

Significance
This paper addresses a fundamental question: Can natural chaperones fold mirror-image proteins? Mirror-image proteins (composed of d-amino acids) are only accessible by chemical synthesis, but are protease resistant and therefore have tremendous potential as long-lived drugs. Many large/complex proteins depend on chaperones for efficient folding. Here we describe the total chemical synthesis of a 312-residue chaperone-dependent protein (DapA) in natural (l-) and mirror-image (d-) forms, the longest fully synthetic proteins yet reported. Using these proteins we show that the natural bacterial GroEL/ES chaperone is “ambidextrous”—i.e., it can fold both natural and mirror-image proteins via nonspecific hydrophobic interactions. Our study also provides proof-of-concept for the use of natural GroEL/ES to fold d-proteins for mirror-image drug discovery and synthetic biology applications.
Keywords: peptide synthesis, protein folding
Abstract
Mirror-image proteins (composed of d-amino acids) are promising therapeutic agents and drug discovery tools, but as synthesis of larger d-proteins becomes feasible, a major anticipated challenge is the folding of these proteins into their active conformations. In vivo, many large and/or complex proteins require chaperones like GroEL/ES to prevent misfolding and produce functional protein. The ability of chaperones to fold d-proteins is unknown. Here we examine the ability of GroEL/ES to fold a synthetic d-protein. We report the total chemical synthesis of a 312-residue GroEL/ES-dependent protein, DapA, in both l- and d-chiralities, the longest fully synthetic proteins yet reported. Impressively, GroEL/ES folds both l- and d-DapA. This work extends the limits of chemical protein synthesis, reveals ambidextrous GroEL/ES folding activity, and provides a valuable tool to fold d-proteins for drug development and mirror-image synthetic biology applications.
All known living organisms use proteins composed of l-amino acids. Mirror-image proteins (composed of d-amino acids) are not found in nature and are promising therapeutic agents due to their resistance to degradation by natural proteases (1, 2). d-peptide inhibitors that target particular protein interfaces can be identified by mirror-image phage display (3, 4), in which a library of phage bearing l-peptides on their surface is screened against a mirror-image (d-) protein target. By symmetry, d-peptide versions of the identified sequences will bind to the natural l-target. Because d-protein targets must be chemically synthesized, this discovery method has thus far been limited to relatively small targets.
Through rigorous application of recent advances in chemical protein synthesis (reviewed in ref. 5), the production of larger synthetic d-proteins is becoming increasingly feasible [e.g., 204-residue d-VEGF dimer (6) and 84-residue d-MDM2/MDMX (7)]. However, many proteins are prone to misfolding, especially as their size and complexity increase (8). Molecular chaperones, such as the extensively studied GroEL/ES, mediate folding by preventing aggregation of many cellular proteins (9, 10). GroEL/ES is thought to interact with these diverse substrates via nonspecific hydrophobic interactions, but it is unknown whether it can fold mirror-image proteins. If natural chaperones cannot fold mirror-image proteins, then the folding of large/complex d-proteins into their active conformations will be a major challenge (in the absence of mirror-image chaperones, which are currently inaccessible).
The binding of substrates by GroEL is an intriguing instance of promiscuous molecular recognition. GroEL has been shown to interact transiently with ∼250 cytosolic proteins in Escherichia coli under normal growth conditions (8, 11). A subset of these proteins exhibit an absolute requirement for GroEL and its cochaperone GroES to avoid aggregation and fold into their native state (8, 12). Interestingly, sequence analysis of known GroEL/ES obligate substrates reveals no obvious consensus binding sequence (11), although structurally they are enriched in aggregation-prone folds (12).
Several lines of evidence suggest the predominant interactions between GroEL/ES and substrate proteins are hydrophobic. Protein substrates trapped in nonnative states have been shown to present hydrophobic surfaces that are otherwise buried in the core of the correctly folded protein, and a hydrophobic binding model is supported by the thermodynamics of binding of these nonnative states to GroEL (13). Additionally, the GroEL apical domain residues implicated in substrate binding are largely hydrophobic (14). Finally, previous studies on the basis of substrate interaction with GroEL using short model peptides have concluded that the most important determinant of substrate binding is the presentation of a cluster of hydrophobic residues (15–17).
The only evidence addressing the chiral specificity of GroEL/ES comes from a study that qualitatively demonstrated binding of a short d-peptide to GroEL (16). However, this NMR study required peptide concentrations that greatly exceed physiologic levels and did not localize the interaction to the substrate-binding region of GroEL. Only recently has it become feasible to directly test the stereospecificity of the GroEL/ES folding reaction by synthesizing the mirror-image version of a chaperone-dependent protein.
