

Significance
The rationale of targeting specific genetic dependencies for the treatment of cancer has been validated by the promising clinical responses obtained with oncogene-targeted therapies. However, in most cases, the development of resistance remains a major obstacle toward achieving long-term or complete disease remission. Here, we identified mutant β-catenin as a common secondary oncogene addiction pathway in MYC-addicted lymphoma. We demonstrate that, although withdrawal or inhibition of either oncogene is sufficient to induce initial tumor regression, only combined inhibition of both oncogene addiction pathways results in sustained tumor regression. Our results suggest clinical outcomes can be dramatically improved through the simultaneous targeted inhibition of multiple oncogenic addiction pathways.
Keywords: oncogene addiction, combination targeted therapy, splice site mutations
Abstract
Many cancers exhibit sensitivity to the inhibition of a single genetic lesion, a property that has been successfully exploited with oncogene-targeted therapeutics. However, inhibition of single oncogenes often fails to result in sustained tumor regression due to the emergence of therapy-resistant cells. Here, we report that MYC-driven lymphomas frequently acquire activating mutations in β-catenin, including a previously unreported mutation in a splice acceptor site. Tumors with these genetic lesions are highly dependent on β-catenin for their survival and the suppression of β-catenin resulted in marked apoptosis causally related to a decrease in Bcl-xL expression. Using a novel inducible inhibitor of β-catenin, we illustrate that, although MYC withdrawal or β-catenin inhibition alone results in initial tumor regression, most tumors ultimately recurred, mimicking the clinical response to single-agent targeted therapy. Importantly, the simultaneous combined inhibition of both MYC and β-catenin promoted more rapid tumor regression and successfully prevented tumor recurrence. Hence, we demonstrated that MYC-induced tumors are addicted to mutant β-catenin, and the combined inactivation of MYC and β-catenin induces sustained tumor regression. Our results provide a proof of principle that targeting multiple oncogene addicted pathways can prevent therapeutic resistance.
Cancer cells are highly sensitive to the targeted inhibition of single driver mutations, eliciting a phenomenon known as “oncogene addiction” (1). The identification of genetic dependencies in multiple tumor types has resulted in the development of several molecularly targeted therapeutics, including the BCR-ABL kinase inhibitor imatinib for the treatment of chronic myelogenous leukemia (CML), the EGFR kinase inhibitor gefitinib for the treatment of non–small cell lung cancer (NSCLC), and the BRAF kinase inhibitor vemurafenib for the treatment of advanced melanoma (2–4). Although these oncogene-targeted agents have provided promising clinical responses, many patients ultimately experience a recurrence of their disease due to the development of drug resistance (4–6). Thus, it has become evident that monotherapy with targeted drugs is insufficient for achieving sustained tumor regression.
Resistance to targeted therapy can arise through multiple mechanisms, depending on the tumor type and the targeted oncogenic pathway (7). Cells frequently acquire resistance through mutations in the targeted oncogene itself that disrupt drug binding, as in the case of BCR-ABL and EGFR (8, 5, 6). In addition, resistance to EGFR inhibition in NSCLC and BRAF inhibition in melanoma has been found to occur through a variety of mechanisms that activate downstream signaling proteins or alternative pathways, which can functionally substitute for loss in activity of the targeted oncogene (9–11). Although significant progress has been made in the identification and inhibition of resistance pathways, it may prove challenging to anticipate and suppress all of the potential mechanisms of resistance for each oncogene-addicted cancer and targeted therapeutic agent.
Combination therapy has been successfully applied to prevent resistance in the treatment of infectious diseases such as HIV (12, 13) and tuberculosis (14). In the context of oncogene-targeted therapy for cancer, it has been proposed that a similar strategy, using combinations directed against multiple dependencies, is the most likely to prevent resistance (7). Indeed, mathematical modeling indicates that targeting at least two independently required pathways may be sufficient to prevent tumor recurrence (15). However, there exists little experimental evidence directly testing such an approach and it remains unclear which combinations of targets would be most effective at inducing long-term remissions.
MYC is one of the most frequently amplified oncogenes in human cancer (16). In the Eμ-tTA/tetO-MYC conditional mouse model, overexpression of MYC results in the development of aggressive T-cell lymphoma, and MYC inactivation in established tumors is sufficient to induce tumor regression through processes such as proliferative arrest, cellular senescence, apoptosis, and the shutdown of angiogenesis (17–19). The extent of regression is dependent on both cell-intrinsic and host-dependent contexts, and in particular, tumors frequently recur following MYC inactivation in the absence of an intact adaptive immune system (20). Recurring tumors restore expression of the MYC transgene or up-regulate expression of endogenous Myc, demonstrating that resistance occurs primarily through reactivation of the MYC pathway (21). Thus, MYC oncogene addiction and tumor recurrence in the Eμ-tTA/tetO-MYC lymphoma model resembles the clinical course of human cancers treated with single agent targeted therapy.
Here, we demonstrate that the combined inactivation of two oncogene addiction pathways can result in sustained tumor regression. Moreover, we describe a previously unidentified splice acceptor site mutation in β-catenin that is associated with MYC-induced lymphomagenesis. Tumors with mutations in β-catenin are also highly addicted to this mutant gene product for their survival. We demonstrate that in MYC-induced lymphomas, combined addiction to both MYC and β-catenin can be exploited in a rational manner to prevent the emergence of therapeutic resistance.
Results
MYC-Induced Lymphomas Frequently Acquire Stabilizing Mutations in β-Catenin.
