

Abstract
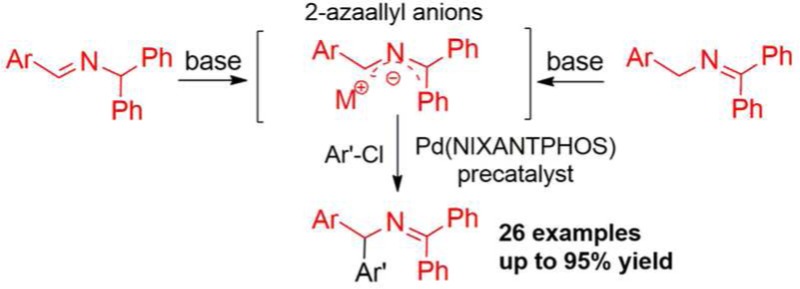
A regioselective arylation of 1,1,3-triaryl-2-azaallyl anions with aryl chlorides is described. The palladium-NIXANTPHOS-based catalyst affords diarylmethylamine derivatives in good yield and without product isomerization. A gram scale sequential one-pot ketimine synthesis/arylation protocol was also developed.
Diarylmethylamines are important structural motifs in medicinal chemistry and are present in several medications.1 Their synthesis has, therefore, attracted much attention. One strategy for the synthesis of diarylmethylamines, and related amines, relies on generation and reactions of 2-azaallyl anions. 2-Azaallyl anions have been shown to be viable substrates for palladium-catalyzed cross-coupling reactions with aryl halides (Scheme 1), although only limited success has been reported with more readily available aryl chloride coupling partners.2
Scheme 1. Previous Reports of Arylation of 2-Azaallyl Anions with Aryl Halides and Triflates.

In 2008 and 2009, Oshima and co-workers2a,2b reported pioneering studies on coupling aryl chlorides with N-benzyl ketimine derivatives via in situ deprotonation with CsOH at 140 °C. Unfortunately, the high reaction temperature caused isomerization of the product in every case (Scheme 1a and b). Earlier this year, Buchwald and Zhu2c reported an elegant enantioselective arylation of 9-aminofluorene-derived aldimines with aryl halides at room temperature (Scheme 1c). Although the reaction worked very well for aryl bromides, chlorobenzene underwent coupling in only 38% yield. Very recently, we described the in situ generation and regioselective arylation of 1,1,3-triaryl-2-azaallyl anions, from ketimine and aldimine precursors (Scheme 2), with aryl bromides.2d In this study we identified that a palladium complex3 of van Leeuwen’s bidentate NIXANTPHOS,4 together with a hindered base [NaN(SiMe3)2], facilitated arylation under mild conditions and eliminated product isomerization.
Scheme 2. Arylation of 2-Azaallyl Anions Using a Pd(NIXANTPHOS)-Based Catalyst.
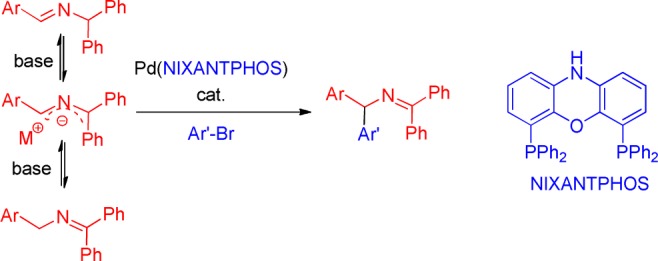
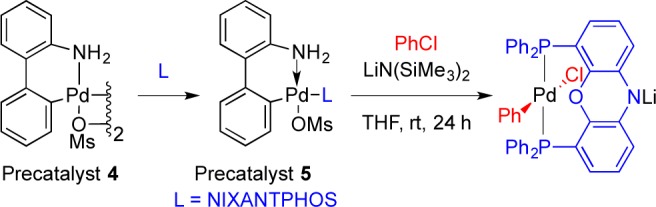
In our initial report, we avoided aryl chloride substrates, because it is well-known that oxidative addition of aryl chlorides to palladium complexes with bidentate phosphines requires temperatures of ∼100 °C, at which temperature isomerization of the type observed by Oshima (Scheme 1a,b) would likely be problematic. However, we subsequently discovered that the Pd(NIXANTPHOS)-based catalyst will oxidatively add aryl chlorides at room temperature (Scheme 3).5 Room temperature oxidative addition of aryl chlorides by palladium generally occurs with bulky monodentate phosphine complexes (PCy3, PtBu3, Q-Phos, Buchwald’s phosphines, etc.),6 because the mechanism proceeds via a Pd–L1 intermediate.7
Scheme 3. Oxidative Addition Using NIXANTPHOS Precatalysts 4 and 5 at 23 °C.
