

Abstract
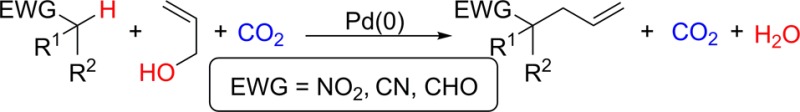
The direct coupling of allyl alcohols with nitroalkanes, nitriles, and aldehydes using catalytic Pd(PPh3)4 has been accomplished via activation of C–OH bonds with CO2. The in situ formation of carbonates from alcohols and CO2 facilitates oxidative addition to Pd to form reactive π-allylpalladium intermediates. In addition, the formation of a strong base activates nucleophiles toward the reaction with the π-allylpalladium electrophile. Overall, this atom economical reaction provides a new C–C bond without the use of an external base and generates water as the only byproduct.
Allylic alcohols are intriguing surrogates to traditional activated electrophiles used in transition-metal-catalyzed allylation reactions due to their wide availability, facile synthesis, and low toxicity. The condensation of an alcohol with a C–H bond to eliminate water and form a new C–C bond is topologically obvious in addition to being step and atom economical.1 However, alcohols often possess low reactivity because the C–O bond is strong and hydroxide is a poor leaving group.2 This challenge has resulted in the development of methods utilizing Lewis3,4 or Brønsted acids5 to activate allylic alcohols toward coupling with a variety of substrates. We sought to find a straightforward method using relatively benign reagents to activate allyl alcohols directly toward the allylation of weakly acidic pronucleophiles.6 Herein we report an in situ activation of allylic alcohols with CO2 that simultaneously generates active electrophiles and a requisite base to activate pronucleophiles (Scheme 1).
Scheme 1. CO2-Catalyzed Allylation.
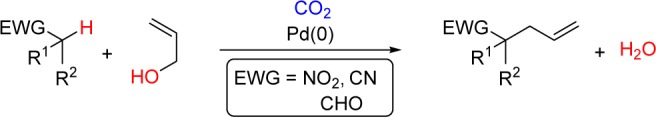
Since it is known that alcohols react reversibly with carbon dioxide to form carbonic acids and carbonates in situ,7 CO2 should activate alcohols toward transition-metal-catalyzed allylic alkylation reactions. Importantly, Yamamoto has demonstrated that CO2 can activate allyl alcohols toward substitution; however, the C–C bond formation was limited to highly stabilized malonate-like enolates (pKa ∼11–14 in DMSO).8,9 Using these initial results as inspiration, we sought to develop a synthetically useful method for the intermolecular allylation of weakly acidic nucleophiles such as nitroalkanes, nitriles, and aldehydes.
Based on Yamamoto’s findings we imagined that nucleophilic attack of the allyl alcohol on CO2 would form an allyl carbonate that is activated toward oxidative addition (Scheme 2). Decarboxylation of the resulting bicarbonate to generate CO2 and hydroxide might allow deprotonation of even weakly acidic C–H bonds to activate pronucleophiles.10 Subsequent nucleophilic attack on the palladium π-allyl complex is expected to form a new C–C bond and regenerate the active catalyst while producing water as the only byproduct.
Scheme 2. Proposed CO2-Catalyzed C–O Bond Activation.

An initial screen of reaction conditions revealed that the use of a polar aprotic solvent, Pd(PPh3)4, and 1 atm of CO2 heated in a sealed vial overnight afforded 1a in good yield (eq 1). When the CO2 was replaced with argon no appreciable amount of 1a was detected by 1H NMR spectroscopy of the crude reaction mixture.
