

Abstract
Conformationally constrained bithiazoles were previously found to have improved efficacy over nonconstrained bithiazoles for correction of defective cellular processing of the ΔF508 mutant cystic fibrosis transmembrane conductance regulator (CFTR) protein. In this study, two sets of constrained bithiazoles were designed, synthesized, and tested in vitro using ΔF508–CFTR expressing epithelial cells. The SAR data demonstrated that modulating the constraining ring size between 7- versus 8-membered in these constrained bithiazole correctors did not significantly enhance their potency (IC50), but strongly affected maximum efficacy (Vmax), with constrained bithiazoles 9e and 10c increasing Vmax by 1.5-fold compared to benchmark bithiazole corr4a. The data suggest that the 7- and 8-membered constrained ring bithiazoles are similar in their ability to accommodate the requisite geometric constraints during protein binding.
Introduction
Cystic fibrosis (CF) is a recessive genetic disease found primarily in Caucasians.1 Of the ∼2000 known mutations in the CF gene, deletion of phenylalanine at position 508 (ΔF508) in the cystic fibrosis transmembrane conductance regulator (CFTR) protein is the most common mutation, present in at least one allele in ∼90% of patients.2−4 Due to this deletion, the ΔF508−CFTR protein is misfolded, retained at the endoplasmic reticulum, and rapidly degraded.4 CFTR protein is a cyclic AMP-regulated chloride channel expressed in the plasma membrane of secretory epithelia in the airways, intestines, pancreas, testes, and exocrine glands, as well as some nonepithelial cell types.4,5 CF patients suffer from chronic lung infections leading to deterioration of lung function, which is the main cause of morbidity and mortality.4 Currently, there is only one drug (potentiator) that targets mutant CFTR (KalydecoTM; VX-770), but it is only helpful to the small number of patients with the G551D mutation (4.3% of CF patients) and some other gating mutations.3,6
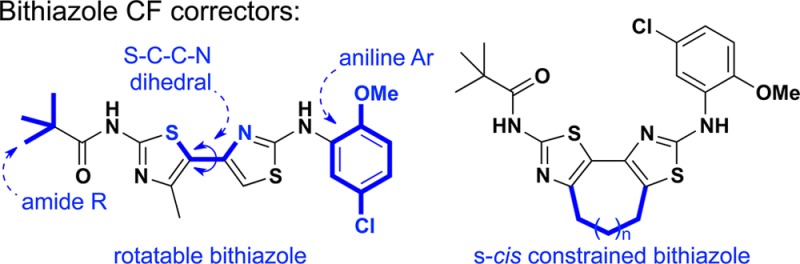
Correctors, first recognized by Verkman et al.,5a are believed to address the misfolding and trafficking of defective ΔF508–CFTR protein and are being investigated by several groups.6 The investigational correctors VX-809 and VX-661 are in clinical trials.7 However, the limited understanding of how small molecules modulate the trafficking of ΔF508–CFTR creates a challenge in efforts to discover effective corrector drug candidates. We previously reported that bithiazoles (cf., corr4a and corr15jf, Figure 1) can correct ΔF508–CFTR, and these studies showed that the conformation of the bithiazole core structure is an important determinant of corrector activity. Corrector 9a (Figure 1), with its constrained bithiazole substructure compared to lead bithiazole corr15jf, was identified in these previous studies.8
Figure 1.
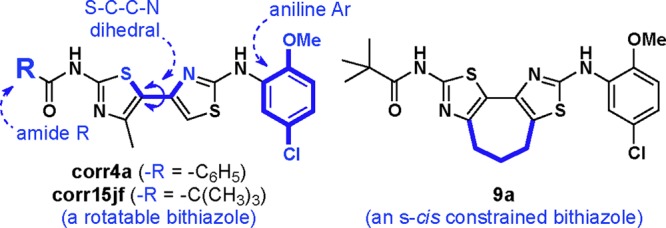
Constrained bithiazole corrector 9a, lead corrector corr15jf, and benchmark corrector corr4a.
The present study was designed to explore the constrained bithiazole core further by (1) defining the optimum size of the ring constraining the bithiazole core and (2) refining the peripheral groups in these constrained correctors using in vitro evaluation with ΔF508–CFTR FRT epithelial cells. It is hoped that refined bithiazole structural features may lead to useful insight into the mechanism of small molecule ΔF508–CFTR protein binding and interactions.8,9
Constrained bithiazoles are of particular interest because the bithiazole moiety is found in some natural products.10 Also, bithiazole 9a, with its constrained conformational advantage over freely rotating bithiazoles corr4a and corr15jf, can be expected to have a more favorable entropy of binding in formation of the corrector-bound-CFTR complex.11 Herein, we report the synthesis and biological evaluation of a set of novel constrained bithiazoles.
Chemistry
On the basis of our previous bithiazole studies,8,12 we were interested in exploring different constraining ring sizes in the bithiazole core substructure. Indeed, as the ring size increases or decreases, the bithiazole substructure becomes more or less rigid, and the interatomic distances and dihedral angles between the thiazole rings modulate. To fully probe these structural features, the constraining carbocycle (red highlight in 1; Figure 2) would, ideally, range from five to eight-membered rings (i.e., m = 0–3 in generalized bithiazole 1).
Figure 2.
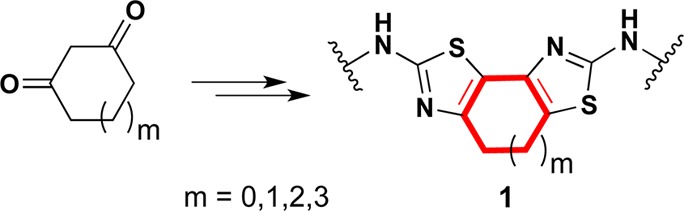
Target analogues (1) of constrained bithiazole corrector 9a.
For 1m = 0 (5-membered constrained bithiazole 2; Figure 3), a variety of routes were explored without success. For instance, α-bromination of 1,3-cyclopentanedione followed by displacement with thiourea cleanly gave 2-amino-4H-cyclopenta[d]thiazol-6(5H)-one. The introduction of the second thiazole ring by a second α-bromination and subsequent condensation with a substituted thiourea failed, even at elevated temperatures in a microwave reactor, to give the targeted bithiazole 2. Presumably the 5,5,5-ring system in 2 is too strained to form under these conditions. In contrast, the first and second thiazole-forming steps for 1m = 1 (6-membered constraining ring) proceeded smoothly, but during the final N-acylation step, the central cyclohexadiene ring spontaneously aromatized (even at −4 °C) to give 3 (Figure 3). To avoid this aromatization, we employed 5,5-dimethyl-1,3-cyclohexanedione and found that the first bromination/thiazole formation sequence worked well. However, as with 1,3-cyclopentanedione, the second bromination/thiazole formation sequence failed to deliver the targeted bithiazole because SN2 reaction on the neopentyl bromide failed under all conditions attempted (for example, using AgBF6 to activate the C–Br bond). Consequently, the only constrained bithiazole 1m = 1 evaluated in vitro (vide supra) was aromatized analogue 3.
Figure 3.
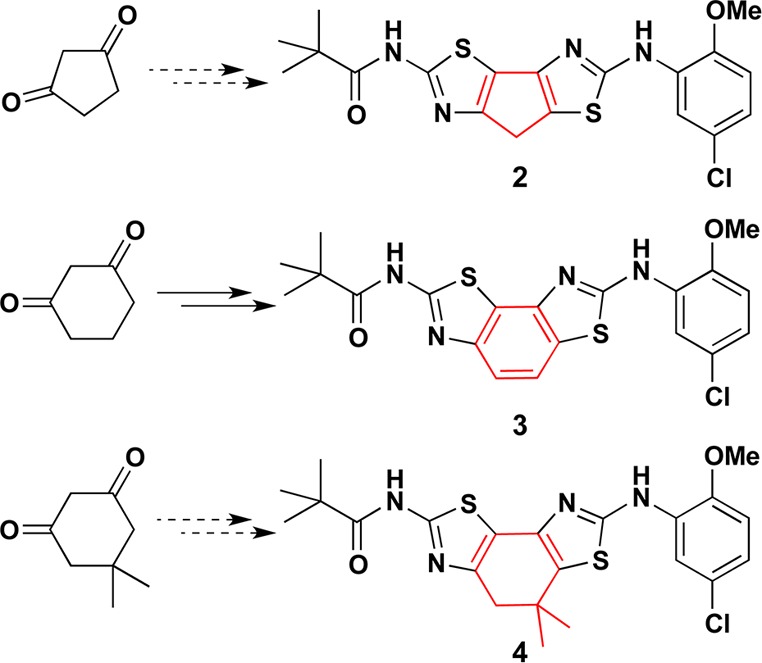
Constrained bithiazole analogues 2–4.