The discovery that our NIXANTPHOS-based palladium catalyst oxidatively adds to aryl chlorides5 at rt inspired us to revisit the arylation of 2-azaallyl anions with more abundant and economical aryl chlorides. Herein, we report the first highly regioselective arylation of 1,1,3-triaryl-2-azaallyl anions with aryl chlorides.
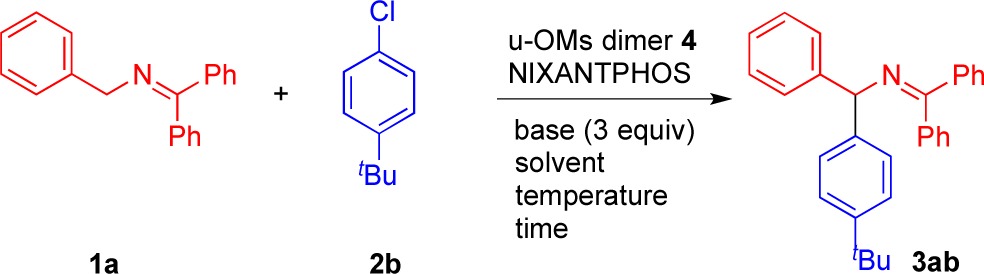
Based on our previous reported arylation with bromoarenes,2d we initiated our studies with a 2:1 ratio of ketimine 1a and aryl chloride 2b, 3 equiv of LiN(SiMe3)2, 5 mol % Buchwald’s μ-OMs dimer 4,3 and 10 mol % NIXANTPHOS. The addition of base was done portionwise to minimize the concentration of the azaallyl anion, which reacts with the imine to give byproducts.2d We tested five solvents [CPME (cyclopentyl methyl ether), THF, toluene, DME (dimethoxyethane), and DCE (1,2-dichloroethane)] at 23 °C (Table 1, entries 1–5). THF was the best of these with a 68% assay yield (Table 1, entry 2). With THF as solvent, a screen of bases MN(SiMe3)2 (M = Na K, Li, entries 6–8) indicated that the reaction with LiN(SiMe3)2 outperformed the others. This result was consistent with our oxidative addition studies (Scheme 3),5 but in contrast to the reactions with aryl bromides where NaN(SiMe3)2 was optimal.2d Raising the temperature to 60 °C resulted in an increase in the yield with shorter reaction times (Table 1, entries 8 and 9). Reducing the catalyst loading from 10 to 5 mol % at 60 °C resulted in a slight drop in the yield from 92% to 86% (Table 1, entry 10). Optimization of the arylation at 23 °C led to generation of 3ab in 81% yield with 10 mol % NIXANTPHOS precatalyst 5 in 14 h (Table 1, entry 11).
