

Abstract
The development of inhibitors for protein–protein interactions frequently involves the mimicry of secondary structure motifs. While helical protein–protein interactions have been heavily targeted, a similar level of success for the inhibition of β-strand and β-sheet rich interfaces has been elusive. We describe an assessment of the full range of β-strand interfaces whose high-resolution structures are available in the Protein Data Bank. This analysis identifies complexes where a β-stand or β-sheet contributes significantly to binding. The results highlight the molecular recognition complexity in strand-mediated interactions relative to helical interfaces and offer guidelines for the construction of β-strand and β-sheet mimics as ligands for protein receptors. The online data set will potentially serve as an entry-point to new classes of protein–protein interaction inhibitors.
Protein–protein interactions (PPIs) regulate a wide array of cellular processes and are attractive targets for drug design.1−3 Successful approaches to the design of PPI inhibitors include high throughput screening of compound libraries, fragment-based screening, and the mimicry of interfacial protein segments that promote complex formation.4−6 Rational design of protein domain mimetics requires access to high-resolution structures of protein complexes and understanding of the features of protein interfaces.5,7−9 Mimicry of helical domains has successfully yielded inhibitors of intractable protein–protein interactions.4,10−13 This success has required knowledge of the helical regions key to the interactions as well as the design of synthetic scaffolds that can mimic the attributes of protein helices. To guide the design of helix mimics, we previously described analysis of protein complexes whose high-resolution structures have been deposited in the Protein Data Bank and cataloged interfacial helices that feature clusters of residues that contribute significantly to binding.14−17 The hot spot residues were characterized using computational alanine scanning mutagenesis analysis.18−20 Here, we describe a similar effort to decode protein interfaces that contain β-strand and β-sheet segments. To our knowledge, one similar effort to identify β-sheet interfaces has been reported.21 In this survey, Nowick, Baldi, and co-workers created a database of β-strands that form sheets with β-strands from another protein chain.
We identified roughly 15 000 β-strand motifs that make valuable contributions to the overall stability of the complex; mimicry of these strands will potentially lead to potent inhibitors. Our analysis shows that, as with α-helical interfaces, aromatic and hydrophobic hot spots are critical for strand-mediated protein–protein interactions.16 Backbone hydrogen bonding, which is utilized by strands but not α-helices for recognition, introduces unsystematic diversity to β-strand binding interactions.22 Single β-strands can be localized in nonenzymatic protein pockets much as is seen in canonical enzyme–substrate complexes.23 Strands participating in PPIs can also exist as part of a β-sheet where the partner protein recognizes side chains on one or multiple strands. β-strands may utilize side chain functionality on one face or both faces of a single strand for interaction with the partner protein. Lastly, β-strands may form hydrogen-bonding interactions with the partner or they may interact exclusively with side chain functionality. Our results offer impetus for the construction of synthetic strand scaffolds but suggest that multiple design strategies will be required to match the diversity present in β-strand-mediated protein–protein interactions.24
Results and Discussion
Peptidomimetics have a rich history as β-strand mimetics to inhibit enzyme activity.24,25 For example, the HIV-1 protease has been successfully targeted with a diverse set of clinically useful strand mimics.24,26,27 Similarly, synthetic strand and sheet mimics have been described as modulators of protein aggregation.28 Cell surface β-strand and β-sheet proteins have been targeted by macrocyclic peptides and synthetic antibodies. AP33 is a neutralizing antibody that binds a β-hairpin peptide epitope on the E2 envelope glycoprotein of hepatitis C virus, pertuzumab binds the receptor tyrosine kinase ErbB2, and cetuximab binds to EGFR’s extracellular domain.29−31 Grb2’s SH2 domain and the E3 ubiquitin ligase E6AP have both been the subjects of peptide or peptidomimetic macrocycle development.32−34 Many successful efforts to develop β-strand, β-hairpin, and β-sheet mimetic scaffolds have been undertaken,22,24,35−45 although the applications of these scaffolds to the disruption of PPIs remains limited.28,46−49
Here, we present a comprehensive analysis of β-strands in PPIs to construct a list of suitable targets. Our studies are centered on the identification of clusters of interfacial residues that contribute to ΔGbinding, with the hypothesis that mimicry of the disposition and orientation of these hot spot residues will lead to successful inhibitors.15,16 Several methods to evaluate the relative importance of different residues to a binding interaction have been described, with alanine scanning mutagenesis20 and ΔSASA50−52 calculations the most commonly employed. Alanine scanning consists of the serial generation (either computational or experimental) of alanine point mutants; point mutants leading to a large change in binding energy upon mutation to alanine (ΔΔG) are likely to be important wild-type residues.18,19,53 Commonly, residues whose mutation to alanine results in a decrease in binding energy of ΔΔG ≥ 1 kcal/mol are designated hot spot residues. In contrast, ΔSASA describes the amount of solvent-accessible surface area that is buried by the residue in question upon binding; the more surface area buried, the greater the entropy decrease upon binding due to desolvation. For this study, much as for our recent work on helical interfaces,17 both ΔΔG and ΔSASA are evaluated, though ΔΔG alone was used to determine if a strand interacts strongly with a binding partner.54
Procedure for Identifying β-Strand Interfaces in Protein–Protein Interactions
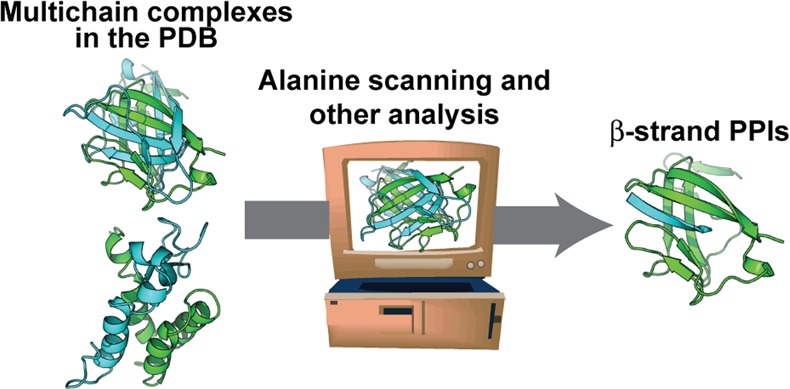
An overview of the approach is depicted in Figure 1. Crystal and NMR structures of protein complexes were obtained from the Protein Data Bank.55 For crystal structures, the multimodel biological assembly files were acquired. Each individual model was refined using the Rosetta “relax” protocol, which involves iterations of all-atom minimization with restraints followed by side-chain repacking. Following this procedure, the best-scoring model was retained for further analysis. Each pair of protein chains was extracted and separately processed by computational alanine scanning using RosettaScripts56 and by the ΔSASA calculation utility NACCESS.57 The secondary structure content of the chains in question was evaluated using the Dictionary of Secondary Structure Prediction (DSSP).58 Three or more consecutive residues with a hydrogen bonding pattern characteristic of β-strands, assigned “E” by DSSP, were considered to be β-strands for the purpose of this study. The online data set contains a listing of all high affinity β-strand complexes. For each high affinity strand entry, we recorded the total, per-residue average, and percent contribution of both ΔΔG and ΔSASA. We also recorded parameters such as length, distance between terminal hot spots, and the relative hot spot positions. The “MimeticScore” represents the sum of the three highest-ΔΔG hot spot residues, and may be used to compare entries in the entire data set.
