

Abstract
Cell surfaces represent a platform through which extracellular signals that determine diverse cellular processes, including migration, division, adhesion, and phagocytosis, are transduced. Techniques to rapidly reconfigure the surface properties of living cells should thus offer the ability to harness these cellular functions. Although the molecular mechanism of phagocytosis is well-characterized, the minimal molecular players that are sufficient to activate this elaborate process remain elusive. We developed and implemented a technique to present a molecule of interest at the cell surface in an inducible manner on a timescale of minutes. We simultaneously induced the cell-surface display of the C2 domain of milk fat globule-EGF factor 8 (MFG-E8) and activated the intracellular small guanosine triphosphatase Rac, which stimulates actin polymerization at the cell periphery. The C2 domain binds to phosphatidylserine, a lipid exposed on the surface of apoptotic cells. By integrating the stimulation of these two processes, we converted HeLa cells into a phagocytic cell line that bound to and engulfed apoptotic human Jurkat cells. Inducing either the cell-surface display of the C2 domain or activating Rac alone was not sufficient to stimulate phagocytosis, which suggests that attachment to the target cell and actin reorganization together constitute the minimal molecular events that are needed to induce phagocytosis. This cell-surface display technique might be useful as part of a targeted, cell-based therapy in which unwanted cells with characteristic surface molecules could be rapidly consumed by engineered cells.
Introduction
The outer surface of cells presents various biomolecules, including lipids, sugars, and proteins, which are exposed to, and potentially interact with, the extracellular environment. These surface molecules are vital to stimulate specific cellular functions, such as migration, division, adhesion, and phagocytosis (1, 2). The goal of synthetic cell biology is not only to understand the molecular mechanisms underlying these cell functions, but also to manipulate them in a predictable manner (3, 4). Rapid reengineering of cell-surface properties should enable us to achieve these goals. General methods, such as protein overexpression and RNA interference (5, 6), enable the modification of molecular constituents at the cell surface, but the effects of these techniques are often too slow to affect rapid biological events, such as adhesion and phagocytosis, for example (7). There are emerging techniques to rapidly manipulate protein constituents specifically at the surface of intracellular organelles (8–12); however, these techniques cannot be readily applied to the cell surface because of the technical challenges involved. Although direct chemical modification of the cell surface has been previously reported (13), this method is limited to the use of small molecules. Here, we developed a technique to modify the cell surface not only with small molecules but also with proteins on a timescale of minutes. We then applied this technique to investigate the minimal signaling events required for phagocytosis. Phagocytosis is a biological process through which cells engulf other cells, including bacteria. The process was first discovered by a Russian immunologist, Élie Metchnikoff, in 1895. A macrophage is one of the main phagocytes in the body, and it engulfs different types of cells, including spleen B cells, apoptotic cells, and nuclei enucleated from red blood cells. The molecular mechanisms underlying phagocytosis have been intensely studied, especially in the case of engulfment of apoptotic cells (14). Macrophages recognize target apoptotic cells through protein-lipid interactions. To bind to phosphatidylserine (PS) residues exposed on the surface of apoptotic cells, macrophages use two surface receptors: T cell immunoglobulin- and mucin domain–containing molecule (Tim4) (15) and the integrin αVβ3 bound to milk fat globule epidermal growth factor (EGF) factor 8 (MFG-E8, also known as lactadherin) (16). Overexpression of these two PS-binding proteins in non-phagocytic cells, such as NIH3T3 fibroblasts, stimulates the engulfment of apoptotic cells (17). However, signaling downstream of these two PS-binding proteins is complex, with multiple molecular players and feedback and crosstalk regulation (18). In addition, it is unclear which of these downstream machineries constitutes the minimal mechanism of phagocytosis. Here, we used our cell-surface display technique to rapidly engineer interactions between a non-phagocytic cell and an apoptotic cell, and found that concurrent Rac activation suffices to render the engineered non-phagocyte phagocytic.
Results
Design and development of dimerization-induced surface display (DISplay)
We begin by explaining the design principle for our cell-surface engineering method. A cell-surface property should be effectively altered by changing the in situ concentration of biomolecules, which can be achieved by inducing the display of biomolecules at the extracellular face of the plasma membrane that were previously confined elsewhere inside cells. However, it is challenging to mediate this type of translocation by simple molecular diffusion because of membrane boundaries between cellular compartments. Alternatively, biomolecules initially contained inside the lumen of the Golgi can be delivered to the plasma membrane and displayed at its outer surface upon binding to a protein present at secretory vesicles (19) (Fig. 1A). To test this, we used the process of chemically inducible dimerization (20), through which a chemical dimerizer, such as rapamycin, induces the formation of a complex of two different proteins, such as FK-506–binding protein (FKBP) and FKBP-rapamycin binding protein (FRB) (21).
Fig. 1. DISplay, a chemically induced, cell-surface engineering technique.