Table 1. Optimization of Arylation of Ketimine 1a with Aryl Chloride 2ba,b.
| entry | 4/L (mol %) | base | solv | temp (°C) | time (h) | yield (%) |
|---|---|---|---|---|---|---|
| 1 | 5/10 | LiN(SiMe3)2 | CPME | 23 | 12 | N.R. |
| 2 | 5/10 | LiN(SiMe3)2 | THF | 23 | 12 | 68 |
| 3 | 5/10 | LiN(SiMe3)2 | Tol. | 23 | 12 | N.R. |
| 4 | 5/10 | LiN(SiMe3)2 | DME | 23 | 12 | 62 |
| 5 | 5/10 | LiN(SiMe3)2 | DCE | 23 | 12 | N.R. |
| 6 | 5/10 | NaN(SiMe3)2 | THF | 23 | 12 | 50 |
| 7 | 5/10 | KN(SiMe3)2 | THF | 23 | 12 | 37 |
| 8 | 5/10 | LiN(SiMe3)2 | THF | 60 | 12 | 92 |
| 9 | 5/10 | LiN(SiMe3)2 | THF | 60 | 6 | 92 |
| 10 | 2.5/5 | LiN(SiMe3)2 | THF | 60 | 6 | 86 |
| 11 | 10c | LiN(SiMe3)2 | THF | 23 | 14 | 83(81)d |
Reactions conducted on a 0.1 mmol scale using 2 equiv of ketimine, 3 equiv of LiN(SiMe3)2, and 1 equiv of ArCl at 0.1 M. Base was added portionwise with speed 0.05 mL/30 min. N.R. is no reaction.
Yield determined by 1H NMR spectroscopy of the crude reaction mixture on a 0.1 mmol scale.
NIXANTPHOS precatalyst 5, base was added portionwise with speed 0.1 mL/30 min.
Isolated yield after chromatographic purification.
The scope of the aryl chloride–ketamine cross-coupling was determined (Table 2) at 60 °C (5 mol % cat.). Chlorobenzene gave 3aa in 90% yield. Aryl chlorides bearing alkyl groups (4-tBu 2b, 3-Me, 2c) provided products 3ab and 3ac in 86% and 77% yield, respectively. Electron-rich 4-chloroanisole (2d) was coupled with 1a in 60% yield at a 10 mol % catalyst loading. Aryl chlorides with electron-withdrawing 4-F, 4-CF3, and 3-CN groups were also well tolerated, providing products in 77% (3ae), 87% (3af), and 91% (3ag) yield, respectively. It is notable that the base-sensitive nitrile-bearing substrate gave an excellent yield. 3-Chloro-N,N-dimethylaniline reacted with 1a in 61% yield (3ah) at a 10 mol % catalyst loading. With little drop in the yields, coupling at 23 °C with a 10 mol % palladium loading (under conditions of Table 1, entry 11) was also successfully employed, affording electronically diverse products 3aa, 3ab, 3ae, 3af in 71%–85% yield (entries 9–12).
Table 2. Scope of Aryl Chlorides in the Arylation of Ketimine 1aa,b.

| entry | Ar | product | Pd (mol %) | temp (°C) | yield (%) |
|---|---|---|---|---|---|
| 1 | Ph | 3aa | 5 | 60 | 90 |
| 2 | 4-tBu-C6H4 | 3ab | 5 | 60 | 86 |
| 3 | 3-Me-C6H4 | 3ac | 5 | 60 | 77 |
| 4 | 4-OMe-C6H4 | 3ad | 10 | 60 | 60 |
| 5 | 4-F-C6H4 | 3ae | 5 | 60 | 77 |
| 6 | 4-CF3-C6H4 | 3af | 5 | 60 | 87 |
| 7 | 3-NC-C6H4 | 3ag | 10 | 60 | 91c |
| 8 | 3-Me2N-C6H4 | 3ah | 10 | 60 | 61 |
| 9 | Ph | 3aa | 10 | 23 | 83d |
| 10 | 4-tBu-C6H4 | 3ab | 10 | 23 | 81d |
| 11 | 4-F-C6H4 | 3ae | 10 | 23 | 71d |
| 12 | 4-CF3-C6H4 | 3af | 10 | 23 | 85d |
Reactions conducted on a 0.1 mmol scale using 2 equiv of 1a, 3 equiv of LiN(SiMe3)2, and 1 equiv of ArCl at 0.1 M. Base was added portionwise with speed 0.05 mL/30 min.
Isolated yield after chromatographic purification.
DME as solvent.