Figure 1.

(a) Schematic for identification of protein interfaces in the Protein Data Bank (PDB) where a β-strand contributes significantly to complex formation. (b) Protocol for extracting β-strand complexes from multichain entries in the PDB. Biological assemblies from the PDB were analyzed using computational alanine scanning mutagenesis to create the data set hosted at www.nyu.edu/projects/arora/sippdb/. The data set allows division of β-strand interfaces into high-affinity and low-affinity interactions.
Classification of Interfacial β-Strands
We classified interfacial strands as strongly interacting or high affinity β-strands if they contained two or more hot spot residues. Hot spot residues are defined as residues whose mutation to alanine results in a concomitant reduction in the binding affinity of the complex by ΔΔG ≥ 1 Rosetta Energy Unit (or REU). Apparent “hot spots” of glycine and proline were omitted from consideration, since such point mutations likely lead to nonlocal effects that are poorly modeled by a fixed-backbone protocol; in particular, glycine to alanine mutations can result in clashes only present in the bound complex that could be relieved by backbone perturbations. ΔΔG values exceeding 8.0 REU were reduced to 8.0. We obtained an initial data set of 17,182 multiprotein entries, clustered to ensure <95% sequence identity, from the PDB. These entries resulted in 37 574 pairs of two-chain complexes; using these criteria, 14 940 high affinity β-strands were found. Roughly 38 000 interface β-strands feature zero or one hot spot residues and are classified as weakly interacting β-strands.
The average length of β-strands in protein complexes that are part of the PDB is five residues, and each strand averages at least one hot spot residue (Table 1). Comparison of the strongly and weakly interacting β-strands suggests that high affinity β-strands are critical to the overall stability of the complex; β-strands in these interfaces contribute 22% of the overall binding interactions, which is significant given the low number of interacting residues. The strongly interacting strands feature 2.5 hot spot residues with a high average per residue ΔΔG of 2.4 REU, implying that mimicry of these strands will lead to potent inhibitors of protein–protein interactions. Because backbone hydrogen bonds are a critical component of β-strand complexes,59 the alanine scanning ΔΔG values likely underrate the overall interaction energy but remain a good measure for assessing the sequence-specific contribution of each residue. Indeed, seminal work on β-sheet interactions, in which both strands are hydrogen bonded, has included in vitro alanine scanning and has found substantial sequence-specific effects.60,61 We have compiled the results in an online database hosted at www.nyu.edu/projects/arora/sippdb/. The database provides descriptions of each high affinity strand, including number of hot spot residues and per residue ΔΔG, with links to the PDB entry for each strand.
Table 1. Comparison of Statistics for Strongly and Weakly Interacting Interfacial β-Strands.
| strongly interacting β-strands (14 940) | weakly interacting β-strands (38 027) | all interfacial β-strands in PPIs (52 967) | |
|---|---|---|---|
| interface strand length, residues | 5.9 ± 2.3 | 4.4 ± 1.6 | 4.8 ± 2.0 |
| no. of hot spot residues | 2.5 ± 0.87 | 0.53 ± 0.49 | 1.1 ± 1.1 |
| per-residue ΔΔG, REU | 1.2 ± 0.65 | 0.47 ± 0.40 | 0.67 ± 0.57 |
| per-residue ΔSASA, Ǻ2 | –41 ± 20 | –22 ± 16 | –27 ± 19 |
| hot spot residue ΔΔG, REU | 2.4 ± 1.6 | 2.0 ± 0.96 | 2.1 ± 1.1 |
| % contribution to ΔΔGcomplex | 22 ± 15 | 9.7 ± 12 | 13 ± 14 |
| % contribution to ΔSASAcomplex | 17 ± 13 | 9.4 ± 11 | 11 ± 12 |
| distance between first and last hot spot residue, Å | 3.9 ± 1.8 | n/a | n/a |
Functional Distribution of β-Strands in All Multiprotein Complexes
β-Strand interactions are often associated with enzymes because protease substrates adopt extended conformations.23 In the construction of this database, we removed all enzyme/β-strand substrate interactions to have the results reflect participation of strands mediating protein–protein interactions. β-Strand interfaces in fact participate widely in protein–protein interactions that involve enzyme partners where the strand is not manipulated by the enzyme action. Only such β-strand interactions are included in our data set. β-strand interfaces are also involved in a diverse set of other biological processes, including transcriptional regulation and protein folding (Figure 2a). Comparison of β-strand and helix mediated interactions reveals that enzymes such as oxidoreductases, transferases, hydrolases, and lyases amount to 55% of β-strand interactions and 67% of α-helix mediated interactions, suggesting that protein–protein interactions represented in the PDB broadly involve at least one enzyme partner. This finding indicates that β-strand and helix mimetics may both serve as inhibitors of enzymatic function not by direct targeting of the catalytic pocket but by interrupting signal transduction pathways mediated by certain enzymes.
Figure 2.