Due to great interest in mirror-image proteins as targets for drug discovery (6, 7, 18, 19) and mirror-image synthetic biology (20, 21), we were intrigued by the possibility that natural (l-) GroEL/ES could assist in the folding of d-proteins. Thus, we synthesized a d-version of a substrate protein and evaluated its folding by GroEL/ES. Furthermore, because most GroEL/ES substrate proteins are large (>250 residues), this project provided an excellent opportunity to demonstrate the power of chemical synthesis methodologies for producing previously inaccessible synthetic proteins.
Results
Selection of DapA as Model Protein.
We began our investigation by searching for the smallest model protein that requires GroEL/ES for folding under physiologic conditions and has a robust activity assay that does not depend on complex chiral reagents (e.g., cofactors or other enzymes that would also require mirror-image synthesis). The E. coli DapA protein [4-hydroxy-tetrahydrodipicolinate synthase (EC 4.3.3.7)] (Fig. 1) meets these criteria. DapA is a 31-kDa protein that forms a homotetramer (22) and catalyzes the stereoselective (23) condensation of l-aspartate-β-semialdehyde and pyruvate to (4S)-4-hydroxy-2,3,4,5-tetrahydro-(2S)-dipicolinic acid (24–26), a key step in the biosynthesis of lysine and diaminopimelic acid, a cell-wall precursor.
Fig. 1.
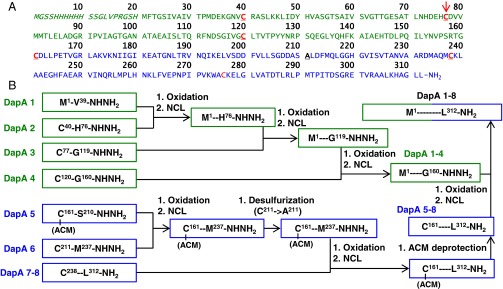
Total chemical synthesis of 312-residue DapA. (A) Target amino acid sequence, including N-terminal half (DapA 1–4, green), C-terminal half (DapA 5–8, blue), N-terminal His tag and thrombin cleavage site (italics), cysteines (red), ligation sites (bold and underline), and A77C mutation (red arrow). (B) Final synthetic strategy, including peptide segments (DapA 1–4, green and DapA 5–8, blue) with ligation hydrazide and cysteine residues indicated. Oxidation (hydrazide to azide), native chemical ligation (NCL), and acetamidomethyl (ACM) cysteine deprotection steps are also indicated.
DapA is highly enriched in GroEL/ES complexes under normal growth conditions (8) and is aggregated or degraded in GroEL-depleted cells (8, 12). In E. coli GroEL-depletion strains, cell death occurs via lysis due to loss of DapA activity, further demonstrating the dependence of DapA on GroEL/ES to adopt its native structure (27). Indeed, in vitro, DapA is absolutely dependent on GroEL/ES for proper folding in physiologic buffer at 37 °C (8). An added benefit of DapA as a model protein is that although it depends on chaperones for folding under physiological conditions, a chemical-folding procedure has been reported using 0.5 M arginine (8), providing an important positive control for enzyme activity independent of chaperone-mediated folding. Although a chemical refolding protocol for DapA was reported, many complex proteins cannot be folded by known chemical means, e.g., E. coli METK (28) and METF (8, 29).
Synthesis of l- and d-DapA.
Because d-proteins can only be accessed through chemical synthesis, a synthetic route to DapA was devised. Synthetic peptides are routinely made using solid-phase peptide synthesis (SPPS), but a project of this magnitude (312 residues) is well outside the capability of current SPPS technology (generally ∼50 residues). To access larger synthetic assemblies, chemoselective ligation techniques, especially native chemical ligation (30), are used to assemble peptide segments into larger constructs (reviewed in refs. 5, 31, and 32). Recent noteworthy synthetic proteins include tetraubiquitin [alone (33) and as part of a semisynthetic tetraubiquitinated α-synuclein (34)], covalent HIV protease dimer (35), l-/d-snow flea antifreeze protein (36), glycosylated EPO (37, 38), and the γ-subunit of F-ATPase (39).
For synthesizing DapA, we used a recently developed method to join peptide segments via native peptide bonds formed between a peptide with a C-terminal hydrazide (for selective conversion to a thioester) and a peptide with an N-terminal Cys (40–43). We selected this chemistry because of the convenient route to peptide hydrazides via Fmoc SPPS (less hazardous and more compatible with acid-sensitive modifications than Boc chemistry), the robustness of the native chemical ligation reaction (30, 44), and the ease of carrying out convergent protein assembly (vs. linear C- to N-assembly).