Lymphomas initiated by MYC overexpression often acquire loss-of-function mutations in tumor suppressor genes such as p53 and p19ARF (22). However, it remains less well known whether MYC overexpression also selects for the activation of additional oncogenic drivers during lymphomagenesis (Fig. 1A). We discovered that a subset of MYC-induced lymphomas and lymphoma-derived cell lines from the Eμ-tTA/tetO-MYC model expressed elevated levels of β-catenin, with both full-length and shorter forms of β-catenin protein detectable by Western blot (Fig. 1C). To determine the mechanism of β-catenin up-regulation in our panel of MYC-induced lymphomas, we isolated genomic DNA and sequenced the entire β-catenin exon 3 region and portions of the upstream and downstream introns. We detected missense mutations in 7 of 30 samples, at or adjacent to the phosphorylation sites required for β-catenin degradation (Fig. 1D). Interestingly, in 4 of 30 samples, we also discovered novel mutations at the exon 3 splice acceptor site that are predicted to result in skipping of exon 3 during processing of the primary transcript and an in-frame deletion of the region required for β-catenin degradation (Fig. 1E). Skipping of exon 3 was confirmed at the mRNA level by sequencing of β-catenin cDNA from lymphoma samples with splice site mutations (Fig. S1). In total, we detected β-catenin mutations in ∼37% of primary lymphomas from Eμ-tTA/tetO-MYC mice (11 of 30; Fig. 1B and Table S1). The type of mutation correlated with the size of β-catenin protein that was detected, with elevated levels of full-length protein in samples with exon 3 missense mutations and expression of a shorter-length protein in samples with splice site mutations (Fig. 1C). Mutations in β-catenin also correlated with significant up-regulation of the β-catenin/T-cell factor (TCF) target genes Axin2 (ninefold higher in mutant vs. WT, P < 0.01) and Enc1 (21-fold higher in mutant vs. WT, P < 0.001; Fig. 1F) (23, 24). Taken together, our results demonstrate that mutations leading to stabilization of β-catenin occur frequently during MYC-induced lymphomagenesis.
Fig. 1.

MYC-induced lymphomas frequently harbor stabilizing mutations in β-catenin. (A) In Eμ-tTA/tetO-MYC transgenic mice, T-cell lymphomas are proposed to arise as a result of MYC overexpression and alterations in unidentified cooperating pathways. (B) Distribution of β-catenin mutations detected by sequencing of DNA from Eμ-tTA/tetO-MYC primary lymphoma specimens. (C) Western blot analysis for β-catenin protein in primary MYC-induced lymphomas and lymphoma-derived cell lines. β-actin is shown as a loading control. Closed and open triangles denote full-length and Δexon3 β-catenin protein, respectively. Tumors with mutant β-catenin are labeled in red. (D) Schematic of β-catenin depicting the phosphorylation residues encoded by exon 3 that are critical for protein degradation. Also shown are sequence chromatograms of mutations in phosphorylation residues identified in primary MYC-induced lymphomas. (E) Diagram of conserved splice donor and acceptor sites required for proper splicing of the β-catenin primary transcript. Also shown are sequence chromatograms of splice acceptor site mutations identified in primary MYC-induced lymphomas. (F) Relative expression levels of transgenic MYC and the β-catenin/TCF target genes Axin2 and Enc1 across a panel of MYC-induced primary lymphomas with WT (n = 8) or mutant (n = 6) β-catenin, as analyzed by qRT-PCR. Shown are means ± SEM with **P < 0.01.
MYC-Induced Lymphomas Exhibit Addiction to Mutant β-Catenin.
Due to the frequency of stabilizing β-catenin mutations in MYC-induced lymphomas, we speculated that β-catenin might be required for tumor maintenance. We used a retroviral vector expressing β-catenin–specific shRNAs and a GFP reporter to infect MYC lymphoma cell lines known to be mutant or WT for β-catenin. Efficient knockdown was observed with three different shRNAs targeting β-catenin (Fig. 2A), and suppression of β-catenin levels resulted in significant depletion of GFP+ cells over time compared with a control nontargeting shRNA (Fig. 2 C and D). Sensitivity to β-catenin knockdown was also observed for an additional 13 MYC lymphoma lines that were characterized as mutant for β-catenin (Fig. S2A). In contrast, MYC lymphoma lines identified as WT for β-catenin were completely resistant to β-catenin suppression (Fig. 2E), confirming a strong correlation between β-catenin mutation status and dependency. Therefore, MYC-induced lymphomas are highly dependent on continued expression of mutant β-catenin.
Fig. 2.

MYC-induced lymphomas exhibit addiction to mutant β-catenin. (A) Western blot analysis of MYC lymphoma cells retrovirally infected with either control shRNA (shControl) or three independent shRNAs targeting β-catenin (shßcat-2, shßcat-3, and shßcat-4). (B) Relative mRNA expression levels of transgenic MYC, β-catenin, and three β-catenin target genes (Axin2, Runx2, and Enc1) in MYC lymphoma cells infected with either control or β-catenin shRNA, as analyzed by qRT-PCR. (C) Representative histograms and (D) quantification of GFP fluorescence in MYC lymphoma cells after infection with either control or β-catenin shRNA coexpressing GFP. (E and F) Quantification of GFP fluorescence over time of MYC lymphoma cell lines mutant or WT for β-catenin and infected with retrovirus expressing either (E) β-catenin shRNA or (F) the β-catenin inhibitor ICAT. Values represent means ± SD of two cell lines with WT β-catenin and two cell lines with mutant β-catenin and three independent experiments per cell line. Data are normalized to the percentage of GFP+ cells at day 1 after infection.