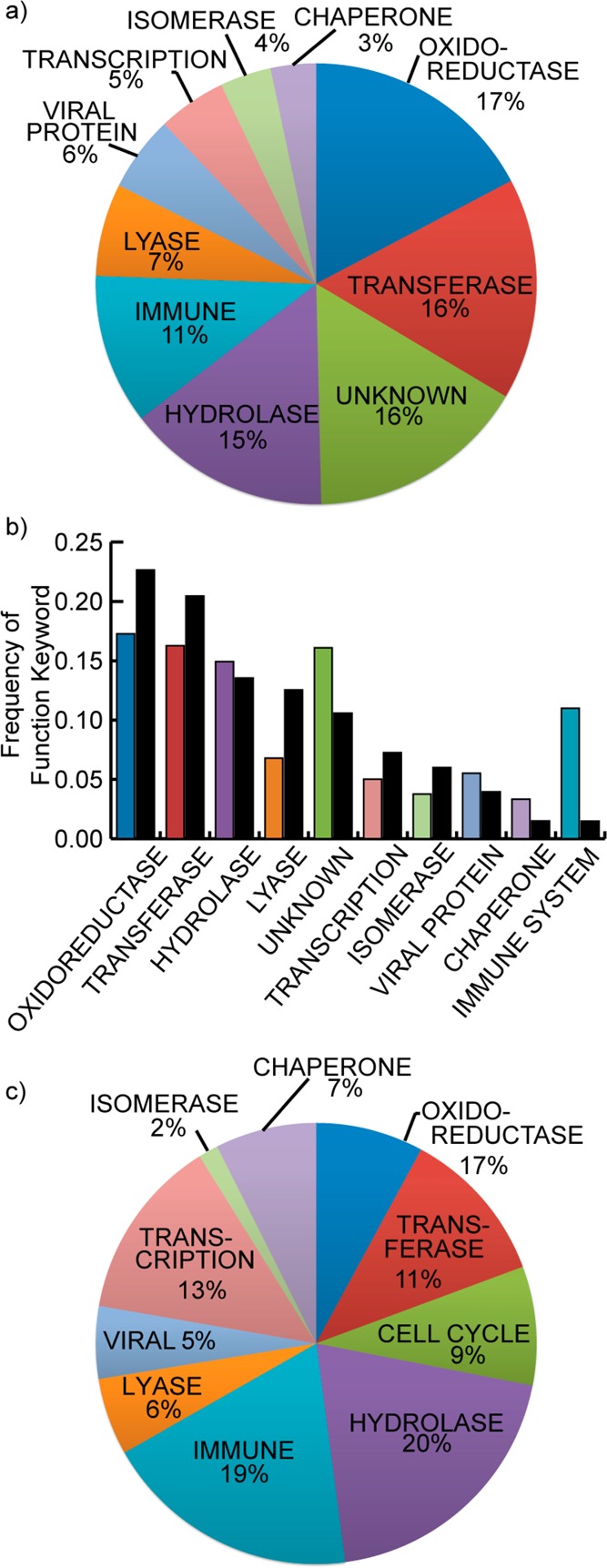
(a) Analysis of high affinity β-strand interfaces shows that these complexes participate in a wide-range of biological functions, but protein–protein interactions where one partner is an enzyme are the dominant category. (b) Comparison of the functions associated with the helix and strand data sets (helix in black; strand in colors) shows that the distribution of functions is similar across these two classes of secondary structure-mediated PPIs, with strands dominating interactions related to the immune system. (c) The functional distribution of strand-mediated nonimmunoglobulin heterodimers shows that PPIs with enzymatic function play a prominent role in different subsets of protein complexes, but other categories such as transcription are better represented in heterodimers without the influence of immunoglobin dimers.
We compared the functional distribution of β-strands in all multiprotein complexes in the PDB to a subset consisting of only heterodimeric β-strand interfaces to determine if different classes of strand-mediated complexes (i.e., homodimers and heterodimers) diverge in function. We considered a set of 2330 heterodimers and further eliminated entries whose protein names contained “heavy chain” and “light chain” to diminish the influence of immunoglobulin interactions, in which β-sheets are dominant secondary structures. The pruned data set contains 1221 complexes. Comparisons of functions associated with all biological assemblies (Figure 2a) and heterodimeric complexes only (Figure 2c) indicates that enzymatic complexes dominate β-strand PPIs across different types of complexes, but categories such as transcriptional complexes are better represented in the pruned set of heterodimers.
Contribution of Individual Residues to β-Strand Interactions
We analyzed the high affinity strand interfaces to assess the relative contribution of individual residues to protein complex formation (Figure 3). Hydrophobic and aromatic residues dominate the binding energy landscape, accounting for roughly 40% of the hot spot residues (Figure 3a). When normalized for natural abundance,62 we find that nonpolar aromatic residues, histidine, and arginine are overrepresented as hot spots at strand interfaces in comparison to polar residues (Figure 3b). These results correlate with the types of amino acids appearing as hot spot residues in protein interfaces,53,63,64 although our data set is considerably larger than those previously examined. The normalized distribution for β-strand hot spots can be compared to those of α-helices (Figure 3c). Interestingly, the energetic contribution of individual residues to helical or strand interfaces is roughly the same for individual amino acids. The comparison suggests that complexation of strand and helical interfaces share similar side chain recognition principles.
Figure 3.
