 22 altogether with massive experiments performed in various in-vitro, ex-vivo and in-vivo systems on DMD 23, 24 CF 18, Rett syndrome 25, 26, HS 27, nephrogenic diabetes insipidus 28, nephropathic cystinosis 29, retinitis pigmentosa 30, ataxia-telegiectasia 31, spinal muscular atrophy (SMA) 32, severe epilepsy syndrome 33 and several genetically induced cancer types 34–36. The resulting data supported the previous findings and highlighted AGs as possible candidates for PTC suppression therapy. However, it also emphasized the complexity of the mechanisms of which these agents induce their therapeutic effects. For example, it has been noticed that the identity of the PTC affects the efficacy of PTC suppression (TGA > TAG > TAA). In addition, the nucleotides located immediately downstream of the stop codon in the mRNA template (+4 position), also seem to determine the read-through potential (C > U > A ≥ G) as well as those located one residue upstream to the codon triplet (-1 position), where U seemed to induce the higher read-through levels. The local sequence context around the PTC and its position within the gene sequence where also shown to affect the induced read-through levels 19, 37, 38. In addition, recent publications demonstrated the ability of AGs to interfere with the nonsense mediated decay (NMD) system, implying that AGs not only induce read-through events on mutant mRNA upon translation, but also stabilize the survival of mutant mRNA species that are generally destined to degradation by the NMD apparatus. These activities are thoroughly discussed in recent review papers by Bedwell and coworkers 14, and by Bidou and et al 15, adding some more complexity to the already complex PTC read-through mechanism.
The chemical structure of AGs also seems to play a major role in their ability to interfere with translation termination. In fact, not all AGs are capable of inducing termination suppression, and the explanation for these facts yet remains rather obscure. In general, AGs containing a 6'-OH group in their first ring (such as G418 and paromomycin, Fig. 1) are superior to those enclosing an NH2 moiety at the same position 19, 39. These observations are usually subjected to one of the main differences between their putative binding sites in eukaryotic vs. prokaryotic systems: the 1408 position (E. coli numbering). It has been well documented that an A1408G mutation in bacteria confers resistance to AGs, and that higher levels of antibacterial resistance are usually observed towards AGs containing an amino moiety in their 6' position 40, 41. To explain these observations it has been suggested that, while both 6’-OH and 6’-NH2 can form H-bond interactions with A1408 in bacterial ribosomes, the 6’-NH2 derivatives are prevented of such interactions with G1408 in eukaryotic ribosomes due to electrostatic repulsion between the positively charged nitrogen atom of the guanine residue and the 6’-NH3+ of AGs.
The fact that eukaryotic ribosomes contain a guanine residue in the corresponding position might explain the higher activity observed in the 6'-OH derivatives. Very recent crystal structures of complexes of G418 with native vs. mutant bacterial binding sites suggested a structural explanation for the 6'-OH selectivity at the molecular level (Fig. 3) 42. In addition, a recent paper published by Baasov and coworkers 43 supplemented the structural basis for AG selectivity to Leishmania, a eukaryotic parasite of which AGs binding site differs only in one nucleotide when compared to human (U1409 in Leishmania vs. C1409 in humans, Fig. 4). This work did not only highlight the differential selectivity of 6'-OH and 6’-NH2 derivatives to eukaryotes, but also indicated for the first time that not all AGs are capable of inducing the same conformational change upon binding; implying that unlike in bacterial species, in eukaryotes the binding affinity and the actual binding to the A-site does not necessary result in biological activity (a more detailed explanation will be presented in section 6). However, despite recent progress in the structural understanding of AGs structure-activity relationships in eukaryotes some questions still remain unanswered, and it is not yet understood, for example, why gentamicin, a 6'-NH2 derivative confers some rather high read- through activities in eukaryotes. Such questions might indicate that we are not yet able to fully understand AG activities in eukaryotes, thus add some more complexity to the effort towards rationally designing some new and improved derivatives.
Figure 3.

The molecular mechanism in which 6'-OH aminoglycosides may interact with guanine 1408. A 2D representation of ring I of G418 interactions with A/G residue in 1408 position.
Figure 4.
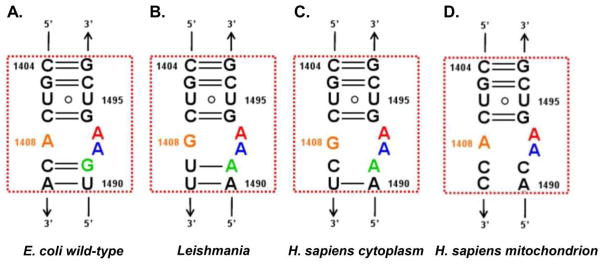
Secondary structures of ribosomal A-sites of (A) E. coli, (B) Leishmania, (C) H. sapiens cytoplasm and (D) H. sapiens mitochondrion. The conserved adenine residues, A1492 and A1493, are highlighted in blue and red, respectively.
3. Aminoglycosides toxicity
Despite the promising results indicating the great potential of using AGs to treat genetic disorders, their use for such therapeutic purposes is quite restricted nowadays. These limitations are mainly due to the relatively high toxicity values prescribed upon their administration. Prolonged uses of AGs are often associated with ototoxicity and/or nephrotoxixty events 44. Such events are sometimes irreversible and in the ototoxicity case might result in a substantial hearing loss 44. Unfortunately, the mechanisms by which AGs induce their toxic effects in eukaryotes are not fully deciphered yet; the current information in this field is very little, and is mainly attributed to their positively charged nature that makes them prone to interact with various negatively charged cellular components such as phospholipids, phospholipases and various metal ions. These interactions are believed to eventually lead the generation of reactive oxygen species (ROS) 45 which are known to enhance cell toxicity. Over the last few years, many efforts have been directed in attempt to overcome the toxicity associated upon AGs administration. Those included co-administration with a variety of compounds including several antioxidants such as vitamin E 46, N-acetyl-cistein-cholins (NAC) 47 and salicylic acid 48–51, iron chelators 50, 52, and some negatively charged agents such as poly-L-aspartate 53, 54 and daptomycin 55, 56, which are considered to limit AGs unspecific interactions with negatively charged cellular components.
AGs attributions to the inhibition of eukaryotic translation machineries are also considered to play an importance in their induced toxicity. In this context, two different mechanisms have recently been suggested to explain AG-induced ototoxicity, a major drawback limiting their potential for suppression therapy. One model suggests that AGs exert their ototoxicity by inhibiting mitochondrial protein synthesis machinery; such inhibition leads to oxidative stress events causing mitochondrial dysfunction which ultimately leads to cell death 57–60; these recent works demonstrated the ability of some natural and synthetic derivatives to inhibit mitochondrial protein synthesis; apparently, due to the relatively high similarity the mitochondrial ribosome shares with their primary target site, the bacterial ribosome (Fig. 4). An alternative model highlights cytoplasmic protein synthesis inhibition as a potential trigger of a cellular pathway, similar to ribotoxic stress response, leading to hair cell apoptosis 61. The two suggested mechanisms were recently demonstrated in various models explaining how AGs induced ototoxicity might result from inhibiting either the mitochondrial or cytoplasmic protein machineries, however, it still remains unclear whether both mechanisms act in concert or one mechanism predominates over other in-vivo. A recent in-vivo work performed in guinea pigs and mice cochlear explants by Shulman et. al. 62 indicated that ototoxicity exerted by a particular AG correlates primarily with its ability to block mitochondrial rather than cytoplasmic protein synthesis. This study focused on four natural and synthetic AG derivatives that were shown to markedly differ in their abilities to block cytoplasmic and mitochondrial protein synthesis. The observed data indicated a strong correlation between AGs interactions with mitoribosomes to cell respiration perturbation resulting in mitochondrial aconitase damaging, enhanced superoxide radical production and free ferrous iron accumulation. This work linked AGs mitochondrial activities to the activation of Fenton reaction, eventually leading to cell apoptosis and highlighted a few AG synthetic derivatives earlier suggested by Baasov and coworkers as promising therapeutic agents showing some enhanced read-through activities altogether with reduced mitoribosome inhibition. Such works give some great hopes to fighting AGs toxic effects suggesting that by selective targeting of AGs to their cytoplasmic site, one should diminish their deleterious side effects and enhance their therapeutic value. In addition they gave rise to the development of some new and improved synthetic AG derivatives that selectivity target the cytoplasmic eukaryotic ribosome 59, avoiding mitochondrial inhibition.