We next directed our attention to the preparation of constrained bithiazoles 9 (1m = 2; 7-membered constraining ring) and 10 (1m = 3; 8-membered constraining ring). This series contained constrained corrector 9a plus several amide (e.g., “R” in 9a–f/10a–f; Scheme 2a) and aniline (e.g., “Ar” in 9g–j/10g–j; Scheme 2b) analogues. These various constrained bithiazoles were synthesized by modification of our previously published procedures8,12 starting from 1,3-cycloheptadione [prepared from (cyclopent-1-en-1-yloxy)trimethylsilane as outlined in Scheme 1a]8 and 1,3-cyclooctadione (prepared from diethyl heptanedioate as outlined in Scheme 1b).13
Scheme 2. (a) Synthesis of Constrained Bithiazole Amide Analogues 9a–f and 10a–f. (b) Synthesis of Constrained Bithiazole Aniline Analogues of 9g–j and 10g–j.

Scheme 1. (a) Synthesis of 1,3-Cycloheptadione. (b) Synthesis of 1,3-Cyclooctadione.

With the requisite diones in hand, α-bromination [CHCl3/H2O (1:1) for 1,3-cycloheptadione; CCl4/H2O (1:1) for 1,3-cyclooctadione] by the dropwise addition of bromine at 0 °C followed by treatment of the crude 2-bromo-1,3-dione with thiourea in ethanol overnight delivered thiazoles 5 and 6 in 38% and 51% yield, respectively, over two steps. As a result of the difficulty in workup, thiazole 6 required basification (sat. NH4OH to pH 8–9) before purification followed by reacidification (HBr in HOAc) ahead of the second bromination. Following a second α-bromination by dropwise addition of bromine to 5 or 6 in HOAc, the resulting α-bromoketone was refluxed in EtOH overnight with 1-(5-chloro-2-methoxyphenyl)thiourea to yield 7a and 8a in 79% and 65% yield, respectively. Bithiazoles 7a and 8a were stirred in DCM and Et3N at 60 °C overnight with various acid chlorides to give the final constrained bithiazoles 9a–f and 10a–f. Constrained bithiazoles 9g–j and 10g–j, wherein the amide moiety was maintained as a pivalamide and the aniline aryl moiety was varied, were prepared in similar fashion, except that different aryl substituted thioureas were used to prepare 7g–j and 8g–j.
ΔF508–CFTR Correction Bioassay
The screening assay utilized Fischer rat thyroid (FRT) epithelial cells coexpressing ΔF508–CFTR and the yellow fluorescent protein (YFP) halide indicator YFP-H148Q/I152L at 37 °C in a 96-well-plate format.5,8,12 Correctors at various concentrations (and negative/positive controls) were added to each well and incubated for 18–24 h. ΔF508–CFTR-facilitated iodide influx was determined from the kinetics of YFP-H148Q/I152L quenching in response to iodide addition in cells treated with a cAMP agonist forskolin and the potentiator genistein.
Results and Discussion
Prior bithiazole-based structure–activity relationship studies supported the premise that the conformation about the bithiazole moiety is an important determinant of ΔF508–CFTR corrector activity.8 Although bithiazoles that are free to rotate around the thiazole–thiazole tethering C,C-bond can accommodate many conformations, we went on to demonstrate that constrained bithiazoles—where the bithiazole moiety was locked into an s-cis conformation (see Figure 1)—led to improved corrector activity.8 With this backdrop, the objectives of this investigation were 3-fold: (i) correlate corrector activity with bithiazole S–C–C–N (see corr4a in Figure 1) dihedral angle; (ii) correlate corrector activity with amide R-group (see corr4a in Figure 1) structural features; and (iii) correlate corrector activity with aniline Ar (see corr4a in Figure 1) structural features.
Our inability to construct the 7H-cyclopenta[1,2-d:3,4-d′]bis(thiazole) analogue of 1 (m = 0) coupled with the fact that the 4,5-dihydrobenzo[1,2-d:3,4-d′]bis(thiazole) analogue of 1 (m = 1) spontaneously aromatizes to 3 limited the probe of lower value S–C–C–N dihedral angle to 0°. Moreover, fully aromatized bithiazole 3 proved to be toxic, killing the ΔF508–CFTR transfected FRT cells employed in the assay. These outcomes evolved objective (i) of this study to a direct comparison of constrained bithiazoles 9a (1; m = 2) and 10a (1; m = 3) to benchmark bithiazole corrector corr4a (these three correctors each has the aniline Ar = 5-chloro-2-methoxyphenyl). The IC50 values of 9a and 10a (2.3 and 4.1 μM, respectively) were improved relative to corr4a (6.0 μM), and the 8-membered constrained corrector 10a was found to have a higher Vmax value (Table 1; 512 μM/s) than either the 7-membered analogue 9a (Table 1; 336 μM/s) or corr4a (Vmax = 475 μM/s).
Table 1. Corrector Activities of Amide-Varied Constrained Bithiazoles 7a/8a and 9a–f/10a–f Relative to Bithiazole corr4a.
|
corr4a:a Vmax = 475 μM/s, IC50 = 6.0 μM | |||||
|---|---|---|---|---|---|
|
9a–f; n = 1 |
10a–f; n = 2 |
||||
| compound | Vmax (μM/s) | IC50 (μM) | compound | Vmax (μM/s) | IC50 (μM) |
| 7a | 351 | 11.0 | 8a | 592 | 9.2 |
| 9a | 336 | 2.3 | 10a | 512 | 4.1 |
| 9b | 473 | 8.9 | 10b | 247 | 4.5 |
| 9c | 329 | 5.8 | 10c | 717 | 8.7 |
| 9d | 447 | 6.6 | 10d | 286 | 2.9 |
| 9e | 678 | 8.8 | 10e | 481 | 8.3 |
| 9f | 505 | 4.8 | 10f | 467 | 6.6 |
Corrector activity of benchmark bithiazole corr4a.
With comparable IC50 values for 9a, 10a, and corr4a and a higher Vmax value for 10a, we were encouraged to continue the exploration of these 7- and 8-membered constrained bithiazole analogues. On the basis of previous research into the bithiazole class of compounds,8,12 we knew that the amide moiety of the bithiazole had improved activity as a pivalamide group compared to a benzamide. A series of six pivalamide-like constrained bithiazoles (Scheme 2a; 9b–e and 10b–e) as well as four pivalamide-unlike constrained bithiazoles (Scheme 2a; ethyl propanoates 9e/10e and isoxazoles 9f/10f)—all with 5-chloro-2-methoxyphenyl as the aniline moiety—were prepared and evaluated for ΔF508–CFTR corrector activity. Although the IC50 values for this set of amides did not reveal any significant increase or decrease in activity relative to corr4a, there were significantly improved Vmax values for 9e (678 μM/s) and 10c (717 μM/s) relative to corr4a (475 μM/s). Importantly, these increases in Vmax are indicative of better chloride cell permeability in the rescued ΔF508–CFTR protein. Finally, although their IC50 values were highest in the Table 1 series, we were surprised to find that free amine analogues 7a and 8a were modest correctors with Vmax values approximately similar to amides 9a–f and 10a–f.
Our next objective was to explore modification of the aniline aryl moiety in these constrained bithiazoles. Previous SAR data8,12 suggested that the electron-withdrawing groups (EWG) on the phenyl ring, like fluorine, reduce corrector activity, whereas electron-donating groups (EDG) on the phenyl ring, like methoxy, improve corrector activity. However, we noted that the aryls substituted simultaneously with EWG and EDG like -Cl and -OMe generally were among the better correctors. Also, LogP is an important feature in drug design, and we reasoned that N-alkylated aniline or pyridine moieties could form ammonium salts to facilitate hydrophilicity. Therefore, we set out to prepare constrained bithiazole analogues 9a,g–j and 10a,g–j to probe these two issues. As outlined in Table 2, all of the constrained bithiazoles in this series were active correctors. Of the set, 7-membered bithiazole 9i and 8-membered bithiazoles 10g and 10j had the lowest IC50 values. Only 7-membered constrained bithiazoles 9h and 9j afforded a significant Vmax improvement relative to 9a.
Table 2. Corrector Activities of Aniline-Varied Constrained Bithiazoles 9a/10a and 9g–j/10g–j Relative to Bithiazole corr4a.
|
corr4a:a Vmax = 475 μM/s, IC50 = 6.0 μM | |||||
|---|---|---|---|---|---|
|
9a,g–j n = 1 |
10a,g–j n = 2 |
||||
| compound | Vmax (μM/s) | IC50 (μM) | compound | Vmax (μM/s) | IC50 (μM) |
| 9a | 336 | 2.3 | 10a | 512 | 4.1 |
| 9g | 366 | 6.9 | 10g | 198 | 2.1 |
| 9h | 509 | 7.2 | 10h | 280 | 5.2 |
| 9i | 336 | 2.2 | 10i | 291 | 5.2 |
| 9j | 485 | 3.6 | 10j | 243 | 0.9 |
Corrector activity of benchmark bithiazole corr4a.
In an effort to understand the relationship between ring size and activity, a search of possible conformations of each of the bithiazole analogues (including full optimizations with density functional theory) was carried out. Figure 4 shows the two lowest energy conformations of bithiazoles 3, 9a, and 10a. Not surprisingly, more conformations of 9a and 10a are accessible compared to 3. Constrained bithiazoles 9a and 10a are not forced into a flat U-shaped conformation (i.e., a central dihedral angle of 0°), which is perhaps related to the cytotoxicity of 3 (although this may also be a result of increased unsaturation). Because the size of the fused ring (7-membered vs 8-membered) does not significantly affect activity, it seems that the target active site can accommodate nonpolar moieties of varying size and shape.
