

Summary
Epidemiological evidence links chronic bacterial infections to the increased incidence of certain types of cancer but the molecular mechanisms by which bacteria contribute to tumour initiation and progression are still poorly characterized. Here we show that chronic exposure to the genotoxin cytolethal distending toxin (CDT) of Gram-negative bacteria promotes genomic instability and acquisition of phenotypic properties of malignancy in fibroblasts and colon epithelial cells. Cells grown for more than 30 weeks in the presence of sublethal doses of CDT showed increased mutation frequency, and accumulation of chromatin and chromosomal aberrations in the absence of significant alterations of cell cycle distribution, decreased viability or senescence. Cell survival was dependent on sustained activity of the p38 MAP kinase. The ongoing genomic instability was associated with impaired activation of the DNA damage response and failure to efficiently activate cell cycle checkpoints upon exposure to genotoxic stress. Independently selected sublines showed enhanced anchorage-independent growth as assessed by the formation of colonies in semisolid agarose. These findings support the notion that chronic infection by CDT-producing bacteria may promote malignant transformation, and point to the impairment of cellular control mechanisms associated with the detection and repair of DNA damage as critical events in the process.
Introduction
Epidemiological evidence links chronic bacterial infections with the increased risk of certain human cancers. Helicobacter pylori infection is a recognized cause of gastric carcinoma, while intestinal microbiota dysbiosis and chronic infection with Salmonella enterica serovar Typhi are associated with increased risk of colon cancer and gallbladder carcinoma respectively (Dutta et al., 2000; Vogelmann and Amieva, 2007; DuPont and DuPont, 2011; Maloy and Powrie, 2011). However, very little is known about the molecular mechanisms by which these pathogens induce the genetic changes associated with tumour initiation and progression.
Chronic inflammation has been proposed as a key event in bacterial oncogenesis (Rakoff-Nahoum and Medzhitov, 2009; Honda and Littman, 2012). Bacterial products are recognized by pattern recognition receptors, such as Toll-like receptors (TLRs) (Carvalho et al., 2011), whose involvement in cancer is highlighted by the strongly reduced incidence of spontaneous colorectal carcinoma (CRC) in ApcMin/+ mice following knockdown of the common TLR transducer MyD88 (Rakoff-Nahoum and Medzhitov, 2007). Sustained TLR signalling activates the NF-κB/IL-6/STAT3 signalling cascade that orchestrates the cellular and host response to infection (Karin et al., 2006; Grivennikov et al., 2010; He and Karin, 2011). The inflammatory environment may act as a tumour initiator via induction of oxidative stress that is conducive to DNA damage (Touati, 2010), while survival and proliferation signals delivered by the activated NF-κB and STAT3 are required for tumour progression, as confirmed in in vivo models of inflammation-associated CRC and hepatocellular carcinoma (Greten et al., 2004; Pikarsky et al., 2004; Grivennikov et al., 2009; He et al., 2010).
Whether bacteria have the capacity to enhance the tumour-promoting effect of chronic inflammation via the production of genotoxins that directly induce DNA damage (Guerra et al., 2011a) is still a matter of debate. The best-characterized genotoxins are the cytolethal distending toxins (CDTs) produced by several Gram-negative bacteria, such as Escherichia coli, Aggregatibacter actinomycetemcomitans, Haemophilus ducreyi, Shigella dysenteriae, Campylobacter sp. and Helicobacter sp., and Salmonella enterica serovar Typhi that establish acute and chronic infections in the liver, oral, intestinal and genital mucosa (Guerra et al., 2011b). Active CDTs consist of three subunits: CdtA, CdtB and CdtC (Scott and Kaper, 1994; Lara-Tejero and Galan, 2001). The enzymatically active subunit, CdtB, shares structural and functional homology with mammalian deoxyribonuclease I (DNase I) (Lara-Tejero and Galan, 2000; Elwell et al., 2001; Nesic et al., 2004), while the CdtA and CdtC subunits contain ricin-like lectin domains (Nesic et al., 2004) that are involved in toxin internalization (Hassane et al., 2003; McSweeney and Dreyfus, 2004; 2005). Translocation of CdtB to the nucleus of intoxicated cells is accompanied by the induction of DNA double-strand breaks and triggering of an ATM-dependent DNA damage response (DDR) characterized by the formation of nuclear foci of phosphorylated histone 2AX (γH2AX), relocalization of the Mre11/Nbs1/Rad50 complex and activation of Chk2 and p53 (Cortes-Bratti et al., 2001; Li et al., 2002; Frisan et al., 2003). It is noteworthy that although some CDTs exhibit host specificity, members of this toxin family induce comparable DNA damage and DDR in a broad spectrum of cell lines of different tissue origin (Guerra et al., 2011b). The DDR provides an efficient barrier to tumorigenesis through induction of cell death or senescence (Bartkova et al., 2006; Halazonetis et al., 2008). However, we have earlier reported that depending on the cell type and dose of toxin, the DDR is counteracted by the concomitant triggering of survival signals mediated by activation of the guanine nucleotide exchange factor Net1, the RhoA GTPase and its downstream effector p38 MAP kinase (MAPK) (Guerra et al., 2008).
Chronic infection of mouse liver and intestine with CDT-producing Helicobacter hepaticus or Campylobacter jejuni, respectively, is associated with dysplasia (Fox et al., 2004; Ge et al., 2007), confirming the capacity of CDT-producing bacteria to induce preneoplastic lesions in vivo. However, these studies did not directly assess the role of CDTs in promoting the molecular changes that may lead to acquisition of a tumorigenic phenotype, including the type of genetic alterations associated with chronic infection and identification of the signalling pathway that promotes cell survival in vivo. To address these issues, we have monitored the occurrence of genomic instability and tumour progression in chronically intoxicated rat fibroblasts and human colon epithelial cells. We report that cells exposed to sublethal doses of the CDTs from H. hepaticus or H. ducreyi exhibit increased frequency of mutations (mainly transversions), accumulation of chromosomal aberrations and enhanced anchorage-independent growth. This effect was associated with impairment of the DDR, partial failure to activate cell cycle checkpoints in response to genotoxic stress and activation of p38 MAPK-dependent survival signals. Collectively, these data suggest that long-term exposure to CDT allows the selection of cells that have bypassed the tumorigenesis barrier, thus favouring tumour progression.
Results
Chronic intoxication with CDT induces signs of malignant transformation
The effects of chronic CDT intoxication on genome integrity and on the acquisition of malignant traits were investigated using as model Big Blue rat fibroblasts, where the occurrence of random mutations can be conveniently monitored based on the rate of functional inactivation of the cII gene encoded by the integrated Big Blue λLIZ shuttle vector (Watson et al., 1998). Since infection experiments with live bacteria are not compatible with the long-term experimental design of the study, Big Blue fibroblasts were exposed to lysates of H. hepaticus strain 3B1 that express the wild-type CDT (wtCDT) and an isogenic strain that carries a deletion of the cdtB gene (ΔCdtB) (Ge et al., 2005) as source of active and nonfunctional toxins respectively.
We first endeavoured to find a sublethal dose of the toxin that could induce minimal cellular damage without halting cell proliferation, thus allowing long-term selection. This choice was based on the observation that chronic infection with CDT-producing H. hepaticus and C. jejuni is associated with dysplasia (Fox et al., 2004; Ge et al., 2007). Thus, although the amount of toxin produced in vivo is unknown, we can safely assume that it must be sufficient to promote cell damage without causing the lethal consequence of intoxication. To this end, Big Blue fibroblasts were treated for 48 h with decreasing amounts of the active toxin or inactive control. As previously reported (Chien et al., 2000), exposure to a 1:1000 dilution of wtCDT-containing lysates induced histone 2AX phosphorylation, a sensor of DNA damage (Ciccia and Elledge, 2010) and arrest in the G2 phase of the cell cycle, while the ΔCdtB control had no effect, thus excluding non-specific toxic effects that might be associated with the use of crude bacteria lysates (Figs 1A and S1A). G2 arrest was not observed in cells exposed to 1:20 000 dilution of wtCDT but the intoxicated cells still exhibited signs of genotoxic stress, as assessed by a significant twofold increase in the frequency of micronuclei (Fig. 1A and B) that remained constant during the first 6 days of intoxication (Fig. 1C). Inactivation of the wild-type CDT by heat treatment of the lysate, assessed by abrogation of histone 2AX phosphorylation (Fig. S1A), prevented the formation of micronuclei in cells exposed to a 1:20 000 dilution (Fig. S1B), confirming the requirement of an active CDT for induction of genotoxic stress.
Fig. 1.
