

Abstract
Cocaine esterase (CocE) is known as the most efficient natural enzyme for cocaine hydrolysis. The major obstacle to the clinical application of wild-type CocE is the thermoinstability with a half-life of only ∼12 min at 37 °C. The previously designed T172R/G173Q mutant (denoted as enzyme E172–173) with an improved in vitro half-life of ∼6 h at 37 °C is currently in clinical trial Phase II for cocaine overdose treatment. Through molecular modeling and dynamics simulation, we designed and characterized a promising new mutant of E172–173 with extra L196C/I301C mutations (denoted as enzyme E196–301) to produce cross-subunit disulfide bonds that stabilize the dimer structure. The cross-subunit disulfide bonds were confirmed by X-ray diffraction. The designed L196C/I301C mutations have not only considerably extended the in vitro half-life at 37 °C to >100 days, but also significantly improved the catalytic efficiency against cocaine by ∼150%. In addition, the thermostable E196–301 can be PEGylated to significantly prolong the residence time in mice. The PEGylated E196–301 can fully protect mice from a lethal dose of cocaine (180 mg/kg, LD100) for at least 3 days, with an average protection time of ∼94h. This is the longest in vivo protection of mice from the lethal dose of cocaine demonstrated within all studies using an exogenous enzyme reported so far. Hence, E196–301 may be developed to become a more valuable therapeutic enzyme for cocaine abuse treatment, and it demonstrates that a general design strategy and protocol to simultaneously improve both the stability and function are feasible for rational protein drug design.
Cocaine overdose and addiction have resulted in serious medical and social problems in modern society.1 So far, there is no anticocaine medication approved by the Food and Drug Administration (FDA).2,3 Cocaine causes its physiological effects by binding with the dopamine transporter and, thus, blocking dopamine reuptake. The disastrous medical and social consequences of cocaine abuse have made a high priority the development of an anticocaine medication. However, despite decades of efforts, the classical pharmacodynamic approach has failed to yield a truly useful small-molecule receptor/transporter antagonist. The alternative pharmacokinetic approach is to interfere with the delivery of cocaine to its receptors and/or accelerate its metabolism in the body.2,4−8 It would be an ideal anticocaine medication to develop an exogenous enzyme which can accelerate cocaine metabolism and produce biologically inactive metabolites.
Bacterial cocaine esterase (CocE) was recognized as the most efficient natural enzyme for hydrolyzing the naturally occurring (−)-cocaine.9 No any other natural esterase has a catalytic activity for cocaine comparable to that of CocE. Studies have shown that CocE can help to prevent extreme cocaine toxicity and even from the lethal effects of cocaine.10 However, a major obstacle to the clinical application of CocE is the thermoinstability of wild-type CocE with a half-life of only ∼12 min at physiological temperature (37 °C).11 It is highly desirable to develop thermostable mutants of CocE for therapeutic treatment of cocaine abuse (overdose and addiction). In fact, thermal stability is a well-known common problem in protein drug development.11 In general, the more thermally stable a protein drug, the longer shelf half-life the protein drug can have.
Generally speaking, the thermal stability of a protein could be improved by enhancing the weak interactions inside the enzyme through either noncovalent forces, such as hydrogen bonds,12 or covalent linkage, such as disulfide bonds.13 Particularly for an enzyme, besides improving its stability, it is also important to maintain the catalytic activity of the enzyme. However, it is much more challenging to engineer an enzyme with an improved stability without decreasing the catalytic activity.14−16 In general, according to the commonly recognized “stability-function trade-off” theory/hypothesis,14 protein residues that contribute to catalysis or ligand binding are not optimal for protein stability and, thus, there is a balance between the stability and function. Indeed, extensive studies14,17−22 demonstrated that thermostabilizing mutations of enzymes decreased the catalytic activities, and that mutations improving the catalytic activities decreased the thermal stability. Nevertheless, some CocE mutants with an improved thermal stability have successfully been designed and discovered in recently reported studies,11,23−26 and these thermostable mutants did not decrease, or only slightly decreased, the catalytic efficiency (kcat/KM) of CocE against cocaine. Further animal behavior studies27−29 revealed that these CocE mutants are promising in development of an enzyme therapy for cocaine abuse.