Figure 4.

Lowest energy conformers of 3, 9a, and 10a. Geometries and energies were computed with M06-2X/6-31+G(d,p) (selected distances are shown in Å; dihedral angles are shown for the central S–C–C–N substructure; see the Computational Methods section in Supporting Information for values and details). Top structures are the lowest energy conformers, whereas bottom structures are the second lowest energy conformers; free energies for the latter relative to the former (0.0 kcal/mol) are shown in kcal/mol.
On the basis of these structural insights, future discovery on constrained bithiazoles could address how structural manipulation of the 7- and 8-membered constraining ring might improve compound potency, pharmacokinetics, and pharmacodynamics. Two broadly cast “constrained-X” options (9x and 10x) are depicted in Figure 4. Although 7-membered analogue 9x is appealing from a symmetry perspective, Scheme 2 chemistry may be complicated by regiochemical issues imposed by the nonsymmetrical starting 1,3-dicarbonyl. The 8-membered analogue 10x, which could be prepared from a symmetrical 1,3-dicarbonyl starting material, would avoid these complications. Finally, in addition to various constrained-X modifications, manipulation of the peripheral groups (i.e., the amide “R” and aniline “Ar” moieties; see Scheme 2) in constrained-X analogues 9x and 10x may prove insightful.
Conclusions
Twenty-three analogues of constrained bithiazole 9a were designed and synthesized to probe how the constrained bithiazole core affects the rescue and chloride channel function of ΔF508–CFTR. We found that modulating the constraining ring size (7- vs 8-membered) in these constrained bithiazole correctors did not significantly increase or decrease the potency (IC50). However, the cell chloride permeability (Vmax) of corrected ΔF508–CFTR was significantly influenced by bithiazole peripheral groups; most notably, bithiazoles 9e and 10c had Vmax 200 μM/s higher than benchmark bithiazole corr4a while maintaining comparable IC50 values. This suggests that the 7- and 8-membered ring sizes studied accommodate the requisite flexibility in the bithiazole core during protein binding.
Experimental Section
General Procedures
All chemicals were purchased from commercial suppliers and used without further purification. Analytical thin layer chromatography was carried out on precoated plates (silica gel 60 F254, 250 μm thickness) and visualized with UV light. Flash chromatography was performed using 60 Å, 32–63 μm silica gel (Scientific Adsorbents). Concentration in vacuo refers to rotary evaporation under reduced pressure. The chemical purity of all compounds was determined by HPLC and HRMS or LC–MS and confirmed to be ≥95%. 1H NMR spectra were recorded at 300, 400, 600, or 800 MHz at ambient temperature with acetone-d6, DMSO-d6, CDCl3, CD3CN, or CD3OD as solvents. 13C NMR spectra were recorded at 75, 100, 150, or 200 MHz at ambient temperature with acetone-d6, DMSO-d6, CDCl3, CD3CN, or D3OD as solvents. Chemical shifts are reported in parts per million (ppm) relative to the residual solvent peak. Infrared spectra were recorded on an ATI-FTIR spectrometer. The specifications of the Waters LC/MS are as follows: electrospray (+) ionization, mass range 100–1500 Da, 20 V cone voltage, and Xterra MS C18 column (2.1 mm × 50 mm × 3.5 μm), 0.2 mL/min; eluents were water/0.1% HCOOH and MeCN/0.1% HCOOH in gradient, and Waters 996 PDA. Preparative HPLC specifications are as follows: 15 mL/min flow rate, Xterra Prep MS C18 OBD column (19 mm × 100 mm); eluents were water/0.1% HCOOH and MeCN/0.1% HCOOH in gradient, and dual wavelength absorbance detector. High-resolution mass spectra were acquired on an LTQ Orbitrap XL mass spectrometer equipped with an electrospray ionization source (ThermoFisher, San Jose, CA), operating in the positive ion mode. Samples were introduced into the source via loop injection at a flow rate of 200 μL/min, in a solvent system of 1:1 acetonitrile/water with 0.1% formic acid. Mass spectra were acquired using Xcalibur, version 2.0.7 SP1 (ThermoFinnigan). The spectra were externally calibrated using the standard calibration mixture and then calibrated internally to <2 ppm with the lock mass tool.
Reagent Preparations
1,3-Cycloheptadione was prepared following a literature procedure, and spectral data were in agreement with literature data.8 1,3-Cyclooctadione was prepared following a literature procedure, and spectral data were in agreement with literature data.13 Substituted thioureas were also prepared following literature procedures, and spectral data were in agreement with literature data.8
ΔF508–CFTR Corrector Activity Assay
The assay was performed using FRT epithelial cells stably coexpressing human ΔF508–CFTR, and the high-sensitivity halide-sensing fluorescent protein YFP-H148Q/I152L were used as described previously.5,8,12 Cells were grown at 37 °C (95% air/5% CO2) for 24 h and then incubated for 16–20 h with 50 μL of medium containing the test compound. At the time of the assay, cells were washed with PBS and then incubated with PBS containing forskolin (20 μM) and genistein (50 μM) for 20 min. Measurements were carried out using a M1000 plate reader (Tecan Trading AG, Switzerland) equipped with a monochromator (excitation: 500 ± 10 nm, emission: 535 ± 10 nm). Each well was assayed individually for I– influx by recording fluorescence continuously (200 ms per point) for 2 s (baseline) and then for 12 s after rapid (<1 s) addition of 165 μL of PBS in which 137 mM Cl– was replaced by I–. I– influx rate was computed by fitting the final 11.5 s of the data to an exponential for extrapolation of initial slope and normalizing for background-subtracted initial fluorescence. All experiments contained negative control (DMSO vehicle) and positive control [N-(2-(5-chloro-2-methoxyphenylamino)-4′-methyl-4,5′-bithiazol-2′-yl)benzamide]. EC50 and Vmax were determined by four-parameter logistic nonlinear regression from concentration–activity data using GraphPad Prism, version 5.01. Initial iodide influx rates were fitted to the Hill equation: activity (initial slope) = A0 + VmaxCiH/(CiH + IC50H), where Ci is compound concentration, H is coefficient, and A0 is background signal.
Computational Methods: All calculations were performed using Gaussian 09.14 Conformational analyses were performed using the Spartan10 software suite.15 Geometries were optimized at the M06-2X/6-31+G(d,p)16,17 level. The identities of all minima were verified by frequency analysis. Structural drawings were produced using CYLView.18
Synthesis of Constrained Bithiazole Analogues
N8-(5-Chloro-2-methoxyphenyl)-5,6-dihydro-4H-cyclohepta[1,2-d:3,4-d′]bis(thiazole)-2,8-diamine (7a)
1,3-Cycloheptadione (1.6 g, 12.7 mmol)
was dissolved in CHCl3 (21 mL) and H2O (21 mL).
The biphasic mixture was stirred in an ice bath, and bromine (2.24
g, 12.0 mmol) was added dropwise via addition funnel. The mixture
was stirred for 1 h in an ice bath, then extracted with DCM (3 ×
10 mL), dried over Na2SO4, and concentrated
under reduced pressure to yield a yellow oil, 2-bromo-1,3-cycloheptadione
(3.1 g crude material). This oil was dissolved in EtOH (28 mL), thiourea
was added, stirred at room temperature overnight, concentrated, and
recrystallized from DCM/hexane (decanted cloudy white solution from
yellow oil that separated during addition of hexane and stored in
freezer) to yield colorless crystals of 2-amino-4,5,6,7-tetrahydro-8H-cyclohepta[d]thiazol-8-one (1.83 g, 38% yield, two steps).
The yellow oil can be purified via column chromatography (100% EtOAc)
to obtain additional 2-amino-4,5,6,7-tetrahydro-8H-cyclohepta[d]thiazol-8-one. These crystals were carried forward
with only a crude 1H and 13C NMR for characterization,
which was in agreement with previously published results.8 To 2-amino-4,5,6,7-tetrahydro-8H-cyclohepta[d]-thiazol-8-one (0.5 g, 1.9 mmol) in HOAc (18 mL) was
added bromine (0.34 g, 2.1 mmol) dropwise and stirred at rt for 30
min. The solid was filtered off, rinsed with cold acetone, and dried
to yield 2-amino-7-bromo-4,5,6,7-tetrahydro-8H-cyclohepta[d]thiazol-8-one
as an off-white solid (0.48 g, 74%). To 2-amino-7-bromo-4,5,6,7-tetrahydro-8H-cyclohepta[d]thiazol-8-one (0.48 g, 1.4 mmol) in EtOH
(10 mL) was added 1-(5-chloro-2-methoxyphenyl)thiourea (0.32 g, 1.5
mmol) and refluxed overnight. The reaction mixture was concentrated
and recrystallized from EtOH to yield 7a as an off-white
powdery solid (0.42 g, 79%). 1H NMR (600 MHz, CDCl3): δ 9.83 (s, 1H), 9.11 (s, 2H), 8.47 (d, J = 2.5 Hz, 1H), 7.07 (d, J = 8.8 Hz, 1H) 6.97 (dd, J = 8.8, 2.5 Hz, 1H), 3.85 (s, 3H), 2.92 (t, J = 5.5 Hz, 2H), 2.89 (t, J = 5.7 Hz, 2H), 2.05–1.98
(m, 2H). HRMS (m/z): [M + H]+ calcd for C16H15ClN4OS2, 379.0449; found, 379.0468.