The challenges and development in designing some new and improved AG derivatives are discussed in the following section (section 4).
4. Challenges in the design of novel nonsense read-through inducers
Synthetic derivatives carrying an AG scaffold have been extensively investigated over the last few decades as potential therapeutic agents to be used for the treatment of bacterial infections 63. These efforts resulted in the development of improved derivatives with reduced toxicity and enhanced ability to delay the emergence of resistance among bacterial species 64, 65. These compounds were massively investigated in both biochemical and structural manners, and along with the molecular understanding of AGs mechanisms of action and resistance in bacterial species, opened a new avenue in the field of antibacterial therapy.
However, due to the limited information regarding their mechanism of action in eukaryotes as well as the higher complexity of these systems, when compared to the prokaryotic ones, only a few novel semi-synthetic derivatives have been suggested as efficient read-through inducers over the last few years. These agents have usually shown an improved read-through activity as well as decreased toxicity when compared to the natural derivatives 59, 66–70. In 2006, Lorson and coworkers 67 demonstrated the ability of 6 semi-synthetic neamine and kanamycin derivatives, to promote read-through of PTC in survival motor neuron-1 (SMN) in fibroblasts derived from SMA patients. One derivative, TC007, was shown to induce up to a 30 fold increase in normal protein production compared to the untreated baseline in a dose dependent manner. Some compounds have shown to induce better activity than the well documented histone deacetylase inhibitor, valoparic acid (VPA) known to induce SMN expression in mutant cells 71, 72. TC007 has later been reported to induce beneficial effects on muscle fiber size and gross motor function in SMN mice model 73, 74. However, the synthetic derivatives tested within this assay were initially designed as antibacterial agents 75 therefore contained an amino group in their 6' position. Unfortunately, no direct comparison to the natural aminoglycoside scaffold has been reported in the subjected work. Comparison of the reported results with earlier work of the same authors indicated similar ability of natural derivatives such as tobramycin and amikacin to induce read-through in SPA patient's fibroblasts 32.
In 2006, our lab first reported the development of a semi-synthetic paromomycin derivative, compound 1 (also named NB30, Fig. 5) 68, to be used as a prototype read-through inducer and a first-generation lead compound. Compound 1 has been shown to induce PTC suppression in-vitro and ex-vivo against DNA fragments that mimic genes with disease causing nonsense mutations such as DMD, HS, USH and CF 68, 76, and recently on mutant retinal explants derived from mice 77. Comparison of the obtained results with those obtained with natural derivatives such as G418, paromomycin and gentamicin, indicated lower activity of this synthetic derivative. However, cell toxicity assays 76, altogether with ototoxicity assays in cochlear explants and acute toxicity experiments in mice 69 indicated reduced toxicity of the synthetic derivative (compound 1) when compared to the natural inducers (paromomycn and gentamicin).
Figure 5.
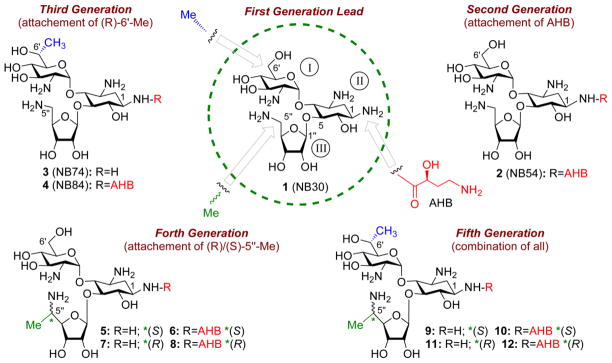
Chronological development of lead compounds on the pseudo-trisaccharide (compound 1) scaffold by systematic optimization of structure-activity-toxicity relationship as follow: first-generation lead (compound 1, also called NB30) 68 was developed by attaching 5-amino ribose at C5 position of the paromamine (rings I and II of paromomycin); second-generation compound 2 (also called NB54), 69 by further attaching (S)-4-amino-2-hydroxybutanoyl (AHB) group at N-1 position of the compound 1; third-generation compounds 3 and 4 (also named NB74 and NB84, respectively), 70 by installing (R)-6'-Me on 1 and 2, respectively; fourth-generation compounds 5–8, 66 by attaching (S)/(R)-5''-Me on either 1 or 2; fifth-generation compounds 9–12, 59 by installing (S)/(R)-5''-Me on either 3 or 4. The identity of each pharmacophore and its attachment site are highlighted: AHB, red; (R)-6'-Me, blue; (S)/(R)-5''-Me, green.
Encouraged by these results we further reported in 2009 the development of the new and improved paromomycin based synthetic derivate, compound 2, as a second-generation lead structure (also named NB54), containing an (S)-4-amino-2-hydroxybutanoyl (AHB) moiety at N-1 position (Fig. 5) 69. The new structure was designed to have lower toxicity values comparing to its prototype (compound 1) based on previously documented results indicating decreased lethal toxicity values (LD50) in mice treated with natural derivatives containing an AHB moiety in their N-1 positions (such as amikacin and butirosin) 69, 78, 79. The resulting structure, compound 2, indeed exhibited much reduced toxicity values when compared to its prototype, compound 169, and has also been shown to induce an enhanced read-trough activity in-vitro 69, ex-vivo 25, 26 and in-vivo in CF mouse model 80. NB54’s activity also exceeded paromomycin and gentamicin's activities.
Approximately one year later, our group reported the development of two new synthetic derivatives, as third-generation leads in which first two rings of G418 (ring I and ring II) were used as an AG scaffold (Fig. 5; compounds 3 and 4, also named NB74 and NB84, respectively) 70. These derivatives were designed to contain a 6'-(R)-methyl that we hypothesized to act as a pharmacophore in G418, but nevertheless, did not contain the garosamine 3rd ring, which has been shown to enhance the toxicity of both G418 and gentamicin, the two structurally highly related AGs (Fig. 1). Evaluation of the resulting compounds, 4 and 5, indicated better read-trough activity in-vitro and ex-vivo, along with significantly reduced cell toxicity compared to the first and second generation leads and the natural AGs paromamycin and gentamicin 25, 26, 70. Compound 4 was later shown to suppress Mecp2-R168X stop mutation in fibroblasts isolated from male Mecp2R168X knock-in mice, an animal model for the study of suppression therapy in Rett syndrome 25. Compound 4 was found to suppress the Mecp2-R168X stop mutation more efficiently than compound 2 and gentamicin. The potential of compound 4 was further tested to attenuate the lysosomal storage disease mucopolysaccharidosis type I-Hurler (MPS I-H), the severe form of α-L-iduronidase deficiency. α-L-iduronidase participates in glycosaminoglycan (GAG) catabolism and its insufficiency causes progressive GAG accumulation and onset of the MPS I-H phenotype, which consists of multiple somatic and neurological defects. Interestingly, 60–80% of MPS I-H patients carry a nonsense mutation in the IDUA gene. Initial study in this regard has shown that 2-weeks treatment with compound 4 restored enough α-L-iduronidase function via PTC suppression to reduce tissue GAG accumulation in the Iduatm1Kmke MPS I-H mouse model, which carries a PTC homologous to the human IDUA-W402X nonsense mutation 81. Most recently 82, it has been shown that long-term administration of compound 4 maintains α-L-iduronidase activity and GAG reduction in Iduatm1Kmke mice throughout a 28-week treatment period. An examination of more complex MPS I-H phenotypes in Iduatm1Kmke mice following 28-weeks treatment with compound 4 revealed significant moderation of the disease in multiple tissues, including the brain, heart and bone, which are resistant to current MPS I-H therapies. It is noteworthy to mention that this study, performed by Bedwell and co-workers, represents the first demonstration that long-term nonsense suppression therapy can moderate progression of a genetic disease.