Our retrosynthetic analysis began by locating all Cys residues (potential ligation junctions) in DapA (Fig. 1A), all of which are located at acceptable ligation junctions (see refs. 40 and 45 for discussions of unacceptable junctions). This information allowed us to break the protein into six segments, leaving two segments >50 residues. To expand the range of potential ligation junctions, we used a free radical-based desulfurization reaction that enables selective conversion of unprotected Cys to Ala (46, 47). This technique allows one to substitute a Cys for a native Ala residue during peptide synthesis (for use in ligation) and then convert the Cys back to the native Ala following assembly. Using this method, we introduced two additional junction sites at A77 and A211, resulting in eight segments overall, ranging in size from 27 to 50 residues (DapA 1–8; Fig. S1). Using optimized SPPS reaction conditions and RP-HPLC column selection (Materials and Methods), we synthesized and purified all eight peptides.
Our initial strategy for the assembly of these eight segments required 12 steps (seven ligations, two desulfurizations, and three Acm removals; Fig. S1) and their associated purifications. Acm was used as an orthogonal Cys protecting group that prevents cyclization/polymerization of peptides containing both an activated C-terminal hydrazide and an N-terminal Cys, and also prevents Cys desulfurization. Following this scheme, we assembled the C-terminal segments (DapA 5–8), but were unable to assemble the N-terminal segments (DapA 1–4). A significant complication was the His thioester on DapA 2 (H76), which was highly susceptible to hydrolysis, leading to low reaction yields during the DapA 2/3 ligation step (Fig. S2). This difficulty, coupled with the large number of manipulations (and concomitant sample losses), resulted in a failure to assemble DapA 1–4 in usable yield.
We reasoned that we could simplify the assembly if we eliminated the desulfurization step necessary to convert the Cys to native Ala at the DapA 2–3 junction. Toward this end, we determined locations in our protein that would likely tolerate permanent mutation to Cys. BLAST analysis of the E. coli DapA identified the 1,000 most-similar homologs (>69% conservation, >49% identity), which were aligned to determine positions where Cys residues naturally occur. Fortuitously, 12% of the aligned sequences contained Cys at position 77, site of the DapA 2–3 junction (Fig. 2A). Next, we analyzed the DapA crystal structure to determine the likelihood of the A77C mutation to disrupt protein structure/function. The side chain of residue 77 is surface-exposed and not in close proximity to the active site or any native Cys residues (>12 Å to the nearest Cys; Fig. 2B). This analysis suggested that introduction of the A77C mutation would likely be well tolerated. Indeed, this mutation affected neither recombinant protein activity (Fig. 2C) nor its dependence on GroEL/ES for folding under physiological conditions (Fig. 2D and Table S1). We ultimately selected a final assembly strategy that incorporated both the A77C mutation and a unified DapA 7–8 segment (we were not able to produce high-quality DapA 1–2, 3–4, or 5–6 unified peptides). This final strategy yielded a seven-segment assembly scheme (Fig. 1B) that removed four synthetic steps (and associated purifications) from the initial scheme.
Fig. 2.
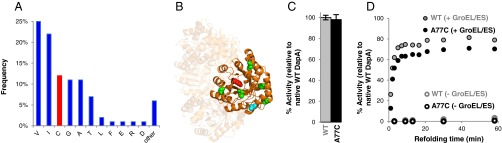
Validation of the DapA A77C mutation. (A) Natural sequence diversity at position 77 from Protein BLAST analysis. (B) Structure of DapA tetramer (PDB ID code 1DHP) showing, on one subunit, the surface-exposed alanine at position 77 (cyan), natural cysteine residues (green), and catalytic lysine at position 181 in the active site (red). (C) Enzyme activity of recombinant native WT and A77C DapA. Error bars indicate SD of at least three measurements. (D) GroEL/ES-mediated refolding of recombinant WT and A77C DapA.
Following this simplified strategy, we successfully assembled the 312-residue synthetic DapA A77C (hereafter referred to as “DapA”) in both l- and d- chiralities (Fig. 3 and SI Text), the longest synthetic peptides reported to date. The peptides were synthesized at milligram scale (1.1 and 1.7 mg of l- and d-DapA, respectively; Figs. S3 and S4). The synthetic l- and d-peptides behave identically to recombinant DapA on a C4 RP-HPLC column (Fig. 3A), and the major products possess the correct mass (Fig. 3 B and C).
Fig. 3.
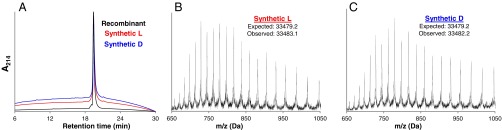
Analysis of synthetic unfolded l- and d-DapA. (A) Analytical RP-HPLC of recombinant (black), synthetic l- (red), and synthetic d-DapA (blue) on C4 column (linear gradient 5–100% buffer B over 30 min; buffer A, 0.1% TFA in water; buffer B, 0.1% TFA in 10% water/90% acetonitrile). (B and C) LC-MS analysis of the synthetic l- and d-DapA, respectively. Observed masses were calculated using the Bayesian Protein Reconstruct tool in Analyst 1.5.1 software (AB Sciex) over the charge states covering 650–1,050 Da. See SI Appendix for larger, detailed mass spectra of the final synthetic products and HPLC and LC-MS characterization of all synthetic intermediates.