NIXANTPHOS precatalyst 5, base was added portionwise with speed 0.1 mL/30 min, 14 h.
Encouraged by the results of the ketimine arylation with aryl chlorides, we next investigated translation of the optimized conditions from Table 1 to the arylation of aldimines (Table 3). Aldimine substrates have the advantage that there are many commercially available aldehyde precursors. Coupling reactions at 60 °C with a 5 mol % catalyst loading and 4-tert-butyl chloro benzene (2b) gave 3ab in 91% yield (Table 3, entry 1). Chlorobenzene (2a) and alkyl substituted 4-methyl chlorobenzene (2i) gave product 3aa and 3ai in 95% and 86% yield, respectively (entries 2–3). Coupling of 1a′ with 4-trifluoromethyl chlorobenzene 2f provided 3af in 85% yield (entry 4). 4-Fluoro chlorobenzene reacted smoothly with aldimine 1a′ to give the product 3ae in 84% yield (entry 5). 1-Chloro-3,5-difluorobenzene (2j, entry 6) coupled in 81% yield at a 10 mol % catalyst loading. Coupling of aldimine 1a′ with 4-tert-butyl chlorobenzene (2b) at 23 °C gave 3ab in 85% yield at a 10 mol % catalyst loading, which is similar to the ketimine isomer (Table 2, entry 10, 81% yield). In a similar fashion, syntheses of 3aa, 3ai, and 3af were also conducted at 23 °C in 84%, 86%, and 89% yield (entries 8–10), respectively.
Table 3. Scope of Aryl Chlorides in the Arylation of Aldimine 1a′a,b.
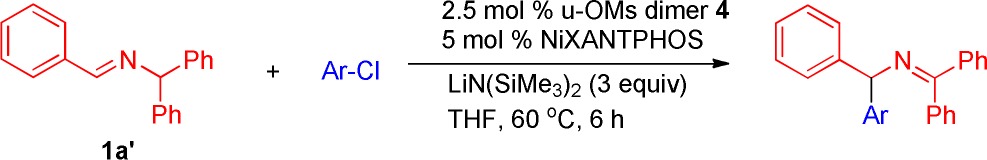
| entry | Ar | prod | Pd (mol %) | temp (°C) | yield (%) |
|---|---|---|---|---|---|
| 1 | 4-tBu-C6H4 | 3ab | 5 | 60 | 91 |
| 2 | Ph | 3aa | 5 | 60 | 95 |
| 3 | 4-Me-C6H4 | 3ai | 5 | 60 | 86 |
| 4 | 4-CF-C6H4 | 3af | 5 | 60 | 85 |
| 5 | 4-F-C6H4 | 3ae | 5 | 60 | 84 |
| 6 | 3,5-F2-C6H3 | 3aj | 10 | 60 | 81 |
| 7 | 4-tBu-C6H4 | 3ab | 10 | 23 | 85c |
| 8 | Ph | 3aa | 10 | 23 | 84c |
| 9 | 4-Me-C6H4 | 3ai | 10 | 23 | 86c |
| 10 | 4-CF3-C6H4 | 3af | 10 | 23 | 89c |
Reactions conducted on a 0.1 mmol scale using 2 equiv of 1a′, 3 equiv of LiN(SiMe3)2, and 1 equiv of ArCl at 0.1 M. Base was added portionwise with speed 0.05 mL/30 min.
Isolated yield after chromatographic purification.
NIXANTPHOS precatalyst 5, base was added portionwise with speed 0.1 mL/30 min, 14 h.
The substrate scope of ketimines with different substituents on the N-benzyl group was then studied (Table 4). The reaction of the 4-methyl benzyl ketimine derivative (1b) proceeded efficiently under a 10 mol % loading in 86% yield (entry 1). Coupling of the 3,5-difluoro benzyl ketimine derivative (1c) furnished the product in excellent yield (88%, entry 3) at a 5 mol % catalyst loading. Heterocyclic 3-pyridyl and 4-pyridyl ketimine derivatives coupled with chlorobenzene (2a) in 60% (3da) and 91% (3ea) yield. The 2-furyl ketimine derivative led to a satisfactory yield of product 3fa (61% yield). It is noteworthy that no substrate in Tables 2–4 exhibited product isomerization.