N-(8-((5-Chloro-2-methoxyphenyl)amino)-5,6-dihydro-4H-cyclohepta[1,2-d:3,4-d′]bis(thiazole)-2-yl)pivalamide (9a)
Bithiazole 7a (20.0 mg, 0.053
mmol, 1.0 equiv) was dissolved in dry THF (1 mL). Triethylamine (18.4
μL, 0.132 mmol, 2.5 equiv) and pivaloyl chloride (10.1 μL,
0.0688 mmol, 1.3 equiv) were added sequentially. The reaction mixture
was purged with nitrogen, heated to 60 °C, and stirred overnight.
Reaction progress was monitored by TLC in 50/50 ethyl acetate/hexanes.
The reaction mixture was concentrated, and the product was isolated
by HPLC as a white solid (12.5 mg, 51%). mp 225–229 °C.
IR (neat): vmax 2966, 2929, 2856, 1678,
1597, 1533, 1244, 1124, 1024 cm–1. 1H
NMR (600 MHz, CDCl3): δ 8.24 (s, 1H), 8.07 (d, J = 2.4 Hz, 1H), 7.57 (s, 1H), 6.92 (dd, 2.4 and 8.4 Hz,
1H), 6.78 (d, J = 8.4 Hz, 1H), 3.90 (s, 3H), 3.06
(at, 5.7 Hz, 2H), 2.98 (at, 5.6 Hz, 2H), 2.17–2.13 (m, 2H),
1.34 (s, 9H). 13C NMR (150 MHz, CDCl3): δ
184.2, 176.2, 158.9, 157.0, 145.5, 145.0, 138.6, 130.8, 126.3, 120.8,
120.1, 115.9, 110.6, 56.0, 39.2, 32.0, 27.2, 27.1, 22.6. HRMS (m/z): [M + H]+ calcd for C21H23ClN4O2S2,
463.1024; found, 463.1021.
N-(8-((5-Chloro-2-methoxyphenyl)amino)-5,6-dihydro-4H-cyclohepta[1,2-d:3,4-d′]bis(thiazole)-2-yl)cyclopropanecarboxamide (9b)
The procedure for 9a was followed,
except cyclopropanecarbonyl chloride (0.14 mmol, 2.6 equiv)
(prepared freshly from cyclopropane carboxylic acid and oxalyl chloride
1:1.2 eq in 50 μL of DCM and a drop of DMF) was substituted,
yielding a yellow solid (12 mg, 51%). 1H NMR (600 MHz,
CDCl3): δ 8.05 (d, J = 2.3 Hz, 1H),
7.57 (s, 1H), 6.91 (dd, J = 8.6, 2.3 Hz, 1H), 6.77
(d, J = 8.6, 1H), 3.89 (s, 3H), 3.09 (at, J = 5.5 Hz, 2H), 2.98 (at, J = 5.5 Hz,
2H), 2.16–2.12 (m, 2H), 1.70–1.64 (m, 1H), 1.23–1.20
(m, 2H), 0.98–0.95 (m, 2H). 13C NMR (150 MHz, CDCl3): δ 171.4, 159.1, 156.8, 145.6, 143.5, 138.1, 130.7,
126.3, 122.7, 120.9, 120.6, 116.0, 110.7, 56.0, 31.9, 29.7, 27.0,
22.4, 15.1, 9.3. HRMS (m/z): [M
+ H]+ calcd for C20H19ClN4O2S2, 447.0711; found, 447.0708.
N-(8-((5-Chloro-2-methoxyphenyl)amino)-5,6-dihydro-4H-cyclohepta[1,2-d:3,4-d′]bis(thiazole)-2-yl)isobutyramide (9c)
The procedure for 9a was followed,
except isobutyryl chloride (7.2 μL, 0.069 mmol, 1.3 equiv) was
substituted, yielding an orange solid (5.9 mg, 25%). mp 208–215
°C. IR (neat): vmax 2964, 2924, 1597,
1531, 1417, 1268, 1248, 1126, 1026, 796 cm–1. 1H NMR (600 MHz, CDCl3): δ 8.21 (s, 1H), 8.06
(d, J = 2.5 Hz, 1H), 7.59 (s, 1H), 6.92 (dd, J = 8.6, 2.5 Hz, 1H), 6.78 (d, J = 8.6
Hz, 1H), 3.90 (s, 3H), 3.04 (at, J = 5.7 Hz, 2H),
2.98 (at, J = 5.6 Hz, 2H), 2.73 (h, J = 6.9 Hz, 1H), 2.18–2.12 (m, 2H), 1.29 (d, J = 6.9 Hz, 6H). 13C NMR (150 MHz, CDCl3): δ
175.1, 166.5, 159.3, 158.0, 145.6, 137.9, 130.6, 126.3, 122.2, 121.0,
120.7, 116.1, 110.7, 56.0, 35.5, 31.0, 26.8, 22.3, 19.0. HRMS (m/z): [M + H]+ calcd for C20H21ClN4O2S2,
449.0867; found, 449.0870.
N-(8-((5-Chloro-2-methoxyphenyl)amino)-5,6-dihydro-4H-cyclohepta[1,2-d:3,4-d′]bis(thiazole)-2-yl)-2-methylpentanamide (9d)
The procedure for 9a was followed,
except 2-methylbutyryl chloride (8.54 μL, 0.069 mmol, 1.3 equiv)
was substituted, yielding a yellow solid (11.8 mg, 48%). mp 138–144
°C. IR (neat): vmax 3207, 2031, 2011,
1969, 1531, 1261, 1178, 1090, 1016, 897, 798, 644 cm-1. 1H NMR (600 MHz, CDCl3): δ 8.22 (s, 1H), 8.05 (d, J = 2.4 Hz, 1 H), 7.57 (s, 1H), 6.92 (dd, J = 8.6, 2.5 Hz, 1H), 6.78 (d, J = 8.7 Hz, 1H), 3.90
(s, 3H), 3.05 (at, J = 5.7 Hz, 2H), 2.99 (at, J = 5.6 Hz, 2H), 2.52–2.46 (m, 1H), 2.17–2.13
(m, 2H), 1.84–1.76 (m, 1H), 1.61–1.54 (m, 1H), 1.26
(d, J = 7.0 Hz, 3H), 0.96 (t, J =
7.4 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ
175.1, 166.5, 159.3, 158.0, 145.6, 137.9, 130.6, 126.3, 122.2, 121.0,
120.8, 116.1, 110.7, 56.0, 38.7, 35.5, 32.3, 31.0, 26.8, 22.3, 19.0.
HRMS (m/z): [M + H]+ calcd
for C21H23ClN4O2S2, 463.1024; found, 463.1024.
Methyl 4-((8-((5-chloro-2-methoxyphenyl)amino)-5,6-dihydro-4H-cyclohepta[1,2-d:3,4-d′]bis(thiazole)-2-yl)amino)-4-oxobutanoate (9e)
The procedure for 9a was followed,
except 3-(carbomethoxy)-propionyl chloride (28.7 μL, 0.233 mmol,
2.2 equiv) was substituted, yielding a yellow solid (7.9 mg, 15%). 1H NMR (600 MHz, CDCl3): δ 8.07 (d, J = 2.3 Hz, 1H), 7.56 (s, 1H), 6.91 (dd, J = 8.6, 2.3 Hz, 1H), 6.77 (d, J = 8.6, 1H), 3.89
(s, 3H), 3.71 (s, 3H), 3.09 (at, J = 5.5 Hz, 2H),
2.98 (at, J = 5.5 Hz, 2H), 2.81–2.73 (m, 2H),
2.17–2.12 (m, 2H). 13C NMR (150 MHz, CDCl3): δ 173.1, 168.7, 159.0, 155.4, 145.7, 138.5, 130.8, 126.3,
123.1, 120.8, 120.2, 115.9, 113.1, 110.6, 56.0, 52.1, 32.6, 31.0,
28.9, 27.2, 22.6. HRMS (m/z): [M
+ H]+ calcd for C21H21ClN4O4S2, 493.0766, found: 493.0758.
N-(8-((5-Chloro-2-methoxyphenyl)amino)-5,6-dihydro-4H-cyclohepta[1,2-d:3,4-d′]bis(thiazole)-2-yl)isoxazole-5-carboxamide (9f)
The procedure for 9a was followed,
except isoxazole 5-carbonyl chloride (6.64 μL, 0.069 mmol, 1.3
equiv) was substituted, yielding a bright yellow solid (11.7 mg, 47%).
mp 115–120 °C. IR (neat): vmax 2924, 1597, 1531, 1415, 1294, 1244, 1124, 1020, 750 cm–1. 1H NMR (400 MHz, DMSO-d6): δ 9.72 (s, 1H), 8.78 (d, J = 2.0 Hz, 1H),
8.61 (d, J = 2.4 Hz, 1H), 8.11 (s, 1H), 7.36 (s,
1H), 7.0 (d, J = 8.7 Hz, 1 H), 6.96 (dd, J = 8.6, 2.5 Hz, 1H), 3.85 (s, 3H), 3.04 (at, J = 5.6 Hz, 2H), 2.94 (at, J = 5.5 Hz, 2H), 2.05–2.00
(m, 2H). HRMS (m/z): [M + H]+ calcd C20H16ClN5O3S2, 474.0.456; found, 474.0462.