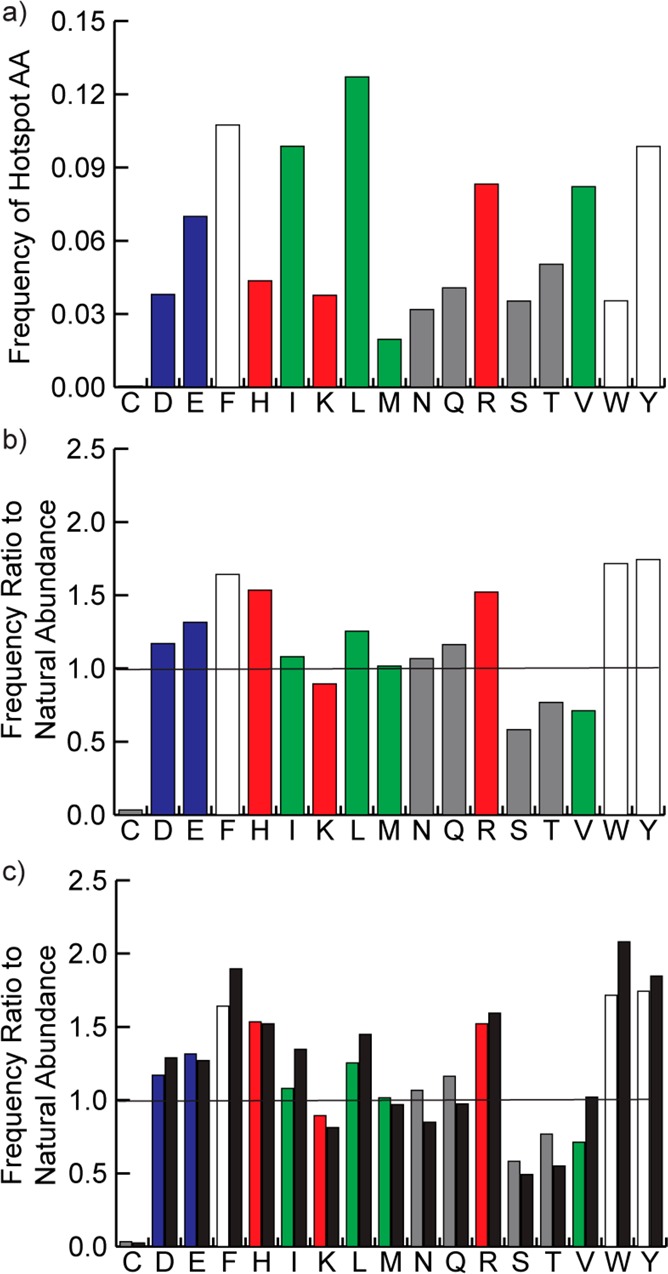
(a) Distribution of hot spot residues in high affinity β-strands. (b) The data from part a normalized to the natural abundance in strands of each amino acid. (c) The normalized distribution of strand hot spots compared to the normalized distribution of helical hot spots (black bars).
We analyzed the relative contribution of each hot spot amino acid to β-strand binding interactions (Figure 4). The relative binding contribution (ΔΔG) of different hot spot amino acids correlates well with the normalized occurrence shown in Figure 3b but not with the raw data in Figure 3a. While leucine is the most prevalent hot spot residue in β-strands, its contribution to binding is less significant when it does appear. In contrast, the aromatic residues and arginine are both more prevalent as hot spots compared to their natural abundance, and they serve as strong hot spot residues when they do appear.
Figure 4.
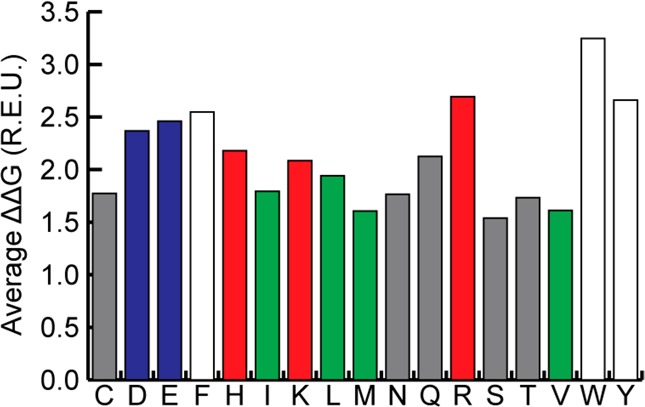
Average ΔΔG of different hot spot amino acids found in strongly interacting β-strands. Aromatic residues and arginine make for the strongest interactions, while small polar and aliphatic residues are the weakest.
Geometrical Diversity in β-Strand Interfaces
β-Strand-mediated interfaces are geometrically diverse,22 especially in comparison to the order presented by helical PPIs, which primarily differ in the number of helical faces involved in binding interactions.16 The contribution of backbone hydrogen bonding to strand interactions, but not helical motifs, is an important factor that enhances diversity of the recognition motifs available to strands (Figure 5).
Figure 5.
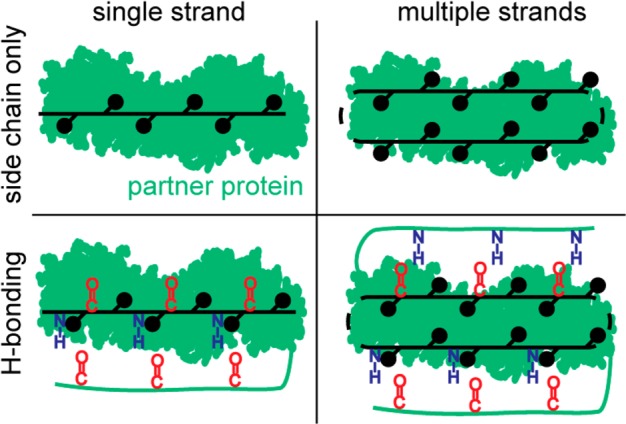
Schematic representing simplified β-strand interactions with a complementary protein receptor shown as green surface. β-Strands utilize a diverse range of binding strategies; for example, β-strands may interact alone or as part of a β-sheet of two or more strands, and β-strands may form hydrogen bonding interactions with the partner or they may interact exclusively with the side chain functionality. Additionally, a β-strand may interact with the side chains of only one face, or it may employ both faces for interaction with the partner protein.
To categorize this diversity, we began by examining the binding energetics of β-strands on different faces to determine if hot spots are largely featured on one face, as with α-helices. The data reveals that high affinity β-strands can be divided into three categories (Figure 6): those with two faces contributing equally to the interaction, those with one face that contributes almost all of the ΔΔG (90% or more), and those where both faces contribute unequally but significantly to the interaction. The results indicate that only a quarter of the β-strands employ only one face for interactions. Although hot spot residues are typically unequally divided on the two strand faces, the analysis suggests that synthetic scaffolds would typically need to mimic both faces to be broadly useful as inhibitors of PPIs.
Figure 6.

Hot spot residues are unevenly distributed among the two faces of a β-strand, with the higher affinity face on average controlling 70% of the total ΔΔG. (a) A pie chart describing three approximate classifications of β-strands. (b) A histogram of the percentage of the total ΔΔG associated with the stronger-interacting face, providing a higher-resolution account of the data summarized by the pie chart.
We evaluated the different types of interactions β-strands participate in with binding partners in the context of transcriptional complexes. Manually filtering for unique strands with three or more hot spot residues resulted in a set of 172 entries describing 133 distinct protein–protein interactions. Two-thirds of these entries described a β-sheet-formation interaction, 95% of which were antiparallel in orientation. Across the PDB, antiparallel β-sheet orientations are more prevalent, on the order of 3:1, as compared to parallel sheets.65 Antiparallel arrangements are thought to possess better hydrogen bonding geometry66 and overall energetics.67 The greater prevalence of antiparallel strand orientations in protein–protein interactions may reflect the more stringent energetic criteria for quaternary versus tertiary structure association.