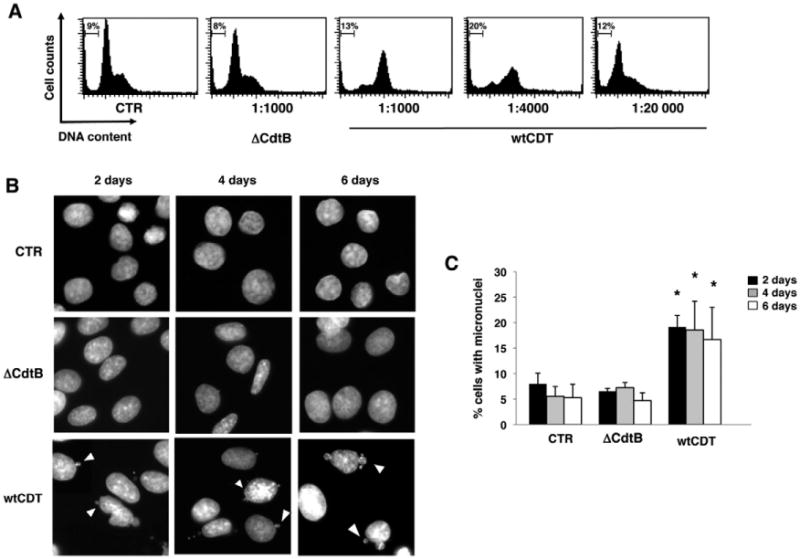
Definition of the CDT sublethal dose. A. Big Blue rat fibroblasts were left untreated (CTR), or exposed to bacteria lysates containing the inactive (ΔCdtB) or wild-type (wtCDT) toxin at the indicated dilutions for 48 h in complete medium. Cell cycle distribution was assessed by PI staining and flow cytometry analysis. Less than 10% of dead cells (sub-G1 population) were found in the untreated cultures and cultures exposed to the ΔCdtB. B. Big Blue rat fibroblasts were left untreated (CTR) or exposed to bacterial lysates containing the inactive (ΔCdtB) or the wild-type (wtCDT) toxin at a 1:20 000 dilution in complete medium, and incubated for the indicated periods of time. Nuclei were stained with DAPI, and visualized by fluorescence microscopy. Magnification 63×. Micronuclei are indicated with head arrows. C. Quantification of cells carrying micronuclei (mean ± SD of three experiments). Statistical analysis was performed using the Student's t-test.
In spite of persistent genotoxic stress, approximately 50% of the cells exposed to 1:20 000 dilution of the wtCDT-containing lysates remained viable, allowing the expansion of 14 chronically intoxicated sublines (CDT1-14) for up to 220 days. The growth properties of eight randomly selected sublines and one representative out of seven control lines, grown in the presence of the same concentration of ΔCdtB, are summarized in Fig. 2. The chronically intoxicated cultures were viable, as detected by trypan blue exclusion (not shown), and presented a cell cycle distribution comparable to that of the controls (Fig. 2A). Their proliferation rate was slightly reduced (Fig. 2B), but this did not correlate with cellular senescence as assessed by staining with β-galactosidase (Fig. 2C), a known property of senescent cells that is not found in presenescent, quiescent or immortal cells (Dimri et al., 1995). Cytogenetic analysis of seven randomly chosen wtCDT-selected sublines revealed a higher frequency of nuclear abnormalities, such as anaphase bridges, micronuclei or nuclei connected by thin chromatin threads in six sublines (Fig. 2D). Thus, the chronically intoxicated cells appeared to have overcome the growth barrier imposed by the DDR (Bartkova et al., 2006; Halazonetis et al., 2008). In order to assess whether this may correlate with the acquisition of traits of malignant transformation, a representative set of five wtCDT-selected and three control sublines were compared for their capacity for anchorage-independent growth measured in soft agar colony assays. Four of the five wtCDT-selected sub-lines formed a significantly higher number of large colonies compared to the controls (Fig. 3).
Fig. 2.
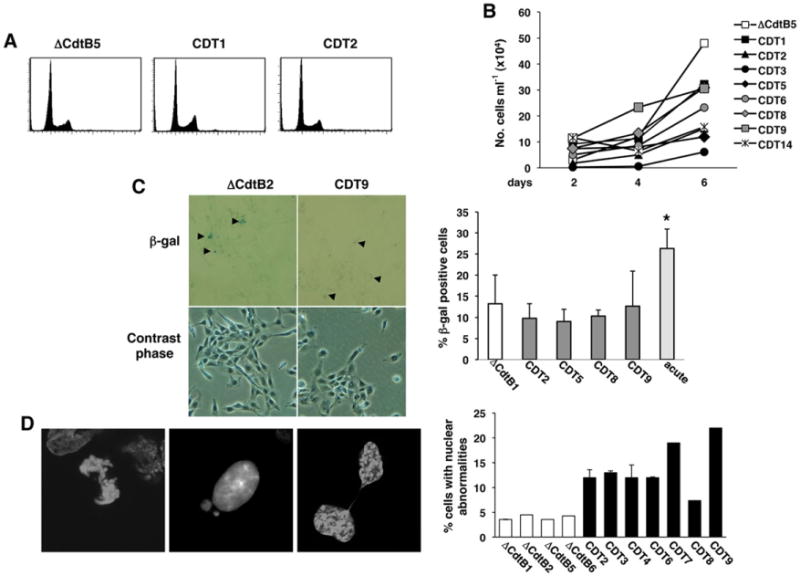
Cells carrying enhanced genomic instability present normal cell cycle distribution and do not undergo senescence. A. Cell cycle distribution of Big Blue sublines selected in the presence of the mutant (DCdtB) or wild-type (CDT) toxin for 220 days, assessed by PI staining and flow cytometry analysis. B. Growth curve of Big Blue rat fibroblasts sublines selected as described in A, and further incubated in toxin-free medium for 3 weeks prior to the assessment of the cell growth to avoid any confounding effect due to the DNA-damaging activity of the toxin. C. Detection of senescent cells. Big Blue rat fibroblasts sublines were selected as described in A. Cells were further incubated for 3 weeks in toxin-free medium prior to the β-galactosidase staining. β-Galactosidase-positive cells are indicated with black arrows in the micrographs, taken at magnification 20×. The right panel shows the quantification of β-galactosidase-positive cells. As positive control, cells were exposed to lysates containing the wtCDT at a 1:1000 dilution for 6 days (acute intoxication). Mean ± SD of three independent experiments. Statistical analysis was performed using the Student t-test. D. Nuclei of the Big Blue rat fibroblasts sublines selected with the mutant (ΔCdtB) or the wild-type (CDT) toxin for 220 days were stained with DAPI and visualized by confocal microscopy. Quantification of the number of cells carrying nuclear abnormalities is presented in the right panel (mean ± SEM of three experiments).
Fig. 3.
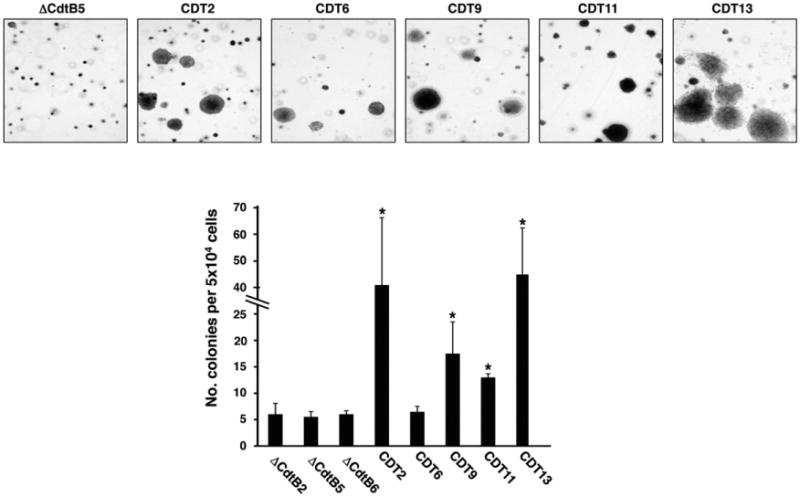
Chronic exposure to CDT promotes anchorage-independent growth. Big Blue sublines were selected with lysates of H. hepaticus containing the mutant or the wild-type toxin for 220 days. Fifty thousand cells per subline were plated in 60 mm dishes in triplicate and colonies were visualized after culture for 3 weeks by staining with crystal violet. The upper panel shows a representative micrograph of the colonies (Nikon TE300, 4 × magnification), while the quantification of the colonies number is presented in the lower panel (mean ± SEM of three experiments). Statistical analysis was performed using the Mann–Whitney test.
Chronic exposure to CDT enhances the frequency of mutations
We next tested the effect of chronic intoxication on genomic integrity. To this end, cellular DNA was extracted at the indicated times and the rate of functional inactivation of the cII gene was evaluated as schematically illustrated in Fig. 4A. The mutation frequency of the seven control sublines did not significantly change over a period of 220 days, while a time-dependent increase in the frequency of mutation was observed in 11 of 14 sublines exposed to wtCDT (Fig. 4B). Depending on the time of appearance of the mutations, the sublines could be classified into three groups: (i) sublines where a high frequency of mutations occurred early and was maintained over time (CDT2); (ii) sublines where the increased frequency of mutations was observed only after ≥ 200 days of selection (CDT3, CDT6, CDT11, CDT12 and CDT14) and (iii) unstable sublines where repeated peaks of increased mutation frequency occurred over time (CDT4, CDT5, CDT8, CDT9 and CDT13).
Fig. 4.