Continuous research in our lab has recently led to the development of new generations of synthetic derivatives which have not only been shown to be potent read-through inducers with relatively low toxicity, but also to be highly selective towards the eukaryotic ribosome, lacking any antibacterial activity (Fig. 5; compounds 5–12) 59, 66. These derivatives, fourth- and fifth-generation leads, were developed by introducing of a new pharmacophore - 5''-(R/S)-methyl. In general, compounds with 5”-(S)-methyl were more active than the corresponding diastereomers with 5”-(R)-methyl. Interestingly, compounds 10 and 12 were shown to have read-trough activity similar to G418 in both in-vitro and ex-vivo systems, but exhibited a significantly reduced cell toxicity. Furthermore, one of these lead structures, compound 9 (also named NB124) restored CFTR function to ~7% of wild type activity in primary human bronchial epithelial (HBE) CF cells (G542X/delF508), a highly relevant preclinical model with endogenous CFTR expression, and rescued CFTR function in a CF mouse model expressing a human CFTR-G542X transgene; efficacy was superior to gentamicin and exhibited favorable pharmacokinetic properties, suggesting in vitro results translated to clinical benefit in vivo 83.
The reduced toxicity of the developed leads shown in Figure 5 was recently attributed to their high selectivity towards the cytoplasmic ribosome altogether with the reduced selectivity towards the mitoribosome 59. In particular, this study provided for the first time the proof of principle that antibacterial activity and toxicity of AGs can be dissected from their PTC suppression activity. Furthermore, using a series of biochemical assays including protein translation inhibition tests in prokaryotic, eukaryotic and mitochondrial systems, it has been shown that the increased specificity towards cytoplasmic ribosome correlates with the increased PTC suppression activity and that the decreased specificity towards mitochondrial ribosome confers the lowered toxicity.
The above mentioned progress in designing synthetic derivatives (compounds 1–12, Fig. 5) with potent PTC suppression activity and low toxicity emerged from a very careful inspection of the structural elements which are highlighted as important for the biological activity and toxicity of AGs. Thus, the design and development of these new lead structures were realized by systematic fine-tuning structure-activity-toxicity relationship studies, and were based mainly on the previously reported biochemical and toxicity data on standard AGs. The results obtained are indeed very encouraging, but unfortunately, “rational design” of synthetic AGs for suppression therapy is still far from being well established, mainly due to the lack of detailed information on the molecular mechanisms of AGs activity and toxicity in eukaryotic systems. As an example, our recent works demonstrated the importance of high selectivity towards the eukaryotic cytoplasmic ribosome for the development of therapeutic agents with reduced toxicity 59, 62. However, compounds that exhibit an enhanced selectivity towards eukaryotic systems do not always exceed better read-through activity. The natural AG apramycin and the synthetic derivative NB33 (Fig. 6) were shown to exhibit a rather high selectivity towards eukaryotic systems; 84, 85 nevertheless, their measured read-through activity had appeared to be rather negligible 84. These results are now better understood from the molecular point of view by the differences inspected in the binding site conformations upon binding of read-trough inducers such as G418 vs. non-read-through inducers such as apramycin to their putative binding site in Leishmania 43 highlighting the importance of AG’s ring I in binding eukaryotic species (a more detailed analysis will follow in section 6). Over the last several years a few more AG based synthetic derivatives have been evaluated as selective potent binders of eukaryotic systems 85–88. However, the estimation of their read-through abilities is yet to be determined.
Figure 6.

Structures of natural (apramycin) and semi-synthetic (NB33) aminoglycosides with high selectivity towards eukaryotic systems. The 2-deoxystreptamine (2-DOS) ring is highlighted in blue.
Further improvement and rational development of new read-through inducers requires the molecular understanding of the read-through mechanism in mammalian cells, and a better characterization of their putative binding sites. Unfortunately, very few indications regarding this field are available nowadays, and this experimental field still remains largely obscure. The following sections will highlight the structural information available up to date in regarding to AGs and their putative binding sites within the eukaryotic and prokaryotic ribosomes.
5. Non-aminoglycoside read-through inducers
AGs are the most extensively investigated low molecular weight compounds, which are capable of enhancing read-through activities in eukaryotic systems 89. However, a few additional non-AG compounds were recently highlighted as potential candidates for the suppression of PTCs in mammalian cells. Some of these agents were discovered in high-throughput screening (HTS) experiments that aimed at screening for non-AG- read-through inducers.
PTC124 (Ataluren, Fig. 7A) is an example of such candidate, that is now tested in several clinical trials for the treatment of various PTC induced genetic disorders such as CF, DMD, hemophilia A and B, and methylmalonic acidemia 90. PTC124 has been discovered at PTC Therapeutics Inc. by screening a library of low molecular weight compounds using a luciferase based reporter system 90. Out of nearly 1,000,000 compounds tested, PTC124 indicated the highest efficiency in suppressing PTCs in-vitro. PTC124 has later been shown to induce read-through at nanomolar concentrations in various ex-vivo experimentations altogether with in-vivo experiments in DMD and CF mice models, demonstrating no detectable toxic effects at the read-through inducing levels 91. Foot-printing experimentation indicated the binding of PTC124 in adjacent to the peptidyl trasferase center of the large ribosomal subunit 92. However, recent evidences indicated a strong stabilization of the firefly luciferase protein used as reporter protein in the initial screen performed by PTC Therapeutics Inc.93, 94. These findings suggested that PTC124 enhanced the basal luciferase activity in-vitro, rather than enhancing the read-through of PTC containing transcript; therefore, implied that the PTC124 elevated read-through levels were resulting from an experimental artifact. Nevertheless, the well documented in-vivo evidences described above, along with more recent ex-vivo and in-vivo works relating to nonsense-mediated congenital Aniridia 95 and Usher syndrome 1c, 96 still highlight the great potential of using PTC124 for suppression therapy.
Figure 7.
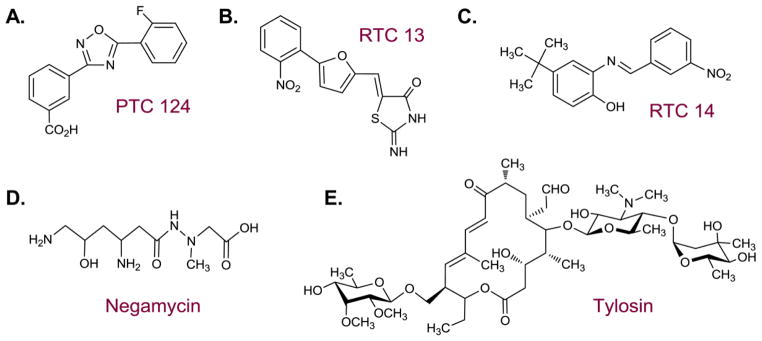
Chemical structures of non-aminoglycoside read-through inducers.
An additional screen performed by Du et al. 97 highlighted the use of a few more nonAG compounds in PTC suppression. The initial screen has been performed using 34,000 compounds, of which 12 were shown to enhance read-through levels in an enzyme-linked immunosorbent assay (ELISA)-based reporter system. Two compounds, RTC13 and RTC14 (Fig. 7B, 7C) were further identified as leading compounds that were used at micromolar concentrations to enhance read-through levels in both AT patients derived cell lines and DMD mice models 97. The mechanism of which these compounds act to suppress PTCs is currently unknown. However, based on the structural similarity of these agents to PTC124, it is assumed that they share similar mechanism of action. RTC13 and RTC14 did not show any measurable toxic side effects upon their administration at read-through inducing levels to mammalian cells.
Two additional compounds, which are currently used as antibacterial agents, were recently evaluated as potential read-through inducers: negamicin (Fig. 7D) and tylosin (Fig. 7E). The dipeptide antibiotic negamycin is known to bind both the prokaryotic A-site, and the wall of the nascent chains exit tunnel in the large ribosomal subunit 98. Negamycin was recently demonstrated to induce read-through, thus restoring the biological activity, of PTC containing adenomatous polyposis coli (APC) gene ex-vivo, involved in the development of colorectal cancer 34. Earlier experimentations demonstrated the ability of negamycin to enhance dystrophin protein levels up to 10% in mdx mice model for DMD 99. These experimentations reported some lower toxicity values when compared to the AG gentamicin .
Tylosin, a member of the macrolide antibiotic family, has also been recently demonstrated to enhance read-through levels of mutant APC construct ex-vivo 36. Further experimentations demonstrated its ability to reduce oncogenic phenotypes in mutant cells and reduce tumor growth in mice, in-vivo 36. Tylosin, as all macrolides is known to bind the ribosome at the protein exit tunnel, near the peptidyl transferase center at the large ribosomal subunit.