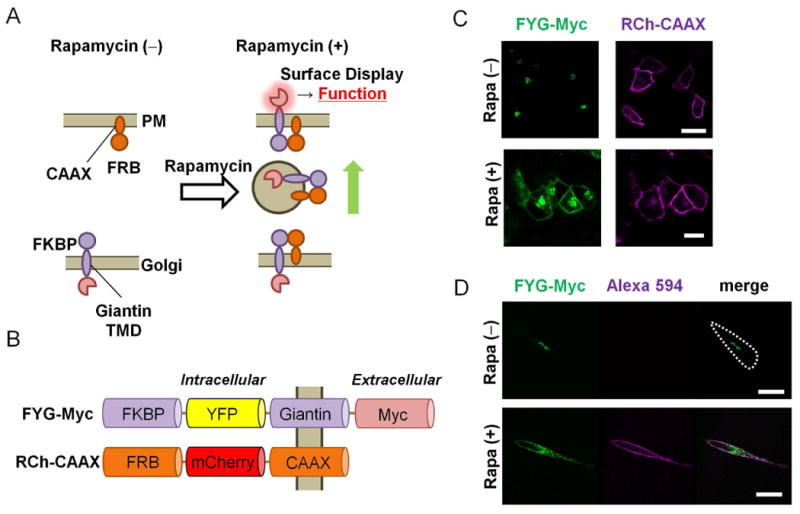
(A) Schematic presentation of the DISplay technique. Upon addition of rapamycin, a transmembrane protein at the Golgi (FKBP-fusion) traps another protein that cycles between the plasma membrane and Golgi (FRB-fusion). The dimerized complex then travels to the PM by vesicular trafficking. See the Supplementary Materials for the detailed mechanisms. (B) Schematic representation of the FKBP- and FRB-based fusion constructs (FYG-Myc and RCh-CAAX). (C) Fluorescence micrographs of HeLa cells transfected with plasmids encoding FYG-Myc (green) and RCh-CAAX (magenta). Images were captured before [Rapa (-)] and 60 min after [Rapa (+)] treatment with 200 nM rapamycin (Rapa). (D) HeLa cells transfected with plasmids encoding FYG-Myc (green) and RCh-CAAX were left untreated or were treated with rapamycin for 60 min before being incubated with an anti-Myc antibody (magenta) and analyzed by fluorescence microscopy. Scale bar: 30 μm. Images in (C) are representative of five independent experiments, and images in (D) are representative of three independent experiments.
We first prepared a series of dimerization proteins: an FKBP protein fused to one of three different Golgi-targeting sequences (Giantin, glucosylceramide synthase, and TGN38) (1, 9, 22), as well as an FRB protein fused to one of two different plasma membrane–targeting sequences, in this case, the N-terminal sequence of the kinase Lyn and C-terminal CAAX motif of K-Ras protein (23). We then introduced a pair of these FKBP and FRB fusion proteins into cells to monitor the location of FKBP proteins before and after the addition of rapamycin (fig. S1). Of all of the combinations tested, FKBP fused to yellow fluorescent protein (YFP), the transmembrane domain of Giantin, and a Myc tag (FYG-Myc), together with FRB fused to red fluorescent protein (mCherry) and the CAAX motif of K-Ras (RCh-CAAX) demonstrated translocation of FKBP proteins from the Golgi to plasma membrane after the addition of rapamycin (Fig. 1B, and Supplementary Text). The observed translocation was not a result of a side effect of rapamycin, because the addition of rapamycin to cells missing either one of the dimerization components did not lead to translocation of FKBP to the plasma membrane (fig. S2). The rapamycin-dependent accumulation of FYG-Myc at the plasma membrane was detected as early as 15 min after the addition of rapamycin (Fig. 1C), and reached a maximum within 60 min (fig. S3 and Movie S1). Subsequent immunostaining of transfected HeLa cells with antibody specific for Myc without permeabilizing cell membranes confirmed that the C-terminus of FYG-Myc was extracellularly exposed (Fig. 1D). This result is consistent with the topology of Giantin as a type II transmembrane protein (9). This dimerization strategy was readily generalized, because we also achieved equally efficient cell-surface display in experiments with different cell types, such as COS-7, NIH3T3, and MDCK cells (fig. S4), or through the use of a different chemical dimerizer, such as gibberellin and its binding proteins, gibberellin insensitive (GAI) and gibberellin insensitive dwarf 1 (GID1) (24) (fig. S5). We coined the term DISplay technique for dimerization-induced surface display.
Characterization of the DISplay mechanism
Next, we determined the translocation mechanism behind the DISplay technique. We first confirmed that the observed cell-surface display of FYG-Myc was not because of damaged organellar membranes (fig. S6). In support of the involvement of vesicular trafficking in the DISplay technique, rapamycin-dependent dimerization of FKBP and FRB resulted in the formation of intracellular vesicles (figs. S7 and S8). To test whether the conventional secretory pathway mediated the translocation process, we repeated the dimerization experiment in the presence of pharmacological inhibitors of Golgi-mediated protein transport, such as Brefeldin A (BFA) and monensin (25, 26). Whereas neither drug blocked rapamycin-induced dimerization of the fusion proteins, the cell-surface display of the fusion protein was markedly impaired (fig. S9 and S10, and Supplementary Text). We observed a similar block in membrane translocation of the fusion of FYG-Myc and RCh-CAAX when the experiment was performed at 4°C (fig. S11). In addition, the K-Ras CAAX tail was essential for the translocation of FYG-Myc to the cell surface (fig. S12). These results suggest that translocation of the heterodimerized complex through the conventional secretory pathway was at least partially involved in the display process. This was somewhat unexpected, because full-length K-Ras is reportedly delivered to the plasma membrane through an unconventional secretory pathway (27).
Presenting small molecules at the cell surface with DISplay
We next explored the ability of the DISplay technique to present small molecules at the cell surface by constructing a fusion protein of FKBP, Giantin, and a SNAP tag (FG-SNAP). The SNAP tag enables the covalent labeling of a protein of interest with O6-benzylguanine derivatives (28). After adding rapamycin to cells xpressing FG-SNAP and RCh-CAAX, we treated the cells with membrane-impermeant O6-benzylguanine conjugated to the fluorescent molecule Alexa Fluor 488 (SNAP-surface 488 (29), fig. S13A). We observed the green fluorescence of SNAP-surface 488 at the plasma membrane of cells only in the presence of rapamycin (fig. S13B), suggesting that FG-SNAP was present at the cell surface. In theory, a long list of commercially available SNAP tag molecules should be presentable at the cell surface with this approach.