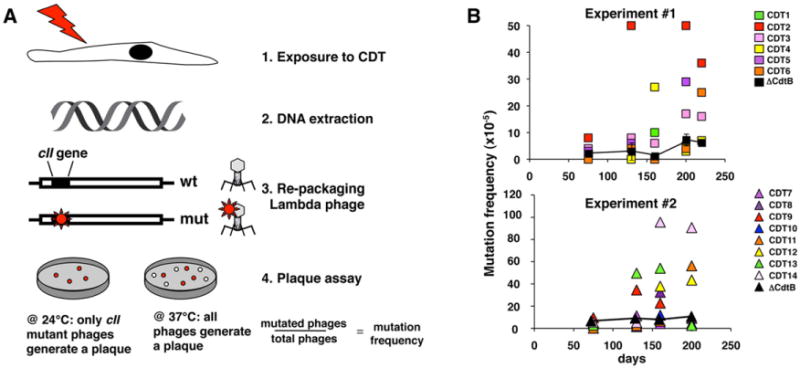
Chronic exposure to CDT enhances mutation frequency. A. Schematic illustration of the mutation analysis. The Big Blue λLIZ shuttle vector was rescued from the genomic DNA of Big Blue rat fibroblasts exposed to bacterial lysates containing the mutant (ΔCdtB) or wild-type (CDT) toxin for the indicated times. Packaged phages were used to infect the G1250 E. coli host strain and allowed to grow at 24°C to select for phages carrying inactivating mutations within the cII gene. To determine the total number of phages recovered, the infected bacteria were grown at 37°C. B. Mutation frequency of the cII gene in seven Big Blue sublines exposed to the ΔCdtB and 14 Big Blue sublines selected in the presence of wtCDT for the indicated periods of time. The sublines were generated in two independent selection processes, indicated as experiments #1 and #2. For each selection, the mutation frequency of cells exposed to the inactive CDT is presented as mean ±SD of four and three sublines respectively.
To identify the type of mutations induced by chronic intoxication, the cII gene was sequenced in phage DNA extracted from 20 plaques derived from four wtCDT-selected sublines 220 days after toxin exposure (CDT2, group I; CDT6, CDT11, CDT14, group II). These cell lines were derived from two independent experiments and presented the highest mutation frequency at the end of selection. The majority of the mutations identified (81%) were point mutations leading to amino acid substitution, while 19% of the mutations were deletion of a single nucleotide leading to a translational frameshift (Table 1). It is noteworthy that transversions (exchange of a purine for a pyrimidine or a pyrimidine for a purine) were the most common mutations observed (53%), while transitions (exchange of a purine for a purine or a pyrimidine for a pyrimidine) were less frequent.
Table 1.
Type of mutations within the cII coding sequence detected in phages rescued from the Big Blue sublines selected with wild-type CDT for 220 days.
| Phages # | Transitiona | Transversiona | Deletions | Total |
|---|---|---|---|---|
| 1 | 2 | 3 | 5 | |
| 2 | 1 | 1 | 2 | |
| 3 | 1 | 4 | 1 | 6 |
| 4 | 1 | 1 | ||
| 5 | 1 | 1 | ||
| 6 | 1 | 1 | ||
| 7 | 1 | 1 | ||
| 8 | 1 | 1 | ||
| 9 | 1 | 1 | ||
| 10 | 1 | 1 | ||
| 11 | 1 | 1 | 2 | |
| 12 | 2 | 1 | 3 | |
| 13 | 3 | 1 | 4 | |
| 14 | 1 | 1 | ||
| 15 | 1 | 1 | ||
| 16 | 1 | 1 | ||
| 17 | 1 | 1 | ||
| 18 | 1 | 1 | ||
| 19 | 1 | 1 | ||
| 20 | 1 | 1 | ||
| Total | 10 | 19 | 7 | 36 |
| Percentage | 28% | 53% | 19% |
All mutations identified are missense.
Cytolethal distending toxin intoxication selects for cells with hampered DDR and constitutive activation of p38 MAPK signalling
Genomic instability, escape from the tumorigenesis barrier and tumour progression are often associated with impairment of the DDR (Bartkova et al., 2006; Negrini et al., 2010). In order to test whether chronic CDT intoxication alters the DDR, four wtCDT sublines representative for the three groups and two control sublines were grown in toxin-free medium for 4 weeks, to avoid any confounding effect due to the DNA-damaging activity of the toxin, and then tested for their response to DNA damage induced by ionizing radiation (8 Gy) or treatment for 4 h with the topoisomerase II inhibitor etoposide (3 μM). The efficiency of the DDR was assessed by immunofluorescence analysis using a phospho-specific H2AX antibody. All sublines showed low basal level of phosphorylated histone H2AX (γH2AX) and the percentage of cell carrying γH2AX foci increased in both wtCDT-selected and control cells (Fig. 5A and B). However, the response was significantly weaker in the wtCDT-selected sublines. In line with the possibility that chronic intoxication may cause impairment of the DDR, treatment of the wtCDT-selected cells with etoposide for 24 h resulted only in a partial arrest in the G2 phase of the cell cycle, and approximately 48% of cells were still able to cycle through the G1 phase, compared to 8% in the control subline, indicating an altered checkpoint response (Fig. 5C).
Fig. 5.
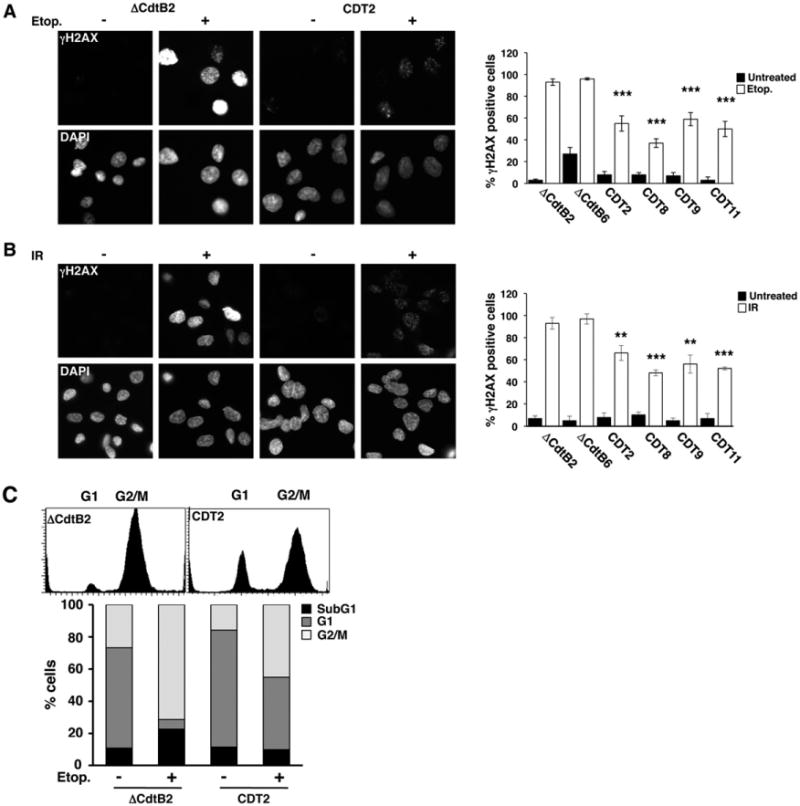
Chronic exposure to CDT results in impaired DNA damage response and DNA repair. A and B. Big Blue sublines selected with bacterial lysates of H. hepaticus containing mutant (ΔCdtB) or wild-type (CDT) toxin for 220 days were incubated for 4 weeks in toxin-free medium, and subsequently left untreated or treated with etoposide (3 μM) for 4 h (A) or irradiated (8 Gy) and further incubated in complete medium for 4 h (B). Phosphorylated H2AX (γH2AX) was detected by indirect immunofluorescence, and nuclei were counterstained with DAPI. Magnification 63×. The quantification of cells positive for γH2AX (mean ± SEM of three experiments) is shown in the right panel. Statistical analysis was performed using the Mann–Whitney test. C. The Big Blue ΔCdtB2 and CDT2 sublines were incubated for 4 weeks in toxin-free medium, and subsequently left untreated or treated with etoposide (3 μM) for 24 h in complete medium. Cell cycle distribution was assessed by PI staining and flow cytometry analysis. The lower panel shows the quantification of dead cells (sub-G1) and cells in different phases of the cell cycle. One representative experiment out of two is shown.
We have previously shown that activation of the p38 MAPK signalling pathways is a key survival factor in cells exposed to high doses of ionizing radiation or CDT (Guerra et al., 2008). We therefore asked whether this pathway is equally important for the survival and proliferation of chronically intoxicated cells. To this end, three representative control and five wtCDT-selected sublines were kept in CDT-free medium for 4 weeks, and the activation of p38 MAPK was then assessed in Western blots using a phosphor-specific antibody (Fig. 6A). Surprisingly, the total levels of p38 MAPK were consistently lower in the wtCDT-selected sublines compared to controls. However, the low levels of p38 were accompanied by a two to fivefold relative increase of the phosphorylated kinase, suggesting hyperactivation of the p38 MAPK signalling. To assess whether the active p38 MAPK is critical for the survival of chronically intoxicated cells, the kinase was blocked by treatment with 20 μM of the specific inhibitor SB203580 for 30 min followed by incubation for 72 h before assessment of cell viability. Inhibition of p38 MAPK did not affect the viability of control sublines, whereas a significant increase in the percentage of dead cells was observed in wtCDT-selected sublines (Fig. 6B).
Fig. 6.