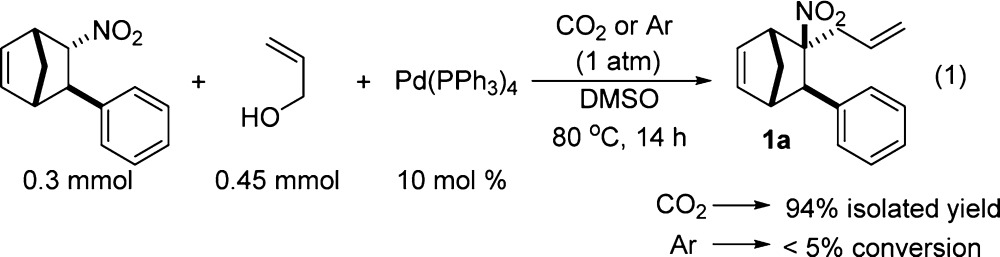 |
1 |
To investigate the substrate scope, the aforementioned reaction conditions were applied to the reactions of other nitroalkanes (Scheme 3). Under these conditions, a variety of substrates that were readily derived from Diels–Alder,11 Baylis–Hillman,12 and Henry13 reactions were allylated in good to excellent yields.
Scheme 3. Allylation of Nitroalkanes via CO2 Activation,b.

Nitroalkane (0.3 mmol) and allyl alcohol (0.45 mmol) in 1.75 mL of DMSO under 1 atm of CO2. b Isolated yields. c Ar replaced CO2 (% conversion via crude 1H NMR). d Allyl alcohol (0.9 mmol), 160 °C, 20 min in a microwave reactor.
Both cyclic and acyclic nitroalkanes were activated by the standard reaction conditions. A variety of functional groups including olefins (1a,c,h), an α,β-unsaturated ester (1l), ethers (1e,f), and indole (1d) were compatible with the reaction conditions. Good to excellent diastereoselectivities were observed with cyclic nitroalkanes (1a,c,h,j). In addition, 1,2-stereocontrol was good even with acyclic nitroalkanes bearing β-ethers (1e,f). Allylation also occurred in high yield directly from β-methallyl alcohol (1j).
Unfortunately, allyl alcohols such as prenyl and crotyl alcohol which bear alkyl substituents were not compatible with the standard reaction conditions.
Lastly, the allylation could be performed in just 20 min under microwave irradiation, although additional equivalents of alcohol were required (1k). Again, replacing CO2 with Ar under microwave conditions did not produce significant quantities of the desired product.
While it was exciting that nitroalkanes could be allylated in high yield under conditions of CO2 activation, nitroalkanes are relatively acidic (pKa ∼17 in DMSO).14 To probe the strength of the base generated in situ under our reaction conditions, we turned our attention toward the allylation of tertiary nitriles (pKa ∼23–25 in DMSO).15
To begin, the reaction scope was explored utilizing commercially available nitriles as well as those synthesized via methylation, benzylation, or Knoevenagel condensation (Scheme 4). As with the nitroalkanes, CO2 activation allowed the allylation of a variety of nitriles in high yield. The method was found to tolerate halogens (2b,c), aryl ether (2d), ketone (2f), and indole substituents (2h). However, allylation of a sterically congested ortho-disubstitued nitrile was unsuccessful under the given reaction conditions (2g). Further, a more acidic diarylalkyl nitrile substrate was successfully allylated at a lower reaction temperature (80 °C) and resulted in a higher isolated yield (2i). Lastly, when argon replaced the CO2 atmosphere, a significant decrease in yield was observed (0%–28%, entries 2b–2f and 2i) thus supporting the requirement of CO2 as an in situ activator of allyl alcohol for nucleophilic allylation.
Scheme 4. Allylation of Nitriles with Allyl Alcohol Activated by CO2,b.

Nitrile (0.3 mmol) and allyl alcohol (0.6 mmol) in 0.5 mL of DMSO under 1 atm of CO2. b Isolated yields. c 2 mL of DMSO used. d Ar replaced CO2 (% conversion via GC/MS). e Reaction at 80 °C.