In these β-sheet interactions, strands made an average of ten hydrogen bond acceptors and donors available to the partner protein, with two-thirds of the carbonyls and amide N–H’s participating in hydrogen bonding. Hydrogen bonding potential between partner carbonyl C=O and amide N–H groups was judged using the standard distance and angle criteria: a hydrogen–oxygen distance less than 2.7 Å (i.e., their van der Waals radii are in contact) and a N–H–O angle greater than 110°. In contrast, hydrogen bonding potential was not fulfilled in interface strands that do not form sheets, with only 19% of the exposed hydrogen bond donors and acceptors forming hydrogen bonds. Multivalent interactions are also common in this set, with roughly two-thirds of the interfaces using more than one high affinity strand for binding interactions. This result suggests that hairpin and β-sheet mimetics will have a substantial role as protein–protein interaction inhibitors.22
Survey of the Data Set
Table 2 depicts five representative examples present in the database that display the diversity of β-strand interfaces. For each entry, a cartoon representation of the complex with the strand(s) highlighted in magenta is provided alongside a representation of the strands in sticks and the partner protein in cartoon with pertinent side-chains displayed in sticks. Entry 1 depicts a high affinity β-strand found in the ternary complex between IKBβ and NFkappaB p65 homodimer, at the p65 homodimer interface. Except for a carbonyl-histidine hydrogen bond, the entire interaction is mediated by side-chain packing interactions and a glutamate-arginine charged interaction. Entry 2 depicts the bacillus transcriptional regulator C-125, as an example of a β-strand that forms a sheet at the interface. Entry 3 depicts part of Bcl6 corepressor making a unique interaction with RING finger protein 1. Exactly one face of hydrogen bonds (alternating amides and carbonyls) is occupied by a strand in the partner protein. Meanwhile, one face of the strand (an F, F, and L) pack in a hydrophobic groove formed by a helix of RING, while the other face (M, E, S) interact with one face of the aforementioned partner strand. Bcl6 contains an additional high affinity strand, antiparallel to the one depicted, which does not form hydrogen bonds with the partner; the two may be mimicked with a β-hairpin. Entry 4 shows a β-hairpin found in the Rap30/Rap74 complex of human transcription initiation factor IIF. Entry 5 shows a pair of nonhairpin strands from the HTH-type transcriptional regulator LRPC, as an example of a complex in which one chain possesses multiple high affinity strands without that do not participate in a hairpin relationship.
Table 2. Selection of the Diverse β-Strands Catalogued by SIPPDBa.
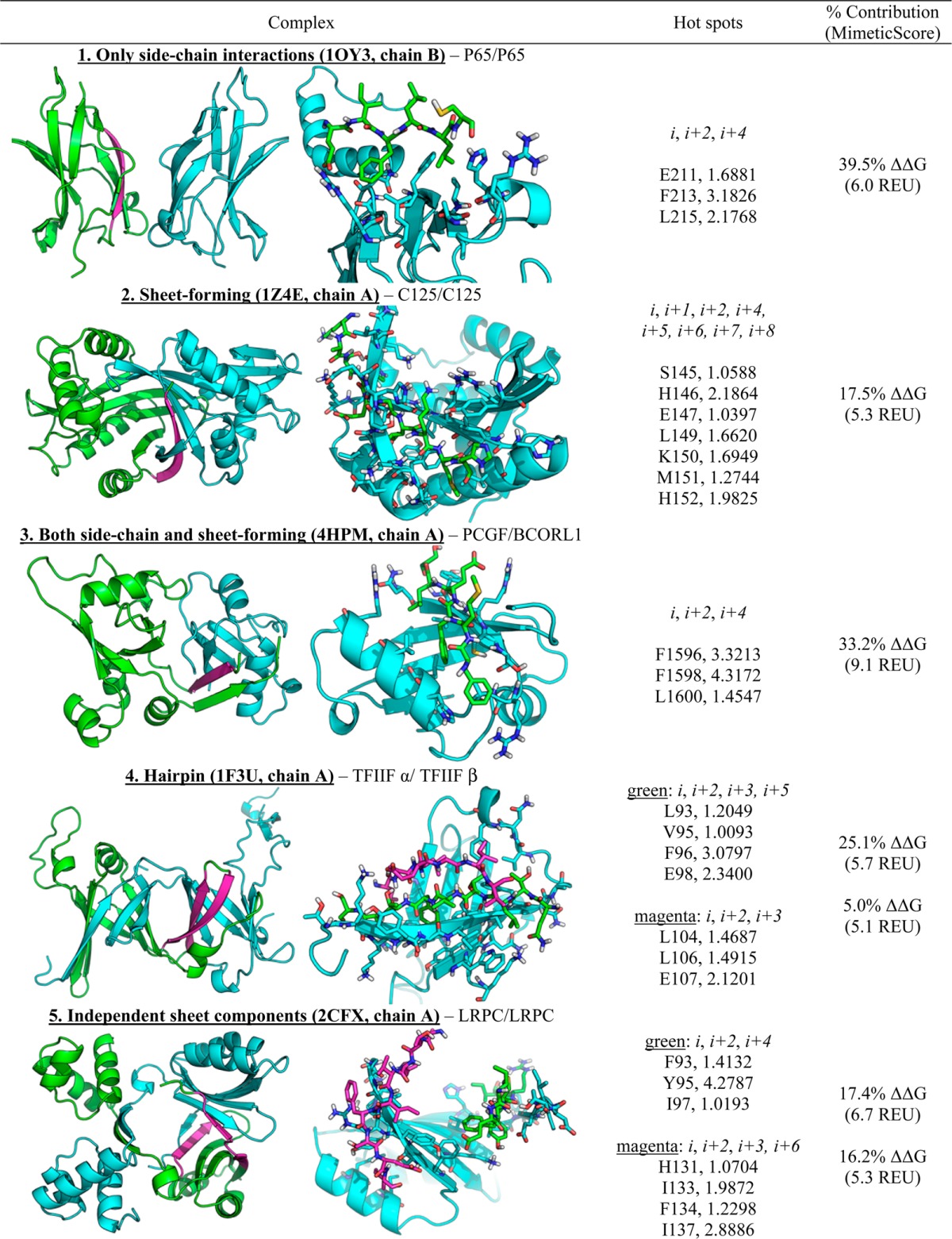
MimeticScore, in the final column, is the sum of the ΔΔG values of the three highest ΔΔG hot spot residues.