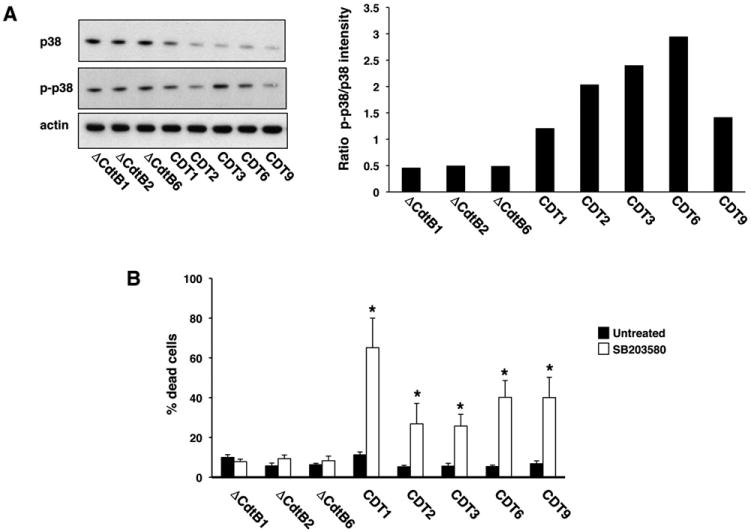
Chronically CDT-intoxicated cells depend on p38 MAPK signalling for their survival. A. Big Blue sublines were selected for 220 days with bacterial lysates of H. hepaticus containing the mutant (ΔCdtB) or the wild-type (CDT) toxin, and further kept in toxin-free medium for 4 weeks. Total cell lysates were analysed by Western blot, using p38-specific (p38) and phospho-p38-specific (p-p38) antibodies. The quantification of p-p38 levels expressed as ratio between the intensity of the phospho-p38-specific and the p38-specific bands is shown in the right panel (mean of two experiments). Actin was used as loading control. B. Big Blue sublines selected as described in A were left untreated or treated with the specific p38 MAPK inhibitor SB203580 (20 μM) for 30 min, then further incubated in complete medium for 72 h. Cell viability was assessed by trypan blue exclusion. Mean ± SD of three experiments. Statistical analysis was performed using the Student t-test.
Cytolethal distending toxin intoxication promotes genomic instability in human colon carcinoma cells
In the final set of experiments, we asked whether chronic CDT intoxication has similar effects in human cells that grow in a microenvironment that can be colonized by CDT-producing bacteria, such as the gastrointestinal tract (Guerra et al., 2011b). To this end, chronic intoxication experiments were performed with the human colon carcinoma line HCT116 (Bunz et al., 1998). Since the CDT from H. hepaticus could not be used in these experiments due to host restriction, we employed a recombinant CDT derived from H. ducreyi (rCDT) that was tested alongside an inactive holotoxin, where the CdtB subunit carries an Asp to Arg mutation in amino acid position 273 of the Mg2+ binding site (mCdtB) (Guerra et al., 2005). The sublethal dose of rCDT was identified in titration experiments (Fig. 7A). HCT116 cells exposed for 48 h to 60 ng ml−1 of rCDT showed a slight increase of cells with ≥ 4n DNA content, but the majority of the cells remained viable, as detected by trypan blue exclusion (not shown).
Fig. 7.
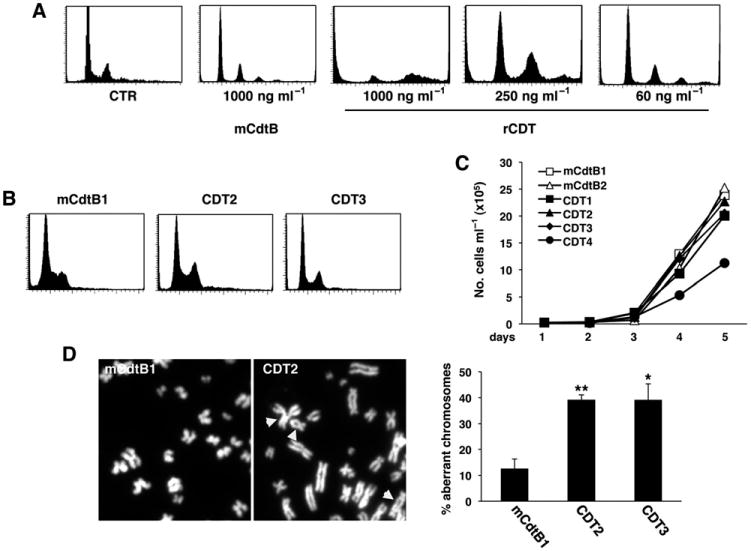
Chronic exposure to active CDT promotes genomic instability in the HCT116 colon cell line. A. The HCT116 cells were left untreated (CTR), or exposed to the inactive toxin (mCdtB) or functional CDT (rCDT) at the indicated concentrations for 48 h in complete medium. Cell cycle distribution was assessed by PI staining and flow cytometry analysis. B. Cell cycle distribution of three representative HCT116 sublines selected in the presence of mutant (mCdtB) or functional (CDT) toxin for 60 days. C. Growth curve of HCT116 sublines selected as described in B, and further incubated in toxin-free medium for 3 weeks. D. Metaphase plates of HCT116 sublines exposed to 60 ng ml−1 with the mutant (mCdtB) or the functional (CDT) toxin in complete medium for 60 days. Magnification 100×. Head arrows indicate abnormal chromosomes. The quantification of mitosis with aberrant chromosomes is shown in the right panel (mean ± SEM of three experiments). Statistical analysis was performed using the Student t-test.
Four sublines were selected by culture in the presence of 60 ng ml−1 rCDT for 60 days. The chronically intoxicated cells showed cell cycle distribution and growth rates comparable to that of control cells exposed to mCdtB (Fig. 7B and C). However, chronic intoxication was associated with a fourfold increase in the number of cells carrying chromosomal aberrations (Fig. 7D), significantly reduced capacity to accumulate γ-H2AX foci following exposure to DNA-damaging agents (Fig. 8A) and partial failure to activate the G2 checkpoint upon etoposide treatment (Fig. 8B). To note, the levels of H2AX phosphorylation in the etoposide-treated HCT116 cells were lower compared to those observed in the Big Blue fibroblasts (cp. Figs 5A and 8A). These results are in agreement with the fact that the HCT116 cells present low levels of the MRN complex (Mre11, Nbs1 and Rad50), resulting in reduced H2AX phosphorylation and Chk2 activation (Habraken et al., 2003).
Fig. 8.
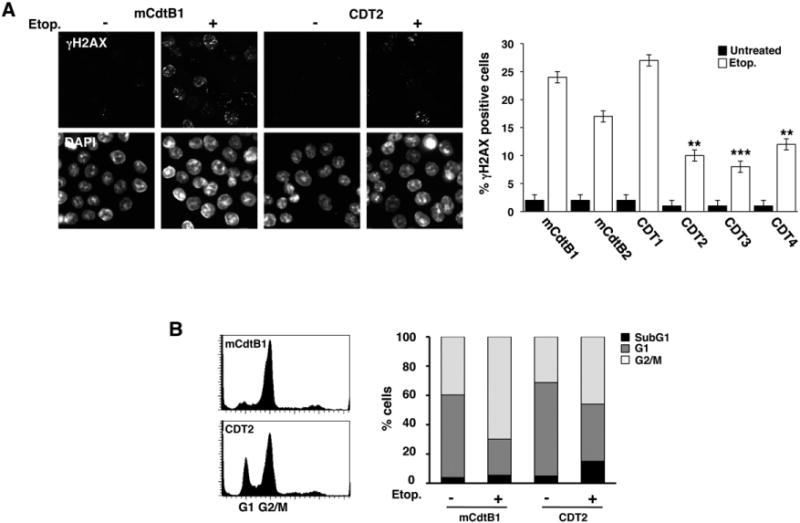
Chronic exposure to CDT results in altered DNA damage response and repair in HCT116 cells. A. HCT116 sublines selected in the presence of mutant (mCdtB) or functional (CDT) toxin for 60 days were incubated in toxin-free medium for 3 weeks and the response to genotoxic stress was assess by treatment with etoposide (3 μM) for 4 h in complete medium. γH2AX was detected by indirect immunofluorescence. The right panel shows the quantification of cells positive for γH2AX (mean ± SEM of three experiments). Statistical analysis was performed using the Mann–Whitney test. B. Cell cycle distribution assessed by PI staining and flow cytometry analysis in the HCT116 sublines mCdtB1 and CDT2 selected as described in A and then treated with etoposide (15 μM) for 24 h. The right panel shows the quantification of dead cells (sub-G1) and cells in different phases of the cell cycle. One representative experiment out of two is shown.
Collectively, these data demonstrate that chronic exposure to sublethal doses of different CDTs promotes genomic instability and altered DDR, two properties that are commonly associated with tumour progression.
Discussion
The tumour-promoting activity of the human microbiome has recently become the focus of intense research (Candela et al., 2011; Plottel and Blaser, 2011). Chronic inflammation and the production of endogenous DNA-damaging agents, such as reactive oxygen species (ROS), have been implicated as key events in microbe-associated carcinogenesis (Touati, 2010). Several Gram-negative bacteria, including E. coli and S. enterica, produce DNA-damaging CDTs that may contribute to the oncogenic process, but the mechanisms by which chronic exposure to the toxin may promote malignancy have not been assessed. Our results begin to fill this knowledge gap by demonstrating that chronic exposure to sublethal doses of CDT promotes genomic instability. This favours the selection of cells that overcome the tumorigenesis barrier imposed by the DDR, while activation of p38 MAPK-dependent survival signals allows the fixation of phenotypic properties that are commonly associated with malignant transformation and tumour progression.