Nevertheless, despite the promising results in the field of PTC repair by non-AG agents, many of these agents suffer from major drawbacks limiting the probability of using them in the clinic. Macrolides, for example, are known to induce severe toxic effects in eukaryotes, which might limit their approval as chronic medications. In addition, both macrolides and Negamicin are highly active against bacterial species and this lack of selectivity to eukaryotes might induce some massive changes in the patient’s flora on the long run. Furthermore, to our knowledge, no recent work was aimed at exploring the molecular mechanisms by which such compounds imply their therapeutic effects on PTC related illnesses, nor alternative synthetic derivatives were designed to try and improve the activity or reduce the toxicity of such agents. Such improvements will have to be further performed in such compounds to enable their usability for the treatment of genetic diseases.
6. The read-through mechanism
6.1 General aspects and gained knowledge from structural investigations of bacterial ribosomes
In general, a read-through event is defined as encoding of a PTC by near cognate aminoacylated tRNA. Read-through events spontaneously occur in basal levels of less than 1% of translating PTC containing transcripts 89. Therapeutic compounds inducing higher read-through activity levels are believed to be able to enhance the chance of near-cognate aminoacylated tRNAs to bind PTCs; therefore advantaging their binding over the binding of release factors (RFs), enabling the synthesis of full length transcripts. These events can rarely occur on natural stop codons as extensively explained by Bedwell and coworkers 14, 89, and are only limited to PTCs.
Recent progress in structural exploration of ribosomes revealed the mechanisms of which translation and translation termination occurs at the atomic level. These findings altogether with the elucidation of AGs mechanism of action in bacterial cells might help in understanding how these therapeutic agents can induce read-through of PTCs.
Overall, the termination of translation greatly differs from the translation of sense codons. Sense codon translation is performed by careful selection of cognate aminoacylated tRNAs containing an appropriate anticodon sequence that can form Watson-Crick pairs with all 3 codon nucleotides located in the mRNA template 100, 101. Translation termination, on the other hand, is encoded by only 3 consensus nucleotide triplets that can only be recognized by proteins termed class I RFs 100–104. Structural studies indicated an overall structural similarity between the structures of RFs and aminoacylated tRNAs. These findings indicated that both factors occupy an ‘L’ shape conformation while bound to the ribosome 100–104. Moreover, both factors have been shown to be rather flexible. These features are believed to enable the simultaneous interaction with both the ribosomal decoding center - the A-site, located in the small ribosomal subunit, and the peptidyl transferase center, located in the large ribosomal subunit .
At the A-site, both factors (tRNAs and RFs) are able to recognize the relevant codon triplet (sense codon/termination codon). tRNAs recognition is performed by tight monitoring of specific hydrogen bonding between the anti-codon triplet located on the tRNA molecule and the codon triplet embedded in the mRNA template. RFs recognition is performed by specific interactions of evolutionary conserved amino-acid motives in the RF that tightly interact with the 3 stop codon nucleotides. Upon recognition of the relevant codon triplets, conformational changes occur in both the decoding center and the recognition factor itself. These changes enable the direction of relevant domains of either tRNA or RF towards the peptidyl transferase center, where the synthesis of emerging polypeptide chain occurs. These changes are irreversible and encompass the GTP to GDP transfer of several extrinsic protein factors such as elongation factors (EFs) or additional RFs (e.g. eRF3). In the peptidyl transferase center tRNAs catalyze the formation of a peptide bond between the relevant amino-acid and the nascent polypeptide chain. RFs, on the other hand, catalyze the hydrolysis of peptide chain, followed by its release, and the beginning of ribosome recycling towards the next translational round (Fig. 8).
Figure 8.

Schematic view of translation elongation and translation termination with magnified elements from ribosome crystal structures. (A) Molecular glance at the bacterial decoding site upon cognate tRNA binding – 'ON' state. The 30S ribosomal subunit is showed at the left side with three tRNA molecules bound (A-site – green; P-site – Yellow; E-site – Blue) and mRNA highlighted in black. The A-site tRNA is bound to an EF (elongation factor – light pink) which is an intrinsic protein participating in the translation elongation process. The actual decoding site is highlighted in red, and is enlarged in the right side of the figure. The mRNA in the enlarged view is highlighted in yellow; A-site bound tRNA in green; and the two conserved adenine residues, A1492 and A1493, flipping out from the helical core, are highlighted in blue and red, respectively. The PDB entry for the presented structure is 2WRQ. (B) Structural view of the bacterial decoding site during termination, with mRNA highlighted in black; P-site and E-site tRNA molecules highlighted in green and blue, respectively; and RF highlighted in red. The decoding site is highlighted in yellow and an enlarged representation is presented in the right side of the figure. In the enlarged object RF is highlighted in red; mRNA in gold; and the conserved adenine residues A1492 and A1493 in blue and purple, respectively. The PDB entry for the presented structure is 2X9R.
At the atomic level, the above mentioned structural rearrangements upon codon recognition greatly differ while comparing the two translational factors. The binding of cognate tRNA to the decoding center in bacterial ribosomes induces the flip out of two evolutionary conserved adenine residues, A1492 and A1493 (E. coli numbering), located in the internal loop of Helix-44 (H44). These bindings also cause the universally conserved base G530 to switch from syn to anti conformations 105. In their new conformations, these residues, A1492 and A1493, can tightly interact with the first two bases of the codon-anticodon helix and are therefore able to monitor and discriminate between Watson-Crick base pairing and mismatches (Fig. 8). However, structural studies on RF binding to bacterial ribosomes 100–104, demonstrated that unlike in the sense codon case, in termination complexes, only A1492 flips out of H44, leaving A1493 inside the helical core to establish stacking interactions with an adenine residue, A1913, located at Helix69 (H69) in the large ribosomal subunit (Fig. 8). These interactions are considered to be highly important for the proper termination of translation process 106, 107.
AGs where long shown to stabilize the flipping out of the two conserved adenine residues upon binding to bacterial ribosomes (Fig. 9) and more recently to the eukaryotic leishmanial A-site 43. These interactions where shown to increase the affinity of the A-site for near-cognate tRNAs; therefore preventing the ribosome from efficiently discriminating between near-cognate and cognate complexes. The above mentioned interactions reduce the fidelity of normal translational processes leading to the accumulation of miscoded or nonfunctional proteins in bacterial and leishmanial cells, eventually leading to cell death 43. AGs have also been shown to interfere with RF based peptide release and induce read through activity in prokaryotes quite long ago 10, 108. However, only recent kinetic indications have linked these interactions to RF inhibition in a competitive manner 109. Such experimentations were designed to measure peptide release efficiency in the absence and presence of paromomycin, a natural AG derivative. The experiments were performed using purified ribosomal complexes containing programmed mRNA with UAA stop codons. The kinetic analysis indicated a strong correlation between paromomycin's concentration and RF K1/2 values, while no change in kcat was observed. In addition, the authors indicated that upon RF binding paromomycin's binding to ribosomes in-vitro was hampered (IC50 values were elevated).
Figure 9.

The read-through mechanism. (A) Schematic representation of the read-through mechanism at the molecular level as indicated from recent X-ray structures (top panel). The structure at the bottom panel presents the A-site conformation upon AG (paromomycin) binding with the two conserved adenine residues (A1492-blue and A1493-red) bulging out from the helical core. PTC denotes Premature Termination Codon. (B) Superposition of the bacterial ribosome structure with the AG paromomycin (yellow sticks) bound to the A-site (PDB ID: 3IBL) and RF bound to the bacterial ribosome (PDB ID: 2X9R). The structure demonstrates the clashing of A1493 (red) with the Cα backbone of the RF (green), therefore suggests for a steric hindrance of RF to the A-site upon the induced conformation when AG is bound to the bacterial A-site.