Our initial efforts to fold these synthetic peptides using GroEL/ES resulted in measurable enzymatic activity, albeit at relatively low levels (∼20–40%; Table S1), likely due to microheterogeneity in the synthetic peptides (SI Appendix). Because active DapA assembles as a tetramer, we reasoned that we could enrich for “foldable” protein by using a chemical refolding procedure followed by size-exclusion chromatography (SEC).
Chemical-Mediated Folding of DapA.
Chaperone-independent folding of DapA has been described using 0.5 M l-arginine (8), a protein refolding additive (48). This method was validated at 13 °C using recombinant DapA and works equally well with d-arginine (Fig. S5). Thus, l-arginine can be used to fold both l- and d-DapA. Importantly, this procedure also provides a chaperone-independent means to evaluate the activity of our synthetic constructs.
After arginine-assisted folding of synthetic l- and d-DapA, we isolated tetrameric protein using SEC (Fig. S6). Following SEC, both the l- and d-DapA synthetic proteins have the expected CD spectra (Fig. 4A) and are enzymatically active (Fig. 4B), demonstrating that both l- and d-synthetic proteins are correctly folded and functional. As hoped, the SEC purification generated synthetic proteins with high specific activity (∼80% compared with recombinant protein). However, the Arg-assisted refolding/SEC purification resulted in a substantial (>10-fold) yield loss, largely due to precipitation during dialysis and concentration steps.
Fig. 4.

Structural and functional characterization of synthetic folded l- and d-DapA. (A) Circular dichroism spectra of Arg-folded and SEC-purified recombinant (black), synthetic l- (red), and synthetic d-DapA (blue). (B) Enzyme activity of Arg-folded and SEC-purified synthetic l- and synthetic d-DapA compared with native recombinant DapA. Error bars indicate SD of at least three assays.
Chaperone-Mediated Folding of DapA.
With folded and equally active synthetic l- and d-DapA in hand, we were poised to perform the definitive experiment comparing the refolding of our synthetic l- and d-DapA by GroEL/ES. This experiment answers the question of whether GroEL/ES is ambidextrous (i.e., Can it fold a mirror-image protein?). The SEC-purified proteins were denatured for 1 h in denaturation buffer (containing 6 M GuHCl) and then diluted 100-fold into refolding buffer with or without GroEL/ES at 37 °C to initiate refolding. At specific time points, refolding was quenched by Mg chelation [1,2-diaminocyclohexanetetraacetic acid (CDTA)] followed by measurement of enzyme activity using a colorimetric assay (8). Interestingly, GroEL/ES refolded both synthetic l- and d-DapA, as demonstrated by the recovery of significant enzymatic activity (Fig. 5 and Table S1).
Fig. 5.

GroEL/ES-mediated refolding of synthetic l- and d-DapA. Refolding of recombinant (black), synthetic l- (red), and synthetic d-DapA (blue) (250 nM) in the presence (closed circles) or absence (open circles) of 7 μM GroEL/ES. Data are normalized to the maximum point in the GroEL/ES refolding of recombinant DapA.
Discussion
The results presented here demonstrate that GroEL/ES is able to fold a d-protein and therefore does not manifest strict stereospecificity in folding its substrates. This result supports a substrate binding mechanism via nonspecific hydrophobic interactions followed by sequestration in the GroEL/ES cage (9, 10). Our study also provides proof-of-concept for the use of natural (l-) GroEL/ES to fold d-proteins for mirror-image drug discovery and synthetic biology applications.
To determine if the ability of GroEL/ES to fold d-proteins is universal, the most definitive approach would be the total chemical synthesis of d-GroEL (548 residues) and d-GroES (97 residues), followed by screening of a suite of well-characterized recombinant l-substrates in refolding assays. Though we observed no difference in the activity of chemically refolded synthetic l- vs. d-DapA, there was a noticeable difference in their chaperone-mediated refolding. More detailed folding studies (49) requiring additional material will be needed to determine whether this difference reflects a general chiral preference in the recognition and/or extent/rate of folding.
Although the synthetic proteins show high specific activity (∼80% of recombinant protein; Fig. 4B), it will be important to improve their quality and yield to expand application of this work to even larger synthetic proteins. We speculate that subtle synthetic defects in our proteins include single-residue deletions, racemization (50), and aspartamide formation (51, 52).
Ultimately, the ability to chemically synthesize proteins of interest not only serves to advance mirror-image drug discovery efforts by making larger targets available, but also provides alluring possibilities for mirror-image synthetic biology (20) and complements efforts to synthesize other large biomolecules (e.g., synthetic genomes) (53). An intriguing prospect is the assembly of a mirror-image in vitro translation apparatus (including mirror-image ribosomal proteins in combination with mirror-image rRNAs; all but one of the 70S subunits are <300 residues), an effort that we have dubbed the “D. coli” project (18). Such a tool would not only provide a facile route to the production of mirror-image biomolecules for drug discovery, but would also facilitate the structural/biochemical study of highly toxic agents in (nontoxic) mirror-image form.