Notably, one of the reported thermostable mutants of CocE, i.e. the T172R/G173Q mutant (known as drug RBP-8000, with ClinicalTrials.gov Identifier of NCT01846481 in clinical development) designed through our computational modeling and simulations,11 has been advanced to the randomized, double-blind, placebo controlled clinical trial phase II (http://www.clinicaltrials.gov/ct2/show/NCT01846481) for cocaine overdose treatment. The T172R/G173Q mutant (denoted as enzyme E172–173 here for convenience) was designed through introducing favorable noncovalent forces including a hydrogen bond between domains I and II of the protein.11 This CocE mutant has an in vitro half-life of ∼6 h at 37 °C without decreasing the catalytic activity of CocE against cocaine.11 The half-life of ∼6 h at 37 °C is long enough for cocaine overdose treatment, because one just needs to use the enzyme to rapidly detoxify cocaine. However, for cocaine addiction treatment, one would like to have a highly efficiently cocaine-metabolizing enzyme in the body with a residence time as long as possible. With a highly efficiently cocaine-metabolizing enzyme in the body, whenever a cocaine abuser uses cocaine again, the enzyme would rapidly metabolize cocaine so that the cocaine abuser would not feel the stimulate effects of cocaine.
To further develop an improved therapeutic enzyme for cocaine abuse treatment, one would like to both extend the half-life of E172–173 at 37 °C and improve the catalytic efficiency against cocaine. It has been shown11,25,26 that the thermal stability of E172–173 at 37 °C can be enhanced by extra mutations on E172–173. However, none of the reported extra mutations on E172–173 improved the catalytic efficiency against cocaine. Here we report a rationally designed new mutant of E172–173, which has not only considerably extended the in vitro half-life at 37 °C, but also significantly improved the catalytic efficiency against cocaine. The new CocE mutant (i.e. the T172R/G173Q/L196C/I301C mutant of CocE, denoted as enzyme E196–301 for convenience) was modified further via PEGylation in order to extend the in vivo residence time of the enzyme. The PEGylated E196–301 was used to fully protect mice from a lethal dose of cocaine (180 mg/kg, LD100) for at least 3 days, indicating that it might be a more promising enzyme candidate for development of novel anticocaine therapeutics.
Results and Discussion
Mutant Design: Insights from Molecular Modeling
We first aimed to stabilize the dimer structure of E172–173 and then analyzed the possibly stabilized dimer structure and estimated how the activity of the dimer would change. Our computational design strategy relied on the molecular dynamics (MD)-simulated dimer structure of E172–173 and the idea that the dimer structure can be stabilized by introducing disulfide bonds between the two subunits of the dimer. So, we carried out a sufficiently long MD simulation (50 ns) on the E172–173 dimer structure in order to obtain a dynamically stable dimer structure. To search for appropriate mutational sites to introduce the cross-subunit disulfide bonds, a self-developed script was used to scan the key internuclear distances between the Cα atoms of the residues on the dimer interface from the collected snapshots of the MD trajectory. Essentially, each pair of residues from different subunits was evaluated computationally for the simulated Cα–Cα distance. If the simulated Cα–Cα distance was within 7 Å, the pair of the residues would be checked manually for further evaluation of the detailed interactions. The most hopeful pair of residues may be mutated to cysteine for introducing possible cross-subunit disulfide bond(s).
Based on the analysis of the MD trajectory, L196C/I301C mutations satisfied all of the structural requirements. Summarized in Table 1 are the maximum, minimum, and average values of the Cα-Cα distances associated with the stable MD trajectory (10 to 50 ns) in comparison with the corresponding Cα-Cα distances in the X-ray crystal structure. As seen in Table 1, the detailed analysis of the MD trajectory predicted that extra L196C/I301C mutations on E172-173 may introduce the desirable cross-subunit disulfide bonds between the two subunits of the E172-173 dimer. Depicted in Figure 1A are the simulated time-dependent Cα–Cα distances for the pair of residues. Depicted in Figure 1B is the MD-simulated E172-173 dimer structure consisting of subunits a and b. Figure 1C is the detailed information about the MD-simulated E172-173 dimer structure concerning this important pair of residues. The interface between the two subunits is mainly composed of some residues from all of the three domains (I, II, and III) in the form of α-helices, β-sheets, and loops. L196 is located on one α-helix (E184-N197) in domain II, I301 is located on a loop in domain I.