ICAT (also known as CTNNBIP1) is an endogenously expressed gene that was first identified in a screen of β-catenin interacting proteins and found to inhibit the interaction between β-catenin and its transcriptional partner TCF (25). Similar to β-catenin knockdown by shRNA, overexpression of ICAT also resulted in specific depletion of MYC lymphoma cells expressing mutant, but not WT, β-catenin (Fig. 2F). In contrast, expression of dominant-negative TCF4 (TCF4ΔN31) was growth suppressive to both β-catenin WT and mutant MYC lymphomas (Fig. S2B). Several reported chemical inhibitors of β-catenin also did not exhibit selective activity when used to treat either β-catenin WT or mutant MYC lymphomas (Fig. S2C), demonstrating that these compounds have significant off-target effects in MYC lymphoma cells. Collectively, these results indicate that MYC lymphomas are sensitive to inhibition of mutant β-catenin, either through shRNA-mediated knockdown or by expression of the inhibitory protein ICAT.
Inducible Inhibition of β-Catenin Through a Chemical-Genetic Approach.
In lymphoma cells from the Eμ-tTA/tetO-MYC model, expression of the MYC transgene is driven by the tetracycline-regulatory system (tet-system) and is suppressed in the presence of tetracycline or tetracycline analogs such as doxycycline (DOX). To suppress β-catenin activity in a similarly inducible manner, we took advantage of a recently developed expression system in which protein stability can be controlled by the presence or absence of a specific chemical ligand (26). A gene of interest can be fused in-frame to a mutant destabilized form of Escherichia coli dihydrofolate reductase (ecDHFR), resulting in constitutive degradation of the entire protein product. Addition of the DHFR ligand trimethoprim (TMP) leads to rapid stabilization of the fusion protein (Fig. 3A). TMP is highly selective for bacterial DHFR and has favorable pharmacological properties, making the ecDHFR-TMP conditional expression system well suited for both in vitro and in vivo studies in mammalian cells. To generate an inducible inhibitor of β-catenin, we fused the destabilizing ecDHFR domain (DD) to the N terminus of ICAT (Fig. 3A) and stably expressed the DD-ICAT fusion in a MYC lymphoma cell line. Levels of DD-ICAT were low in the absence of TMP and were rapidly induced with addition of TMP (Fig. 3B).
Fig. 3.

Inducible inhibition of β-catenin through a chemical-genetic approach. (A) Schematic outlining the inducible DD-ICAT β-catenin inhibitor consisting of a degradation domain (DD) and the β-catenin inhibitor ICAT. DD-ICAT is constitutively degraded and becomes stabilized in the presence of the ligand TMP. Once stabilized, DD-ICAT prevents the interaction between β-catenin and TCF and effectively inhibits β-catenin transcriptional activity. (B) Western blot analysis of MYC lymphoma cells expressing DD-ICAT and treated with TMP for 0–8 h. (C) Western blot analysis of MYC lymphoma cells expressing DD-ICAT treated with either DOX alone, TMP alone, or both DOX and TMP. (D) Relative expression of β-catenin target genes Axin2, Enc1, Mecom/Evi1, and Runx2 in MYC lymphoma cells expressing DD-ICAT and treated with either DMSO or TMP for 2 d. (E) Quantification of luciferase signals in MYC lymphoma cells infected with a 7× TCF binding site luciferase reporter and DD-ICAT, treated with either DMSO or TMP for 24 h. Values are means ± SD of normalized firefly/renilla luciferase ratios from three independent experiments.
We first confirmed orthogonal regulation of MYC and DD-ICAT by treating cells with DOX alone, TMP alone, or a combination of DOX and TMP. MYC levels were only suppressed in the presence of DOX, whereas DD-ICAT expression was only induced in the presence of TMP (Fig. 3C). Next, we investigated the effects of DD-ICAT induction on β-catenin activity. Treatment with TMP and stabilization of DD-ICAT resulted in down-regulation of the β-catenin/TCF target genes Axin2, Enc1, Mecom, and Runx2 (Fig. 3D) (23, 24, 27, 28), as well as down-regulation of a luciferase reporter driven by a 7× repeat of the canonical TCF binding sequence (Fig. 3E) (29). Our results demonstrate the activity of a novel inducible inhibitor of β-catenin that can be temporally controlled and effectively suppresses β-catenin–dependent transcription.
Inhibition of β-Catenin Induces Apoptosis Through Regulation of Bim and Bcl-xL Expression.
We next sought to take advantage of the DD-ICAT inducible system to characterize the dependence of MYC-induced lymphomas on the β-catenin pathway. MYC lymphoma cells expressing DD-ICAT were treated with TMP, and levels of cell death over time were measured by staining for the cell surface apoptotic marker Annexin-V and the DNA dye 7-aminoactinomycin D (7-AAD). Inhibition of β-catenin activity via induced expression of DD-ICAT resulted in an increase in the total percentage of apoptotic cells, from a basal level of ∼13% Annexin-V+ cells before treatment to 64% Annexin-V+ cells after 3 d of treatment with TMP (Fig. 4A). In contrast to MYC inactivation, which induces both cell cycle arrest and apoptosis (17), we found that β-catenin inhibition had no significant effect on the percentage of proliferating cells, as determined by staining for DNA content with propidium iodide (PI) (Fig. 4E). As a control, TMP-induced expression of a DD-YFP fusion protein had no effect on the survival of MYC lymphoma cells both in vitro and in vivo (Fig. S3 A–D), demonstrating the lack of any nonspecific toxicity due to expression of the ecDHFR destabilizing domain or treatment with TMP.