Presenting a phosphatidylserine-binding domain at cell surface with DISplay
Finally, we used the DISplay technique to rapidly reconstitute a cellular function, namely phagocytosis. Although HeLa cells internalize particles when the particle size, shape, and surface chemistry meet strict criteria (30, 31), they are not professional phagocytic cells. This prompted us to explore a synthetic approach to render HeLa cells into phagocytes of apoptotic cells. Phagocytosis is composed of several processes, including recognition, binding, and tethering, as well as internalization of the target cell (17). The first step is primarily achieved by a milk fat globule–epidermal growth factor (EGF)–factor 8 (MFG-E8) (16), which binds to phosphatidylserine (PS) exposed on the surface of target apoptotic cells through its C-terminal C2 domain. Through an Arg-Gly-Asp (RGD)-motif in the EGF domain, MFG-E8 also binds to the integrin αVβ3, which is present on the surface of phagocytes, such as macrophages (18), thus serving as a bridge between the two cells. In other words, PS lipids serve as an “eat me” signal on the surface of apoptotic cells, whereas integrin-bound MFG-E8 serves as an “eat you” counterpart on the cell surface of macrophages. For engulfment of cells, this initial interaction between PS, MFG-E8, and integrin αVβ3 needs to coincide with tethering to the target cell through the plasma membrane protein Tim4 and its downstream effectors (17).
We first tested whether the interaction between PS and the C2 domain was sufficient for HeLa cells to phagocytose apoptotic cells (Fig. 2A). We constructed a fusion protein consisting of FKBP, YFP, the Giantin transmembrane domain, and the C2 domain of MFG-E8 (FYG-C2), and this was co-expressed with RCh-CAAX in transfected HeLa cells. We found that FYG-C2 was localized to Golgi in these cells, as expected (figs. S14 and S15). We also observed the translocation of FYG-C2 to the plasma membrane in response to rapamycin (figs. S14 and S15), which indicated that the C2 domain was displayed at the cell surface. We then added Jurkat cells that we had previously confirmed to be apoptotic (fig. S16). Before the addition of rapamycin, only 25% (9 out of 35 cells) HeLa cells that were transfected with plasmid encoding FYG-C2 bound to the apoptotic Jurkat cells (Fig. 2, B and C). In contrast, in the presence of rapamycin, there was a statistically significant increase in the percentage of HeLa cells that bound to the apoptotic cells (63%, 22 of 35 cells, P < 0.005 by Mann-Whitney U-test; Fig. 2, B and C, and fig. S17). When we repeated the experiment by adding non-apoptotic Jurkat cells instead of apoptotic cells, there was little binding of HeLa cells (21%, 7 of 34 cells, Fig. 2, B and C). In another control, we expressed FYG-Myc instead of FYG-C2 in the HeLa cells, and we did not observe binding of the HeLa cells to the apoptotic Jurkat cells (Fig. 2, B and C). The extent of the attachment of FYG-C2–expressing HeLa cells to apoptotic cells was comparable to that exhibited by RAW264.7 cells, a mouse macrophage cell line. In the absence of MFG-E8, 13 of 156 RAW264.7 cells (8%) bound to apoptotic Jurkat cells, whereas in the presence of MFG-E8, 48 of 109 RAW264.7 cells (44%) bound to the apoptotic cells (fig. S18). Of note, HeLa cells displaying FYG-C2 did not engulf the apoptotic Jurkat cells despite binding to the target cells, which suggests that cell-cell interactions alone were insufficient for the engulfment of target cells.
Fig. 2. Rapidly induced display of the C2 domain of MFG-E8 at the cell-surface stimulates binding to apoptotic cells.
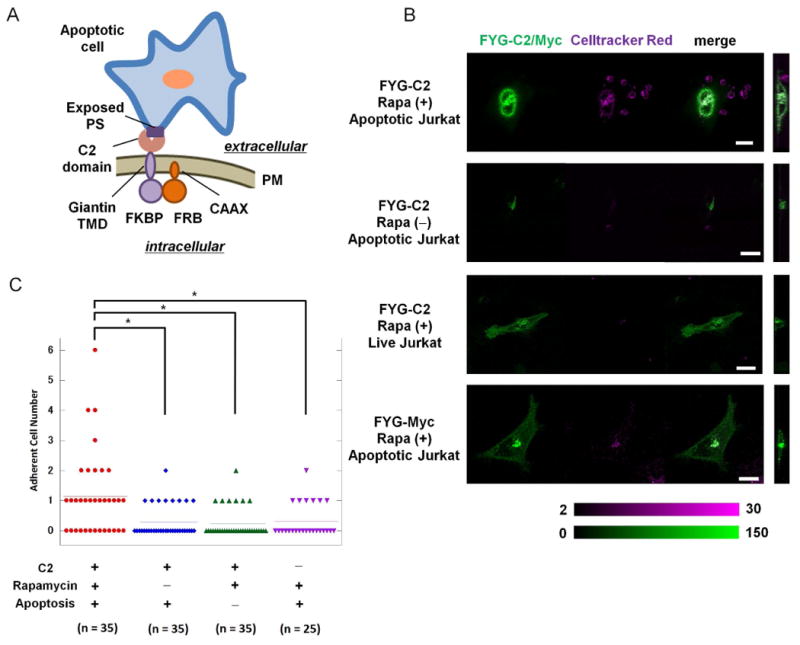
(A) Schematic presentation of an engineered HeLa cell bound to an apoptotic Jurkat cell through the interaction between the C2 domain of MFG-E8 displayed on the surface of the HeLa cell and phosphatidylserine (PS) on the surface of the Jurkat cell. (B) HeLa cells transfected with plasmid encoding RCh-CAAX and with plasmids encoding either FYG-C2 or FYG-Myc (green) were left untreated or were treated with 200 nM rapamycin for 60 min before being incubated for 3 hours with either apoptotic or live Jurkat cells labeled with CellTracker Red (magenta). Cells were then analyzed by fluorescence microscopy to examine cell-cell adherence. Color bars indicate arbitrary fluorescence intensities for YFP (green) and CellTracker Red (magenta). Scale bar: 20 μm. Data are representative of the analysis of 25 to 35 cells from two independent experiments. (C) Scatter plots from the adhesion assays described in (B). *P < 0.05 by Mann-Whitney U-test. Each of the symbols used corresponds to the indicated experimental condition. Horizontal lines indicate the average score.