Table 4. Scope of Ketimine in Arylation of Chlorobenzenea.

| entry | Ar | product | yield (%) |
|---|---|---|---|
| 1 | 4-Me-C6H4 | 3ai | 86 |
| 2 | 3,5-F2-C6H3 | 3aj | 88 |
| 3 | 3-Py | 3da | 60 |
| 4 | 4-Py | 3ea | 91 |
| 5 | 2-furyl | 3fa | 61 |
Reactions conducted on a 0.1 mmol scale using 2 equiv of ketimines, 3 equiv of LiN(SiMe3)2, and 1 equiv of PhCl at 0.1 M. Base was added portionwise with speed 0.05 mL/30 min.
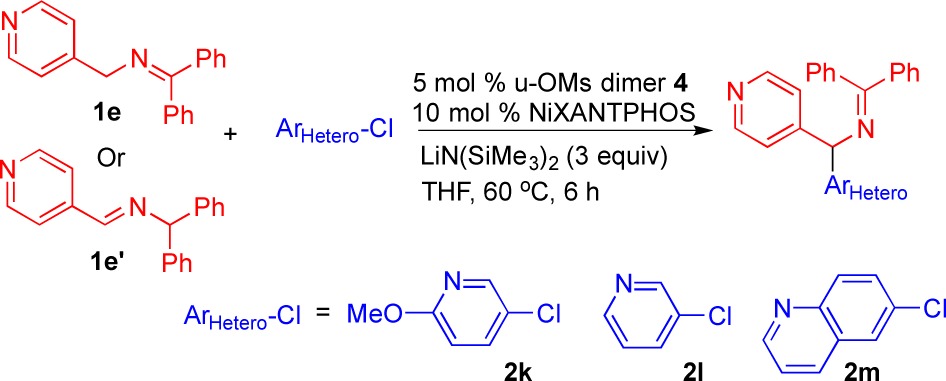
Synthesis of bis-heterocyclic diarylmethylamine derivatives via coupling of 4-pyridyl ketimine and aldimine with heterocyclic aryl chlorides was next investigated. Good-to-excellent yields were observed with ketimine and aldimine isomers. At a 10 mol % catalyst loading, electron-rich 5-chloro-2-methoxypyridine (2k) exhibited good reactivity with 3ek generated in 71% yield from ketimine 1e and 70% yield from aldimine 1e′ (Table 5, entries 1–2). Employing 3-chloropyridine (2l) led to a 78% yield with ketamine 1e and an 82% yield with aldimine 1e′ (Table 5, entries 3–4). Excellent yields were achieved with 6-chloroquinoline (2m) at a 5 mol % catalyst loading. Product 3em was obtained in 90% yield from both couplings with 1e and 1e′ (Table 5, entries 5–6).
Table 5. Bis-heterocyclic Diarylmethylamine Derivativea,b.
| entry | imine | ArHetero–Cl | prod. | Pd (mol %) | yield (%) |
|---|---|---|---|---|---|
| 1 | 1e | 2k | 3ek | 10 | 70 |
| 2 | 1e′ | 2k | 3ek | 10 | 71 |
| 3 | 1e | 2l | 3el | 10 | 78 |
| 4 | 1e′ | 2l | 3el | 10 | 82 |
| 5 | 1e | 2m | 3em | 5 | 90 |
| 6 | 1e′ | 2m | 3em | 5 | 90 |
Reactions conducted on a 0.1 mmol scale using 2 equiv of imine, 3 equiv of LiN(SiMe3)2, and 1 equiv of ArCl at 0.1 M. Base was added portionwise with speed 0.1 mL/30 min.
Isolated yield after chromatographic purification.