Materials and Methods
Peptide Synthesis and Ligation.
Peptides were synthesized via Fmoc–SPPS on a commercial peptide synthesizer (Prelude; Protein Technologies, Inc). Peptide hydrazides were prepared on 2-hydrazine chlorotrityl resin (ChemPep). Peptide hydrazides were activated in 6 M GuHCl, 100 mM sodium phosphate, pH 3.0 (5–20 mM NaNO2) at −20 °C for 20 min. Peptides were then ligated in 6 M GuHCl, 200 mM 4-mercaptophenylacetic acid (MPAA), 200 mM sodium phosphate, pH adjusted to 7.0–7.2, at 25 °C for 5–20 h. Ligation reactions were quenched by addition of freshly prepared tris(2-carboxyethyl)phosphine to ∼130 mM and incubated for >10 min.
Peptide Purification and Characterization.
Analytical reverse-phase HPLC was performed using Phenomenex Jupiter 4-μm Proteo C12 90 Å (150 × 4.6 mm) or Phenomenex Jupiter 5-μm C4 300 Å (150 × 4.6 mm) columns. Preparative reverse-phase HPLC of crude peptides was performed on either Phenomenex Jupiter 4-μm Proteo C12 90 Å (250 × 21.2 mm) or Phenomenex Jupiter 10-μm C4 300 Å (250 × 21.2 mm) column. Semipreparative reverse-phase HPLC of ligation products was performed on a Phenomenex Jupiter 10-μm C4 300 Å (250 × 10 mm) column. Purified peptides were analyzed by LC/MS on a Phenomenex Aeris WIDEPORE 3.6-μm C4 (50 × 2.1 mm) column on an AB Sciex API 3000 LC/MS/MS system. The major observed deconvoluted masses from mass spectrometry were calculated using Bayesian Peptide and Protein Reconstruct Tools in Analyst 1.5.1 Software (AB Sciex). See SI Appendix for full characterization of all peptides.
Enzyme Activity Assay.
Ten-microliter samples of DapA (250 nM) were added to 240 μL of DapA assay buffer [200 mM imidazole (pH 7.4), 35 mM Na pyruvate, 4 mM dl-aspartate-β-semialdehyde, 0.5 mg/mL o-aminobenzaldehyde, 12.5 mM CDTA] to initiate the enzyme activity assay (10 nM final enzyme concentration). The assay is quenched after 15 min of agitation at room temperature on a microplate shaker (800 rpm) by the addition of 50 μL 2 M HCl, developed by continuing the agitation for 1 h at room temperature, followed by measuring absorbance at 562 nm. Under these conditions, this assay demonstrates good linearity (A562 < 0.4 for WT recombinant DapA; saturation occurs at A562 >1.5).
Arginine-Assisted Folding.
DapA constructs (both recombinant and synthetic) were dissolved in denaturation buffer [6 M GuHCl, 20 mM MOPS (pH 7.4), 100 mM KCl, 10 mM MgCl2, 10 mM DTT] with 0.5 M arginine and diluted to final concentration of ∼37 µM. Samples were incubated at room temperature for 40 min, 13 °C for 20 min, and then dialyzed [Slide-A-Lyzer minidialysis cassettes 3500 molecular weight cutoff (MWCO)] against 100× volume of refolding buffer [20 mM MOPS (pH 7.4), 100 mM KCl, 10 mM MgCl2, 10 mM sodium pyruvate, 1 mM DTT] with 0.5 M arginine for 2.5 h at 13 °C. Samples were then further dialyzed against 100× volume 100 mM ammonium bicarbonate (pH 8) for 1 h. The dialyzed sample was used directly in functional assays (post-Arg and pre-SEC) or concentrated by Vivaspin 500 10,000 MWCO centrifugal concentrators and further purified by SEC (Superdex 200 10/30; GE Healthcare) in 100 mM ammonium bicarbonate (pH 8) running buffer with a flow rate of 0.75 mL/min (post-Arg and post-SEC). Following SEC, samples were again concentrated and prepared for structural (CD spectroscopy) and functional assays (direct activity and GroEL/ES refolding).
Chaperone Refolding Assay.