In summary, we have cataloged β-strand interfaces whose structures are available in the Protein Data Bank according to the relative contribution of individual strands to complex formation. Our evaluations suggest that there are roughly 15 000 high affinity β-strands mediated protein–protein interactions. Mimics of these strands would lead to potent inhibitors as has been illustrated for helical PPIs; however, the variety and complexity of β-strand-based interface structures is staggering and suggests that a range of synthetic approaches will be required for inhibition. A large proportion of β-strands participate in sheet interactions, which points to an important role for β-hairpin and β-sheet mimics.
Methods
Given a PDB file with <95% sequence homology to those already in the database, the following general procedure was employed. First, the PDB was “cleaned,” removing HETATM entries and any crystal water. Then, the PDB was separated into its distinct MODEL entries. One to four models are common for biological assemblies derived from X-ray structures, while 20 are typical for NMR structures; viral capsid structures can have 60. Each model was subjected to refinement using Rosetta’s relax protocol, with atoms restrained to their initial coordinates and with extra side chain rotamers generated for the first and second χ-dihedral angles. Five refined structures were generated for each model, and the lowest-energy model (using Rosetta’s talaris2013 scoring function) was selected for further processing. This relaxation procedure identifies the model from a multimodel PDB file that is best compatible with the Rosetta scoring function. Moreover, it helps to correct structural errors like steric clashes, misassigned side chain amides in glutamine and asparagine, and problematic rotamers. Correcting these errors in the starting structure prior to alanine scanning avoids an asymmetrical comparison between a poor input structure and a repaired alanine point mutant.
We applied the DSSP program to generate secondary structure assignments for that best model and then split it into individual files for each chain and for each pair of chains. We performed alanine scanning using the RosettaScripts AlaScan filter, averaging the result of 20 applications for better convergence and again using talaris2013 for scoring.
The subsequent data were processed with Perl scripts to identify strands (i.e., sequences of three or more residues long) with two or more hot spot residues (ΔΔG > 1.0 REU). Those interfaces with such qualifying strands were processed using the SASA calculation program NACCESS (the program was run on the files for each individual chain and on the complex file) and further analyzed to produce the data set available online at http://www.nyu.edu/projects/arora/sippdb/.
Acknowledgments
This work was financially supported by the National Institutes of Health (R01GM073943) and the National Science Foundation (CHE-1151554). A.M.W. thanks New York University for the Kramer Fellowship.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- London N.; Raveh B.; Schueler-Furman O. (2013) Druggable protein–protein interactions—From hot spots to hot segments. Curr. Opin. Chem. Biol. 17, 952–959. [DOI] [PubMed] [Google Scholar]
- Ryan D. P.; Matthews J. M. (2005) Protein–protein interactions in human disease. Curr. Opin. Struct. Biol. 15, 441–446. [DOI] [PubMed] [Google Scholar]
- Zinzalla G.; Thurston D. E. (2009) Targeting protein–protein interactions for therapeutic intervention: A challenge for the future. Future Med. Chem. 1, 65–93. [DOI] [PubMed] [Google Scholar]
- Azzarito V.; Long K.; Murphy N. S.; Wilson A. J. (2013) Inhibition of α-helix-mediated protein–protein interactions using designed molecules. Nat. Chem. 5, 161–173. [DOI] [PubMed] [Google Scholar]
- Arkin M. R.; Wells J. A. (2004) Small-molecule inhibitors of protein–protein interactions: Progressing towards the dream. Nat. Rev. Drug. Discovery 3, 301–317. [DOI] [PubMed] [Google Scholar]
- Cesa L. C.; Patury S.; Komiyama T.; Ahmad A.; Zuiderweg E. R. P.; Gestwicki J. E. (2013) Inhibitors of difficult protein–protein interactions identified by high-throughput screening of multiprotein complexes. ACS Chem. Biol. 8, 1988–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. D.; Dugan A.; Gestwicki J. E.; Mapp A. K. (2012) Fine-tuning multiprotein complexes using small molecules. ACS Chem. Biol. 7, 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj M.; Bullock B. N.; Arora P. S. (2013) Plucking the high hanging fruit: A systematic approach for targeting protein–protein interactions. Bioorg. Med. Chem. 21, 4051–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells J. A.; McClendon C. L. (2007) Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature 450, 1001–1009. [DOI] [PubMed] [Google Scholar]
- Verdine G. L.; Hilinski G. J. (2012) Stapled peptides for intracellular drug targets. Methods Enzymol. 503, 3–33. [DOI] [PubMed] [Google Scholar]