Aldehydes, which are prone to aldol dimerization, were also allylated in high yield utilizing similar conditions (Scheme 5).16 Acyclic (3a–3g) and cyclic (3h, 3i) α-aryl aldehydes bearing various electron-donating (p-Me, p-OMe) and -withdrawing (o-F, p-OCHF3) groups reacted quickly (1–2 h) and gave high yields of allylated products. Substrates with α-benzyl substituents (3k, 3l), including a protected amino aldehyde (3j), required longer reaction times but still provided good yields of product. Control reactions again revealed that a CO2 atmosphere was required for good reactivity.
Scheme 5. Allylation of Aldehydes,b.

Aldehyde (0.30 mmol) and alcohol (0.45 mmol) in 2.0 mL of DMSO under 1 atm of CO2. b Isolated yields. c Ar replaced CO2 (% conversion via GC/MS).
Next, the scope of the reaction of nitriles and aldehydes with substituted allylic alcohols was examined. Gratifyingly, except for the prenyl alcohol (4i), isolated reaction yields with substituted allylic alcohols were found to be comparable to those utilizing the unsubstituted allyl alcohol (Table 1). Both alkyl and aryl substituted allylic alcohols provided the allylated products in good to excellent yields with generally good selectivity for formation of the linear allyl regioisomer.17 One exception is crotyl alcohol, which showed only slight selectivity for the linear trans product (4c);18 this selectivity was significantly enhanced upon increasing the substituent size to ethyl or propyl (4d, 4e). The linear selectivity of the CO2-activated intermolecular variant is thought to arise from an outer-sphere C-allylation mechanism, and the observed increase in trans selectivity with substituent size may be attributed to a higher bias for the syn-palladium π-allyl intermediate.19