The DapA refolding assay (to evaluate GroEL/ES chaperone activity) was adapted from ref. 8. Twenty-five micromolar stocks of DapA were prepared from lyophilized powder (pre-SEC) or buffer exchanged (post-SEC) into denaturation buffer [20 mM MOPS (pH 7.4), 100 mM KCl, 10 mM MgCl2, 10 mM DTT, 6 M GuHCl] and allowed to denature for 1 h at 25 °C. Refolding was initiated by diluting 100× into 37 °C refolding buffer [20 mM MOPS (pH 7.4), 100 mM KCl, 10 mM MgCl2, 10 mM sodium pyruvate, 5 mM ATP] with or without 7 μM GroEL monomer and 7 μM GroES monomer. Final DapA concentrations used in refolding assays were 250 nM. At specific time points, 10-μL aliquots of the refolding reaction were added to 240 μL of DapA assay buffer, which simultaneously quenches chaperone-mediated refolding and initiates assay of enzyme activity (measured as described above).
Supplementary Material
Acknowledgments
We thank Costa Georgopoulos and Debbie Ang for their helpful discussions, provision of protocols, gift of our initial stock of GroEL, and critical reading of the manuscript; Rob Marquardt for his mass spectrometry advice and training; the Gary Keck laboratory for use of its ozone generator for preparing dl-aspartate-β-semialdehyde; Janet Iwasa for advice on figure design; and Debra Eckert for critical review of the manuscript. Funding for this work was provided by National Institutes of Health (NIH) Predoctoral Training Grant F31CA171677 (to M.T.J.) and NIH Grants AI076168 and AI102347 (to M.S.K.).
Footnotes
Conflict of interest statement: M.S.K. is a Scientific Director, consultant, and equity holder of the d-Peptide Research Division of Navigen, which is commercializing d-peptide inhibitors of viral entry.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1410900111/-/DCSupplemental.
References
- 1.Zawadzke LE, Berg JM. A racemic protein. J Am Chem Soc. 1992;114(10):4002–4003. [Google Scholar]
- 2.Milton RC, Milton SC, Kent SB. Total chemical synthesis of a D-enzyme: The enantiomers of HIV-1 protease show reciprocal chiral substrate specificity. Science. 1992;256(5062):1445–1448. doi: 10.1126/science.1604320. [DOI] [PubMed] [Google Scholar]
- 3.Schumacher TN, et al. Identification of D-peptide ligands through mirror-image phage display. Science. 1996;271(5257):1854–1857. doi: 10.1126/science.271.5257.1854. [DOI] [PubMed] [Google Scholar]
- 4.Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. Potent D-peptide inhibitors of HIV-1 entry. Proc Natl Acad Sci USA. 2007;104(43):16828–16833. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verzele D, Madder A. Patchwork protein chemistry: A practitioner’s treatise on the advances in synthetic peptide stitchery. ChemBioChem. 2013;14(9):1032–1048. doi: 10.1002/cbic.201200775. [DOI] [PubMed] [Google Scholar]
- 6.Mandal K, et al. Chemical synthesis and X-ray structure of a heterochiral D-protein antagonist plus vascular endothelial growth factor protein complex by racemic crystallography. Proc Natl Acad Sci USA. 2012;109(37):14779–14784. doi: 10.1073/pnas.1210483109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu M, et al. A left-handed solution to peptide inhibition of the p53-MDM2 interaction. Angew Chem Int Ed Engl. 2010;49(21):3649–3652. doi: 10.1002/anie.201000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kerner MJ, et al. Proteome-wide analysis of chaperonin-dependent protein folding in Escherichia coli. Cell. 2005;122(2):209–220. doi: 10.1016/j.cell.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 9.Horwich AL, Fenton WA. Chaperonin-mediated protein folding: using a central cavity to kinetically assist polypeptide chain folding. Q Rev Biophys. 2009;42(2):83–116. doi: 10.1017/S0033583509004764. [DOI] [PubMed] [Google Scholar]
- 10.Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem. 2013;82:323–355. doi: 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- 11.Houry WA, Frishman D, Eckerskorn C, Lottspeich F, Hartl FU. Identification of in vivo substrates of the chaperonin GroEL. Nature. 1999;402(6758):147–154. doi: 10.1038/45977. [DOI] [PubMed] [Google Scholar]
- 12.Fujiwara K, Ishihama Y, Nakahigashi K, Soga T, Taguchi H. A systematic survey of in vivo obligate chaperonin-dependent substrates. EMBO J. 2010;29(9):1552–1564. doi: 10.1038/emboj.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin Z, Schwartz FP, Eisenstein E. The hydrophobic nature of GroEL-substrate binding. J Biol Chem. 1995;270(3):1011–1014. doi: 10.1074/jbc.270.3.1011. [DOI] [PubMed] [Google Scholar]