Using the accumulated structural evidences presented here we are now capable of understanding how such inhibition is possible at molecular levels. As indicated above, AGs binding to the A-site induces the flipping out of A1492 and A1493. Such conformational change promotes the binding of near-cognate tRNAs, thus enabling the incorporation of such tRNA species to the nascent protein chain instead of releasing it by the incorporation of a RF in the ribosomal A-site (Fig. 8A). As indicated from recent X-ray structures, the conformation obtained upon RF binding is far remoted, where only A1492 is partially flipped out from the helical core and A1493 maintains stacking interactions with A1913 of H69 (Fig. 8B). The flipping out of A1493 upon AG binding might induce a steric clash with the RF backbone, thus limit its actual binding to the A-site (Fig. 9B). In addition, the flipping out of A1493 prevents the stacking interactions with A1913 of H69 110. Since the stacking interactions between A1493 and A1913 have been highlighted as important components of RF recognition by the ribosome, the prevention of such interaction and/or the suggested steric clash between A1493 and RF backbone due to AG binding, might add an additional explanation for AG-induced inhibition of RF binding. These structural indications suggest that the induced conformation upon AG binding acts simultaneously to inhibit RF binding while facilitating near-cognate tRNA incorporation to the A-site; therefore shifting the near-cognate tRNA-RF competitive equilibrium towards a read-through event. These mechanistic implications might also explain the ability of some aforementioned non-AG agents to enhance the basal read-through events. Indeed, PTC124 and tylosin where shown to bind relatively to the peptidyl transferase center, which can introduce steric hindrance for RF binding, but not for tRNA binding; meaning that read-through events might result from enhancing the probability of basal read-through via enlarging the tRNA chances to occupy the A-site upon RF competition.
6.2 Eukaryotic ribosomes: status and read-through implications
As mentioned in the previous section, the recent structural insights in bacterial ribosomes shed light on the read-through mechanism in bacterial systems. However, unfortunately, up to this date, a little is known about the read-through mechanism in eukaryotes. Recent structural work revealed the three dimensional structures of the 80S yeast ribosome 111, 112 and the 40S, 60S subunits of the protozoa Tetrahymena thermophila 113, 114 at relatively high resolutions. More recent papers supplied the X-ray structures of 40S ribosomal subunits purified from rabbit reticulocytes at ~8Å resolutions 115. This recent structural information opens a variety of opportunities underling the variance in eukaryotic vs. prokaryotic translation mechanisms altogether with unlimited possibilities to explore the interactions of small molecules, such as AGs, with eukaryotic ribosomes. Nevertheless, one mustn't forget that this field is still very fresh and the study of complex molecules such as ribosomes still remains very complicated. In addition no structure of eukaryotic ribosome in complex with active small molecules such as AGs is presently available.
The recent work published by Shalev et. al. 43 demonstrated the binding of two AGs (G418 and apramycin) to their putative binding site in Leishmania ribosomes. Leishmania is a eukaryotic parasite sharing a great A-site similarity with human ribosomes (Fig. 4). In this work, it has been shown for the first time that AGs exhibiting misreading activities in bacteria and read-through activities in human, such as G418, are potent inhibitors of Leishmanial growth and are able to induce the flip out of the two conserved adenine residues, A1492 and A1493, in the leishmanial A-site. The obtained conformation was identical to the one observed upon G418 binding to bacterial species (Fig. 10). These results support the notion that AGs read-through activity in eukaryotes might be similar to those observed in prokaryotes. In addition, the paper highlights that upon apramycin's binding to the same A-site construct, only one of the two conserved adenines (A1493) is directed outside the helical core, whereas the other adenine residue (A1492) is maintained into the helical core and forms some electrostatic interactions with the G1408 residue. These results correlate well with additional structural work presented by Kondo et. al 84, 116 and Hermann et. al. 86 demonstrating similar conformations of two different human A-site models upon apramycin binding or upon NB33, a semi-synthetic derivative generated in our lab, binding (Fig. 10). Since both apramycin and NB33 are known to lack any read-through or misreading activity and in addition, apramycin was found not to cause leishmanial growth inhibition, this data might supply an indirect support for the suggested readthrough mechanism of AGs action in eukaryotes, implying for the importance of the full flipping out of the two conserved adenine residues (A1492 and A1493) in order to achieve a read-through and/or misreading activities.
Figure 10.
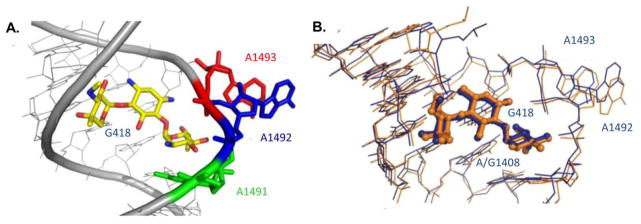
Structural information gained from structural studies of eukaryotic A-sites in complex with AGs. (A) Crystal structure of G418 (yellow) bound to leishmanial A-site indicating the flipping out of the two conserved adenine residues 1492 (blue) and 1493 (red). PDB code 4K32. (B) Superimposition of G418 bound to the bacterial A-site (orange) and the leishmanial A-site (blue). G418 is highlighted in ball-and stick representation. Superimposition was performed using the PyMol software align algorithm using all identical atoms (r.m.s.d. 0.9 Å). PDB codes are 1MWL and 4K32 for the bacterial and Leishmania structures, respectively. In all figures A/G1491, A1492, A1493 are highlighted in ball and stick representation colored green, blue and red, respectively.
To conclude this section, despite no actual evidence exists, the evolutional conservancy of active ribosomal locations such as the decoding center and the peptidyl transferase center imply for a rather high similarity between the read-through mechanisms in prokaryotes to those observed in eukaryotes; therefore, the structural insights available in prokaryotes might serve as a putative base for understanding the molecular mechanisms by which AGs induce read-through events in eukaryotes.
7. Summary and Outlook
This abbreviated overview highlights that AGs hold great potential to serve as therapeutic agents for the treatment of PTC induced genetic disorders. Even though such potential of AGs was demonstrated nearly 20 years ago, their induced toxic effects in mammals introduced a rather high barrier for their further development as potential drugs to treat genetic diseases. These limitations gave rise to search for alternative approaches aimed at identifying novel, non-aminoglycoside structures with improved PTC readthrough activity and lower toxicity. HTS methodologies that were used over the last few years to scan large libraries of chemicals indeed resulted in some promising outcomes 117, 118. However, in parallel to the HTS approach, recent progress in AGs research resulted in the development of new and improved derivatives that overcome some of their toxicity drawbacks, making them feasible candidates for such therapeutic manners. Those recent studies have also provided significant insights into our current understanding in molecular mechanisms of AGs-induced toxic side-effects and pave the way for the design and development of more potent and less toxic derivatives. While these recent achievements are indeed very encouraging, they also have demonstrated the current need in underling the mechanisms of which AGs and other non-AG structures exert their biological activity in eukaryotic cells for rational development of new and improved therapeutic agents.
Up to date, no structures of the human A-site in complex with any of the AGs that induce read-through is yet available, and the elucidation of read-through mechanism in eukaryotic systems is yet to be determined. Nevertheless, the recent achievements in structural ribosomal research shed light on AGs mechanism of action as miscoding agents and read-through inducers in bacterial systems. Based on the relatively high similarity of AGs binding sites in bacterial ribosomes to their putative binding sites in the eukaryotic ribosome, altogether with the recent development in eukaryotic ribosome structures it is likely to assume that these mechanisms share great similarity. However, further deep investigations are still needed to be performed in order to establish their mechanism of action in eukaryotic system and make it practicable for rational drug design.
Acknowledgments
This work was supported by the NIH/NIGMS grant (1 R01 GM094792-01 A1) and GIF research grant (G-1048-95.5/2009). M.S. acknowledges the Schulich Fellowship for Ph.D students. We gratefully thank all students, postdocs and co-workers that contributed to this area of research at our and collaborators’ laboratories.