Fig. 4.

Mutant β-catenin is required for survival of MYC lymphomas through regulation of apoptotic factors. (A) MYC lymphoma cells expressing DD-ICAT were treated with TMP and levels of apoptosis were tracked by staining for Annexin-V-PE and 7AAD. (B) Western blot analysis for apoptotic proteins in cells treated with TMP for 0, 1, 2, and 3 d. (C) Levels of proapoptotic Bim and antiapoptotic Bcl-xL mRNA in cells treated with TMP for 0, 1, 2, and 3 d, as determined by qRT-PCR. (D) Quantification of apoptosis as measured by staining with Annexin-V after treatment with TMP in cells expressing Bim shRNA or Bcl-xL cDNA compared with empty vector control. All values represent means of at least three independent experiments ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. (E) MYC lymphoma cells expressing DD-ICAT were either untreated, treated with TMP (1 μM), or treated with DOX for 24 h. Cells were fixed and stained with PI to measure DNA content. Histogram events are gated on viable cells.
To further characterize the mechanism of apoptosis due to inhibition of β-catenin, we collected cells that were treated with TMP for 0, 1, 2, and 3 d and screened the expression of several pro- and antiapoptotic factors by Western blot (Fig. 4B). Inhibition of β-catenin led to an increase in levels of cleaved caspase-3, which correlated with an induction in levels of Bim and a reduction in levels of Bcl-xL at both the mRNA and protein levels (Fig. 4 B and C). We observed no changes in the levels of Birc5, an antiapoptotic factor shown to be a transcriptional target of β-catenin/TCF in colon carcinoma cells (30) (Fig. 4B). In addition, shRNA-mediated knockdown of Bim or ectopic expression of Bcl-xL was sufficient to significantly dampen the apoptotic response to β-catenin inhibition (Fig. 4D). These results demonstrate that β-catenin is required for the survival of MYC-induced lymphomas by promoting the expression of Bcl-xL and suppressing the expression of Bim.
Combined Inactivation of MYC and β-Catenin Synergizes in Tumor Regression and Is Sufficient to Prevent Tumor Recurrence.
As we have previously shown, MYC-induced lymphomas fail to undergo sustained tumor regression upon MYC inactivation in the absence of an intact host immune system (20). We speculated that targeting a secondary addiction pathway in addition to MYC might be sufficient to prevent tumor recurrence. To test this hypothesis, a cohort of SCID mice was transplanted s.c. with MYC-induced lymphoma cells expressing DD-ICAT and a firefly luciferase reporter (LUC2). Tumors were allowed to establish, and mice were treated with either DOX alone, TMP alone, or a combination of DOX and TMP. Tumor burden was monitored over time by bioluminescence imaging (Fig. 5A). Although inhibition of a single oncogene induced significant short-term regression, all tumors eventually recurred, with resistant tumor cells emerging after ∼2 wk of continuous treatment. Strikingly, the combined inhibition of both MYC and β-catenin resulted in more rapid tumor regression and long-term recurrence-free survival, despite cessation of treatment after only 16 d (Fig. 5 A–D). Control DD-YFP–expressing tumors developed resistance to combination treatment with DOX+TMP (Fig. S3E), confirming the absence of nonspecific toxicity due to TMP treatment or expression of the DD domain. Our findings demonstrate that the outgrowth of resistant cells can be effectively suppressed by the combined inhibition of both MYC and β-catenin.
Fig. 5.
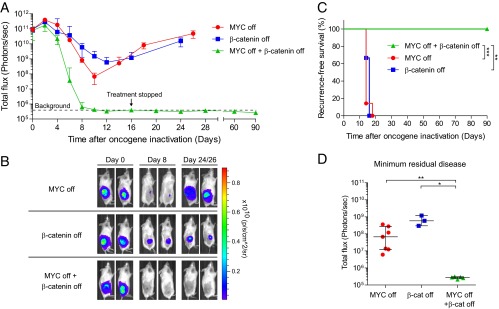
Combined targeting of MYC and β-catenin prevents tumor recurrence. (A) MYC lymphoma cells expressing LUC2 and DD-ICAT were transplanted s.c. into SCID recipients. Bioluminescence signal was quantified over time after initiation of treatment to inactivate MYC (+DOX), β-catenin (+TMP), or both MYC and β-catenin (+DOX/TMP). Values are means ± SEM (DD-ICAT +DOX, n = 7; DD-ICAT +TMP, n = 3; DD-ICAT +DOX/TMP, n = 5). (B) Representative bioluminescence images of tumor-bearing mice at 0, 8, and 24 (+TMP, +DOX/TMP) or 26 (+DOX) d after initiation of treatment. (C) Recurrence-free survival of mice transplanted with MYC lymphomas and treated with DOX, TMP, or both DOX and TMP as in A. Tumors were scored as recurrences on the day when the bioluminescence signal increased after initial tumor regression. Survival curves were compared by the log-rank test. (D) Quantification of minimum residual disease in transplanted mice treated with DOX, TMP, or both DOX and TMP. Values are means ± 95% CIs. *P < 0.05, **P < 0.01, ***P < 0.001.