Inducing cell engulfment through Rac activation
The small guanonsine triphosphatase (GTPase) Rac reorganizes the actin cytoskeleton and plays a key role in the internalization of apoptotic cells (32). Therefore, we repeated the C2 DISplay assay in the presence of active Rac. This was achieved by expressing a constitutively active form of Rac, Rac (CA), in HeLa or COS-7 cells. The effect of Rac (CA) on actin polymerization at the cell periphery was confirmed by confocal microscopy (fig. S19). The addition of rapamycin not only stimulated the formation of interactions between the HeLa or COS-7 cells and the apoptotic Jurkat cells, but also led to the engulfment of the apoptotic Jurkat cells (Fig. 3, A and B). In particular, 38% (23 of 61 cells) of the HeLa cells consumed the apoptotic cells (Fig. 3, A and B). In contract, very few of the cells that were not subjected to the DISplay technique engulfed the apoptotic cells (5 of 54 cells, P < 0.05 by Mann-Whitney U-test) (Fig. 3, A and B). Statistical analysis of the synthetic engulfment assays were summarized (Table 1 and table S1). The synthetic induction of cell engulfment was not limited to HeLa cells, because inert, generic COS-7 cells also rapidly became phagocytic when they were subjected to the DISplay procedure [18 out of 54 cells (33%), Fig. 3C, fig. S20, and tables S2 and S3]. Macrophages derived from chemically stimulated mice have previously been used for the engulfment of apoptotic Jurkat cells (33). Compared to the physiological phagocytosis by macrophages, our synthetic phagocytosis by non-phagocytes appears to be less efficient. While the difference may be the result of different experimental conditions, such as assay duration, further improvement of our synthetic approach may lead to highly efficient phagocytosis by non-phagocytes.
Fig. 3. The rapidly induced display of the C2 domain of MFG-E8 at the cell-surface coupled with the activation of intracellular Rac1 stimulates the engulfment of apoptotic cells.
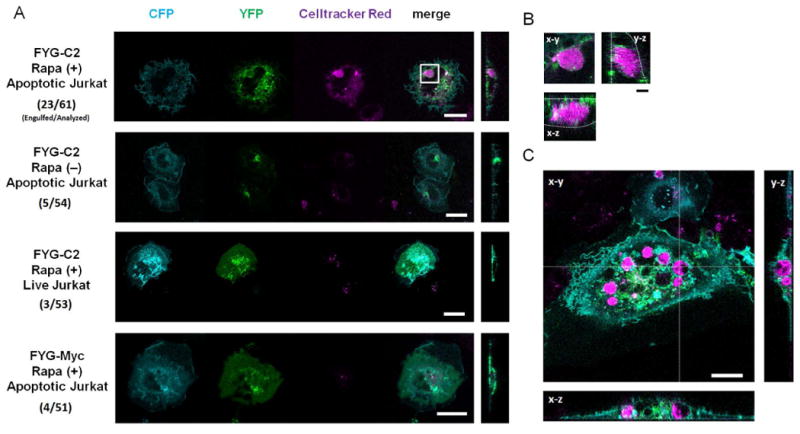
(A) HeLa cells transfected with plasmids encoding CFP-Rac1 (CA), FRB-CFP-CAAX (RCh-CAAX), and either FYG-C2 or FYG-Myc were left untreated or were treated with 200 nM rapamycin for 60 min, and then were incubated for 12 hours in the presence of CellTracker Red–labeled apoptotic or live Jurkat cells before being analyzed by fluorescence microscopy. Signals from CFP, YFP, and CellTracker Red are presented in cyan, green, and magenta, respectively. Scale bar: 20 μm. Right: micrographs are y-z plane images of the merged fluorescence. The numbers of cells engulfed versus those analyzed are shown. Statistical analysis is summarized in Table 1 and table S1. (B) Magnified view of an engulfed Jurkat cell indicated by the white square in (A). Right and bottom micrographs indicate y-z and x-z plane images, respectively. White dotted lines indicate the cell periphery of the HeLa cells. Images in (A) and (B) are representative of at least 51 cells from two independent experiments. Scale bar: 4 μm. (C) Representative fluorescence images of COS7 cells (cyan) with engulfed apoptotic cells (magenta) shown in three confocal planes (x-y, x-z, and y-z). The COS7 cells were transfected with plasmids encoding CFP-Rac1 (CA), FRB-CFP-CAAX (RCh-CAAX), and FYG-C2, treated with 200 nM rapamycin for 60 min, and incubated for 12 hours in the presence of apoptotic Jurkat cells labeled with CellTracker Red. Images in (C) are representative of 54 cells from two independent experiments. Statistical analysis is summarized in tables S2 and S3. Scale bar, 20 μm.
Table 1.