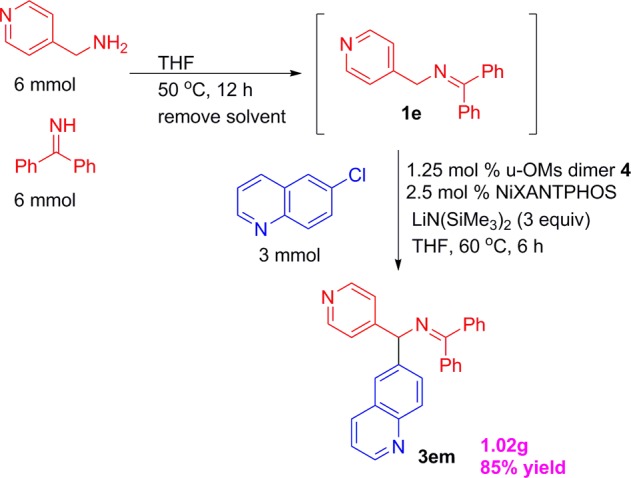
The arylation is also amenable to a sequential one-pot imine synthesis/arylation protocol (Scheme 4), which streamlines the generation of diarylmethylamine derivatives. Thus, we conducted a 3 mmol scale sequential one-pot synthesis of 4-pyridyl ketimine 1e, followed by arylation with 6-chloroquinoline 2m at a 2.5 mol % catalyst loading. The bis-heterocylic product 3el was isolated in 85% yield (1.02 g).
Scheme 4. Gram Scale Sequential One-Pot Ketimine Synthesis/Arylation Protocol.
In conclusion, we have developed the first synthetically useful arylation of 2-azaallyl anions with aryl chlorides. The key to the success of this transformation is use of the Pd-NIXANTPHOS precatalyst that efficiently generates catalytically active species and facilitates the coupling with aryl chlorides. The reaction can be used with either ketimine or aldimine substrates, has good generality, and can be applied to a telescoped ketimine synthesis/arylation reaction on gram scale.
Acknowledgments
We thank the National Science Foundation (CHE-1152488) and National Institutes of Health (NIGMS 104349) for financial support. We thank Tiezheng Jia, Dr. Jiadi Zhang, and Dr. Sonia Montel of UPenn for helpful discussions.
Supporting Information Available
Procedures, characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Li J. J.Contemporary Drug Synthesis; Wiley–Interscience: 2004; p 221. [Google Scholar]; b Grant J. A.; Riethuisen J.-M.; Moulaert B.; DeVos C. Ann. Allergy. Asthma. Immunol. 2002, 88, 190. [DOI] [PubMed] [Google Scholar]; c Doggrell S. A.; Liang L. C. N-S Arch. Pharmacol. 1998, 357, 126. [DOI] [PubMed] [Google Scholar]; d Plobeck N.; Delorme D.; Wei Z.-Y.; Yang H.; Zhou F.; Schwarz P.; Gawell L.; Gagnon H.; Pelcman B.; Schmidt R.; Yue S. Y.; Walpole C.; Brown W.; Zhou E.; Labarre M.; Payza K.; St-Onge S.; Kamassah A.; Morin P.-E.; Projean D.; Ducharme J.; Roberts E. J. Med. Chem. 2000, 43, 3878. [DOI] [PubMed] [Google Scholar]
- a Niwa T.; Suehiro T.; Yorimitsu H.; Oshima K. Tetrahedron 2009, 65, 5125. [Google Scholar]; b Niwa T.; Yorimitsu H.; Oshima K. Org. Lett. 2008, 10, 4689. [DOI] [PubMed] [Google Scholar]; c Zhu Y.; Buchwald S. L. J. Am. Chem. Soc. 2014, 136, 4500. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Li M.; Yücel B.; Adrio J.; Bellomo A.; Walsh P. J. Chem. Sci. 2014, 5, 2383. [DOI] [PMC free article] [PubMed] [Google Scholar]; For additional references related to functionalization of 2-azaallyl anions, see:; e Yeagley A. A.; Chruma J. J. Org. Lett. 2007, 9, 2879. [DOI] [PubMed] [Google Scholar]; f Fields W. H.; Khan A. K.; Sabat M.; Chruma J. J. Org. Lett. 2008, 10, 5131. [DOI] [PubMed] [Google Scholar]; g Yeagley A. A.; Lowder M. A.; Chruma J. J. Org. Lett. 2009, 11, 4022. [DOI] [PubMed] [Google Scholar]; h Fields W. H.; Chruma J. J. Org. Lett. 2010, 12, 316. [DOI] [PubMed] [Google Scholar]; i Li Z.; Jiang Y. Y.; Yeagley A. A.; Bour J. P.; Liu L.; Chruma J. J.; Fu Y. Chem.