To determine the mechanism of enhanced tumor regression we observed with combined targeting of MYC and β-catenin, we stained tumors treated with DOX alone, TMP alone, or DOX+TMP, for the proliferation marker Ki-67 and the apoptosis marker cleaved caspase-3 (Fig. 6). Inactivation of MYC or combined inactivation of both MYC and β-catenin for 4 d resulted in a greater reduction in Ki-67+ cells compared with inhibition of β-catenin alone (Ki-67+ cells: MYC off = 2.0% and MYC+β-cat off = 0.7% vs. β-cat off = 32.8%, P < 0.001; Fig. 6B). Inhibition of β-catenin or combined inactivation of both MYC and β-catenin resulted in more apoptosis after 2 d of treatment compared with withdrawal of MYC alone (cleaved caspase-3+ cells: β-cat off = 10.1% and MYC+β-cat off = 8.8% vs. MYC off = 1.7%, P < 0.01; Fig. 6C). Thus, the simultaneous inactivation of MYC and β-catenin promotes a greater decrease in proliferation and increase in apoptosis compared with inhibition of either oncogene alone. Taken together, our results demonstrate that combined inhibition of MYC and β-catenin in MYC-induced lymphomas synergizes to promote tumor regression and is sufficient to prevent the development of resistance.
Fig. 6.
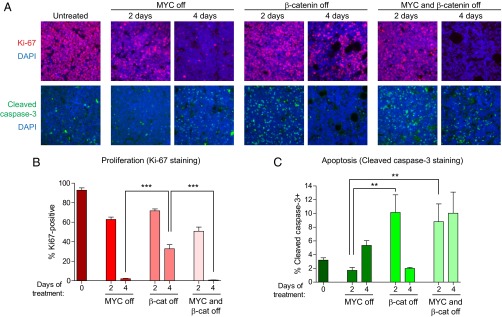
Combined inactivation of MYC and β-catenin synergizes to inhibit proliferation and promote apoptosis in vivo. (A) MYC lymphoma cells expressing DD-ICAT were transplanted s.c. into recipient SCID mice. Mice were either left untreated or treated with DOX (MYC off), TMP (β-catenin off), or both DOX and TMP (MYC and β-catenin off) for 2–4 d. Tumors were harvested and stained for the proliferation marker Ki-67 and the apoptotic marker cleaved caspase-3. Nuclei were stained using DAPI. Representative images are shown. (B and C) Quantification of Ki-67 and cleaved caspase-3 staining, in two to three independent tumors from each time point and five fields per tumor. Values represent means ± SEM. **P < 0.01, ***P < 0.001.
Discussion
The emergence of therapeutic resistance represents a significant obstacle toward achieving long-term remissions with oncogene-targeted therapy. Although rational combinations of targeted agents offer the greatest potential for preventing tumor recurrence, it remains unclear which specific combinations would provide the greatest therapeutic benefit. Here, we took advantage of a transgenic mouse model system in which tumors frequently recur after MYC inactivation to investigate the efficacy of simultaneously inhibiting two driver oncogenic events. We discovered that MYC-induced lymphomas frequently acquire activating mutations in a splice acceptor site of β-catenin resulting in its stabilization. In turn, this mutation correlated with marked sensitivity of tumors to β-catenin inhibition. By generating a novel inducible inhibitor of β-catenin, we were able to study the in vivo consequences of targeting either MYC or β-catenin independently vs. combined inhibition of both oncogenes. The inhibition of either oncogene alone uniformly gives rise to resistance, whereas targeting two oncogenes results in more rapid and sustained tumor regression. Our results specifically identify β-catenin activation through a mutation in a splice acceptor site as an additional driver event associated with MYC-induced lymphomagenesis. Further, we illustrate that cancers can exhibit addiction to multiple oncogenic drivers and that the combined targeting of two independently required oncogenic dependencies is sufficient to prevent the outgrowth of resistant cells.
β-catenin is tightly regulated at the posttranslational level through phosphorylation of residues in exon 3 (Fig. 1D), leading to ubiquitination and proteasome-mediated degradation (31). Mutations that abrogate β-catenin phosphorylation and result in protein stabilization have been found in a variety of human cancers (32). We discovered a previously unidentified mechanism of β-catenin stabilization through point mutations in the exon 3 splice acceptor site. Although splice site mutations have been associated with loss of function in several tumor suppressor genes, such as TP53, RB1, and BRCA1 (33–36), there have been relatively few reports of splicing mutations resulting in the gain of function of an oncogene. Examples to date include deletions at the intron 10–exon 11 boundary of the receptor tyrosine kinase c-KIT that result in loss of an auto-inhibitory region and constitutive activation of the protein (37, 38), and splice acceptor site mutations in c-MET that lead to skipping of exon 14 and deletion of a juxtamembrane domain important for negative regulation (39). Intragenic deletions in β-catenin that remove all or part of exon 3 have been reported in colorectal carcinoma (40). We conclude that the β-catenin oncogene can also become activated through single nucleotide substitutions occurring at the exon 3 splice acceptor site.
Overexpression of MYC due to genomic amplification, translocation, or via activation of upstream regulators represents one of the most common features in human cancer. In addition, the WNT/β-catenin pathway is also frequently activated in cancer, due to mutations in β-catenin or loss-of-function mutations in negative regulators such as APC and AXIN2 (32). Interestingly, although MYC is a prominent downstream target of WNT/β-catenin signaling (41), our findings suggest a strong selective pressure for β-catenin functions independent of MYC in our lymphoma model. Wnt/β-catenin pathway activation has also been observed in models of MYC-driven liver cancer, suggesting MYC and β-catenin can functionally cooperate during tumorigenesis in diverse tissue contexts (42, 43). It has also been reported that lymphomas initiated by activated β-catenin frequently exhibit overexpression of MYC due to genomic amplification or rearrangement and that MYC is required for β-catenin driven lymphomagenesis (44–46), further supporting a cooperative model for MYC and β-catenin function during tumorigenesis.