Abbreviations
- 2-DOS
2-Deoxystreptamine
- APC
Adenomatous polyposis coli
- AGs
Aminoglycsides
- AT
Ataxia-telangiectasia
- CF
Cystic fibrosis
- DMD
Duchenne muscular dystrophy
- E. coli
Escherichia coli
- ELISA
Enzyme-linked immunosorbent assay
- EF
Elongation factor
- H44
Helix-44
- H69
Helix-69
- HTS
High-throughput-screening
- HS
Hurler syndrome
- LD50
Lethal toxicity values
- NAC
N-acetyl-cistein-cholins
- NMD
Nonsense mediated decay
- PTCs
Premature termination codons
- ROS
Reactive oxygen species
- RF
Release factor
- RRE
Rev-responsive element
- rRNA
Ribosomal RNA
- TAR
Tat-responsive element
- AHB
(S)-4-amino-2-hydroxybutanoyl
- SMA
Spinal muscular atrophy
- SMN
Survival motor neuron-1
- USH
Usher syndrome
- VPA
Valoparic acid
References
- 1.Davis BD. Microbiol Rev. 1987;51:341–350. doi: 10.1128/mr.51.3.341-350.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jana S, Deb JK. Appl Microbiol Biotechnol. 2006;70:140–150. doi: 10.1007/s00253-005-0279-0. [DOI] [PubMed] [Google Scholar]
- 3.Botto R, Coxon BJ. Med Chem. 1993;105:1021–1028. [Google Scholar]
- 4.Kaul M, Barbieri CM, Kerrigan JE, Pilch DS. J Mol Biol. 2003;326:1373–1387. doi: 10.1016/s0022-2836(02)01452-3. [DOI] [PubMed] [Google Scholar]
- 5.von AU, Davies J, Schroeder R. Nature. 1991;353:368–370. doi: 10.1038/353368a0. [DOI] [PubMed] [Google Scholar]
- 6.Waldsich C, Semrad K, Schroeder R. RNA. 1998;4:1653–1663. doi: 10.1017/s1355838298981444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Werstuck G, Green MR. Science. 1998;282:296–298. doi: 10.1126/science.282.5387.296. [DOI] [PubMed] [Google Scholar]
- 8.Mei HY, Cui M, Heldsinger A, Lemrow SM, Loo JA, Sannes-Lowery KA, Sharmeen L, Czarnik AW. Biochemistry. 1998;37:14204–14212. doi: 10.1021/bi981308u. [DOI] [PubMed] [Google Scholar]
- 9.Zapp ML, Stern S, Green MR. Cell. 1993;74:969–978. doi: 10.1016/0092-8674(93)90720-b. [DOI] [PubMed] [Google Scholar]
- 10.Gorini L, Kataja E. Proc Natl Acad Sci U S A. 1964;51:487–493. doi: 10.1073/pnas.51.3.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.OMIM. 2014 http://www.ncbi.nlm.nih.gov/omim.
- 12.Atkinson J, Martin R. Nucleic Acids Res. 1994;22:1327–1334. doi: 10.1093/nar/22.8.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kellermayer R. Eur J Med Genet. 2006;49:445–450. doi: 10.1016/j.ejmg.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 14.Keeling KM, Wang D, Conard SE, Bedwell DM. Crit Rev Biochem Mol Biol. 2012;47:444–463. doi: 10.3109/10409238.2012.694846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bidou L, Allamand V, Rousset JP, Namy O. Trends Mol Med. 2012;18:679–688. doi: 10.1016/j.molmed.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 16.Burke JF, Mogg AE. Nucleic Acids Res. 1985;13:6265–6272. doi: 10.1093/nar/13.17.6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaufman RJ. J Clin Invest. 1999;104:367–368. doi: 10.1172/JCI8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kerem E. Curr Opin Pulm Med. 2004;10:547–552. doi: 10.1097/01.mcp.0000141247.22078.46. [DOI] [PubMed] [Google Scholar]
- 19.Manuvakhova M, Keeling K, Bedwell DM. RNA. 2000;6:1044–1055. doi: 10.1017/s1355838200000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilschanski M, Famini C, Blau H, Rivlin J, Augarten A, Avital A, Kerem B, Kerem E. Am J Respir Crit Care Med. 2000;161:860–865. doi: 10.1164/ajrccm.161.3.9904116. [DOI] [PubMed] [Google Scholar]
- 21.Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, Aviram M, Bdolah-Abram T, Bebok Z, Shushi L, Kerem B, Kerem E. N Engl J Med. 2003;349:1433–1441. doi: 10.1056/NEJMoa022170. [DOI] [PubMed] [Google Scholar]
- 22.Malik V, Rodino-Klapac LR, Viollet L, Mendell JR. Ther Adv Neurol Disord. 2010;3:379–389. doi: 10.1177/1756285610388693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL. J Clin Invest. 1999;104:375–381. doi: 10.1172/JCI7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malik V, Rodino-Klapac LR, Viollet L, Wall C, King W, Al-Dahhak R, Lewis S, Shilling CJ, Kota J, Serrano-Munuera C, Hayes J, Mahan JD, Campbell KJ, Banwell B, Dasouki M, Watts V, Sivakumar K, Bien-Willner R, Flanigan KM, Sahenk Z, Barohn RJ, Walker CM, Mendell JR. Ann Neurol. 2010;67:771–780. doi: 10.1002/ana.22024. [DOI] [PubMed] [Google Scholar]
- 25.Brendel C, Belakhov V, Werner H, Wegener E, Gartner J, Nudelman I, Baasov T, Huppke P. J Mol Med (Berl) 2011;89:389–398. doi: 10.1007/s00109-010-0704-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vecsler M, Ben ZB, Nudelman I, Anikster Y, Simon AJ, Amariglio N, Rechavi G, Baasov T, Gak E. PLoS One. 2011;6:e20733. doi: 10.1371/journal.pone.0020733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keeling KM, Brooks DA, Hopwood JJ, Li P, Thompson JN, Bedwell DM. Hum Mol Genet. 2001;10:291–299. doi: 10.1093/hmg/10.3.291. [DOI] [PubMed] [Google Scholar]
- 28.Sangkuhl K, Schulz A, Rompler H, Yun J, Wess J, Schoneberg T. Hum Mol Genet. 2004;13:893–903. doi: 10.1093/hmg/ddh105. [DOI] [PubMed] [Google Scholar]
- 29.Helip-Wooley A, Park MA, Lemons RM, Thoene JG. Mol Genet Metab. 2002;75:128–133. doi: 10.1006/mgme.2001.3272. [DOI] [PubMed] [Google Scholar]
- 30.Grayson C, Bartolini F, Chapple JP, Willison KR, Bhamidipati A, Lewis SA, Luthert PJ, Hardcastle AJ, Cowan NJ, Cheetham ME. Hum Mol Genet. 2002;11:3065–3074. doi: 10.1093/hmg/11.24.3065. [DOI] [PubMed] [Google Scholar]
- 31.Xi B, Guan F, Lawrence DS. J Am Chem Soc. 2004;126:5660–5661. doi: 10.1021/ja0318939. [DOI] [PubMed] [Google Scholar]
- 32.Wolstencroft EC, Mattis V, Bajer AA, Young PJ, Lorson CL. Hum Mol Genet. 2005;14:1199–1210. doi: 10.1093/hmg/ddi131. [DOI] [PubMed] [Google Scholar]
- 33.Huang X, Tian M, Hernandez CC, Hu N, Macdonald RL. Neurobiol Dis. 2012;48:115–123. doi: 10.1016/j.nbd.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Floquet C, Rousset JP, Bidou L. PLoS One. 2011;6:e24125. doi: 10.1371/journal.pone.0024125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Floquet C, Deforges J, Rousset JP, Bidou L. Nucleic Acids Res. 2011;39:3350–3362. doi: 10.1093/nar/gkq1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zilberberg A, Lahav L, Rosin-Arbesfeld R. Gut. 2010;59:496–507. doi: 10.1136/gut.2008.169805. [DOI] [PubMed] [Google Scholar]
- 37.Floquet C, Hatin I, Rousset JP, Bidou L. PLoS Genet. 2012;8:e1002608. doi: 10.1371/journal.pgen.1002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Howard MT, Shirts BH, Petros LM, Flanigan KM, Gesteland RF, Atkins JF. Ann Neurol. 2000;48:164–169. [PubMed] [Google Scholar]
- 39.Howard MT, Anderson CB, Fass U, Khatri S, Gesteland RF, Atkins JF, Flanigan KM. Ann Neurol. 2004;55:422–426. doi: 10.1002/ana.20052. [DOI] [PubMed] [Google Scholar]