2-Amino-5,6,7,8-tetrahydrocycloocta[d]thiazol-9(4H)-one (A)
To a heterogeneous mixture of cyclooctane-1,3-dione
(2.08 g, 14.8 mmol) in CCl4/water (1:1, 50 mL) was added
a solution of Br2 (2.61g, 0.84 mL, 16.3 mmol) in CCl4 (25 mL) dropwise at 0 °C. The mixture was stirred at
0 °C for 1 h and extracted with DCM. The organic layer was collected,
and DCM was removed under reduced pressure to afford 2-bromocyclooctane-1,3-dione
as a yellow oil which was used without further purification. To a
solution of 2-bromocyclooctane-1,3-dione in anhydrous EtOH (35 mL)
was added thiourea (1.24 g, 16.3 mmol). The mixture was stirred at
room temperature overnight. EtOH was removed under reduced pressure,
and the resulting light brown residue was redissolved in water (50
mL), basified with concentrated NH4OH solution until pH
= 8–9, and extracted with EtOAc (3 × 50 mL). The combined
organics were dried over anhydrous Na2SO4 and
concentrated to give the crude product which was triturated with CHCl3 to afford A as a yellow solid (1.47 g, 51% yield
over two steps. 1H NMR (600 MHz, DMSO-d6): δ 7.88 (s, 2H), 3.01 (t, J =
7.0 Hz, 2H), 2.78 (t, J = 7.0 Hz, 2H), 1.73–1.55
(m, 4H), 1.45–1.33 (m, 2H). 13C NMR (150 MHz, DMSO-d6): δ 191.1, 171.6, 159.9, 125.4, 39.6,
30.0, 24.0, 23.1, 22.6. HRMS (m/z): [M + H]+ calcd C9H12N2OS, 197.0749; found, 197.0749.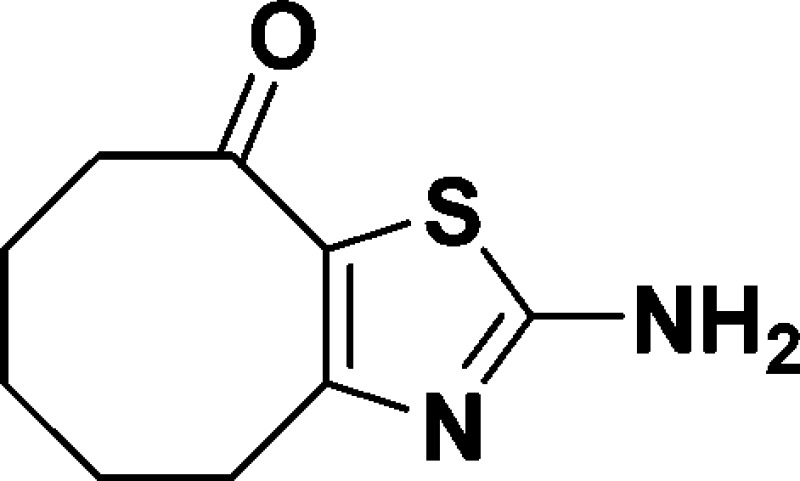
2-Amino-8-bromo-5,6,7,8-tetrahydrocycloocta[d]thiazol-9(4H)-one dihydrobromide salt (B)
Compound A (0.70 g, 3.59 mmol) dissolved in glacial acetic acid (18
mL) was treated dropwise with a solution of HBr in HOAc (18 mL, 33
wt % in acetic acid). The mixture was stirred at room temperature
for 30 min. Br2 (0.573 g, 0.184 mL, 3.59 mmol) was added
dropwise. The reaction mixture was stirred at room temperature for
1 h. Removing solvent under reduced pressure yielded the crude product
which was triturated with DCM to afford B as a tan solid
(1.08 g, 70%). 1H NMR (600 MHz, DMSO-d6): δ 10.26 (s, 1H), 9.35 (s, 3H), 5.84 (dd, J = 11.9, 7.2 Hz, 1H), 3.42–3.26 (m, 1H), 2.97 (dd, J = 15.1, 6.5 Hz, 1H), 2.23 (dtd, J = 13.5,
7.2, 3.3 Hz, 1H), 2.05 (t, J = 12.6 Hz, 1H), 1.75
(dd, J = 13.5, 6.5 Hz, 1H), 1.62 (dd, J = 9.7, 5.5 Hz, 2H), 1.42–1.24 (m, 1H). 13C NMR
(150 MHz, DMSO-d6): δ 184.5, 170.0,
150.0, 121.0, 55.7, 35.6, 27.1, 23.5, 22.4. HRMS (m/z): [M + H]+ calcd for C9H11BrN2OS, 274.9854; found, 274.9867.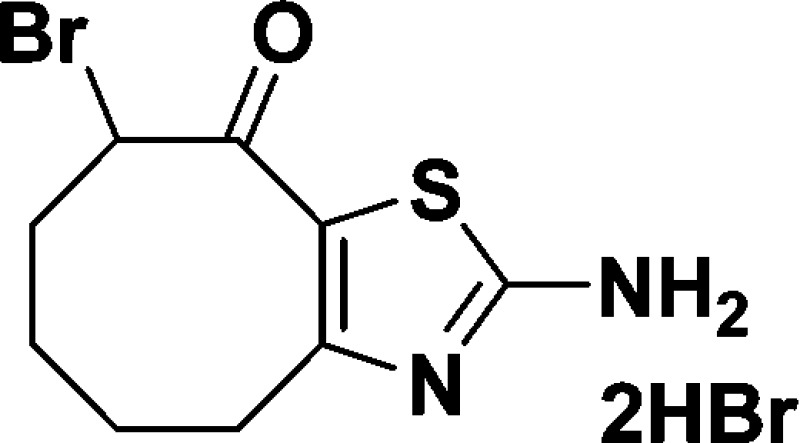
N9-(5-Chloro-2-methoxyphenyl)-4,5,6,7-tetrahydrocycloocta[1,2-d:3,4-d′]bis(thiazole)-2,9-diamine (8a)
A suspension of B (0.451 g,
1.03 mmol, 1.0 equiv) and 1-(5-chloro-2-methoxyphenyl)thiourea (0.224
g, 1.03 mmol, 1.0 equiv) in anhydrous EtOH (10 mL) was heated at reflux
for 24 h. EtOH was removed under reduced pressure, and the residue
was redissolved in water (10 mL), basified with 1 M NaOH (50 mL),
and extracted with EtOAc (2 × 50 mL). The combined organics were
dried over anhydrous Na2SO4, concentrated, and
purified by column chromatography (100% EtOAc, Rf = 0.20) to afford 8a as a light brown solid
(0.266 g, 65%). 1H NMR (600 MHz, CDCl3): δ
8.05 (d, J = 2.3 Hz, 1H), 7.67 (s, 1H), 6.88 (dd, J = 8.6, 2.3 Hz, 1H), 6.75 (d, J = 8.6
Hz, 1H), 6.12 (s, 2H), 3.85 (s, 3H), 2.79 (t, J =
10.5 Hz, 4H), 1.78 (dt, J = 10.5, 5.2 Hz, 4H). 13C NMR (150 MHz, CDCl3): δ 167.3, 159.5,
145.8, 138.8, 130.9, 126.2, 123.2, 121.9, 121.0, 116.3, 115.4, 110.8,
56.1, 29.5, 27.3, 24.5, 23.5. HRMS (m/z): [M + H]+ calcd for C17H17ClN4OS2, 393.0611; found, 393.0593.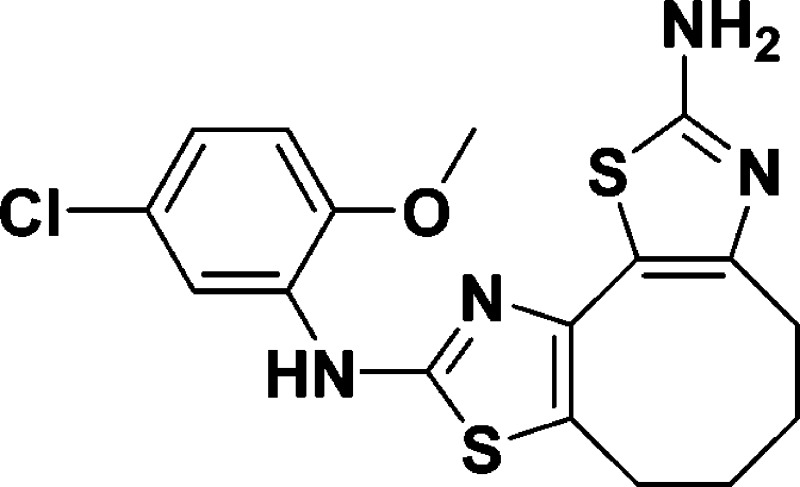
N-(9-((5-Chloro-2-methoxyphenyl)amino)-4,5,6,7-tetrahydrocycloocta-[1,2-d:3,4-d′]bis(thiazole)-2-yl)pivalamide (10a)
To a solution of 8a (0.266
g, 0.676 mmol, 1.0 equiv) in anhydrous DCM (7 mL) was added TEA (188
μL, 1.35 mmol, 2.0 equiv). Pivaloyl chloride (92 μL, 0.743
mmol, 1.1 equiv) was then added in one portion. The reaction mixture
was purged with nitrogen and stirred at 60 °C overnight at which
time LC-MS indicated the completion of reaction. DCM was removed in
vacuo, and the resulting solid was purified by HPLC to yield 10a as a yellow solid (0.183 g, 57%). 1H NMR (600
MHz, CDCl3): δ 9.46 (s, 1H), 8.07 (d, J = 2.5 Hz, 1H), 7.64 (s, 1H), 6.97–6.84 (m, 1H), 6.77 (dd, J = 8.6, 2.5 Hz, 1H), 3.88 (d, J = 3.0
Hz, 3H), 2.93–2.77 (m, 4H), 1.81 (m, 4H), 1.34 (s, 9H). 13C NMR (150 MHz, CDCl3): δ 175.8, 159.4,
157.3, 146.6, 145.7, 139.0, 130.8, 126.3, 122.8, 120.9, 120.9, 116.1,
110.7, 56.1, 39.2, 29.9, 27.3, 27.0, 24.7, 24.3. HRMS (m/z): [M + H]+ calcd for C22H25ClN4O2S2, 477.1186;
found, 477.1176.