Table 1. Maximum, Minimum, and Average Cα–Cα Distances between Key Residues in the E172-173 Dimer Obtained from the 50 ns MD Simulation in Comparison with the Corresponding Cα–Cα Distances in the X-ray Crystal Structure.
| |
Cα–Cα
distances (Å) |
||
|---|---|---|---|
| E172-173 structure | L196a–I301b | I301a–L196b | |
| crystal structure of E172-173a | 7.29 | 7.20 | |
| MD-simulated E172-173 structure (50 ns MD)b | max. | 10.85 | 7.19 |
| min. | 5.16 | 4.51 | |
| avg. | 7.15 | 5.44 | |
T172R/G173Q CocE (E172–173) crystal structure (PDB ID: 3I2F).
Fully relaxed MD simulation of the E172–173 dimer structure starting from the crystal structure.
Figure 1.
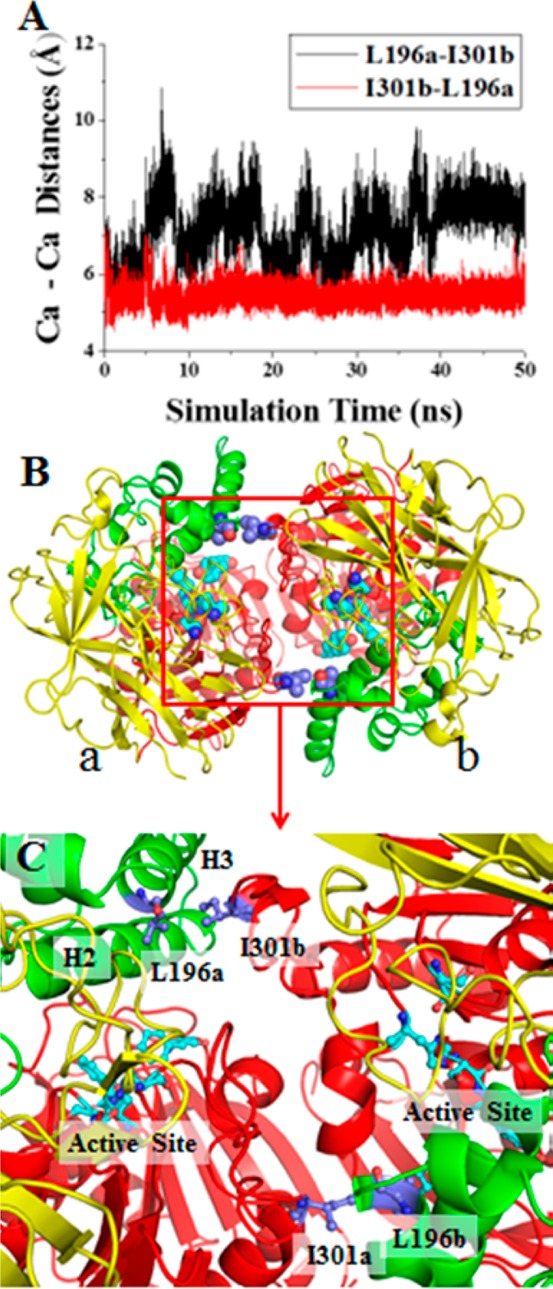
Modeled E172-173 dimer structure. (A) The time-dependent Cα–Cα distances between L196 and I301 in the MD-simulated dimer structure of E172-173. The letters a and b indicated after the residue numbers refer to subunits a and b, respectively. (B) The modeled E172-173 dimer structure shown in ribbons (with a and b referring to subunits a and b, respectively), domain I is shown in red, domain II is shown in green, and domain III is shown in yellow. (C) Key residues L196 (a/b) and I301 (a/b) shown in ball and sticks on the dimer interface.