Statistical risk ratio analysis of engulfment assays in which HeLa cells were used under different conditions.
| Sample conditions | Number of cells engulfed | Number of cells analyzed | Risk1 | Risk Ratio2 C2(+), Rapa(+), apoptosis(+) | 95% CI3 | ||
|---|---|---|---|---|---|---|---|
| FYG-X | Rapa | Jurkat | |||||
| C2 (MFG-E8) | + | Apoptotic | 23 | 61 | 0.38 | – | – |
| C2 (MFG-E8) | - | Apoptotic | 5 | 54 | 0.09 | 4.07 | 1.66-9.97 |
| C2 (MFG-E8) | + | Non-apoptotic | 3 | 53 | 0.06 | 6.66 | 2.12-20.94 |
| Myc | + | Apoptotic | 4 | 51 | 0.08 | 4.81 | 1.78-13.00 |
Risk was determined by dividing the number of cells engulfed by the number of cells analyzed. The data were obtained from two independent experiments (Fig. 3A).
Risk Ratio (RR) was determined by dividing a risk value of the test condition [C2 (+), rapamycin (+), and apoptotic Jurkat (+)] by that of each control condition.
A 95% CI was determined for each control condition versus test condition. The calculation was performed based on the equation: 95% CI = exp (ln RR ± SQRT (1/a + 1/b + 1/c + 1/d), where a = the number of cells engulfed (test condition), b = the number of cells analyzed (test condition), c = the number of cells engulfed (control), d = the number of cells analyzed (control). Because the 95% CI does not include the null value (RR = 1), we concluded that the engulfment occurred with statistical significance under the exposure of the C2 domain (P < 0.05).
Discussion
Our results suggest that specific binding to target cells and actin reorganization are the two signaling events that are necessary and sufficient to stimulate the engulfment of apoptotic cells. It is thought that target cell–binding by a macrophage through two membrane proteins, MFG-E8 and Tim4, and the subsequent activation of downstream effectors of integrins concertedly form a coincidence detector of apoptotic cells (17). This system may serve as a stringent safeguard mechanism for macrophages to prevent them from mistakenly phagocytosing healthy cells. Here, we suggest that not all of these steps are necessary to induce phagocytosis by nonphagocytic cells. The next engineering challenge is to introduce a coincidence function into this synthetic system. For example, integrating DISplay with the AND logic device that executes Rac activation (24) may enable a more precise control of cellular functions rerouting.
With the DISplay technique, we rapidly manipulated the components of the cell surface and engineered a system such that inert cells could be synthetically programmed to exhibit a complex cell function such as phagocytosis. This technique thus offers a potentially useful research tool to gain insights into the causal relationships between cell-surface molecules and cell functions driven by cell-cell and cell-matrix interactions. In the future, the synthetic reconstruction of cell engulfment could be applied for targeted cell-based therapies (34). In particular, elimination of target disease cells that exhibit a characteristic molecular feature on the surface might become possible, for example, by inducing the DISplay of a single-chain Fv antibody against the surface molecule of interest.
Materials and Methods
Plasmid preparation
To generate the FKBP-YFP-Giantin-Myc construct, the complementary DNA (cDNA) encoding FY-Giantin was digested from the FY-Giantin-C1 vector (9) with NheI and BamHI, and then was inserted into the multiple cloning site of the plasmid pcDNA3.1. An oligomer encoding the Myc-tag sequence (GAACAGAAACTGATCTCTGAAGAAGACCTG) was digested with BamHI and XhoI and was inserted into the plasmid FYG-pcDNA3.1. To generate the FG-SNAP construct, a polymerase chain reaction (PCR) product encoding FKBP was digested with NheI and BsrGI and was replaced with the FKBP-YFP-encoding sequence from the plasmid FYG-SNAP, which was prepared in advance. To generate the FYG-C2 construct, PCR was performed with primers [Forward (nucleotides 898 to 928): GAATTCCTCCTGGGCTGTGAGTTGCACGGATGTTCTG; Reverse (nucleotides 1365 to 1485): CCTGCGCCTGGAGCTGCTGGGCTGTTAACTCGAGATGC] to produce a cDNA encoding the C2 domain (corresponding to amino acid residues 300 to 464) of mouse MFG-E8 (GenBank, European Molecular Biology Laboratory and DNA Data Bank of Japan: NM_008594), which was flanked with sites for the restriction enzymes BamHI and XhoI. The fragment was digested with these restriction enzymes and inserted into the plasmid FYG, which had been previously digested with the same enzymes. The plasmid encoding mouse MFG-E8 was a kind gift from Dr. S. Nagata (Kyoto University). To generate the RCh-CAAX construct, a PCR product encoding the membrane-targeting motif of the cytoplasmic tail of K-Ras was digested with BsrGI and XhoI and inserted into the multiple cloning cite of the expression plasmid pECFP-C1. The human K-Ras tail construct was previously described (23). To generate the RCh-CAAX construction, a PCR product encoding mCherry digested with AgeI and BsrGI was replaced with ECFP in the RC-CAAX vector. Construction of FKBP-glucosylceramide synthase and FKBP-TGN38 was described previously (9).
Cell culture and transfections
HeLa, COS-7, NIH3T3, MDCK, and RAW264.7 cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% supplemental fetal bovine serum (FBS, Gibco). Jurkat cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco) supplemented with 10% FBS. Cells were transiently transfected with Lipofectamine LTX (Invitrogen) according to the manufacturer's protocols.
Induction of cell-surface display by small molecules
To induce cell-surface display, cells were treated with 200 nM rapamycin or 10 μM GA3-AM diluted in DMEM containing DMSO as a solvent.
Fluorescence imaging
Imaging was performed with a confocal microscope equipped with a white light laser (TCS SP-5X, Leica). Appropriate excitation wavelengths and emission windows were chosen for the acquisition of fluorescence images in sequential scanning mode. The data in figs. S5 and S6 were acquired with an inverted microscope (Axiovert200M, Zeiss) equipped with a spinning disk confocal unit (CSU10, Yokogawa), as described previously (9). Fluorescence imaging was performed in phenol red–free DMEM containing 10% FBS under a 5% CO2 atmosphere at 37°C.