We found that β-catenin inhibition resulted in the induction of apoptosis, which closely correlated with decreased levels of the antiapoptotic factor Bcl-xL (Fig. 4 A–C). Ectopic expression of Bcl-XL was also sufficient to completely abrogate the cell death associated with loss of β-catenin activity (Fig. 4D). β-catenin has been shown to directly activate the expression of Bcl-xL (45, 47, 48), and we found that Bcl-xL is a critical downstream mediator of the addiction to β-catenin in MYC-induced lymphomas. The phenotypic consequences of β-catenin inhibition may be explained by an “oncogenic shock” model, in which the rapid loss of survival cues such as Bcl-xL and the persistence of proapoptotic signaling results in an imbalance that favors cell death (49). Interestingly, we observed a synergistic effect on the rate of tumor regression from the inhibition of both MYC and β-catenin (Fig. 5A) compared with inhibition of either oncogene alone. The targeting of each oncogene elicited distinct cellular outcomes, with MYC inactivation resulting primarily in cell cycle arrest and β-catenin inhibition leading mainly to the induction of apoptosis. The accelerated tumor regression upon inactivation of both oncogenes can then be attributed to the collective loss of both proliferation and survival signals. Taken together, these results indicate that MYC and β-catenin appear to play complementary roles to promote and maintain tumorigenesis.
The effectiveness of combination therapy in our model system may be explained by the low probability of tumor cells with cross-resistance to inhibition of both oncogenes. A similar effect is believed to be responsible for the success of other types of combination therapy, including highly active antiretroviral therapy (HAART) for HIV infection, in which the use of a mixture of inhibitors targeting different viral enzymes is effective at preventing the emergence of virions that are resistant to any single drug (12, 13). We conclude that the suppression of resistance with combination targeted therapy will be most effective by selecting targets that are each essential for tumor maintenance but are sufficiently independent as to minimize the likelihood of cross-resistance arising from a single mutation.
We provided a proof of concept that the simultaneous targeted inhibition of two mutually required oncogenes may be sufficient to prevent the emergence of therapeutic resistance. Additional factors, such as tumor burden and heterogeneity, overall tumor mutation rate, and tumor-host interactions, will likely play a critical role in determining the number and types of targeted therapies that will be most effective in combination. In addition, the ability to target many known and newly discovered driver events will largely depend on the development of therapeutics against novel classes of drug targets, such as transcription factors and chromatin regulators. However, as we continue to uncover the full landscape of genetic alterations in cancer, the identification and combined targeting of multiple oncogene addiction pathways offers great potential toward preventing resistance and improving therapeutic outcomes.
Materials and Methods
Transgenic Mice and Lymphoma Cell Lines.
All animal experiments were approved by the Institutional Animal Care and Use Committee at Stanford University. MYC lymphomas and lymphoma-derived cell lines were named for the identifying number of the mouse from which they were obtained. Cell lines were grown in RPMI 1640 media supplemented with 10% (vol/vol) FBS, penicillin/streptomycin, and 50 μM β-mercaptoethanol. MYC was inactivated by treatment with 20 ng/mL of doxycycline.
Sequencing of β-Catenin.
Genomic DNA was extracted from primary tumors or cell lines using the Qiagen DNeasy kit or by standard phenol-chloroform purification. A region spanning intron 2 and exon 3 of β-catenin was PCR amplified (see primers in Table S2) and directly sequenced (Sequetech). Mutant peaks were detected by manual inspection of sequencing chromatograms and with Mutation Surveyor (Softgenetics). To sequence β-catenin cDNA, RNA was first isolated from cells using the Nucleospin RNA II kit (Machery-Nagel), and first-strand cDNA synthesis was carried out with SuperScript II (Invitrogen) using oligo-dT primers. The 5′ region of the β-catenin transcript was amplified by PCR (see primers in Table S2) and directly sequenced.
Proliferation and Apoptosis Assays.
MYC lymphoma cells were infected with retrovirus, and the percentage of GFP+ infected cells was tracked over time using a FACScan flow cytometer. Apoptosis was quantified by staining with Annexin-V-PE and 7-AAD (BD Biosciences). To assess cell cycle status, cells were first fixed in 70% (vol/vol) ethanol in PBS at −20 °C for at least 12 h. After washing in PBS, cells were resuspended in PI staining solution (PBS/0.1% TritonX-100, 0.2 mg/mL RNase A, and 20 μg/mL PI) and incubated for 30 min at room temperature while protected from light. DNA content was then measured using a FACScan flow cytometer. All flow cytometric data were analyzed with Flowjo software (Tree Star Inc.).
Plasmids.
For β-catenin shRNA-mediated knockdown, oligos were PCR amplified and cloned into the XhoI/EcoRI sites of the retroviral LMP vector (RNAi Codex/Openbiosystems). Sequences are listed in Table S3. To generate pMSCV-ICAT-PIG, ICAT was PCR amplified from murine brain cDNA and ligated into BglII and XhoI sites. To create pMSCV-TCFΔN31-PIG, dominant-negative TCF was PCR amplified from pPGS-CITEneo-TCFΔN31 (obtained from Karl Sylvester, Stanford University, Stanford, CA) and ligated into BglII and XhoI sites.
Inducible β-Catenin Inhibitor.