Bacteria and genomic instability
Genomic instability is an enabling feature of cancer that allows the accumulation of genetic alterations driving tumour progression (Hanahan and Weinberg, 2011). This ‘mutator phenotype’ can be achieved by impairment of the defence systems that detect and repair DNA damage caused by exogenous and endogenous genotoxic agents (Loeb et al., 2008). Inactivation of the DDR may also contribute to the pathogenesis of sporadic and infection-associated cancers. Downregulation of the mismatch repair (MMR) proteins, MSH2, MSH6, MLH1, PMS2 and PMS1, has been observed in gastric cell lines exposed to H. pylori, the first bacterium recognized as human carcinogen (Kim et al., 2002). Active H. pylori infection is also associated with increased frequency of microsatellite instability in the gastric epithelium (Leung et al., 2000), and eradication of the infection by antibiotic treatment increases the levels of expression of MSH2 and MLH1 (Park et al., 2005). An approximately fourfold increase of mutation frequency has been detected in the gastric epithelium of Big Blue transgenic C57Bl/6 mice chronically infected with H. pylori (Touati et al., 2003). The majority of the mutations identified in this model are transversions that are usually associated with oxidative DNA damage (Sekiguchi and Tsuzuki, 2002), which led to the hypothesis that bacteria-induced inflammation may be the major cause of DNA damage.
Our finding that chronic exposure to CDTs increases the frequency of mutations/genomic aberrations (Figs 2, 4 and 7) and reduces the DDR capacity of the intoxicated cells (Figs 5 and 8) is in line with the synergistic effect of DNA damage and DDR impairment in malignant transformation. Interestingly, mitotic and chromosomal aberrations and a limited increase of mutations were also observed after short-term infection of mammalian cells with E. coli strains producing colibactin, the other known bacterial genotoxin (Cuevas-Ramos et al., 2010). It is noteworthy that we observed a substantial increase in the frequency of mutations and significant changes in chromosomal aberrations after more than 130 days and 60 days of continuous exposure to CDT of the Big Blue fibroblasts and HCT116 cells, respectively (Figs 4B and 7D), which emphasizes the complexity of the selection process that can be easily under- or overestimated in short-term experiments or cocultivation with live bacteria.
We found that the majority of the mutations induced by long-term exposure to the H. hepaticus CDT are transversions (Table 1). This result was unexpected since the type of DNA damage and cellular responses induced by CDT intoxication are similar to those evoked by ionizing radiation (Frisan et al., 2003) which is known to promote large-and small-scale nucleotide deletions as well as transitions and transversions in vitro and in vivo in Big Blue mice (Sankaranarayanan, 1991; Winegar et al., 1994; Nelson et al., 1996; Hoyes et al., 1998), whereas transversions are a mutagenic signature of oxidative stress and UVA radiation (Cheng et al., 1992; Kim et al., 2007; Ikehata and Ono, 2011). Since mutations were only observed in cells exposed to the active CDT, they are likely to be a direct consequence of the DNA-damaging activity of the toxin (Figs 1, 2, 4 and 7). However, part of the damage might be due to a ‘bystander effect’, similar to the radiation-like response of cells that are close to the site of irradiation. This effect is dependent on the capacity of irradiated cells to secrete cytokines such as TGF-β and IL-8 that induce ROS production in non-irradiated cells (Lorimore et al., 2003). Infection with CDT-producing H. hepaticus induces significantly higher mRNA levels of proinflammatory mediators, including TNF-α, IFN-γ and IL-6, in the liver of A/JCr mice compared to control animals infected with bacteria carrying the mutant toxin (Ge et al., 2007). Similarly, acute intoxication with purified CDTs promotes secretion of a broad array of cytokines in vitro (Guerra et al., 2011b). Based on this evidence, we measured the levels of ROS in cells exposed to sublethal doses of active or mutated CDT. Short-term exposure did not significantly influence the levels of intracellular ROS, whereas long-term exposure to the active CDT was associated with an approximately twofold increase of ROS compared to untreated control cells or cells exposed to the mutant CDT (Fig. S2). While in agreement with numerous reports indicating oxidative stress as a signature of cancer cells (Storz, 2005; de Oliveira et al., 2012), this finding excludes ROS production as a primary cause of DNA damage in CDT-intoxicated cells but points to the possible late synergistic effect on tumour progression.
The ultimate validation of our findings will be the demonstration that chronic infection with a relevant CDT-producing bacterium promotes similar levels and type of mutations in association with induction of preneoplastic or neoplastic lesions in immunocompetent animals. Based on the data presented in this study, immunohistochemistry may be used to test several markers of tumour progression. However, the in vivo analysis will not allow monitoring over time the occurrence of molecular changes that lead to the acquisition of a tumorigenic phenotype, such as characterization of the effect of CDT intoxication on the DDR, and identification of the signalling pathways that promote cell survival.
p38 MAPK in bacterial carcinogenesis
The MAP kinases play key roles in inflammation by regulating the expression and activation of proinflammatory cytokines (IL-1, IL-6 and TNFα) and their downstream targets, such as the transcription factors NF-κB and STAT3 (Kishimoto, 2005; Wagner and Nebreda, 2009; Silke, 2011). Constitutive activation of NF-κB in the intestinal mucosa of mice was shown to trigger destructive inflammation only when accompanied by the activation of p38 MAPK, following infection with the intestinal pathogen Citrobacter rodentium, or administration of the bacteria endotoxin lipopolysaccharide (LPS) (Guma et al., 2011). In addition, activation of p38 MAPK was shown to promote the survival of colon and gastric cell lines exposed to chemotherapeutic agents (Fenton et al., 2006; Gout et al., 2006; Yang et al., 2011). A pivotal role of p38 MAPK in the survival of cells exposed to genotoxic stress was also highlighted in our previous studies demonstrating the RhoA-dependent phosphorylation of p38 MAPK in HeLa cells exposed to CDT or ionizing radiation (Guerra et al., 2008). Collectively, these data suggest that p38 MAPK activation could play a dual role in bacterial carcinogenesis. First, the activation of a strong inflammatory response, causing destruction of the intestinal epithelial barrier, may allow invasion of the mucosa by the micro-flora, fuelling a procarcinogenic inflammatory process. Second, the delivery of survival signals may select for cells carrying genomic instability, contributing to overcome the tumorigenesis barrier.
We observed a reproducible decrease in the total level of the p38 MAPK in cells selected with the active holotoxin compared to controls (Fig. 6). One possible explanation for this phenomenon is that the constitutive activation of the kinase, evidenced by the high relative levels of phosphorylation, may trigger a negative feedback that results in downregulation of the protein. It is noteworthy that p38 MAPK may also exert proapoptotic functions (Wagner and Nebreda, 2009), and the total levels must therefore be tightly regulated. In spite of the decreased total levels, the selective toxicity of the SB203580 inhibitor indicates that this pathway is essential for maintaining the viability of the cells selected with the active toxin (Fig. 6).
In conclusion, our data support the possibility that chronic infection by CDT-producing bacteria may play a role in carcinogenesis and provide molecular evidence for the type of genetic and functional alterations that could be involved in the process. CDT shares several well-established features of known carcinogens including: induction of genomic instability, alteration of the DDR, breaking of the tumorigenesis barrier (lack of senescence), constitutive activation of survival signals and tumour progression (anchorage-independent growth). These end-points are similar to other carcinogenic agents that share the same mode of action of CDT (e.g. ionizing radiation); however, the type of mutations and the survival responses activated by chronic CDT exposure presented in this study are not common outcomes.
While the association of chronic infection with S. enterica and increased risk of hepatobiliary carcinoma is well established, the possible contribution of CDT-producing bacteria to other types of human cancers has not been explored. Interestingly, a high prevalence of Campylobacter species, many of which are known to produce CDTs (Pickett et al., 1996; Martinez et al., 2006; Ripabelli et al., 2010), has been detected in patients of Crohn's disease (Mahendran et al., 2011), a type of inflammatory bowel disease (IBD) that is strongly associated with progression to colorectal cancer (Feagins et al., 2009). Our findings point to the importance of comparing the expression profile of bacterial genotoxins (CDT and colibactin) in the microbiota of healthy individuals and IBD patients. In mouse models, chronic infection with CDT-positive C. jejuni is associated with severe gastritis, gastric hyperplasia and dysplasia (Fox et al., 2004). Similarly, a strong inflammatory response was observed in the liver of male A/JCr mice infected with H. hepaticus, and the presence of CDT was associated with progression towards dysplasia (Ge et al., 2007). While these studies did not address whether CDT promoted the molecular changes that led to the acquisition of a tumorigenic phenotype, they confirm that the induction of preneoplastic lesions is a common property of different members of this toxin family.
Experimental procedures
Cell lines
The Big Blue rat fibroblasts (Stratagene, La Jolla, CA, USA) are derived from a Rat2 embryonic fibroblasts transfected with Big Blue λLIZ shuttle vector. Each cell carries 50 to 70 integrated copies of the lambda shuttle vector. The human CRC cell line HCT116 was previously described (Bunz et al., 1998). The cell lines were maintained in Dulbecco's modified eagle medium (DMEM; Sigma-Aldrich, St. Louis, MO, USA) supplemented with medium 2 mM l-glutamine (Sigma-Aldrich), 10% fetal bovine serum (Invitrogen, Grand Island, NY, USA), penicillin 100 U ml−1 and streptomycin 100 mg ml−1 (Sigma-Aldrich) and 10 μg ml−1 ciprofloxacin (Sigma-Aldrich) (complete medium). For the Big Blue rat fibroblasts, the complete medium was supplemented with 200 mg ml−1 G418 sulfate (Invitrogen). Cells were kept in incubators at 37°C in a humid atmosphere of 5% CO2.
Bacteria strains
Helicobacter hepaticus strain ATCC51449, expressing the wild-type CDT, or the control strain HhcdtBm7, expressing a mutant inactive CDT (ΔCdtB), (Ge et al., 2005) was cultured on a rotary shaker to mid-logarithmic growth phase in liquid medium (Brucella broth, supplemented with 10% horse serum) and in a microaerobic atmosphere (10% hydrogen, 10% CO2, 80% nitrogen). Bacteria were harvested by centrifugation, washed twice with phosphate-buffered saline (PBS), resuspended in PBS and sonicated for 5 min in a Branson sonicator. The lysates were filtered, sterilized and stored at −20°C.