- Harrison R. S.; Shepherd N. E.; Hoang H. N.; Ruiz-Gomez G.; Hill T. A.; Driver R. W.; Desai V. S.; Young P. R.; Abbenante G.; Fairlie D. P. (2010) Downsizing human, bacterial, and viral proteins to short water-stable α-helices that maintain biological potency. Proc. Natl. Acad. Sci. U.S.A. 107, 11686–11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushal S.; Lao B. B.; Henchey L. K.; Dubey R.; Mesallati H.; Traaseth N. J.; Olenyuk B. Z.; Arora P. S. (2013) Protein domain mimetics as in vivo modulators of hypoxia-inducible factor signaling. Proc. Natl. Acad. Sci. U.S.A. 110, 15602–15607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma M. D.; Haase H. S.; Peterson-Kaufman K. J.; Lee E. F.; Clarke O. B.; Colman P. M.; Smith B. J.; Horne W. S.; Fairlie W. D.; Gellman S. H. (2012) Evaluation of diverse α/β-backbone patterns for functional α-helix mimicry: Analogues of the Bim BH3 domain. J. Am. Chem. Soc. 134, 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochim A. L.; Arora P. S. (2009) Assessment of helical interfaces in protein–protein interactions. Mol. BioSyst. 5, 924–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochim A. L.; Arora P. S. (2010) Systematic analysis of helical protein interfaces reveals targets for synthetic inhibitors. ACS Chem. Biol. 5, 919–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock B. N.; Jochim A. L.; Arora P. S. (2011) Assessing helical protein interfaces for inhibitor design. J. Am. Chem. Soc. 133, 14220–14223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergey C. M.; Watkins A. M.; Arora P. S. (2013) HippDB: A database of readily targeted helical protein–protein interactions. Bioinformatics 29, 2806–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clackson T.; Wells J. A. (1995) A hot-spot of binding-energy in a hormone–receptor interface. Science 267, 383–386. [DOI] [PubMed] [Google Scholar]
- Cunningham B. C.; Wells J. A. (1989) High-resolution epitope mapping of hGH–receptor interactions by alanine-scanning mutagenesis. Science 244, 1081–1085. [DOI] [PubMed] [Google Scholar]
- Kortemme T.; Kim D. E.; Baker D. (2004) Computational alanine scanning of protein–protein interfaces. Sci. STKE 2004, pl2. [DOI] [PubMed] [Google Scholar]
- Dou Y.; Baisnee P. F.; Pollastri G.; Pecout Y.; Nowick J.; Baldi P. (2004) ICBS: A database of interactions between protein chains mediated by β-sheet formation. Bioinformatics 20, 2767–2777. [DOI] [PubMed] [Google Scholar]
- Cheng P. N.; Pham J. D.; Nowick J. S. (2013) The supramolecular chemistry of β-sheets. J. Am. Chem. Soc. 135, 5477–5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyndall J. D.; Nall T.; Fairlie D. P. (2005) Proteases universally recognize β-strands in their active sites. Chem. Rev. 105, 973–999. [DOI] [PubMed] [Google Scholar]
- Loughlin W. A.; Tyndall J. D. A.; Glenn M. P.; Fairlie D. P. (2004) β-Strand mimetics. Chem. Rev. 104, 6085–6117. [DOI] [PubMed] [Google Scholar]
- Ripka A. S.; Rich D. H. (1998) Peptidomimetic design. Curr. Opin. Chem. Biol. 2, 441–452. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Chapsal B. D.; Weber I. T.; Mitsuya H. (2007) Design of HIV protease inhibitors targeting protein backbone: An effective strategy for combating drug resistance. Acc. Chem. Res. 41, 78–86. [DOI] [PubMed] [Google Scholar]
- Smith A. B.; Hirschmann R.; Pasternak A.; Guzman M. C.; Yokoyama A.; Sprengeler P. A.; Darke P. L.; Emini E. A.; Schleif W. A. (1995) Pyrrolinone-Based HIV Protease Inhibitors. Design, Synthesis, and Antiviral Activity: Evidence for Improved Transport. J. Am. Chem. Soc. 117, 11113–11123. [Google Scholar]
- Cheng P.-N.; Liu C.; Zhao M.; Eisenberg D.; Nowick J. S. (2012) Amyloid β-sheet mimics that antagonize protein aggregation and reduce amyloid toxicity. Nat. Chem. 4, 927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter J. A.; Owsianka A. M.; Jeffery N.; Matthews D. J.; Keck Z.-Y.; Lau P.; Foung S. K. H.; Taylor G. L.; Patel A. H. (2012) Toward a Hepatitis C Virus Vaccine: the Structural Basis of Hepatitis C Virus Neutralization by AP33, a Broadly Neutralizing Antibody. J. Virol. 86, 12923–12932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin M. C.; Carey K. D.; Vajdos F. F.; Leahy D. J.; de Vos A. M.; Sliwkowski M. X. (2004) Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 5, 317–328. [DOI] [PubMed] [Google Scholar]
- Li S.; Schmitz K. R.; Jeffrey P. D.; Wiltzius J. J. W.; Kussie P.; Ferguson K. M. (2005) Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 7, 301–311. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Voigt J.; Wu J. X.; Yang D. J.; Burke T. R. (2001) Macrocyclization in the design of a conformationally constrained Grb2 SH2 domain inhibitor. Bioorg. Med. Chem. Lett. 11, 1889–1892. [DOI] [PubMed] [Google Scholar]
- Phan J.; Shi Z. D.; Burke T. R. Jr.; Waugh D. S. (2005) Crystal structures of a high-affinity macrocyclic peptide mimetic in complex with the Grb2 SH2 domain. J. Mol. Biol. 353, 104–115. [DOI] [PubMed] [Google Scholar]
- Yamagishi Y.; Shoji I.; Miyagawa S.; Kawakami T.; Katoh T.; Goto Y.; Suga H. (2011) Natural product-like macrocyclic N-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library. Chem. Biol. 18, 1562–1570. [DOI] [PubMed] [Google Scholar]
- Rose G. D., Glerasch L. M., and Smith J. A. (1985) Turns in peptides and proteins, in Advances in Protein Chemistry (Anfinsen C.B., Edsall J. T., and Frederic M. R., Eds.), pp 1–109, Academic Press, New York. [DOI] [PubMed] [Google Scholar]
- Freire F.; Gellman S. H. (2009) Macrocyclic design strategies for small, stable, parallel β-sheet scaffolds. J. Am. Chem. Soc. 131, 7970–7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanger H. E.; Gellman S. H. (1998) Rules for antiparallel β-sheet design: d-Pro-Gly is superior to l-Asn-Gly for β-hairpin nucleation. J. Am. Chem. Soc. 120, 4236–4237. [Google Scholar]
- Robinson J. A. (2008) β-Hairpin peptidomimetics: Design, structures, and biological activities. Acc. Chem. Res. 41, 1278–1288. [DOI] [PubMed] [Google Scholar]
- Smith A. B.; Guzman M. C.; Sprengeler P. A.; Keenan T. P.; Holcomb R. C.; Wood J. L.; Carroll P. J.; Hirschmann R. (1994) De-novo design, synthesis, and X-ray crystal-structures of pyrrolinone-based β-strand peptidomimetics. J. Am. Chem. Soc. 116, 9947–9962. [Google Scholar]