The N-terminal ecDHFR-DD-ICAT fusion was first generated using splicing by overlap extension (SOE) PCR. The fused PCR product was then ligated into pMSCV-puro (BglII/XhoI). The N-terminal ecDHFR-DD-YFP fusion was PCR amplified directly from pBMN-DHFR-(R12Y G67S Y100I)-YFP-HA-IRES-HcRed-tandem (obtained from Tom Wandless, Stanford University, Stanford, CA) and ligated into pMSCV-puro (BglII/XhoI). Stabilization of fusion proteins was induced by treatment with 1 μM of TMP (Sigma).
TCF Luciferase Reporter Assay.
The 7TFP lentiviral Wnt reporter (29) was obtained from Addgene (plasmid 24308) and modified to replace the puromycin resistance gene with the neomycin resistance gene, generating 7TFN. MYC lymphoma cells expressing renilla luciferase alone or both renilla luciferase and DD-ICAT were stably transduced with the 7TFN reporter. The Dual-Glo Luciferase assay system (Promega) was used to quantify the relative levels of luciferase activity ± 1 μM TMP. Wnt-responsive firefly luciferase values were normalized to control renilla luciferase values.
Tumor Transplantation and Bioluminescence Imaging.
Lymphoma cells were transplanted s.c. into the flank of immunodeficient SCID mice. Tumors were monitored by caliper measurement and bioluminescence imaging. Treatment of mice with DOX, TMP, or DOX+TMP was initiated when tumors reached similar bioluminescent signals. Bioluminescent signal from tumors was measured by i.p. injection of mice with 100 μL of a solution of d-Luciferin (30 mg/mL) and imaging with the IVIS Spectrum (Perkin-Elmer). Images were collected and analyzed using Living Image software (Perkin-Elmer). To inactivate MYC, mice were injected once i.p. with 10 μg of DOX in PBS and maintained on drinking water containing DOX (100 μg/mL), which was refreshed weekly. To induce DD-ICAT or DD-YFP, mice were treated by daily i.p. injection of 5 mg of pharmaceutical-grade trimethoprim lactate (ABT-590; Alpha Diagnostic International) and maintained on drinking water containing a suspension of 1 mg/mL of TMP (Trimethoprim-Sulfamethoxazole suspension; Qualitest Pharmaceuticals), which was refreshed every 3 d.
Immunofluorescence.
Tumor-bearing mice were killed at 0, 2, and 4 d after MYC inactivation, and transplanted tumors were harvested and fixed in formalin. Paraffin-embedded sections were immunostained with anti–Ki-67 (BD Biosciences, 556003) or anti-cleaved caspase-3 (Cell Signaling Techology, 9661). All antibodies are listed in Table S4. Images were obtained on a Nikon microscope and analyzed using CellProfiler software (www.cellprofiler.org/).
Supplementary Material
Acknowledgments
We thank members of the D.W.F. laboratory for helpful suggestions and guidance, Tom Wandless (Stanford University) for the ecDHFR degradation domain constructs, and Karl Sylvester (Stanford University) for the dominant-negative TCF construct. This work was supported by National Institutes of Health (NIH) Training Grant 5 T32 AI07290 (to P.S.C.), NIH Grants R01CA170378 and U54CA149145 (to D.W.F.), and Leukemia and Lymphoma Society Translational Research Grant R6223-07 (to D.W.F.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. S.R.H. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1406123111/-/DCSupplemental.
References
- 1.Weinstein IB. Cancer. Addiction to oncogenes—The Achilles heal of cancer. Science. 2002;297(5578):63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 2.Druker BJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 3.Paez JG, et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 4.Flaherty KT, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kobayashi S, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 6.Pao W, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glickman MS, Sawyers CL. Converting cancer therapies into cures: Lessons from infectious diseases. Cell. 2012;148(6):1089–1098. doi: 10.1016/j.cell.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah NP, et al. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. J Clin Invest. 2007;117(9):2562. doi: 10.1172/JCI30890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johannessen CM, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nazarian R, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engelman JA, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 12.Gulick RM, et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N Engl J Med. 1997;337(11):734–739. doi: 10.1056/NEJM199709113371102. [DOI] [PubMed] [Google Scholar]
- 13.Hammer SM, et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. N Engl J Med. 1997;337(11):725–733. doi: 10.1056/NEJM199709113371101. [DOI] [PubMed] [Google Scholar]
- 14.Fox W, Ellard GA, Mitchison DA. Studies on the treatment of tuberculosis undertaken by the British Medical Research Council tuberculosis units, 1946-1986, with relevant subsequent publications. Int J Tuberc Lung Dis. 1999;3(10) Suppl 2:S231–S279. [PubMed] [Google Scholar]
- 15.Bozic I, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. eLife. 2013;2:e00747. doi: 10.7554/eLife.00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beroukhim R, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4(2):199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 18.Giuriato S, et al. Sustained regression of tumors upon MYC inactivation requires p53 or thrombospondin-1 to reverse the angiogenic switch. Proc Natl Acad Sci USA. 2006;103(44):16266–16271. doi: 10.1073/pnas.0608017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu CH, et al. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci USA. 2007;104(32):13028–13033. doi: 10.1073/pnas.0701953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rakhra K, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell. 2010;18(5):485–498. doi: 10.1016/j.ccr.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi PS, et al. Lymphomas that recur after MYC suppression continue to exhibit oncogene addiction. Proc Natl Acad Sci USA. 2011;108(42):17432–17437. doi: 10.1073/pnas.1107303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13(20):2658–2669. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujita M, et al. Up-regulation of the ectodermal-neural cortex 1 (ENC1) gene, a downstream target of the beta-catenin/T-cell factor complex, in colorectal carcinomas. Cancer Res. 2001;61(21):7722–7726. [PubMed] [Google Scholar]