Recombinant H. ducreyi CDT
The wild-type and mutant recombinant H. ducreyi CDT subunits (CdtA, CdtB and CdtC) were constructed, expressed and purified as previously described (Guerra et al., 2005). The active (rCDT) or mutant (mCDT) holotoxin was reconstituted by incubation of equal moles of each subunit for 60 min at 37°C and stored in PBS supplemented with 20% glycerol.
Micronuclei and nuclear abnormalities assay
Fifty thousand cells were seeded on 13 mm cover slides in 24-well plates in 1 ml of complete medium and allowed to adhere for 24 h. The slides were then rinsed in PBS and fixed by incubation in ice-cold methanol (Sigma-Aldrich) for 20 min at −20°C and in ice-cold acetone for 10 s. The nuclei were stained by incubation with 1 μg ml−1 Hoechst 33342 (Sigma-Aldrich) in PBS for 20 min at room temperature. Digital images were captured using a LEITZ-BMRB fluorescence microscope (Leica, Wetzlar, Germany, Magnification 63×) equipped with a CCD camera, and analysed using the ImageJ software.
Metaphase plate analysis
Fifty thousand HCT116 cells were plated on six-well plates in 2 ml of complete medium and allowed to adhere for 48 h. Rapidly growing cells were treated with 100 ng ml−1 colcemide (Karyo-MAX Invitrogen, Paisley, UK) for 8 h to induce metaphase arrest. Cells blocked in anaphase were harvested by vigorous pipetting and cell pellets were incubated in 1 ml of hypotonic buffer containing 75 mM KCl for 1 h at 37°C. Forty-five millilitres of ice-cold fixative solution (methanol: acetic acid 3:1) was gently added to the cells that were further incubated at room temperature for 1 h. After centrifuged at 1500 g, the cell pellet was resuspended in 2 ml of fixative solution, dropped onto cold glass slides and mounted in DAPI-containing Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA). Digital images were captured by fluorescence microscope (LEITZ-BMRB; magnification 100×), and analysed using ImageJ software.
Subline selection
The Big Blue rat fibroblasts were cultured in complete medium supplemented with a 1:20 000 dilution of the bacteria lysate containing the active (wtCDT) or the non-functional (ΔCdtB) toxin. Fresh medium was replaced once a week. After approximately 4 weeks of selection, colonies were harvested using a sterile glass Pasteur pipette. The sublines were expanded individually in complete medium supplemented with the indicated lysate at 1:20 000 dilution for up to 220 days. In experiment #1 four ΔCdtB (ΔCdtB1-4) and six wtCDT (CDT1-6) sublines were selected, and in experiment #2 three ΔCdtB (ΔCdtB5-8) and eight wtCDT (CDT7-14) sublines were selected. At the indicated times, pellets of 1 × 107 cells were snap-frozen in liquid nitrogen, and kept at −70°C for extraction of the genomic DNA and the cII mutation assay. The HCT116 cells were selected with 60 ng ml−1 wild-type H. ducreyi toxin (rCDT), or with the inactive holotoxin containing the Asp to Arg mutation in the Mg2+ binding site at the amino acid position 273 of the CdtB subunit (mCdtB) (Guerra et al., 2005) for a maximum of 60 days.
cII mutation assay
DNA isolation, packaging of the phage, plating of the packaged DNA samples and determination of mutants were carried using the λ Select-cII Mutation Detection System for Big Blue Rodents (Stratagene). Briefly, the high molecular weight genomic DNA was extracted from the Big Blue rat fibroblasts using the RecoverEase DNA Isolation Kit (Stratagene). The λLIZ shuttle vector containing the cII target gene was rescued from total genomic DNA with the Transpack Packaging Extract Kit and the resulting phages plated on E. coli host strain G1250. To determine the titre of packaged phages, E. coli G1250 bacteria were mixed with 1:20 and 1:100 dilutions of phage stock, plated on TB1 agar plates and incubated overnight at 37°C (non-selective conditions). For mutant selection, the undiluted packaged phages were mixed with E. coli G1250 bacteria, plated on TB1 agar plates and incubated at 24°C for 46–48 h (selective condition). More than 300 000 rescued phages were screened for each time point and cell line. To validate the selection screening, approximately half of the phages that grew at 24°C were recovered and replated with the E. coli G1250 bacteria on selective condition (24°C). The cII mutation frequency is defined as the number of mutant plaques (determined at 24°C) divided by the total number of plaques screened (determined at 37°C).
DNA sequence analysis
The cII mutant plaques were harvested using sterile toothpicks and placed into 25 μl of H2O in microcentrifuge tubes. Samples were boiled 5 min and spun down at 10 000 g for 10 min at room temperature. Five microlitres of the supernatant were used as template for PCR amplification of the entire 294 bp cII gene according to the instructions of the manufacturer (Stratagene), using the primers: 5′-CCACACCTATGGTGTATG-3′ and 5′-CCTCTGCCGAAGTTGAGTAT-3′. Vent polymerase (New England Biolabs, Ipswich, MA, USA) was used to minimize mutations introduced by PCR.
Cell cycle analysis
Cells, harvested by trypsinization, were fixed for 20 min on ice with 1 ml of ice-cold 70% ethanol, and subsequently resuspended in 0.5 ml of propidium iodide (PI) solution (3.8 mM sodium citrate, 0.3% NP40, 0.05 mg ml−1 PI, 0.02 mg ml−1 RNase) in PBS for 2 h at 4°C. Flow cytometry analysis was performed using a FACSCanto II flow cytometer (Becton and Dickinson, Franklin Lakes, NJ, USA). Pulse processing was used to exclude cell doublets from the analysis. Data from 1 × 104 cells were collected and analysed using the FACSDiva software (Becton and Dickinson).
SB203580 treatment
Seventy-five thousand cells were plated in 12-well plates in 1 ml of complete medium and allowed to adhere for 24 h. Cells were pretreated with the p38 MAPK-specific inhibitor SB203580 (20 μM) (Calbiochem, San Diego, CA, USA) in complete medium for 30 min at 37°C, and further incubated in complete medium for 72 h. Cells were trypsinized, mixed with equal volume of trypan blue solution (Sigma-Aldrich) and counted in a Bürker chamber.
Immunofluorescence
Fifty thousand cells were seeded on 13 mm diameter slides in 24-well plates in 1 ml of complete medium and allowed to adhere for 24 h. Cells were treated with the indicated concentration of etoposide (Sigma-Aldrich) for 4 h or irradiated (8 Gy) and further incubated in complete medium for 4 h. Phosphorylated histone H2AX was detected by immunofluorescence using a specific mouse monoclonal antibody (Millipore, Billerica, MA, USA) as previously described (Guerra et al., 2005). The threshold for γH2AX positivity was calculated as the mean fluorescence intensity (MFI) of untreated cells plus two standard deviations. One hundred cells per subline were counted in each experiment.
Soft agar colony assay
Sixty-millimetre dishes were coated with a bottom layer of 0.9% low temperature melting agarose in complete medium (Sea-Plaque, FMC BioProducts, Rockland, Maine, USA) and let solidify for 30 min at room temperature. A top layer of 2 ml of 0.45% low temperature melting agarose in complete medium containing 5 × 104 cells was spread on the coated dishes and let solidify for 30 min at room temperature. Two millilitres of complete medium was added, and the dishes were then incubated at 37°C in a humid atmosphere of 5% CO2 for 3–4 weeks until colonies were visible. The medium was replaced weekly. Dishes were stained with 0.5 ml of 0.005% Crystal Violet (Sigma-Aldrich) for 2 h at room temperature and then photographed (magnification 4×) by a phase contrast light microscope (Nikon eclipse TE300) equipped with an Olympus Camera C-5060 digital camera. An area in pixel greater than 20 000 measured by the ImageJ software was used as cut-off for the definition of a colony. This value corresponds to the mean plus three standard deviations of the colonies detected in untreated Big Blue fibroblasts.
Growth curve analysis
Twenty-five thousand cells were plated on 12-well plates and cultured in normal medium. At the indicated time points, cells were trypsinized, mixed with equal volume of trypan blue solution (Sigma-Aldrich) and counted in a Bürker chamber.
Cellular senescence assay
The senescence-associated β-galactosidase staining was performed using the Senescence beta-Galactosidase Staining Kit (Cell Signaling Technology, Beverly, MA, USA) according to the manufacturer's instructions. Images were captured with a phase contrast light microscope (Nikon eclipse TE300), equipped with an Olympus C-5060 digital camera.
Western blot analysis
Proteins were fractionated by SDS-polyacrylamide gel electrophoresis using precast 4–12% gradient gels (Invitrogen), transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA) and probed with 1:1000 dilution of the indicated antibodies, followed by the appropriate horseradish peroxidase-conjugated secondary antibody (GE Healthcare, Piscataway, NJ, USA). The blots were developed by enhanced chemiluminescence (GE Healthcare) according to the instructions of the manufacturer. The following antibodies were used: the mouse monoclonal antibody to p38 MAPK and the rabbit polyclonal serum α-phospho-p38 MAPK (Cell Signaling Technology, Beverly, MA, USA), and the mouse monoclonal anti-β actin (Sigma-Aldrich).