N-(9-((5-Chloro-2-methoxyphenyl)amino)-4,5,6,7-tetrahydrocycloocta[1,2-d:3,4-d′]bis(thiazole)-2-yl)cyclopropanecarboxamide (10b)
The procedure for 10a was
followed, except cyclopropane carbonyl chloride (83 μL, 0.917
mmol, 1.1 equiv) was substituted, yielding a yellow solid (0.0359
g, 9%). 1H NMR (600 MHz, CDCl3): δ 11.15
(s, 1H), 8.04 (d, J = 2.2 Hz, 1H), 7.80 (s, 1H),
6.89 (dt, J = 8.6, 2.2 Hz, 1H), 6.75 (dd, J = 8.6, 2.2 Hz, 1H), 3.85 (d, J = 3.4
Hz, 3H), 2.90–2.77 (m, 4H), 1.78 (s, 4H), 1.61 (tt, J = 8.1, 4.6 Hz, 1H), 1.20–1.15 (m, 2H), 0.90 (dt, J = 7.2, 3.4 Hz, 2H). 13C NMR (150 MHz, CDCl3): δ 171.8, 159.7, 158.4, 146.7, 146.0, 139.0, 130.9,
126.2, 122.6, 121.2, 120.6, 116.4, 110.8, 56.1, 29.9, 27.1, 24.8,
24.4, 15.0, 9.2. HRMS (m/z): [M
+ H]+ calcd for C21H21ClN4O2S2, 461.0873; found, 461.0865.
N-(9-((5-Chloro-2-methoxyphenyl)amino)-4,5,6,7-tetrahydrocycloocta [1,2-d:3,4-d′]bis(thiazole)-2-yl)isobutyramide (10c)
The procedure for 10a was followed, except
isobutyryl chloride (87 μL, 0.830 mmol, 1.2 equiv) was substituted,
yielding a yellow solid (0.132 g, 41%). 1H NMR (600 MHz,
CDCl3): δ 9.56 (s, 1H), 8.04 (d, J = 2.5 Hz, 1H), 7.81 (s, 1H), 6.94–6.83 (m, 1H), 6.75 (d, J = 8.6 Hz, 1H), 3.84 (s, 3H), 2.90–2.78 (m, 4H),
2.59 (hept, J = 6.9 Hz, 1H), 1.84–1.74 (m,
4H), 1.23 (d, J = 6.9 Hz, 6H). 13C NMR
(150 MHz, CDCl3): δ 174.5, 159.7, 157.6, 146.9, 145.9,
139.0, 130.9, 126.2, 122.6, 121.1, 120.8, 116.4, 110.8, 56.1, 35.6,
30.0, 27.1, 24.7, 24.4, 19.3. HRMS (m/z): [M + H]+ calcd for C21H23ClN4O2S2, 463.1029; found, 463.1021.
N-(9-((5-Chloro-2-methoxyphenyl)amino)-4,5,6,7-tetrahydrocycloocta[1,2-d:3,4-d′]bis(thiazole)-2-yl)-2-methylbutanamide (10d)
The procedure for 10a was
followed, except 2-methylbutyryl chloride (85 μL, 0.683 mmol,
1.1 equiv) was substituted, yielding a light yellow solid (0.10 g,
34%). 1H NMR (600 MHz, CDCl3): δ 9.72
(s, 1H), 8.02 (d, J = 2.4 Hz, 1H), 7.85 (s, 1H),
6.88 (dd, J = 8.6, 2.4 Hz, 1H), 6.75 (d, J = 8.6 Hz, 1H), 3.84 (s, 3H), 3.04–2.61 (m, 4H),
2.35 (sextet, J = 7.0 Hz, 1H), 1.89–1.66 (m,
5H), 1.50 (dp, J = 14.0, 7.0 Hz, 1H), 1.20 (d, J = 7.0 Hz, 3H), 0.89 (t, J = 7.0 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ 174.1, 159.7,
157.5, 146.9, 146.0, 139.0, 130.9, 126.2, 122.6, 121.2, 120.8, 116.4,
110.8, 56.1, 42.8, 30.0, 27.2, 27.1, 24.7, 24.3, 17.1, 11.8. HRMS
(m/z): [M + H]+ calcd
for C22H25ClN4O2S2, 477.1186; found, 477.1176.
Methyl 4-((9-((5-chloro-2-methoxyphenyl)amino)-4,5,6,7 tetrahydrocycloocta[1,2-d:3,4-d′]bis(thiazole)-2-yl)amino)-4-oxobutanoate (10e)
The procedure for 10a was
followed, except methyl 4-chloro-4-oxobutyrate (76 μL, 0.618
mmol, 1.1 equiv) was substituted, yielding a light yellow solid (0.121
g, 42%). 1H NMR (600 MHz, CDCl3): δ 10.63
(s, 1H), 8.05 (d, J = 2.5 Hz, 1H), 7.91 (s, 1H),
6.85 (dd, J = 8.6, 2.5 Hz, 1H), 6.72 (d, J = 8.6 Hz, 1H), 3.80 (s, 3H), 3.65 (s, 3H), 2.85 (d, J = 7.0 Hz, 2H), 2.80–2.72 (m, 6H), 1.80–1.70
(m, 4H). 13C NMR (150 MHz, CDCl3): δ 173.1,
169.5, 159.7, 157.4, 147.1, 145.9, 138.9, 130.8, 126.0, 122.4, 121.0,
120.6, 116.4, 110.7, 56.0, 52.0, 30.7, 29.9, 28.7, 27.0, 24.5, 24.2.
HRMS (m/z): [M + H]+ calcd
for C22H23ClN4O4S2, 507.0927; found, 507.0918.
N-(9-((5-Chloro-2-methoxyphenyl)amino)-4,5,6,7-tetrahydrocyclo-octa[1,2-d:3,4-d′]bis(thiazole)-2-yl)isoxazole-5-carboxamide (10f)
The procedure for 10a was
followed, except isoxazole-5-carbonyl chloride (76 μL, 0.787
mmol, 1.1 equiv) was substituted, yielding a yellow solid (0.0422
g, 12%). 1H NMR (600 MHz, CDCl3): δ 9.55
(s, 1H), 8.39 (d, J = 1.7 Hz, 1H), 8.07 (d, J = 2.4 Hz, 1H), 7.76 (s, 1H), 7.15–7.11 (m, 1H),
6.90 (dd, J = 8.6, 2.4 Hz, 1H), 6.77 (d, J = 8.6 Hz, 1H), 3.87 (s, 3H), 2.97–2.75 (m, 4H),
1.88–1.75 (m, 4H). 13C NMR (150 MHz, CDCl3): δ 161.6, 159.8, 156.9, 153.7, 151.3, 146.4, 145.9, 138.5,
130.8, 126.3, 123.0, 121.9, 121.2, 116.3, 110.9, 108.3, 56.1, 29.7,
27.0, 24.6, 24.0. HRMS (m/z): [M
+ H]+ calcd for C21H18ClN5O3S2, 488.0618; found, 488.0609.
N-(8-((5-Chloro-2-(dimethylamino)phenyl)amino)-5,6-dihydro-4H-cyclo-hepta[1,2-d:3,4-d′]bis(thiazole)-2-yl)pivalamide (9g)
The procedures for 7a and 9a were followed, except 1-(5-chloro-2-(dimethylamino)-phenyl)thiourea
(0.229 g, 1.1 mmol, 1.1 equiv) was substituted, yielding a yellow
solid (0.015 g, 15%). 1H NMR (400 MHz, CDCl3): δ 8.28 (s, 1H), 7.99 (d, J = 2.3, 1H),
7.07 (d, J = 8.4, 1H), 6.91 (dd, J = 2.3, 8.4, 1H), 3.18–2.85 (m, 4H), 2.64 (s, 6H), 2.15 (s,
2H), 1.35 (s, 9H). 13C NMR (100 MHz, CDCl3):
δ 184.3, 176.2, 159.2, 156.9, 145.3, 140.2, 138.8, 136.7, 130.8,
122.8, 121.4, 120.7, 115.6, 44.9, 39.3, 38.7, 32.3, 27.5, 22.8. HRMS
(m/z): [M + H]+ calcd
for C22H27ClN5OS2, 476.1340;
found, 476.1338.