Table 1. Scope of Allyl Alcoholsa,b.
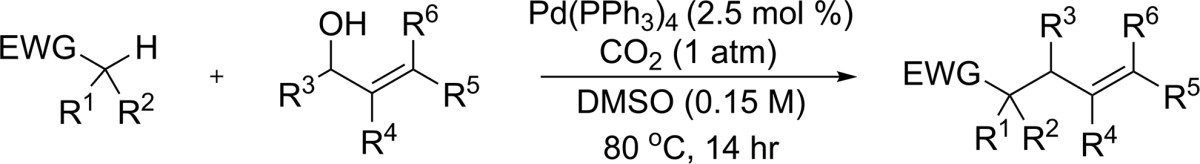
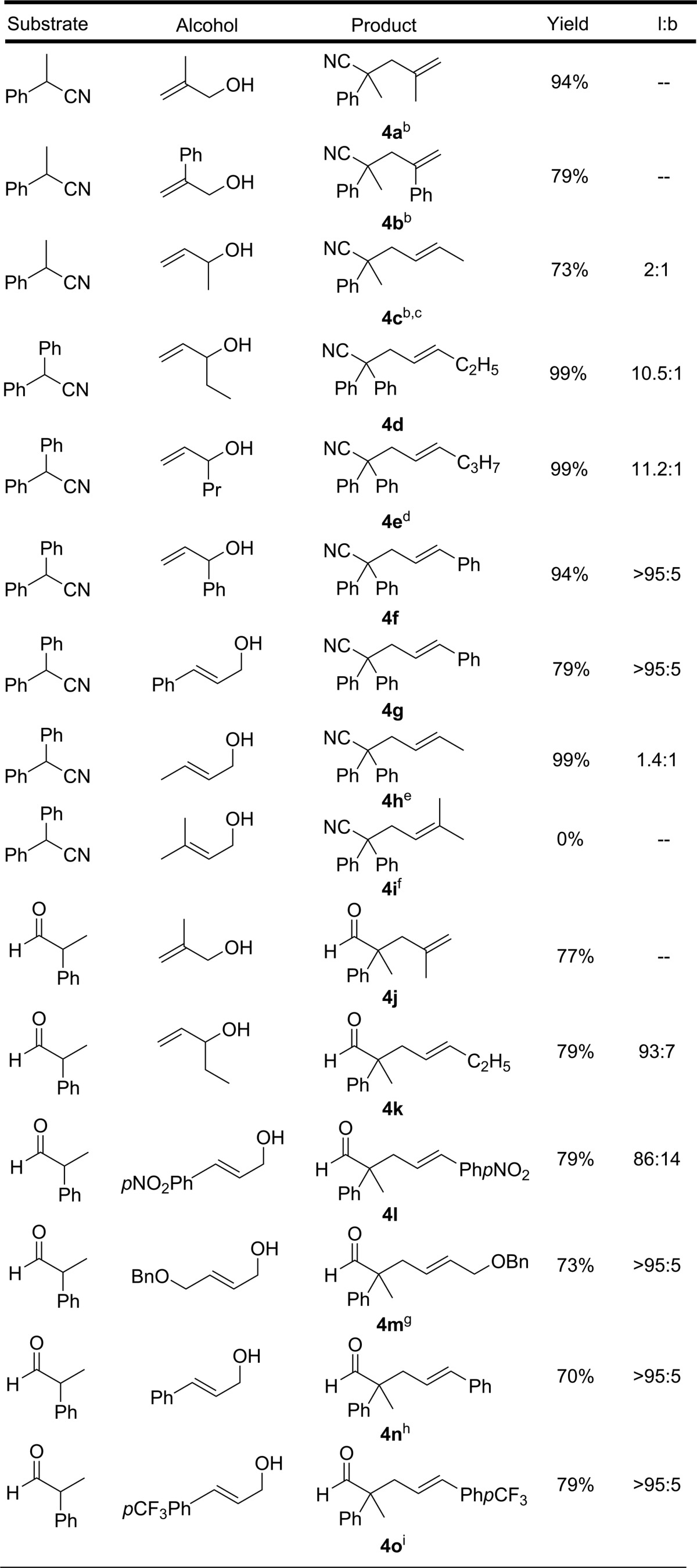
Nitrile (0.3 mmol) and allylic alcohol (0.6 mmol) in 2 mL of DMSO under 1 atm of CO2.
Reaction at 90 °C in 0.5 mL of DMSO.
1:2.5 (cis/trans).
1:14.3 (cis/trans).
1:4.6 (cis/trans).
Reaction time 4 h.
Reaction time 19 h. 1:5.3 (cis/trans).
Reaction time 8 h; a 23% conversion in the control reaction was observed.
Reaction time 7 h.
In conclusion, we have shown that CO2 can activate allyl alcohols toward oxidative addition of palladium. In addition to activating the alcohol, the conditions provide a base strong enough to activate weakly acidic pronucleophiles with a pKa up to ∼25. Thus, CO2 participates in both the activation of the electrophile and the nucleophile toward C–C bond formation. Ultimately, the combination of palladium catalysis with CO2 activation allows the substitution of allylic alcohols by nitroalkanes, nitriles, and aldehydes and produces water as the only byproduct.
Acknowledgments
We thank the National Science Foundation (CHE-1058855), the KU Center for Environmentally Beneficial Catalysis, and the Kansas Bioscience Authority Rising star program for financial support. We thank Dr. Victor Day molecular structures group, University of Kansas for X-ray crystallographic analysis using a diffractometer purchased with NSF-MRI Grant CHE-0923449. Support for the NMR instrumentation was provided by NSF Academic Research Infrastructure Grant No. 9512331, NIH Shared Instrumentation Grant No. S10RR024664, and NSF Major Research Instrumentation Grant No. 0320648.