Immunostaining of the Myc tag
HeLa cells were grown on coverslips in a 6-well plate and were transiently transfected with plasmids encoding FYG-Myc and RCh-CAAX. Twelve hours later, the cells were treated with rapamycin for 60 min, and then were fixed with 4% paraformaldehyde (PFA). All cells were incubated with primary antibody (rabbit anti-myc IgG2 71D10, #2278, Cell Signaling, at a 1:200 dilution) and subsequently with secondary antibody (Alexa Fluor 594–conjugated goat anti-rabbit IgG, #A11012, Life Technologies, at a 1:400 dilution) with or without 0.2% Triton X-100 (Sigma).
SNAP-tag labeling
HeLa cells were transiently transfected with plasmids encoding FG-SNAP and RCh-CAAX. Twelve hours later, the cells were treated with rapamycin for 60 min, and cell-surface SNAP-tag was detected with SNAP-surface 488 (New England Biolabs) according to the manufacturer's protocols.
Phalloidin staining
HeLa or COS7 cells were transiently transfected with plasmid encoding CFP-Rac1(CA). Twelve hours later, cells were fixed with 4% P FA. After permeabilization with PBS containing 0.5% triton X-100 for 5 min, cellular actin was detected with Acti-stain 670 phalloidin (Cytoskeleton).
Preparation of apoptotic cells
After labeling Jurkat cells with CellTracker Red (Invitrogen) according to the manufacturer's instructions, cells were irradiated with UV light (40 mJ/cm3) with the UV Crosslinker (funakoshi), and were incubated for 3 hours in RPMI 1640 medium. Cells were then isolated by centrifugation, and adjusted to a concentration of 5 × 105 cells/ml in Hank's Balanced Salt Solution (HBSS) including calcium and magnesium (Gibco). To confirm whether apoptosis was induced, Jurkat cells were labeled with propidium iodide (PI) and Alexa Fluor 647–conjugated annexin V and then were analyzed by flow cytometry.
Adhesion assay
HeLa cells were grown on 35-mm poly-D-lysine–coated dishes (MatTek) and were transfected with plasmid encoding RCh-CAAX together with plasmids encoding either FYG-C2 or FYG-Myc. After 60 min of treatment with rapamycin, the cells were incubated for 3 hours with apoptotic Jurkat cells that were labeled with CellTracker Red (5 × 105 cells/ml). The cells were then carefully washed with HBSS with a Shake-LR (TAITEC) to remove non-adherent and weakly adherent cells. Those cells containing CellTracker Red that bordered the YFP fluorescence signal of HeLa cells in all x-y, y-z, and x-z planes were considered to be adherent cells. The numbers of adherent Jurkat cells were then determined under a confocal fluorescence microscope (TCS SP-5, Leica).
Engulfment assay
HeLa or COS-7 cells were grown on 35-mm poly-D-lysine–coated dishes (MatTek) and were transfected with plasmids encoding CFP-Rac1 (CA) and RCh-CAAX, together with plasmid encoding either FYG-C2 or FYG-Myc. After 60 min of treatment with rapamycin, apoptotic or non-apoptotic cells labeled with CellTracker Red were added and were incubated for 12 hours. After non-adherent cells were washed out three times, fluorescence images of cells that expressed all three constructs were taken. Whether Jurkat cells were engulfed was determined by searching for vesicles containing CellTracker Red that were larger than 5 μm in diameter and were located in the intracellular region (as judged from analysis of Z-stacks). After counting the cell numbers, both the risk ratio (RR) and the 95% confidence interval (CI) of the RR were calculated for each control condition versus test condition [C2 (+), rapamycin (+), and apoptotic Jurkat cells (+)]. In the analysis, statistical significance (P < 0.05) was tested by checking whether the 95% CI contained the null number (RR = 1) or not. A “risk value” was determined by dividing the number of cells engulfed by the number of cells analyzed. The RR was determined by dividing the risk of the test condition [C2 (+), rapamycin (+), and apoptotic Jurkat cells (+)] by the risk of each control condition. The 95% CI was calculated based on the equation:
where a = the number of cells engulfed (test condition), b = the number of cells analyzed (test condition), c = the number of cells engulfed (control), and d = the number of cells analyzed (control). The odds ratio (OR) and the 95% CI of the OR were calculated as described below for each control condition versus test condition. In this analysis, statistical significance (P < 0.05) was tested by checking whether the 95% CI contained the null number (RR = 1) or not. An “odd value” was determined by dividing the number of cells engulfed by the number of cells analyzed. The OR was calculated based on the equation:
The 95% CI was calculated based on the equation:
where a = the number of cells engulfed (test condition), b = the number of cells that were not engulfed (test condition), c = the number of cells engulfed (control), and d = the number of cells that were not engulfed (control).
Supplementary Material
Fig. S1. Analysis of various constructs for rapamycin-dependent dimerization.
Fig. S2. Rapamycin has no side effects on protein localization.
Fig. S3. Analysis of the dynamics and kinetics of the DISplay procedure.
Fig. S4. The DISplay can be used with different cell types.
Fig. S5. Gibberellin-based dimerization system.
Fig. S6. Analysis of the localization of Golgi-anchored proteins and the effect of rapamycin on organelles.
Fig. S7. Analysis of intracellular vesicles formed de novo after dimerization of FYG-Myc and RC-CAAX.
Fig. S8. Colocalization of FY-Giantin and VSVG protein.
Fig. S9. Localization of Golgi-anchored protein in HeLa cells treated with Brefeldin A.