- 14.Fenton WA, Kashi Y, Furtak K, Horwich AL. Residues in chaperonin GroEL required for polypeptide binding and release. Nature. 1994;371(6498):614–619. doi: 10.1038/371614a0. [DOI] [PubMed] [Google Scholar]
- 15.Chen L, Sigler PB. The crystal structure of a GroEL/peptide complex: Plasticity as a basis for substrate diversity. Cell. 1999;99(7):757–768. doi: 10.1016/s0092-8674(00)81673-6. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z, Feng Hp, Landry SJ, Maxwell J, Gierasch LM. Basis of substrate binding by the chaperonin GroEL. Biochemistry. 1999;38(39):12537–12546. doi: 10.1021/bi991070p. [DOI] [PubMed] [Google Scholar]
- 17.Coyle JE, Jaeger J, Gross M, Robinson CV, Radford SE. Structural and mechanistic consequences of polypeptide binding by GroEL. Fold Des. 1997;2(6):R93–R104. doi: 10.1016/S1359-0278(97)00046-1. [DOI] [PubMed] [Google Scholar]
- 18.Weinstock MT, Francis JN, Redman JS, Kay MS. Protease-resistant peptide design-empowering nature’s fragile warriors against HIV. Biopolymers. 2012;98(5):431–442. doi: 10.1002/bip.22073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Welch BD, et al. Design of a potent D-peptide HIV-1 entry inhibitor with a strong barrier to resistance. J Virol. 2010;84(21):11235–11244. doi: 10.1128/JVI.01339-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forster AC, Church GM. Synthetic biology projects in vitro. Genome Res. 2007;17(1):1–6. doi: 10.1101/gr.5776007. [DOI] [PubMed] [Google Scholar]
- 21.Church GM, Regis E. Regenesis. New York: Basic; 2012. [Google Scholar]
- 22.Mirwaldt C, Korndörfer I, Huber R. The crystal structure of dihydrodipicolinate synthase from Escherichia coli at 2.5 A resolution. J Mol Biol. 1995;246(1):227–239. doi: 10.1006/jmbi.1994.0078. [DOI] [PubMed] [Google Scholar]
- 23.Dobson RC, Gerrard JA, Pearce FG. Dihydrodipicolinate synthase is not inhibited by its substrate, (S)-aspartate beta-semialdehyde. Biochem J. 2004;377(Pt 3):757–762. doi: 10.1042/BJ20031389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blickling S, et al. Reaction mechanism of Escherichia coli dihydrodipicolinate synthase investigated by X-ray crystallography and NMR spectroscopy. Biochemistry. 1997;36(1):24–33. doi: 10.1021/bi962272d. [DOI] [PubMed] [Google Scholar]
- 25.Devenish SR, Blunt JW, Gerrard JA. NMR studies uncover alternate substrates for dihydrodipicolinate synthase and suggest that dihydrodipicolinate reductase is also a dehydratase. J Med Chem. 2010;53(12):4808–4812. doi: 10.1021/jm100349s. [DOI] [PubMed] [Google Scholar]
- 26.Yugari Y, Gilvarg C. The condensation step in diaminopimelate synthesis. J Biol Chem. 1965;240(12):4710–4716. [PubMed] [Google Scholar]
- 27.McLennan N, Masters M. GroE is vital for cell-wall synthesis. Nature. 1998;392(6672):139. doi: 10.1038/32317. [DOI] [PubMed] [Google Scholar]
- 28.Ying BW, Taguchi H, Kondo M, Ueda T. Co-translational involvement of the chaperonin GroEL in the folding of newly translated polypeptides. J Biol Chem. 2005;280(12):12035–12040. doi: 10.1074/jbc.M500364200. [DOI] [PubMed] [Google Scholar]
- 29.Tang YC, et al. Structural features of the GroEL-GroES nano-cage required for rapid folding of encapsulated protein. Cell. 2006;125(5):903–914. doi: 10.1016/j.cell.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 30.Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Synthesis of proteins by native chemical ligation. Science. 1994;266(5186):776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 31.Kent SB. Total chemical synthesis of proteins. Chem Soc Rev. 2009;38(2):338–351. doi: 10.1039/b700141j. [DOI] [PubMed] [Google Scholar]
- 32.Hackenberger CP, Schwarzer D. Chemoselective ligation and modification strategies for peptides and proteins. Angew Chem Int Ed Engl. 2008;47(52):10030–10074. doi: 10.1002/anie.200801313. [DOI] [PubMed] [Google Scholar]
- 33.Kumar KS, et al. Total chemical synthesis of a 304 amino acid K48-linked tetraubiquitin protein. Angew Chem Int Ed Engl. 2011;50(27):6137–6141. doi: 10.1002/anie.201101920. [DOI] [PubMed] [Google Scholar]