Detection of reactive oxygen species
Reactive oxygen species levels were measured by labelling the cells for 45 min with 2′, 7′-dichlorofluorescein diacetate (DCFDA, 5 μM, Invitrogen). Cells were washed once in PBS, and flow cytometry analysis was performed using a FACS Calibur flow cytometer (Becton and Dickinson). Data from 1 × 104 cells were collected and analysed using the CellQuest software (Becton and Dickinson).
Statistical analysis
Statistical analysis was performed using spss Statistics software (IBM). Mann–Whitney test was used for significance of γH2AX and soft agar quantification. Student t-test was used to compare all other experiments. Asterisks were used to indicate different levels of significance: *P < 0.05, **P < 0.01, ***P < 0.001.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Heat inactivation abrogates the DNA-damaging activity of CDT.
A. Big Blue fibroblasts were left untreated (CTR) or exposed to the lysates containing the inactive (ΔCdtB), wild-type (wtCDT) toxin or the wild-type toxin heat inactivated at 90°C for 10 min (hwtCDT) at a 1:1000 dilution for 8 h in complete medium. Phosphorylated H2AX (γH2AX) was detected by indirect immunofluorescence (green), and nuclei were counterstained with DAPI (blue). Magnification 63×. The quantification of cells positive for γH2AX is shown in the right panel.
B. Big Blue fibroblasts were left untreated (CTR) or exposed to the lysates containing the inactive (ΔCdtB), wild-type (wtCDT) toxin or the wild-type toxin heat inactivated (hwtCDT) at a 1:20 000 dilution in complete medium, and incubated for the indicated periods of time. Nuclei were stained with DAPI, and visualized by fluorescence microscopy. Magnification 63×. Micronuclei are indicated with head arrows. The right panel shows the quantification of cells carrying micronuclei (mean ± SD of 50 cells randomly chosen per condition). Statistical analysis was performed using the Student's t-test.
Fig. S2. Chronic exposure to CDT promotes ROS production. Big Blue fibroblasts were left untreated (CTR) or exposed to bacterial lysates containing the inactive (ΔCdtB) or the wild-type (wtCDT) toxin at a 1:20 000 dilution in complete medium for the indicated periods of time. Levels of intracellular ROS were assessed by FACS analysis. The dotted line represents the mean fluorescence intensity (MFI) in untreated cells. The numbers in the histograms represent the MFI of ΔCdtB- or wtCDT-treated cells.
Supplementary Material
Acknowledgments
This work was supported by grants awarded by Swedish Research Council, the Swedish Cancer Society, the Magnus Bergvall Foundation and the Karolinska Institutet to T. F., and by the European Community Integrated Project on Infection and Cancer (INCA) (Project No. LSHC-CT-2005-018704) to M. G. M. R. G. and L. L. are supported by the Karolinska Institutet doctoral funding (KID). T. F. is a fellow of the Swedish Cancer Society.
Footnotes
Author information: The authors declare no conflict of interest.
References
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Candela M, Guidotti M, Fabbri A, Brigidi P, Franceschi C, Fiorentini C. Human intestinal microbiota: cross-talk with the host and its potential role in colorectal cancer. Crit Rev Microbiol. 2011;37:1–14. doi: 10.3109/1040841X.2010.501760. [DOI] [PubMed] [Google Scholar]
- Carvalho FA, Aitken JD, Vijay-Kumar M, Gewirtz AT. Toll-like receptor–gut microbiota interactions: perturb at your own risk! Annu Rev Physiol. 2011;74:177–198. doi: 10.1146/annurev-physiol-020911-153330. [DOI] [PubMed] [Google Scholar]
- Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G→T and A→C substitutions. J Biol Chem. 1992;267:166–172. [PubMed] [Google Scholar]
- Chien CC, Taylor NS, Ge Z, Schauer DB, Young VB, Fox JG. Identification of cdtB homologues and cytolethal distending toxin activity in enterohepatic Helicobacter spp. J Med Microbiol. 2000;49:525–534. doi: 10.1099/0022-1317-49-6-525. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Bratti X, Karlsson C, Lagergard T, Thelestam M, Frisan T. The Haemophilus ducreyi cytolethal distending toxin induces cell cycle arrest and apoptosis via the DNA damage checkpoint pathways. J Biol Chem. 2001;276:5296–5302. doi: 10.1074/jbc.M008527200. [DOI] [PubMed] [Google Scholar]
- Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrede JP. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci USA. 2010;107:11537–11542. doi: 10.1073/pnas.1001261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPont AW, DuPont HL. The intestinal microbiota and chronic disorders of the gut. Nat Rev Gastroenterol Hepatol. 2011;8:523–531. doi: 10.1038/nrgastro.2011.133. [DOI] [PubMed] [Google Scholar]
- Dutta U, Garg PK, Kumar R, Tandon RK. Typhoid carriers among patients with gallstones are at increased risk for carcinoma of the gallbladder. Am J Gastroenterol. 2000;95:784–787. doi: 10.1111/j.1572-0241.2000.01860.x. [DOI] [PubMed] [Google Scholar]
- Elwell C, Chao K, Patel K, Dreyfus L. Escherichia coli CdtB mediates cytolethal distending toxin cell cycle arrest. Infect Immun. 2001;69:3418–3422. doi: 10.1128/IAI.69.5.3418-3422.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feagins LA, Souza RF, Spechler SJ. Carcinogenesis in IBD: potential targets for the prevention of colorectal cancer. Nat Rev Gastroenterol Hepatol. 2009;6:297–305. doi: 10.1038/nrgastro.2009.44. [DOI] [PubMed] [Google Scholar]
- Fenton JI, Hursting SD, Perkins SN, Hord NG. Interleukin-6 production induced by leptin treatment promotes cell proliferation in an Apc (Min/+) colon epithelial cell line. Carcinogenesis. 2006;27:1507–1515. doi: 10.1093/carcin/bgl018. [DOI] [PubMed] [Google Scholar]
- Fox JG, Rogers AB, Whary MT, Ge Z, Taylor NS, Xu S, et al. Gastroenteritis in NF-kappaB-deficient mice is produced with wild-type Camplyobacter jejuni but not with C. jejuni lacking cytolethal distending toxin despite persistent colonization with both strains. Infect Immun. 2004;72:1116–1125. doi: 10.1128/IAI.72.2.1116-1125.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisan T, Cortes-Bratti X, Chaves-Olarte E, Stenerlöw B, Thelestam M. The Haemophilus ducreyi cytolethal distending toxin induces DNA double strand breaks and promotes ATM-dependent activation of RhoA. Cell Microbiol. 2003;5:695–707. doi: 10.1046/j.1462-5822.2003.00311.x. [DOI] [PubMed] [Google Scholar]
- Ge Z, Feng Y, Whary MT, Nambiar PR, Xu S, Ng V, et al. Cytolethal distending toxin is essential for Helicobacter hepaticus colonization in outbred Swiss Webster mice. Infect Immun. 2005;73:3559–3567. doi: 10.1128/IAI.73.6.3559-3567.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Z, Rogers AB, Feng Y, Lee A, Xu S, Taylor NS, Fox JG. Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell Microbiol. 2007;9:2070–2080. doi: 10.1111/j.1462-5822.2007.00939.x. [DOI] [PubMed] [Google Scholar]
- Gout S, Morin C, Houle F, Huot J. Death receptor-3, a new E-selectin counter-receptor that confers migration and survival advantages to colon carcinoma cells by triggering p38 and ERK MAPK activation. Cancer Res. 2006;66:9117–9124. doi: 10.1158/0008-5472.CAN-05-4605. [DOI] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra L, Teter K, Lilley BN, Stenerlow B, Holmes RK, Ploegh HL, et al. Cellular internalization of cytolethal distending toxin: a new end to a known pathway. Cell Microbiol. 2005;7:921–934. doi: 10.1111/j.1462-5822.2005.00520.x. [DOI] [PubMed] [Google Scholar]
- Guerra L, Carr HS, Richter-Dahlfors A, Masucci MG, Thelestam M, Frost JA, Frisan T. A bacterial cytotoxin identifies the RhoA exchange factor Net1 as a key effector in the response to DNA damage. PLoS ONE. 2008;3:e2254. doi: 10.1371/journal.pone.0002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra L, Guidi R, Frisan T. Do bacterial genotoxins contribute to chronic inflammation, genomic instability and tumor progression? FEBS J. 2011a;278:4577–4588. doi: 10.1111/j.1742-4658.2011.08125.x. [DOI] [PubMed] [Google Scholar]