N-(8-((3-Methylpyridin-2-yl)amino)-5,6-dihydro-4H-cyclohepta[1,2-d:3,4-d′]bis(thiazole)-2-yl)pivalamide (10h)
The procedures for 7a and 9a were followed, except 1-(3-methylpyridin-2-yl)thiourea
(0.167 g, 1.1 mmol, 1.1 equiv) was substituted, yielding a yellow
solid (0.024 g, 26%). mp 154–155 °C. 1H NMR
(400 MHz, CDCl3): δ 8.85 (br s, 1H), 8.16 (d, J = 4.0, 1H), 7.97 (br s, 1H), 7.37 (d, J = 7.1, 1H), 6.78 (dd, J = 5.1, 7.2, 1H), 3.13–2.93
(m, 4H), 2.27 (s, 3H), 2.14–2.10 (m, 2H), 1.30 (s, 9H). 13C NMR (100 MHz, CDCl3): δ 175.6, 156.5,
155.2, 149.7, 145.9, 144.4, 138.3, 137.0, 123.5, 123.3, 117.8, 116.5,
39.3, 33.1, 27.5, 27.0, 23.0, 16.8. HRMS (m/z): [M + H]+ calcd for C20H23N5OS2, 414.1417; found, 414.1404.
N-(8-((3-(Dimethylamino)phenyl)amino)-5,6-dihydro-4H-cyclohepta[1,2-d:3,4-d′]bis(thiazole)-2-yl)pivalamide (10i)
The procedures for 7a and 9a were followed, except 1-(2-(dimethylamino)phenyl)thiourea
(0.195 g, 1.1 mmol, 1.1 equiv) was substituted, yielding a yellow
solid (0.010g, 10%). mp 111–112 °C. 1H NMR
(600 MHz, CDCl3): δ 9.09 (s, 1H), 7.21 (s, 1H), 7.18
(t, J = 8.2 Hz, 1H), 7.00 (s, 1H), 6.59 (d, J = 8.2,
1H), 6.45 (d, J = 8.2, 1H), 3.07 (t, J = 5.7 Hz, 2H), 3.01 (s, 3H), 2.95 (t, J = 5.7 Hz,
2H), 2.16–2.12 (m, 2H), 1.32 (s, 9H). 13C NMR (150
MHz, CDCl3): δ 175.8, 161.1, 155.8, 151.6, 145.3,
141.5, 138.4, 130.0, 123.3, 119.5, 107.5, 106.5, 102.5, 40.9, 39.3,
32.6, 29.9, 27.5, 22.9. HRMS (m/z): [M + H]+ calcd for C22H27N5OS2, 442.1730; found, 442.1725.
N-(8-((2-Methoxy-5-nitrophenyl)amino)-5,6-dihydro-4H-cyclohepta[1,2-d:3,4-d′]bis(thiazole)-2-yl)pivalamide (10j)
The procedures for 7a and 9a were followed, except 1-(2-methoxy-5-nitrophenyl)thiourea
(0.227 g, 1.1 mmol, 1.1 equiv) was substituted, yielding a yellow
solid (0.006 g, 6%). 1H NMR (600 MHz, CDCl3):
δ 10.60 (s, 1H), 8.89 (s, 1H), 7.94 (d, J =
9.3, 1H), 7.62 (s, 1H), 6.94 (d, J = 9.3, 1H), 3.97
(s, 3H), 3.13–2.94 (m, 4H), 2.25–2.05 (m, 2H), 1.25
(s, 9H). 13C NMR (150 MHz, DMSO): δ 176.1, 159.1,
155.7, 152.4, 146.2, 140.8, 137.5, 130.4, 121.1, 120.5, 117.4, 111.1,
110.1, 56.6, 38.7, 32.3, 26.7, 26.1, 22.3. HRMS (m/z): [M + H]+ calcd for C21H23N5O4S2, 474.1264;
found, 474.1253.
N-(9-((5-Chloro-2-(dimethylamino)phenyl)amino)-4,5,6,7-tetrahydrocycloocta-[1,2-d:3,4-d′]bis(thiazole)-2-yl)pivalamide (10g)
The procedures for 8a and 10a was followed, except 1-(5-chloro-2-(dimethylamino)-phenyl)thiourea
(0.260 g, 1.13 mmol, 1.0 equiv) was substituted, yielding a tan solid
(0.146 g, 53%). 1H NMR (600 MHz, CDCl3): δ
8.92 (s, 1H), 8.37 (s, 1H), 8.02 (d, J = 2.3 Hz,
1H), 7.02 (d, J = 8.4 Hz, 1H), 6.84 (dd, J = 8.4, 2.3 Hz, 1H), 2.87–2.72 (m, 4H), 2.56 (s,
6H), 1.82–1.67 (m, 4H), 1.27 (s, 9H). 13C NMR (150
MHz, CDCl3): δ 175.6, 159.3, 156.9, 147.3, 140.0,
139.1, 136.5, 130.4, 122.4, 121.2, 121.1, 120.8, 115.6, 44.7, 39.0,
30.0, 27.2, 26.8, 24.7, 24.3. HRMS (m/z): [M + H]+ calcd for C23H28ClN5OS, 490.1502; found, 490.1491.
N-(9-((3-Methylpyridin-2-yl)amino)-4,5,6,7-tetrahydrocycloocta[1,2-d:3,4-d′]bis(thiazole)-2-yl)pivalamide (10h)
The procedure for 8a and 10a was followed, except 1-(3-methylpyridin-2-yl)thiourea
(0.171 g, 1.02 mmol, 1.0 equiv) was substituted, yielding a yellow
solid (0.0845 g, 40%). 1H NMR (600 MHz, CDCl3): δ 8.97 (s, 1H), 8.45–7.82 (m, 2H), 7.35 (d, J = 7.2 Hz, 1H), 6.92–6.59 (m, 1H), 2.81 (s, 4H),
2.21 (s, 3H), 1.76 (s, 4H), 1.26 (s, 9H). 13C NMR (150
MHz, CDCl3): δ 175.5, 156.7, 156.6, 149.6, 147.1,
144.1, 138.1, 137.1, 125.3, 120.9, 117.7, 116.3, 39.0, 30.1, 27.3,
27.2, 24.4, 24.3, 16.7. HRMS (m/z): [M + H]+ calcd for C21H25N5OS2, 428.1579; found, 428.1570.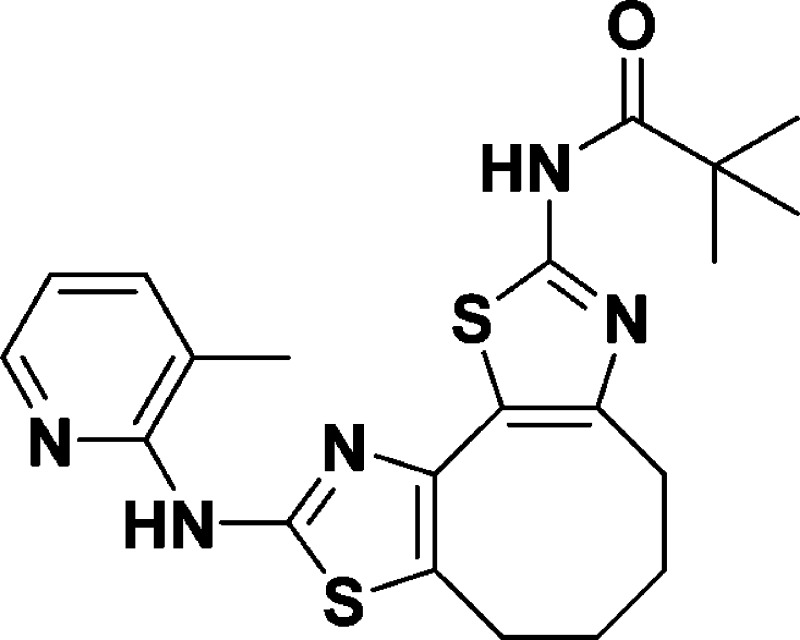
N-(9-((3-(Dimethylamino)phenyl)amino)-4,5,6,7-tetrahydrocycloocta[1,2-d:3,4-d′]bis(thiazole)-2-yl)pivalamide (10i)
The procedure for 8a and 10a was followed, except 1-(3-(dimethylamino)phenyl)thiourea
(0.197 g, 1.01 mmol, 1.0 equiv) was substituted, yielding a tan solid
(0.0817 g, 22%). 1H NMR (600 MHz, CDCl3): δ
8.92 (s, 1H), 8.26 (s, 1H), 7.12 (t, J = 8.1 Hz,
1H), 6.77 (t, J = 2.1 Hz, 1H), 6.60 (dd, J = 7.8, 1.9 Hz, 1H), 6.39 (d, J = 8.3
Hz, 1H), 2.92 (s, 6H), 2.90–2.67 (m, 4H), 1.88–1.69
(m, 4H), 1.26 (s, 9H). 13C NMR (150 MHz, CDCl3): δ 175.6, 161.8, 157.0, 151.5, 146.9, 141.5, 138.5, 129.8,