Supporting Information Available
Experimental procedures, 1H, 13C, 19F NMR spectra, and characterization data of all novel products. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Godula K.; Sames D. Science 2006, 312, 67. [DOI] [PubMed] [Google Scholar]
- Blanksby S. J.; Ellsion G. B. Acc. Chem. Res. 2003, 36, 255. [DOI] [PubMed] [Google Scholar]
- Chuit C.; Felkin H.; Frajerman C.; Roussi G.; Swierczewski G. Chem. Commun. 1968, 1604. [Google Scholar]
- a Itoh K.; Hamaguchi N.; Miura M.; Nomura M. J. Chem. Soc., Perkin Trans. 1 1992, 2833. [Google Scholar]; b Starý I.; Stará I. G.; Koĉovský P. Tetrahedron Lett. 1993, 34, 179. [Google Scholar]; c Masuyama Y.; Kagawa M.; Kurusu Y. Chem. Lett. 1995, 1121. [Google Scholar]; d Satoh T.; Ikeda M.; Miura M.; Nomura M. J. Org. Chem. 1997, 62, 4877. [DOI] [PubMed] [Google Scholar]; e Yang S.-C.; Hung C.-W. J. Org. Chem. 1999, 64, 5000. [DOI] [PubMed] [Google Scholar]; f Tomaru Y.; Horino Y.; Araki M.; Tanaka S.; Kimura M. Tetrahedron Lett. 2000, 41, 5705. [Google Scholar]; g Kimura M.; Horino Y.; Mukai R.; Tanaka C.; Tamaru Y. J. Am. Chem. Soc. 2001, 123, 10401. [DOI] [PubMed] [Google Scholar]; h Tamaru Y. Eur. J. Org. Chem. 2005, 2647. [Google Scholar]; i Trost B. M.; Quancard J. J. Am. Chem. Soc. 2006, 128, 6314. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Yamashita Y.; Gopalarathnam A.; Hartwig J. F. J. Am. Chem. Soc. 2007, 129, 7508. [DOI] [PubMed] [Google Scholar]; k Takahashi M.; McLaughlin M.; Micalizio G. C. Angew. Chem., Int. Ed. 2009, 48, 3648. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Matsubara R.; Masuda K.; Nakano J.; Kobayashi S. Chemm. Commun. 2010, 46, 8662. [DOI] [PubMed] [Google Scholar]; m Yang H.; Fang L.; Zhang M.; Zhu C. Eur. J. Org. Chem. 2009, 666. [Google Scholar]
- a Manabe K.; Kobayashi S. Org. Lett. 2003, 5, 3241. [DOI] [PubMed] [Google Scholar]; b Kinoshita H.; Shinokubo H.; Oshima K. Org. Lett. 2004, 6, 4085. [DOI] [PubMed] [Google Scholar]; c Usui I.; Schmidt S.; Keller M.; Breit B. Org. Lett. 2008, 10, 1207. [DOI] [PubMed] [Google Scholar]; d Mukherjee S.; List B. J. Am. Chem. Soc. 2007, 129, 11336. [DOI] [PubMed] [Google Scholar]; e Jiang G.; List B. Adv. Synth. Catal. 2011, 353, 1667. [Google Scholar]; f Krautwald S.; Sarlah D.; Schafroth M.; Carreira E. M. Science 2013, 340, 1065. [DOI] [PubMed] [Google Scholar]; g Rueping M.; Uria U.; Lin M.-Y.; Atodiresei I. J. Am. Chem. Soc. 2011, 133, 3732. [DOI] [PubMed] [Google Scholar]