- Guerra L, Cortes-Bratti X, Guidi R, Frisan T. The biology of the cytolethal distending toxins. Toxins (Basel) 2011b;3:172–190. doi: 10.3390/toxins3030172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guma M, Stepniak D, Shaked H, Spehlmann ME, Shenouda S, Cheroutre H, et al. Constitutive intestinal NF-kappaB does not trigger destructive inflammation unless accompanied by MAPK activation. J Exp Med. 2011;208:1889–1900. doi: 10.1084/jem.20110242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habraken Y, Jolois O, Piette J. Differential involvement of the hMRE11/hRAD50/NBS1 complex, BRCA1 and MLH1 in NF-kappaB activation by camptothecin and X-ray. Oncogene. 2003;22:6090–6099. doi: 10.1038/sj.onc.1206893. [DOI] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hassane DC, Lee RB, Pickett CL. Campylobacter jejuni cytolethal distending toxin promotes DNA repair responses in normal human cells. Infect Immun. 2003;71:541–545. doi: 10.1128/IAI.71.1.541-545.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Karin M. NF-kappaB and STAT3 – key players in liver inflammation and cancer. Cell Res. 2011;21:159–168. doi: 10.1038/cr.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Yu GY, Temkin V, Ogata H, Kuntzen C, Sakurai T, et al. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell. 2010;17:286–297. doi: 10.1016/j.ccr.2009.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Littman DR. The microbiome in infectious disease and inflammation. Annu Rev Immunol. 2012;30:759–795. doi: 10.1146/annurev-immunol-020711-074937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyes KP, Wadeson PJ, Sharma HL, Hendry JH, Morris ID. Mutation studies in lacI transgenic mice after exposure to radiation or cyclophosphamide. Mutagenesis. 1998;13:607–612. doi: 10.1093/mutage/13.6.607. [DOI] [PubMed] [Google Scholar]
- Ikehata H, Ono T. The mechanisms of UV mutagenesis. J Radiat Res. 2011;52:115–125. doi: 10.1269/jrr.10175. [DOI] [PubMed] [Google Scholar]
- Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–835. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Tao H, Carloni E, Leung WK, Graham DY, Sepulveda AR. Helicobacter pylori impairs DNA mismatch repair in gastric epithelial cells. Gastroenterology. 2002;123:542–553. doi: 10.1053/gast.2002.34751. [DOI] [PubMed] [Google Scholar]
- Kim SI, Pfeifer GP, Besaratinia A. Mutagenicity of ultraviolet A radiation in the lacI transgene in Big Blue mouse embryonic fibroblasts. Mutat Res. 2007;617:71–78. doi: 10.1016/j.mrfmmm.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto T. Interleukin-6: from basic science to medicine – 40 years in immunology. Annu Rev Immunol. 2005;23:1–21. doi: 10.1146/annurev.immunol.23.021704.115806. [DOI] [PubMed] [Google Scholar]
- Lara-Tejero M, Galan JE. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science. 2000;290:354–357. doi: 10.1126/science.290.5490.354. [DOI] [PubMed] [Google Scholar]
- Lara-Tejero M, Galan JE. CdtA, CdtB and CdtC form a tripartite complex that is required for cytolethal distending toxin activity. Infect Immun. 2001;69:4358–4365. doi: 10.1128/IAI.69.7.4358-4365.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung WK, Kim JJ, Kim JG, Graham DY, Sepulveda AR. Microsatellite instability in gastric intestinal metaplasia in patients with and without gastric cancer. Am J Pathol. 2000;156:537–543. doi: 10.1016/S0002-9440(10)64758-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Sharipo A, Chaves-Olarte E, Masucci MG, Levitsky V, Thelestam M, Frisan T. The Haemophilus ducreyi cytolethal distending toxin activates sensors of DNA damage and repair complexes in proliferating and non-proliferating cells. Cell Microbiol. 2002;4:87–99. doi: 10.1046/j.1462-5822.2002.00174.x. [DOI] [PubMed] [Google Scholar]
- Loeb LA, Bielas JH, Beckman RA. Cancers exhibit a mutator phenotype: clinical implications. Cancer Res. 2008;68:3551–3557. doi: 10.1158/0008-5472.CAN-07-5835. discussion 3557. [DOI] [PubMed] [Google Scholar]
- Lorimore SA, Coates PJ, Wright EG. Radiation-induced genomic instability and bystander effects: inter-related nontargeted effects of exposure to ionizing radiation. Oncogene. 2003;22:7058–7069. doi: 10.1038/sj.onc.1207044. [DOI] [PubMed] [Google Scholar]
- McSweeney LA, Dreyfus LA. Nuclear localization of the Escherichia coli cytolethal distending toxin CdtB subunit. Cell Microbiol. 2004;6:447–458. doi: 10.1111/j.1462-5822.2004.00373.x. [DOI] [PubMed] [Google Scholar]
- McSweeney LA, Dreyfus LA. Carbohydrate-binding specificity of the Escherichia coli cytolethal distending toxin CdtA-II and CdtC-II subunits. Infect Immun. 2005;73:2051–2060. doi: 10.1128/IAI.73.4.2051-2060.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahendran V, Riordan SM, Grimm MC, Tran TA, Major J, Kaakoush NO, et al. Prevalence of Campylobacter species in adult Crohn's disease and the preferential colonization sites of Campylobacter species in the human intestine. PLoS ONE. 2011;6:e25417. doi: 10.1371/journal.pone.0025417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- Martinez I, Mateo E, Churruca E, Girbau C, Alonso R, Fernandez-Astorga A. Detection of cdtA, cdtB, and cdtC genes in Campylobacter jejuni by multiplex PCR. Int J Med Microbiol. 2006;296:45–48. doi: 10.1016/j.ijmm.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability – an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- Nelson SL, Parks KK, Grosovsky AJ. Ionizing radiation signature mutations in human cell mutants induced by low-dose exposures. Mutagenesis. 1996;11:275–279. doi: 10.1093/mutage/11.3.275. [DOI] [PubMed] [Google Scholar]
- Nesic D, Hsu Y, Stebbins CE. Assembly and function of a bacterial genotoxin. Nature. 2004;429:429–433. doi: 10.1038/nature02532. [DOI] [PubMed] [Google Scholar]
- de Oliveira MF, Amoedo ND, Rumjanek FD. Energy and redox homeostasis in tumor cells. Int J Cell Biol. 2012;2012:593838. doi: 10.1155/2012/593838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DI, Park SH, Kim SH, Kim JW, Cho YK, Kim HJ, et al. Effect of Helicobacter pylori infection on the expression of DNA mismatch repair protein. Helicobacter. 2005;10:179–184. doi: 10.1111/j.1523-5378.2005.00309.x. [DOI] [PubMed] [Google Scholar]
- Pickett CL, Pesci EC, Cottle DL, Russell G, Erdem AN, Zeytin H. Prevalence of cytolethal distending toxin production in Campylobacter jejuni and relatedness of Campylobacter sp. cdtB gene. Infect Immun. 1996;64:2070–2078. doi: 10.1128/iai.64.6.2070-2078.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- Plottel CS, Blaser MJ. Microbiome and malignancy. Cell Host Microbe. 2011;10:324–335. doi: 10.1016/j.chom.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317:124–127. doi: 10.1126/science.1140488. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer. 2009;9:57–63. doi: 10.1038/nrc2541. [DOI] [PubMed] [Google Scholar]
- Ripabelli G, Tamburro M, Minelli F, Leone A, Sammarco ML. Prevalence of virulence-associated genes and cytolethal distending toxin production in Campylobacter spp. isolated in Italy. Comp Immunol Microbiol Infect Dis. 2010;33:355–364. doi: 10.1016/j.cimid.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan K. Ionizing radiation and genetic risks. III. Nature of spontaneous and radiation-induced mutations in mammalian in vitro systems and mechanisms of induction of mutations by radiation. Mutat Res. 1991;258:75–97. doi: 10.1016/0165-1110(91)90029-u. [DOI] [PubMed] [Google Scholar]
- Scott DA, Kaper JB. Cloning and sequencing of the genes encoding Escherichia coli cytolethal distending toxins. Infect Immun. 1994;62:244–251. doi: 10.1128/iai.62.1.244-251.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi M, Tsuzuki T. Oxidative nucleotide damage: consequences and prevention. Oncogene. 2002;21:8895–8904. doi: 10.1038/sj.onc.1206023. [DOI] [PubMed] [Google Scholar]
- Silke J. The regulation of TNF signalling: what a tangled web we weave. Curr Opin Immunol. 2011;23:620–626. doi: 10.1016/j.coi.2011.08.002. [DOI] [PubMed] [Google Scholar]
- Storz P. Reactive oxygen species in tumor progression. Front Biosci. 2005;10:1881–1896. doi: 10.2741/1667. [DOI] [PubMed] [Google Scholar]
- Touati E. When bacteria become mutagenic and carcinogenic: lessons from H. pylori. Mutat Res. 2010;703:66–70. doi: 10.1016/j.mrgentox.2010.07.014. [DOI] [PubMed] [Google Scholar]
- Touati E, Michel V, Thiberge JM, Wuscher N, Huerre M, Labigne A. Chronic Helicobacter pylori infections induce gastric mutations in mice. Gastroenterology. 2003;124:1408–1419. doi: 10.1016/s0016-5085(03)00266-x. [DOI] [PubMed] [Google Scholar]
- Vogelmann R, Amieva MR. The role of bacterial pathogens in cancer. Curr Opin Microbiol. 2007;10:76–81. doi: 10.1016/j.mib.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- Watson DE, Cunningham ML, Tindall KR. Spontaneous and ENU-induced mutation spectra at the cII locus in Big Blue Rat2 embryonic fibroblasts. Mutagenesis. 1998;13:487–497. doi: 10.1093/mutage/13.5.487. [DOI] [PubMed] [Google Scholar]
- Winegar RA, Lutze LH, Hamer JD, O'Loughlin KG, Mirsalis JC. Radiation-induced point mutations, deletions and micronuclei in lacI transgenic mice. Mutat Res. 1994;307:479–487. doi: 10.1016/0027-5107(94)90258-5. [DOI] [PubMed] [Google Scholar]
- Yang SY, Miah A, Sales KM, Fuller B, Seifalian AM, Winslet M. Inhibition of the p38 MAPK pathway sensitises human colon cancer cells to 5-fluorouracil treatment. Int J Oncol. 2011;38:1695–1702. doi: 10.3892/ijo.2011.982. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.