- 40.Pfister P, Hobbie S, Vicens Q, Bottger EC, Westhof E. Chembiochem. 2003;4:1078–1088. doi: 10.1002/cbic.200300657. [DOI] [PubMed] [Google Scholar]
- 41.Recht MI, Douthwaite S, Puglisi JD. EMBO J. 1999;18:3133–3138. doi: 10.1093/emboj/18.11.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kondo J. Angew Chem Int Ed Engl. 2011;50:1–5. [Google Scholar]
- 43.Shalev M, Kondo J, Kopelyanskiy D, Jaffe CL, Adir N, Baasov T. Proc Natl Acad Sci U S A. 2013;110:13333–13338. doi: 10.1073/pnas.1307365110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schacht J. Ann N Y Acad Sci. 1999;884:125–130. [PubMed] [Google Scholar]
- 45.Nagai J, Takano M. Drug Metab Pharmacokinet. 2004;19:159–170. doi: 10.2133/dmpk.19.159. [DOI] [PubMed] [Google Scholar]
- 46.Kharkheli E, Kevanishvili Z, Maglakelidze T, Davitashvili O, Schacht J. Georgian Med News. 2007;1:14–17. [PubMed] [Google Scholar]
- 47.Feldman L, Sherman RA, Weissgarten J. Semin Dial. 2012;25:491–494. doi: 10.1111/j.1525-139X.2012.01090.x. [DOI] [PubMed] [Google Scholar]
- 48.Li G, Sha SH, Zotova E, Arezzo J, Van de Water T, Schacht J. Lab Invest. 2002;82:585–596. doi: 10.1038/labinvest.3780453. [DOI] [PubMed] [Google Scholar]
- 49.Sha SH, Schacht J. Lab Invest. 1999;79:807–813. [PubMed] [Google Scholar]
- 50.Sha SH, Schacht J. Hear Res. 2000;142:34–40. doi: 10.1016/s0378-5955(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 51.Sha SH, Qiu JH, Schacht J. N Engl J Med. 2006;354:1856–1857. doi: 10.1056/NEJMc053428. [DOI] [PubMed] [Google Scholar]
- 52.Song BB, Sha SH, Schacht J. Free Radic Biol Med. 1998;25:189–195. doi: 10.1016/s0891-5849(98)00037-9. [DOI] [PubMed] [Google Scholar]
- 53.Beauchamp D, Laurent G, Maldague P, Abid S, Kishore BK, Tulkens PM. J Pharmacol Exp Ther. 1990;255:858–866. [PubMed] [Google Scholar]
- 54.Gilbert DN, Wood CA, Kohlhepp SJ, Kohnen PW, Houghton DC, Finkbeiner HC, Lindsley J, Bennett WM. J Infect Dis. 1989;159:945–953. doi: 10.1093/infdis/159.5.945. [DOI] [PubMed] [Google Scholar]
- 55.Thibault N, Grenier L, Simard M, Bergeron MG, Beauchamp D. Antimicrob Agents Chemother. 1994;38:1027–1035. doi: 10.1128/aac.38.5.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thibault N, Grenier L, Simard M, Bergeron MG, Beauchamp D. Life Sci. 1995;56:1877–1887. doi: 10.1016/0024-3205(95)00162-y. [DOI] [PubMed] [Google Scholar]
- 57.Bottger EC, Schacht J. Hear Res. 2013;303:12–19. doi: 10.1016/j.heares.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hobbie SN, Akshay S, Kalapala SK, Bruell CM, Shcherbakov D, Bottger EC. Proc Natl Acad Sci U S A. 2008;105:20888–20893. doi: 10.1073/pnas.0811258106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kandasamy J, Atia-Glikin D, Shulman E, Shapira K, Shavit M, Belakhov V, Baasov T. J Med Chem. 2012;55:10630–10643. doi: 10.1021/jm3012992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matt T, Ng CL, Lang K, Sha SH, Akbergenov R, Shcherbakov D, Meyer M, Duscha S, Xie J, Dubbaka SR, Perez-Fernandez D, Vasella A, Ramakrishnan V, Schacht J, Bottger EC. Proc Natl Acad Sci U S A. 2012;109:10984–10989. doi: 10.1073/pnas.1204073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Francis SP, Katz J, Fanning KD, Harris KA, Nicholas BD, Lacy M, Pagana J, Agris PF, Shin JB. J Neurosci. 2013;33:3079–3093. doi: 10.1523/JNEUROSCI.3430-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shulman E, Belakhov V, Wei G, Kendall A, Meyron-Holtz EG, Ben-Shachar D, Schacht J, Baasov T. J Biol Chem. 2013;289:2318–2330. doi: 10.1074/jbc.M113.533588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Houghton JL, Green KD, Chen W, Garneau-Tsodikova S. Chembiochem. 2010;11:880–902. doi: 10.1002/cbic.200900779. [DOI] [PubMed] [Google Scholar]
- 64.Perez-Fernandez D, Shcherbakov D, Matt T, Leong NC, Kudyba I, Duscha S, Boukari H, Patak R, Dubbaka SR, Lang K, Meyer M, Akbergenov R, Freihofer P, Vaddi S, Thommes P, Ramakrishnan V, Vasella A, Bottger EC. Nat Commun. 2014;5:3112. doi: 10.1038/ncomms4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pokrovskaya V, Baasov T. Expert Opin Drug Discov. 2010;5:883–902. doi: 10.1517/17460441.2010.508069. [DOI] [PubMed] [Google Scholar]
- 66.Kandasamy J, Atia-Glikin D, Belakhov V, Baasov T. Med Chem Comm. 2011;2:165–171. [Google Scholar]
- 67.Mattis VB, Rai R, Wang J, Chang CW, Coady T, Lorson CL. Hum Genet. 2006;120:589–601. doi: 10.1007/s00439-006-0245-7. [DOI] [PubMed] [Google Scholar]
- 68.Nudelman I, Rebibo-Sabbah A, Shallom-Shezifi D, Hainrichson M, Stahl I, Ben-Yosef T, Baasov T. Bioorg Med Chem Lett. 2006;16:6310–6315. doi: 10.1016/j.bmcl.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 69.Nudelman I, Rebibo-Sabbah A, Cherniavsky M, Belakhov V, Hainrichson M, Chen F, Schacht J, Pilch DS, Ben-Yosef T, Baasov T. J Med Chem. 2009;52:2836–2845. doi: 10.1021/jm801640k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nudelman I, Glikin D, Smolkin B, Hainrichson M, Belakhov V, Baasov T. Bioorg Med Chem. 2010;18:3735–3746. doi: 10.1016/j.bmc.2010.03.060. [DOI] [PubMed] [Google Scholar]
- 71.Brichta L, Hofmann Y, Hahnen E, Siebzehnrubl FA, Raschke H, Blumcke I, Eyupoglu IY, Wirth B. Hum Mol Genet. 2003;12:2481–2489. doi: 10.1093/hmg/ddg256. [DOI] [PubMed] [Google Scholar]
- 72.Sumner CJ, Huynh TN, Markowitz JA, Perhac JS, Hill B, Coovert DD, Schussler K, Chen X, Jarecki J, Burghes AH, Taylor JP, Fischbeck KH. Ann Neurol. 2003;54:647–654. doi: 10.1002/ana.10743. [DOI] [PubMed] [Google Scholar]
- 73.Mattis VB, Ebert AD, Fosso MY, Chang CW, Lorson CL. Hum Mol Genet. 2009;18:3906–3913. doi: 10.1093/hmg/ddp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mattis VB, Fosso MY, Chang CW, Lorson CL. BMC Neurosci. 2009;10:142. doi: 10.1186/1471-2202-10-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chang CW, Hui Y, Elchert B, Wang J, Li J, Rai R. Org Lett. 2002;4:4603–4606. doi: 10.1021/ol0269042. [DOI] [PubMed] [Google Scholar]
- 76.Rebibo-Sabbah A, Nudelman I, Ahmed ZM, Baasov T, Ben–Yosef T. Hum Genet. 2007;122:373–381. doi: 10.1007/s00439-007-0410-7. [DOI] [PubMed] [Google Scholar]
- 77.Goldmann T, Rebibo-Sabbah A, Overlack N, Nudelman I, Belakhov V, Baasov T, Ben-Yosef T, Wolfrum U, Nagel-Wolfrum K. Invest Ophthalmol Vis Sci. 2010;51:6671–6680. doi: 10.1167/iovs.10-5741. [DOI] [PubMed] [Google Scholar]
- 78.Fujisawa K, Hoshiya T, Kawaguchi H. J Antibiot (Tokyo) 1974;27:677–681. doi: 10.7164/antibiotics.27.677. [DOI] [PubMed] [Google Scholar]
- 79.Kondo S, Hotta K. J Infect Chemother. 1999;5:1–9. doi: 10.1007/s101560050001. [DOI] [PubMed] [Google Scholar]