Fig. S10. Localization of Golgi-anchored protein in HeLa cells treated with monensin.
Fig. S11. Localization of Golgi-anchored protein in HeLa cells at 4°C.
Fig. S12. Dimerization of Golgi-anchored protein constructs.
Fig. S13. Labeling at the cell surface with the SNAP-tag.
Fig. S14. Localization of C2-fused protein.
Fig. S15. Immunostaining of the Myc tag in cells expressing FYG-Myc-C2.
Fig. S16. Flow cytometric analysis of apoptotic Jurkat cells.
Fig. S17. Fluorescence micrographs of HeLa cells bound to apoptotic Jurkat cells.
Fig. S18. Attachment of apoptotic Jurkat cells to the surface of RAW264.7 cells.
Fig. S19. Actin structures in cells expressing Rac1 (CA).
Fig. S20. Fluorescence micrographs of COS-7 cells engulfing Jurkat cells.
Table S1. Statistical odds ratio analysis of engulfment assays with HeLa cells.
Table S2. Statistical risk ratio analysis of engulfment assays with COS-7 cells.
Table S3. Statistical odds ratio analysis of engulfment assays with COS-7 cells.
Movies 1 to 3.
References (35)
Acknowledgments
We thank C. Machamer, R. DeRose, S. C. Phua, S. Razavi, and Y. Urano for critical comments on the manuscript. We thank S. Nagata (Kyoto University) for the plasmid encoding MFG-E8.
Funding: This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (22000006 to T.N. and 24655147 to T.K.); the Japan Science and Technology Agency (10602 to T.K., 10216 to TI); and the National Institute of Health (NIH) (GM092930 to T.I.). T.K. is a recipient of a research grant from the Mochida Memorial Foundation for Medical and Pharmaceutical Research.
Footnotes
Author contributions: T. K. and T.I. conceived the experimental design; H.O., T.K., and T.I. generated the DNA constructs; H.O. and T.K. performed the cell biology experiments and analyzed the experimental data under the supervision of M.A., K.H., T.U., T.T., T.N., and T.I.; and the manuscript was written by T.K. and T.I.
Competing interests: The authors declare that they have no competing interests.
Contributor Information
Toru Komatsu, Email: tkomatsu@mol.f.u-tokyo.ac.jp.
Takanari Inoue, Email: jctinoue@jhmi.edu.
References and Notes
- 1.Hynes RO. Integrins - a family of cell-surface receptors. Cell. 1987;48:549–554. doi: 10.1016/0092-8674(87)90233-9. [DOI] [PubMed] [Google Scholar]
- 2.Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW. Epidermal growth factor receptor: mechanisms of activation and signaling. Exp Cell Res. 2003;284:31–53. doi: 10.1016/s0014-4827(02)00098-8. [DOI] [PubMed] [Google Scholar]
- 3.Galloway KE, Flanco E, Smolke CD. Dynamically reshaping signaling networks to program cell fate via genetic controllers. Science. doi: 10.1126/science.1235005. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chau AH, Walter JM, Gerardin J, Tang C, Lim WA. Designing synthetic regulatory networks capable of self-organizing cell polarization. Cell. 2012;151:320–332. doi: 10.1016/j.cell.2012.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryding AD, Sharp MG, Mullins JJ. Conditional transgenic technologies. J Endocrinol. 2001;171:1–14. doi: 10.1677/joe.0.1710001. [DOI] [PubMed] [Google Scholar]
- 6.Medema RH. Optimizing RNA interference for application in mammalian cells. Biochem J. 2004;380:593–603. doi: 10.1042/BJ20040260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stefan C, Jansen S, Bollen M. NPP-type ectophosphodiesterases: Unity in diversity. Trends Biochem Sci. 2005;30:542–550. doi: 10.1016/j.tibs.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 8.Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AG, Wandless TJ. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komatsu T, Kukelyansky I, McCaffery JM, Ueno T, Varela LC, Inoue T. Organelle-specific, rapid induction of molecular activities and membrane tethering. Nat Methods. 2010;7:206–208. doi: 10.1038/nmeth.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inoue T, Heo WD, Grimley JS, Wandless TJ, Meyer T. An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat Methods. 2005;2:415–418. doi: 10.1038/nmeth763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boncompain G, Divoux S, Gareil N, de Forges H, Lescure A, Latreche L, Mercanti V, Jollivet F, Raposo G, Perez F. Synchronization of secretory protein traffic in populations of cells. Nat Methods. 2012;9:493–U120. doi: 10.1038/nmeth.1928. [DOI] [PubMed] [Google Scholar]
- 12.Chen D, Gibson ES, Kennedy MD. A light-triggered protein secretion system. J Cell Biol. 2013;201:631–640. doi: 10.1083/jcb.201210119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saxon E, Bertozzi CR. Cell surface engineering by a modified Staudinger reaction. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 14.Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140:619–630. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 15.Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 16.Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417:182–187. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- 17.Toda S, Hanayama R, Nagata S. Two-step engulfment of apoptotic cells. Mol Cell Biol. 2012;32:118–125. doi: 10.1128/MCB.05993-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu Y, Tibrewal N, Birge RB. Phosphatidylserine recognition by phagocytes: a view to a kill. Trends Cell Biol. 2006;16:189–197. doi: 10.1016/j.tcb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 19.de Matteis MA, Luini A. Exiting the Golgi complex. Nat Rev Mol Cell Biol. 2008;9:273–284. doi: 10.1038/nrm2378. [DOI] [PubMed] [Google Scholar]