121.2, 121.1, 107.2, 106.5, 102.4, 40.6, 39.0, 30.0, 27.2, 26.8, 24.7,
24.5. HRMS (m/z): [M + H]+ calcd for C23H29N5OS2, 456.1892; found, 456.1884.
N-(9-((2-Methoxy-5-nitrophenyl)amino)-4,5,6,7-tetrahydrocycloocta[1,2-d:3,4-d′]bis(thiazole)-2-yl)pivalamide (10j)
The procedure for 8a and 10a was followed, except 1-(2-methoxy-5-nitrophenyl)thiourea
(0.229 g, 1.01 mmol, 1.0 equiv) was substituted, yielding a yellow
solid (0.0997 g, 34%). 1H NMR (600 MHz, CDCl3): δ 9.04 (d, J = 2.7 Hz, 1H), 8.90 (s, 1H),
7.77 (dd, J = 9.0, 2.7 Hz, 2H), 6.84 (d, J = 9.0 Hz, 1H), 3.94 (s, 3H), 2.90–2.72 (m, 4H),
1.86–1.67 (m, 4H), 1.28 (s, 9H). 13C NMR (150 MHz,
CDCl3): δ 175.6, 158.6, 157.0, 151.5, 147.2, 141.7,
139.2, 130.1, 122.9, 120.8, 117.6, 110.9, 108.9, 56.5, 39.1, 29.9,
27.2, 26.8, 24.6, 24.3. HRMS (m/z): [M + H]+ calcd for C22H25N5O4S2, 488.1426; found, 488.1415.
Acknowledgments
We thank Professor Makhluf J. Haddadin (American University of Beirut, Beirut, Lebanon) for helpful insights and Kelli Gottlieb for assistance collecting HRMS data. The authors thank the Tara K. Telford Fund for Cystic Fibrosis Research at UC Davis, the National Institutes of Health (DK072517 and GM076151), the National Science Foundation (CHE-0614756, CHE-0443516, CHE-0449845, CHE-9808183 – NMR spectrometers; CHE-030089–computer time from the Pittsburgh Supercomputer Center), and the Petroleum Research Fund of the American Chemical Society (grant 52801-ND4) for their generous support. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation [ACI-1053575].
Glossary
ABBREVIATIONS:
- cystic fibrosis transmembrane conductance regulator
(CFTR)
- cystic fibrosis
(CF)
- Fischer rat thyroid
(FRT)
- yellow fluorescent protein
(YFP)
- electron-withdrawing group
(EWG)
- electron-donating group
(EDG)
Supporting Information Available
1H and 13C NMR spectra as well as a corrector activity figure for compounds 9a, 10a, 9e, and 10c; HRMS data summary for all compounds; and computational data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bobadilla J. L.; Macek M. Jr.; Fine J. P.; Farrell P. M. Cystic fibrosis: a worldwide analysis of CFTR mutations — correlation with incidence data and application to screening. Hum. Mutat. 2002, 19, 575–606. [DOI] [PubMed] [Google Scholar]
- CF mutation database: genet.sickkids.on.ca/cftr/StatisticsPage.html (accessed 5/2/2014).
- Cystic Fibrosis Foundation Patient Registry 2012 Annual Data Report: http://www.cff.org/livingwithcf/qualityimprovement/patientregistryreport/ (accessed 5/2/2014).
- a Amaral M. D.; Kunzelmann K. Molecular targeting of CFTR as a therapeutic approach to cystic fibrosis. Trends Pharmacol. Sci. 2007, 28, 334–341. [DOI] [PubMed] [Google Scholar]; b Amaral M. D. CFTR and chaperones: processing and degradation. J. Mol. Neurosci. 2004, 23, 41–48. [DOI] [PubMed] [Google Scholar]; c Amaral M. D. Therapy through chaperones: sense or anti-sense? Cystic fibrosis as a model disease. J. Inherit. Metab. Dis. 2005, 29, 477–487. [DOI] [PubMed] [Google Scholar]
- a Pedemonte N.; Lukacs G. L.; Du K.; Caci E.; Zegarra-Moran O.; Galietta L. J. V.; Verkman A. S. Small-molecule correctors of defective Delta F508-CFTR cellular processing identified by high-throughput screening. J. Clin. Invest. 2005, 115, 2564–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Verkman A. S.; Galietta L. J. Chloride Channels as Drug Targets. Nat. Rev. Drug Discovery 2009, 8, 153–171. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lukacs G. L.; Verkman A. S. CFTR: Folding, Misfolding and Correcting the DeltaF508 Conformational Defect. Trends Mol. Med. 2012, 18, 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Okiyoneda T.; Veit G.; Dekkers J. F.; Bagdany M.; Soya N.; Xu H.; Roldan A.; Verkman A. S.; Kurth M.; Simon A.; Hegedus T.; Beekman J. M.; Lukacs G. L. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat. Chem. Biol. 2013, 9, 444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wang Y.; Wrennall J. A.; Cai Z.; Li H.; Sheppard D. N. Understanding how cystic fibrosis mutations disrupt CFTR function: From single molecules to animal models. Int. J. Biochem. Cell B 2014, 52, 47–57. [DOI] [PubMed] [Google Scholar]; b Ramsey B. W.; Davies J.; McElvaney N. G.; Tullis E.; Bell S. C.; Dřevínek P.; Griese M.; McKone E. F.; Wainwright C. E.; Konstan M. W.; Moss R.; Ratjen F.; Sermet-Gaudelus I.; Rowe S. M.; Dong Q.; Rodriguez S.; Yen K.; Ordoñez C.; Elborn J. S. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Van Goor F.; Yu H.; Burton B.; Hoffman B. J. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J. Cyst. Fibros. 2014, 13, 29–36. [DOI] [PubMed] [Google Scholar]
- a Deeks E. D. Ivacaftor: A Review of Its Use in Patients with Cystic Fibrosis. Drugs 2013, 73, 1595. [DOI] [PubMed] [Google Scholar]; b Kopeikin Z.; Yuksek Z.; Yang H.-Y.; Bompadre S. G.. Combined effects of VX-770 and VX-809 on several functional abnormalities of F508del-CFTR channels. J. Cystic Fibrosis 2014, DOI: 10.1016/j.jcf.2014.04.003. [DOI] [PubMed] [Google Scholar]
- Yu G. J.; Yoo C. L.; Yang B.; Lodewyk M. W.; Meng L.; El-Idreesy T. T.; Fettinger J. C.; Tantillo D. J.; Verkman A. S.; Kurth M. J. Potent s-cis-Locked Bithiazole Correctors of ΔF508 Cystic Fibrosis Transmembrane Conductance Regulator Cellular Processing for Cystic Fibrosis Therapy. J. Med. Chem. 2008, 51, 6044–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vistoli G.; Pedretti A.; Testa B. Assessing drug-likeness-what are we missing?. Drug Discovery Today 2008, 13, 285–294. [DOI] [PubMed] [Google Scholar]
- Du L.; Chen M.; Zhang Y.; Shen B. BlmIII and BlmIV Nonribosomal Peptide Synthetase-Catalyzed Biosynthesis of the Bleomycin Bithiazole Moiety Involving Both in Cis and in Trans Aminoacylation. Biochemistry 2003, 42, 9731–9740. [DOI] [PubMed] [Google Scholar]
- Dinner A. R.; Sali A.; Smith L. J.; Dobson C. M.; Karplus M. Understanding protein folding via free-energy surfaces from theory and experiment. Trends Biochem. Sci. 2000, 25, 331–339. [DOI] [PubMed] [Google Scholar]
- Yoo C. L.; Yu G. J.; Yang B.; Robins L. I.; Verkman A. S.; Kurth M. J. 4′-Methyl-4,5′-bithiazole-based correctors of defective DF508-CFTR cellular processing. Bioorg. Med. Chem. Lett. 2008, 18, 2610–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrung M. C.; Webste N. J. G. Mechanism of Intramolecular Photocycloadditons of Cyclooctenones. J. Org. Chem. 1987, 52, 3603–3613. [Google Scholar]
- Frisch M. J.; et al. Gaussian 09, revision B.01; Gaussian, Inc., Wallingford, CT, 2009. (full reference in Supporting Information).
- Shao Y.; et al. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191 (full reference in Supporting Information). [DOI] [PubMed] [Google Scholar]
- a Becke A. D. Density–functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]; b Becke A. D. A new mixing of Hartree–Fock and local-density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar]; c Lee C.; Yang W.; Parr R. G. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]; d Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar]
- a Zhao Y.; Truhlar D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]; b Zhao Y.; Truhlar D. G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [DOI] [PubMed] [Google Scholar]
- CYLview, 1.0b; Legault C. Y.Université de Sherbrooke, 2009. (http://www.cylview.org).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.