- Phillips S. T.; Rezac M.; Abel U.; Kossenjans M.; Bartlett P. A. (2002) ″@-tides″: The 1,2-dihydro-3(6H)-pyridinone unit as a β-strand mimic. J. Am. Chem. Soc. 124, 58–66. [DOI] [PubMed] [Google Scholar]
- Wyrembak P. N.; Hamilton A. D. (2009) Alkyne-linked 2,2-disubstituted-indolin-3-one oligomers as extended β-strand mimetics. J. Am. Chem. Soc. 131, 4566–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelo N. G.; Arora P. S. (2005) Nonpeptidic foldamers from amino acids: Synthesis and characterization of 1,3-substituted triazole oligomers. J. Am. Chem. Soc. 127, 17134–17135. [DOI] [PubMed] [Google Scholar]
- Lingard H.; Han J. T.; Thompson A. L.; Leung I. K. H.; Scott R. T. W.; Thompson S.; Hamilton A. D. (2014) Diphenylacetylene-linked peptide strands induce bidirectional β-sheet formation. Angew. Chem., Int. Ed. 53, 3650–3653. [DOI] [PubMed] [Google Scholar]
- Fuller A. A.; Du D.; Liu F.; Davoren J. E.; Bhabha G.; Kroon G.; Case D. A.; Dyson H. J.; Powers E. T.; Wipf P.; Gruebele M.; Kelly J. W. (2009) Evaluating β-turn mimics as β-sheet folding nucleators. Proc. Natl. Acad. Sci. U.S.A. 106, 11067–11072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C. W.; Sun Y.; Del Valle J. R. (2012) Substituted imidazo[1,2-a]pyridines as β-strand peptidomimetics. Org. Lett. 14, 6162–6165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond M. C.; Harris B. Z.; Lim W. A.; Bartlett P. A. (2006) β-Strand peptidomimetics as potent PDZ domain ligands. Chem. Biol. 13, 1247–1251. [DOI] [PubMed] [Google Scholar]
- Fasan R.; Dias R. L.; Moehle K.; Zerbe O.; Vrijbloed J. W.; Obrecht D.; Robinson J. A. (2004) Using a β-hairpin to mimic an α-helix: Cyclic peptidomimetic inhibitors of the p53-HDM2 protein–protein interaction. Angew. Chem., Int. Ed. Engl. 43, 2109–2112. [DOI] [PubMed] [Google Scholar]
- Huang Z.; Zhang M.; Burton S. D.; Katsakhyan L. N.; Ji H. (2014) Targeting the Tcf4 G13ANDE17 binding site to selectively disrupt β-catenin/T-cell factor protein–protein interactions. ACS Chem. Biol. 9, 193–201. [DOI] [PubMed] [Google Scholar]
- Zutshi R.; Franciskovich J.; Shultz M.; Schweitzer B.; Bishop P.; Wilson M.; Chmielewski J. (1997) Targeting the dimerization interface of HIV-1 protease: Inhibition with cross-linked interfacial peptides. J. Am. Chem. Soc. 119, 4841–4845. [Google Scholar]
- Lee B.; Richards F. M. (1971) Interpretation of protein structures—Estimation of static accessibility. J. Mol. Biol. 55, 379–400. [DOI] [PubMed] [Google Scholar]
- Lo Conte L.; Chothia C.; Janin J. (1999) The atomic structure of protein–protein recognition sites. J. Mol. Biol. 285, 2177–2198. [DOI] [PubMed] [Google Scholar]
- Fischer T. B.; Holmes J. B.; Miller I. R.; Parsons J. R.; Tung L.; Hu J. C.; Tsai J. (2006) Assessing methods for identifying pair-wise atomic contacts across binding interfaces. J. Struct. Biol. 153, 103–112. [DOI] [PubMed] [Google Scholar]
- Bogan A. A.; Thorn K. S. (1998) Anatomy of hot spots in protein interfaces. J. Mol. Biol. 280, 1–9. [DOI] [PubMed] [Google Scholar]
- Koes D. R.; Camacho C. J. (2012) Small-molecule inhibitor starting points learned from protein–protein interaction inhibitor structure. Bioinformatics 28, 784–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleishman S. J.; Leaver-Fay A.; Corn J. E.; Strauch E. M.; Khare S. D.; Koga N.; Ashworth J.; Murphy P.; Richter F.; Lemmon G.; Meiler J.; Baker D. (2011) RosettaScripts: A scripting language interface to the Rosetta macromolecular modeling suite. PLoS One 6, e20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard S. J., and Thornton J. M. (1993) NACCESS; Department of Biochemistry and Molecular Biology, University College, London. [Google Scholar]
- Kabsch W.; Sander C. (1983) Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- Pedersen S. W.; Pedersen S. B.; Anker L.; Hultqvist G.; Kristensen A. S.; Jemth P.; Stromgaard K. (2014) Probing backbone hydrogen bonding in PDZ/ligand interactions by protein amide-to-ester mutations. Nat. Commun. 5, 3215. [DOI] [PubMed] [Google Scholar]
- Smith C. K.; Regan L. (1995) Guidelines for protein design—The Energetics of β-sheet side-chain interactions. Science 270, 980–982. [DOI] [PubMed] [Google Scholar]
- Blasie C. A.; Berg J. M. (1997) Electrostatic interactions across a β-sheet. Biochemistry 36, 6218–6222. [DOI] [PubMed] [Google Scholar]
- Nelson D. L., Lehninger A. L., and Cox M. M. (2008) Principles of Biochemistry, 5th ed., W.H. Freeman, New York. [Google Scholar]
- Jones S.; Thornton J. M. (1995) Protein–protein interactions—A review of protein dimer structures. Prog. Biophys. Mol. Biol. 63, 31–65. [DOI] [PubMed] [Google Scholar]
- Guharoy M.; Chakrabarti P. (2007) Secondary structure based analysis and classification of biological interfaces: Identification of binding motifs in protein–protein interactions. Bioinformatics 23, 1909–1918. [DOI] [PubMed] [Google Scholar]
- Caudron B.; Jestin J. L. (2012) Sequence criteria for the anti-parallel character of protein β-strands. J. Theor. Biol. 315, 146–149. [DOI] [PubMed] [Google Scholar]
- Zhao Y. L.; Wu Y. D. (2002) A theoretical study of β-sheet models: Is the formation of hydrogen-bond networks cooperative?. J. Am. Chem. Soc. 124, 1570–1571. [DOI] [PubMed] [Google Scholar]
- Wang Z. X.; Wu C.; Lei H. X.; Duan Y. (2007) Accurate ab initio study on the hydrogen-bond pairs in protein secondary structures. J. Chem. Theory Comput. 3, 1527–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]