- 20.Putyrski M, Schultz C. Protein translocation as a tool: The current rapamycin story. FEBS Lett. 2012;586:2097–2105. doi: 10.1016/j.febslet.2012.04.061. [DOI] [PubMed] [Google Scholar]
- 21.Banaszynski LA, Liu CW, Wandless TJ. Characterization of the FKBP/Rapamycin/FRB ternary complex. J Am Chem Soc. 2005;127:4715–4721. doi: 10.1021/ja043277y. [DOI] [PubMed] [Google Scholar]
- 22.Munro S. An investigation of the role of transmembrane domains in Golgi protein retention. EMBO J. 1995;14:4695–4704. doi: 10.1002/j.1460-2075.1995.tb00151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fivaz M, Meyer T. Reversible intracellular translocation of KRas but not HRas in hippocampal neurons regulated by Ca2+/calmodulin. J Cell Biol. 2005;170:429–441. doi: 10.1083/jcb.200409157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miyamoto T, DeRose R, Suarez A, Ueno T, Chen M, Sun TP, Wolfgang MJ, Mukherjee C, Meyers DJ, Inoue T. Rapid and orthogonal logic gating with a gibberellin-induced dimerization system. Nat Chem Biol. 2012;8:465–470. doi: 10.1038/nchembio.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dinter A, Berger EG. Golgi-disturbing agents. Histochem Cell Biol. 1998;109:571–590. doi: 10.1007/s004180050256. [DOI] [PubMed] [Google Scholar]
- 26.Tartakoff AM. Perturbation of vesicular traffic with the carboxylic ionophore monensin. Cell. 1983;32:1026–1028. doi: 10.1016/0092-8674(83)90286-6. [DOI] [PubMed] [Google Scholar]
- 27.Apolloni A, Prior IA, Lindsay M, Parton RG, Hancock JF. H-ras but not K-ras traffics to the plasma membrane through the exocytic pathway. Mol Cell Biol. 2000;20:2475–2487. doi: 10.1128/mcb.20.7.2475-2487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keppler A, Gendreizig S, Gronemeyerm T, Pick H, Vogel H, Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21:86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 29.Vivero-Pol L, George N, Krumm H, Johnsson K, Johnsson N. Multicolor imaging of cell surface proteins. J Am Chem Soc. 2005;127:12770–12771. doi: 10.1021/ja0533850. [DOI] [PubMed] [Google Scholar]
- 30.El-Shazly S, Ahmad S, Mustafa AS, Al-Attiyah R, Krajci D. Internalization by HeLa cells of latex beads coated with mammalian cell entry (Mce) proteins encoded by the mce3 operon of Mycobacterium tuberculosis. J Med Microbiol. 2007;56:1145–1151. doi: 10.1099/jmm.0.47095-0. [DOI] [PubMed] [Google Scholar]
- 31.Gratton SEA, Ropp PA, Pohlhaus RD, Luft JC, Madden VJ, Napier ME, DeSimone JM. The effect of particle design on cellular internalization pathways. Proc Natl Acad Sci U S A. 2008;105:11613–11618. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Castellano F, Montcourrier P, Chavrier P. Membrane recruitment of Rac1 triggers phagocytosis. J Cell Sci. 2000;113:2955–2961. doi: 10.1242/jcs.113.17.2955. [DOI] [PubMed] [Google Scholar]
- 33.Nishi C, Toda S, Segawa K, Nagata S. Tim4- and MerTK- mediated engulfment of apoptotic cells by mouse resident peritoneal macrophages. Mol Cell Biol. 2014;34:1512–1520. doi: 10.1128/MCB.01394-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stasi AD, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, Liu H, Cruz CR, Savoldo B, Gee AP, Schindler J, Krance RA, Heslop HE, Spencer DM, Rooney CM, Brenner MK. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Komatsu T, Johnsson K, Okuno H, Bito H, Inoue T, Nagano T, Urano Y. Real-time measurements of protein dynamics using fluorescence activation-coupled protein labeling method. J Am Chem Soc. 2011;133:6745–6751. doi: 10.1021/ja200225m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Analysis of various constructs for rapamycin-dependent dimerization.
Fig. S2. Rapamycin has no side effects on protein localization.
Fig. S3. Analysis of the dynamics and kinetics of the DISplay procedure.
Fig. S4. The DISplay can be used with different cell types.
Fig. S5. Gibberellin-based dimerization system.
Fig. S6. Analysis of the localization of Golgi-anchored proteins and the effect of rapamycin on organelles.
Fig. S7. Analysis of intracellular vesicles formed de novo after dimerization of FYG-Myc and RC-CAAX.
Fig. S8. Colocalization of FY-Giantin and VSVG protein.
Fig. S9. Localization of Golgi-anchored protein in HeLa cells treated with Brefeldin A.
Fig. S10. Localization of Golgi-anchored protein in HeLa cells treated with monensin.
Fig. S11. Localization of Golgi-anchored protein in HeLa cells at 4°C.
Fig. S12. Dimerization of Golgi-anchored protein constructs.
Fig. S13. Labeling at the cell surface with the SNAP-tag.
Fig. S14. Localization of C2-fused protein.
Fig. S15. Immunostaining of the Myc tag in cells expressing FYG-Myc-C2.
Fig. S16. Flow cytometric analysis of apoptotic Jurkat cells.
Fig. S17. Fluorescence micrographs of HeLa cells bound to apoptotic Jurkat cells.
Fig. S18. Attachment of apoptotic Jurkat cells to the surface of RAW264.7 cells.
Fig. S19. Actin structures in cells expressing Rac1 (CA).
Fig. S20. Fluorescence micrographs of COS-7 cells engulfing Jurkat cells.
Table S1. Statistical odds ratio analysis of engulfment assays with HeLa cells.
Table S2. Statistical risk ratio analysis of engulfment assays with COS-7 cells.
Table S3. Statistical odds ratio analysis of engulfment assays with COS-7 cells.
Movies 1 to 3.
References (35)