- 24.Jho EH, et al. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22(4):1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tago K, et al. Inhibition of Wnt signaling by ICAT, a novel beta-catenin-interacting protein. Genes Dev. 2000;14(14):1741–1749. [PMC free article] [PubMed] [Google Scholar]
- 26.Iwamoto M, Björklund T, Lundberg C, Kirik D, Wandless TJ. A general chemical method to regulate protein stability in the mammalian central nervous system. Chem Biol. 2010;17(9):981–988. doi: 10.1016/j.chembiol.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong YF, Soung Y, Schwarz EM, O’Keefe RJ, Drissi H. Wnt induction of chondrocyte hypertrophy through the Runx2 transcription factor. J Cell Physiol. 2006;208(1):77–86. doi: 10.1002/jcp.20656. [DOI] [PubMed] [Google Scholar]
- 28.Wei W, et al. Biphasic and dosage-dependent regulation of osteoclastogenesis by β-catenin. Mol Cell Biol. 2011;31(23):4706–4719. doi: 10.1128/MCB.05980-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuerer C, Nusse R. Lentiviral vectors to probe and manipulate the Wnt signaling pathway. PLoS ONE. 2010;5(2):e9370. doi: 10.1371/journal.pone.0009370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang T, et al. Evidence that APC regulates survivin expression: A possible mechanism contributing to the stem cell origin of colon cancer. Cancer Res. 2001;61(24):8664–8667. [PubMed] [Google Scholar]
- 31.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149(6):1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 32.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14(15):1837–1851. [PubMed] [Google Scholar]
- 33.Holmila R, Fouquet C, Cadranel J, Zalcman G, Soussi T. Splice mutations in the p53 gene: Case report and review of the literature. Hum Mutat. 2003;21(1):101–102. doi: 10.1002/humu.9104. [DOI] [PubMed] [Google Scholar]
- 34.Horowitz JM, et al. Frequent inactivation of the retinoblastoma anti-oncogene is restricted to a subset of human tumor cells. Proc Natl Acad Sci USA. 1990;87(7):2775–2779. doi: 10.1073/pnas.87.7.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mazoyer S, et al. A BRCA1 nonsense mutation causes exon skipping. Am J Hum Genet. 1998;62(3):713–715. doi: 10.1086/301768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takahashi T, D’Amico D, Chiba I, Buchhagen DL, Minna JD. Identification of intronic point mutations as an alternative mechanism for p53 inactivation in lung cancer. J Clin Invest. 1990;86(1):363–369. doi: 10.1172/JCI114710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen LL, et al. A mutation-created novel intra-exonic pre-mRNA splice site causes constitutive activation of KIT in human gastrointestinal stromal tumors. Oncogene. 2005;24(26):4271–4280. doi: 10.1038/sj.onc.1208587. [DOI] [PubMed] [Google Scholar]
- 38.Corless CL, et al. KIT gene deletions at the intron 10-exon 11 boundary in GI stromal tumors. J Mol Diagn. 2004;6(4):366–370. doi: 10.1016/S1525-1578(10)60533-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Asaoka Y, et al. Gastric cancer cell line Hs746T harbors a splice site mutation of c-Met causing juxtamembrane domain deletion. Biochem Biophys Res Commun. 2010;394(4):1042–1046. doi: 10.1016/j.bbrc.2010.03.120. [DOI] [PubMed] [Google Scholar]
- 40.Iwao K, et al. Activation of the beta-catenin gene by interstitial deletions involving exon 3 in primary colorectal carcinomas without adenomatous polyposis coli mutations. Cancer Res. 1998;58(5):1021–1026. [PubMed] [Google Scholar]
- 41.He TC, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281(5382):1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 42.Calvisi DF, Factor VM, Loi R, Thorgeirsson SS. Activation of beta-catenin during hepatocarcinogenesis in transgenic mouse models: Relationship to phenotype and tumor grade. Cancer Res. 2001;61(5):2085–2091. [PubMed] [Google Scholar]
- 43.de La Coste A, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci USA. 1998;95(15):8847–8851. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaveri D, et al. β-Catenin activation synergizes with Pten loss and Myc overexpression in Notch-independent T-ALL. Blood. 2013;122(5):694–704. doi: 10.1182/blood-2012-12-471904. [DOI] [PubMed] [Google Scholar]
- 45.Dose M, et al. β-Catenin induces T-cell transformation by promoting genomic instability. Proc Natl Acad Sci USA. 2014;111(1):391–396. doi: 10.1073/pnas.1315752111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo Z, et al. Beta-catenin stabilization stalls the transition from double-positive to single-positive stage and predisposes thymocytes to malignant transformation. Blood. 2007;109(12):5463–5472. doi: 10.1182/blood-2006-11-059071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosenbluh J, et al. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012;151(7):1457–1473. doi: 10.1016/j.cell.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xie H, Huang Z, Sadim MS, Sun Z. Stabilized beta-catenin extends thymocyte survival by up-regulating Bcl-xL. J Immunol. 2005;175(12):7981–7988. doi: 10.4049/jimmunol.175.12.7981. [DOI] [PubMed] [Google Scholar]
- 49.Sharma SV, et al. A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell. 2006;10(5):425–435. doi: 10.1016/j.ccr.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.