- a Sundararaju B.; Achard M.; Bruneau C. Chem. Soc. Rev. 2012, 41, 4467. [DOI] [PubMed] [Google Scholar]; b Atkins K. E.; Walker W. E.; Manyik R. M. Tetrahedron Lett. 1970, 3821. [Google Scholar]; c Haudegond J.-P.; Chauvin Y.; Commereuc D. J. Org. Chem. 1979, 44, 3063. [Google Scholar]; d Ozawa F.; Okamoto H.; Kawagishi S.; Yamamoto S.; Minami T.; Yoshifuji M. J. Am. Chem. Soc. 2002, 124, 10968. [DOI] [PubMed] [Google Scholar]; e Bergbreiter D. E.; Weatherford D. A. J. Chem. Soc., Chem. Commun. 1989, 883. [Google Scholar]; f Kayaki Y.; Koda T.; Ikariya T. J. Org. Chem. 2004, 69, 2595. [DOI] [PubMed] [Google Scholar]; g Tao Y.; Wang B.; Wang B.; Qu L.; Qu J. Org. Lett. 2010, 12, 2726. [DOI] [PubMed] [Google Scholar]
- Weikel R. R.; Hallett J. P.; Liotta C. L.; Eckert C. A. Ind. Eng. Chem. Res. 2007, 46, 5252. [Google Scholar]
- Olmstead W. N.; Bordwell F. G. J. Org. Chem. 1980, 3299. [Google Scholar]
- Sakamoto M.; Shimizu I.; Yamamoto A. Bull. Chem. Soc. Jpn. 1996, 69, 1065. [Google Scholar]
- The hydroxide produced would also be expected to equilibrate with the bicarbonate to generate carbonate.; a Alberty R. A. J. Phys. Chem. 1995, 99, 11028. [Google Scholar]; b Adamczyk K.; Schwarz-Premont M.; Pines D.; Pines E.; Nibbering E. T. J. Science 2009, 1690. [DOI] [PubMed] [Google Scholar]
- Allen C. F. H.; Bell A.; Gates J. W. Jr. J. Org. Chem. 1943, 373. [Google Scholar]
- Rao J. S.; Basavaiah D. Tetrahedron Lett. 2004, 1621. [Google Scholar]
- Heinzelman R. V.; Anthony W. C.; Lyttle D. A.; Szmuszkovicz J. J. Org. Chem. 1960, 1548. [Google Scholar]
- Matthews W. S.; Bares J. E.; Bartmess J. E.; Bordwell F. G.; Cornforth F. J.; Drucker G. E.; Margolin Z.; McCallum R. J.; McCollum G. J.; Vanier N. R. J. Am. Chem. Soc. 1975, 7006. [Google Scholar]
- This is an estimate based on the pKa’s of PhCH2CN (21.9), PhCH(Me)CN (23.0), and p-MeOC6H4CH2CN (23.8).; a Bordwell F. G.; Cheng J. P.; Bausch M. J.; Bares J. E. J. Phys. Org. Chem. 1988, 1, 209. [Google Scholar]; b Bordwell F. G.; Bares J. E.; Bartmess J. E.; McCollum G. J.; Van der Puy M.; Vanier N. R.; Matthews W. S. J. Org. Chem. 1977, 42, 321. [Google Scholar]
- For other various strategies utilizing an allyl alcohol, see refs (5c) , (5e) , (5f), and; a Usui I.; Schmidt S.; Breit B. Org. Lett. 2009, 1453. [DOI] [PubMed] [Google Scholar]; b Jiang G.; List B. Angew. Chem., Int. Ed. 2011, 9471. [DOI] [PubMed] [Google Scholar]; c Krautwald S.; Schaforth M. A.; Carreira E. M. J. Am. Chem. Soc. 2014, 3020. [DOI] [PubMed] [Google Scholar]
- a Recio A. III; Tunge J. A. Org. Lett. 2009, 11, 5630. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Waetzig S. R.; Tunge J. A. J. Am. Chem. Soc. 2007, 129, 4138. [DOI] [PubMed] [Google Scholar]; c Grenning A. J.; Tunge J. A. J. Am. Chem. Soc. 2011, 133, 14785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmaier U.; Pohlman M. Synlett 2004, 623. [Google Scholar]
- a Kazmaier U.; Zumpe F. L. Angew. Chem., Int. Ed. 2000, 39, 802. [DOI] [PubMed] [Google Scholar]; b Consiglio G.; Waymouth R. M. Chem. Rev. 1989, 89, 257. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.