- 80.Rowe SM, Sloane P, Tang LP, Backer K, Mazur M, Buckley-Lanier J, Nudelman I, Belakhov V, Bebok Z, Schwiebert E, Baasov T, Bedwell DM. J Mol Med (Berl) 2011;89:1161–1149. doi: 10.1007/s00109-011-0787-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang D, Belakhov V, Kandasamy J, Baasov T, Li SC, Li YT, Bedwell DM, Keeling KM. Mol Genet Metab. 2012;105:116–25. doi: 10.1016/j.ymgme.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gunn G, Dai Y, Du M, Belakhov V, Kandeasamy J, Schoeb T, Baasov T, Bedwell D, Keeling K. Mol Genet Metab. 2014:111, S50. doi: 10.1016/j.ymgme.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xue X, Mutyam V, Tang L, Biswas S, Du M, Jackson LA, Dai Y, Belakhov YV, Shalev M, Chen F, Schacht J, Bridges R, Baasov T, Hong J, Bedwell DM, Rowe SM. Am J Respir Cell Mol Biol. 2013 doi: 10.1165/rcmb.2013-0282OC. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kondo J, Hainrichson M, Nudelman I, Shallom-Shezifi D, Barbieri CM, Pilch E, Westhof DS, Baasov T. Chembiochem. 2007;8:1700–1709. doi: 10.1002/cbic.200700271. [DOI] [PubMed] [Google Scholar]
- 85.Kythreoti G, Vourloumis DA. Anal Biochem. 2011;412:102–107. doi: 10.1016/j.ab.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 86.Hermann T, Tereshko V, Skripkin E, Patel DJ. Blood Cells Mol Dis. 2007;38:193–198. doi: 10.1016/j.bcmd.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 87.Simonsen KB, Ayida BK, Vourloumis D, Winters GC, Takahashi M, Shandrick S, Zhao Q, Hermann T. Chembiochem. 2003;4:886–890. doi: 10.1002/cbic.200300689. [DOI] [PubMed] [Google Scholar]
- 88.Vourloumis D, Winters GC, Takahashi M, Simonsen KB, Ayida BK, Shandrick S, Zhao Q, Hermann T. Chembiochem. 2003;4:879–885. doi: 10.1002/cbic.200300688. [DOI] [PubMed] [Google Scholar]
- 89.Keeling KM, Bedwell DM. Wiley Interdiscip Rev RNA. 2011;2:837–852. doi: 10.1002/wrna.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL. Nature. 2007;447:89–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 91.Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, Leonard EM, Almstead NG, Ju W, Peltz SW, Miller LL. J Clin Pharmacol. 2007;47:430–444. doi: 10.1177/0091270006297140. [DOI] [PubMed] [Google Scholar]
- 92.Karp GM, Hwang S, Chen G, Almstead NG. 7772259 B2. US patent. 2010
- 93.Auld DS, Thorne N, Maguire WF, Inglese J. Proc Natl Acad Sci U S A. 2009;106:3585–3590. doi: 10.1073/pnas.0813345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Auld DS, Lovell S, Thorne N, Lea WA, Maloney DJ, Shen M, Rai G, Battaile KP, Thomas CJ, Simeonov A, Hanzlik RP, Inglese J. Proc Natl Acad Sci U S A. 2010;107:4878–4883. doi: 10.1073/pnas.0909141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gregory-Evans CY, Wang X, Wasan KM, Zhao J, Metcalfe AL, Gregory-Evans K. J Clin Invest. 2014;124:111–116. doi: 10.1172/JCI70462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Goldmann T, Overlack N, Moller F, Belakhov V, van WM, Baasov T, Wolfrum U, Nagel-Wolfrum K. EMBO Mol Med. 2012;4:1186–1199. doi: 10.1002/emmm.201201438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Du L, Jung ME, Damoiseaux R, Completo G, Fike F, Ku JM, Nahas S, Piao C, Hu H, Gatti RA. Mol Ther. 2013;21:1653–1660. doi: 10.1038/mt.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schroeder SJ, Blaha G, Moore PB. Antimicrob Agents Chemother. 2007;51:4462–4465. doi: 10.1128/AAC.00455-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Arakawa M, Shiozuka M, Nakayama Y, Hara T, Hamada M, Kondo S, Ikeda D, Takahashi Y, Sawa R, Nonomura Y, Sheykholeslami K, Kondo K, Kaga K, Kitamura T, Suzuki-Miyagoe Y, Takeda S, Matsuda R. J Biochem. 2003;134:751–758. doi: 10.1093/jb/mvg203. [DOI] [PubMed] [Google Scholar]
- 100.Korostelev A, Zhu J, Asahara H, Noller HF. EMBO J. 2010;29:2577–2585. doi: 10.1038/emboj.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Laurberg M, Asahara H, Korostelev A, Zhu J, Trakhanov S, Noller HF. Nature. 2008;454:852–857. doi: 10.1038/nature07115. [DOI] [PubMed] [Google Scholar]
- 102.Jin H, Kelley AC, Loakes D, Ramakrishnan V. Proc Natl Acad Sci U S A. 2010;107:8593–8598. doi: 10.1073/pnas.1003995107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Petry S, Brodersen DE, Murphy FV, Dunham CM, Selmer M, Tarry MJ, Kelley AC, Ramakrishnan V. Cell. 2005;123:1255–1266. doi: 10.1016/j.cell.2005.09.039. [DOI] [PubMed] [Google Scholar]
- 104.Weixlbaumer A, Jin H, Neubauer C, Voorhees RM, Petry S, Kelley AC, Ramakrishnan V. Science. 2008;322:953–956. doi: 10.1126/science.1164840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ogle JM, Brodersen DE, Clemons WM, Tarry MJ, Carter AP, Ramakrishnan V. Science. 2001;292:897–902. doi: 10.1126/science.1060612. [DOI] [PubMed] [Google Scholar]
- 106.Borovinskaya MA, Pai RD, Zhang W, Schuwirth BS, Holton JM, Hirokawa G, Kaji H, Kaji A, Cate JH. Nat Struct Mol Biol. 2007;14:727–732. doi: 10.1038/nsmb1271. [DOI] [PubMed] [Google Scholar]
- 107.Pai RD, Zhang W, Schuwirth BS, Hirokawa G, Kaji H, Kaji A, Cate JH. J Mol Biol. 2008;376:1334–1347. doi: 10.1016/j.jmb.2007.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brown CM, McCaughan KK, Tate WP. Nucleic Acids Res. 1993;21:2109–2115. doi: 10.1093/nar/21.9.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Youngman EM, He SL, Nikstad LJ, Green R. Mol Cell. 2007;28:533–543. doi: 10.1016/j.molcel.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 110.Demeshkina N, Jenner L, Westhof E, Yusupov M, Yusupova G. Nature. 2012;484:256–259. doi: 10.1038/nature10913. [DOI] [PubMed] [Google Scholar]
- 111.Ben-Shem A, Jenner L, Yusupova G, Yusupov M. Science. 2010;330:1203–1209. doi: 10.1126/science.1194294. [DOI] [PubMed] [Google Scholar]
- 112.Ben–Shem A, Garreau de LN, Melnikov S, Jenner L, Yusupova G, Yusupov M. Science. 2011;334:1524–1529. doi: 10.1126/science.1212642. [DOI] [PubMed] [Google Scholar]
- 113.Klinge S, Voigts-Hoffmann F, Leibundgut M, Arpagaus S, Ban N. Science. 2011;334:941–948. doi: 10.1126/science.1211204. [DOI] [PubMed] [Google Scholar]
- 114.Rabl J, Leibundgut M, Ataide SF, Haag A, Ban N. Science. 2011;331:730–736. doi: 10.1126/science.1198308. [DOI] [PubMed] [Google Scholar]
- 115.Lomakin IB, Steitz TA. Nature. 2013;500:307–311. doi: 10.1038/nature12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kondo J, Francois B, Urzhumtsev A, Westhof E. Angew Chem Int Ed Engl. 2006;45:3310–3314. doi: 10.1002/anie.200600354. [DOI] [PubMed] [Google Scholar]
- 117.Gonzalez-Hilarion S, Beghyn T, Jia J, Debreuck N, Berte G, Mamchaoui K, Mouly V, Gruenert DC, Deprez B, Lejeune F. Orphanet J Rare Dis. 2012;7:58. doi: 10.1186/1750-1172-7-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Loudon JA. J Bioanal Biomed. 2013;5(4):79–96. [Google Scholar]