- 34.Haj-Yahya M, et al. Synthetic polyubiquitinated α-Synuclein reveals important insights into the roles of the ubiquitin chain in regulating its pathophysiology. Proc Natl Acad Sci USA. 2013;110(44):17726–17731. doi: 10.1073/pnas.1315654110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torbeev VY, Kent SB. Convergent chemical synthesis and crystal structure of a 203 amino acid “covalent dimer” HIV-1 protease enzyme molecule. Angew Chem Int Ed Engl. 2007;46(10):1667–1670. doi: 10.1002/anie.200604087. [DOI] [PubMed] [Google Scholar]
- 36.Pentelute BL, Gates ZP, Dashnau JL, Vanderkooi JM, Kent SBH. Mirror image forms of snow flea antifreeze protein prepared by total chemical synthesis have identical antifreeze activities. J Am Chem Soc. 2008;130(30):9702–9707. doi: 10.1021/ja801352j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kochendoerfer GG, et al. Design and chemical synthesis of a homogeneous polymer-modified erythropoiesis protein. Science. 2003;299(5608):884–887. doi: 10.1126/science.1079085. [DOI] [PubMed] [Google Scholar]
- 38.Wang P, et al. Erythropoietin derived by chemical synthesis. Science. 2013;342(6164):1357–1360. doi: 10.1126/science.1245095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wintermann F, Engelbrecht S. Reconstitution of the catalytic core of F-ATPase (αβ)3γ from Escherichia coli using chemically synthesized subunit γ. Angew Chem Int Ed Engl. 2013;52(4):1309–1313. doi: 10.1002/anie.201206744. [DOI] [PubMed] [Google Scholar]
- 40.Fang GM, et al. Protein chemical synthesis by ligation of peptide hydrazides. Angew Chem Int Ed Engl. 2011;50(33):7645–7649. doi: 10.1002/anie.201100996. [DOI] [PubMed] [Google Scholar]
- 41.Zheng JS, Tang S, Qi YK, Wang ZP, Liu L. Chemical synthesis of proteins using peptide hydrazides as thioester surrogates. Nat Protoc. 2013;8(12):2483–2495. doi: 10.1038/nprot.2013.152. [DOI] [PubMed] [Google Scholar]
- 42.Fang GM, Wang JX, Liu L. Convergent chemical synthesis of proteins by ligation of peptide hydrazides. Angew Chem Int Ed Engl. 2012;51(41):10347–10350. doi: 10.1002/anie.201203843. [DOI] [PubMed] [Google Scholar]
- 43.Zheng JS, Tang S, Guo Y, Chang HN, Liu L. Synthesis of cyclic peptides and cyclic proteins via ligation of peptide hydrazides. ChemBioChem. 2012;13(4):542–546. doi: 10.1002/cbic.201100580. [DOI] [PubMed] [Google Scholar]
- 44.Johnson ECB, Kent SBH. Insights into the mechanism and catalysis of the native chemical ligation reaction. J Am Chem Soc. 2006;128(20):6640–6646. doi: 10.1021/ja058344i. [DOI] [PubMed] [Google Scholar]
- 45.Hackeng TM, Griffin JH, Dawson PE. Protein synthesis by native chemical ligation: Expanded scope by using straightforward methodology. Proc Natl Acad Sci USA. 1999;96(18):10068–10073. doi: 10.1073/pnas.96.18.10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wan Q, Danishefsky SJ. Free-radical-based, specific desulfurization of cysteine: A powerful advance in the synthesis of polypeptides and glycopolypeptides. Angew Chem Int Ed Engl. 2007;46(48):9248–9252. doi: 10.1002/anie.200704195. [DOI] [PubMed] [Google Scholar]
- 47.Yan LZ, Dawson PE. Synthesis of peptides and proteins without cysteine residues by native chemical ligation combined with desulfurization. J Am Chem Soc. 2001;123(4):526–533. doi: 10.1021/ja003265m. [DOI] [PubMed] [Google Scholar]
- 48.Tsumoto K, et al. Role of arginine in protein refolding, solubilization, and purification. Biotechnol Prog. 2004;20(5):1301–1308. doi: 10.1021/bp0498793. [DOI] [PubMed] [Google Scholar]
- 49.Georgescauld F, et al. GroEL/ES chaperonin modulates the mechanism and accelerates the rate of TIM-barrel domain folding. Cell. 2014;157(4):922–934. doi: 10.1016/j.cell.2014.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benoiton NL. Chemistry of Peptide Synthesis. Boca Raton, FL: CRC; 2006. pp. 93–124. [Google Scholar]
- 51.Mergler M, Dick F, Sax B, Stähelin C, Vorherr T. The aspartimide problem in Fmoc-based SPPS. Part II. J Pept Sci. 2003;9(8):518–526. doi: 10.1002/psc.473. [DOI] [PubMed] [Google Scholar]
- 52.Mergler M, Dick F, Sax B, Weiler P, Vorherr T. The aspartimide problem in Fmoc-based SPPS. Part I. J Pept Sci. 2003;9(1):36–46. doi: 10.1002/psc.430. [DOI] [PubMed] [Google Scholar]
- 53.Gibson DG, et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science. 2010;329(5987):52–56. doi: 10.1126/science.1190719. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.