—Eur. J. 2012, 18, 14527. [DOI] [PubMed] [Google Scholar]; j Tunge J. A.; Burger E. C. Eur. J. Org. Chem. 2005, 1715. [Google Scholar]; k Burger E. C.; Tunge J. A. J. Am. Chem. Soc. 2006, 128, 10002. [DOI] [PubMed] [Google Scholar]; l Grenning A. J.; Tunge J. A. J. Am. Chem. Soc. 2011, 133, 14785. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Weaver J. D.; Recio A. III; Grenning A. J.; Tunge J. A. Chem. Rev. 2011, 111, 1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bruno N. C.; Tudge M. T.; Buchwald S. L. Chem. Sci. 2013, 4, 916. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bruno N. C.; Buchwald S. L. Org. Lett. 2013, 15, 2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a van der Veen L. A.; Keeven P. H.; Schoemaker G. C.; Reek J. N. H.; Kamer P. C. J.; van Leeuwen P. W. N. M.; Lutz M.; Spek A. L. Organometallics 2000, 19, 872. [Google Scholar]; b Birkholz M.-N.; Freixa Z.; van Leeuwen P. W. N. M. Chem. Soc. Rev. 2009, 38, 1099. [DOI] [PubMed] [Google Scholar]; c van Leeuwen P. W. N. M.; Kamer P. C. J.; Reek J. N. H.; Dierkes P. Chem. Rev. 2000, 100, 2741. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Bellomo A.; Trongsiriwat N.; Jia T.; Carroll P. J.; Dreher S. D.; Tudge M. T.; Yin H.; Robinson J. R.; Schelter E. J.; Walsh P. J. J. Am. Chem. Soc. 2014, 136, 6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Barder T. E.; Walker S. D.; Martinelli J. R.; Buchwald S. L. J. Am. Chem. Soc. 2005, 127, 4685. [DOI] [PubMed] [Google Scholar]; b Barrios-Landeros F.; Carrow B. P.; Hartwig J. F. J. Am. Chem. Soc. 2009, 131, 8141. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Fu G. C. Acc. Chem. Res. 2008, 41, 1555. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hartwig J. F. Acc. Chem. Res. 2008, 41, 1534. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Martin R.; Buchwald S. L. Acc. Chem. Res. 2008, 41, 1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lewis A. K. de K.; Caddick S.; Cloke F. G. N.; Billingham N. C.; Hitchcock P. B.; Leonard J. J. Am. Chem. Soc. 2003, 125, 10066. [DOI] [PubMed] [Google Scholar]; b Galardon E.; Ramdeehul S.; Brown J. M.; Cowley A.; Hii K. K.; Jutand A. Angew. Chem., Int. Ed. 2002, 41, 1760. [DOI] [PubMed] [Google Scholar]; c Schoenebeck F.; Houk K. N. J. Am. Chem. Soc. 2010, 132, 2496. [DOI] [PubMed] [Google Scholar]; d Senn H. M.; Ziegler T. Organometallics 2004, 23, 2980. [Google Scholar]; e Kozuch S.; Amatore C.; Jutand A.; Shaik S. Organometallics 2005, 24, 2319. [Google Scholar]; f Ahlquist M.; Norrby P.-O. Organometallics 2007, 26, 550. [Google Scholar]; g Barrios-Landeros F.; Hartwig J. F. J. Am. Chem. Soc. 2005, 127, 6944. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.