According to the locations of these residues, it is highly possible to introduce a pair of cross-subunit disulfide bonds (C196a-C301b and C301a-C196b) through the L196C/I301C mutations on E172–173. The computationally designed new mutant, i.e. the T172R/G173Q/L196C/I301C mutant (denoted as enzyme E196–301 for convenience), may have a pair of cross-subunit disulfide bonds: one between C196 of subunit a (C196a) and C301 of subunit b (C301b), and the other between C301 of subunit a (C301a) and C196 of subunit b (C196b). It is also interesting to note that residue I301 is on a loop, implying that the loop flexibility may help to form the desirable cross-subunit disulfide bonds. In addition, molecular modeling and X-ray structural analysis (see below) of the dimer structure of the T172R/G173Q/L196C/I301C mutant also suggested that the extra L196C/I301C mutations could slightly increase the size of the active site cavity of the enzyme and, thus, might improve the catalytic activity.
In Vitro Characterization of the Designed T172R/G173Q/L196C/I301C Mutant
Based on the computational insights, we carried out wet experimental tests, including site-directed mutagenesis, protein expression, purification, and enzyme activity assays on the T172R/G173Q and T172R/G173Q/L196C/I301C mutants of CocE. For comparison, we also prepared and characterized the T172R/G173Q/G4C/S10C mutant which has been known to have a pair of cross-subunit disulfide bonds.26 All of the mutants were expressed similarly well. To minimize the possible systematic experimental errors of the kinetic data, we simultaneously prepared and characterized all of the three mutants under the same experimental conditions, which allowed us to fairly compare their catalytic activity against (−)-cocaine. Michaelis–Menten kinetics of the enzymatic hydrolysis of (−)-cocaine was determined by performing the sensitive radiometric assays using [3H](−)-cocaine (labeled on its benzene ring) with varying concentrations of the substrate. Depicted in Figure 2 are the measured kinetic data, and summarized in Table 2 are the kinetic parameters determined at 37 °C.
Figure 2.

Plots of measured initial reaction rates (represented in μM min–1 per nM enzyme at 37 °C, with error bars) versus the substrate concentration for (−)-cocaine hydrolysis catalyzed by CocE mutants: (A) T172R/G173Q; (B) T172R/G173Q/G4C/S10; (C) T172R/G173Q/L196C/I301C.
Table 2. Kinetic Parameters Determined for (−)-Cocaine Hydrolysis Catalyzed by the T172R/G173Q, T172R/G173Q/G4C/S10C, and T172R/G173Q/L196C/I301C Mutants of CocEa.
| CocE mutant | kcat (min–1) | KM (μM) | kcat/KM (min–1 M–1) |
|---|---|---|---|
| T172R/G173Q | 2600 | 2.9 | 9.2 × 108 |
| T172R/G173Q/G4C/S10C | 2340 | 2.1 | 1.1 × 109 |
| T172R/G173Q/L196C/I301C | 3450 | 1.5 | 2.3 × 109 |
The kinetic analysis was performed at 37 °C.
As seen in Table 2, kcat = 2600 min–1, KM = 2.9 μM, and kcat/KM = 9.2 × 108 min–1 M–1 for the T172R/G173Q mutant under the current clinical development. Compared to the T172R/G173Q mutant, the T172R/G173Q/G4C/S10C mutant has a slightly smaller kcat value (2340 min–1) and a slightly smaller KM value (2.1 μM). Overall, the catalytic efficiency (kcat/KM) changed about 20% (from 9.2 × 108 min–1 M–1 to 1.1 × 109 min–1 M–1). Interestingly, the new mutant (T172R/G173Q/L196C/I301C) designed in the present study has both a significantly increased kcat value (3450 min–1) and a significantly smaller KM value (1.5 μM). As a result, the catalytic efficiency (kcat/KM = 2.3 × 109 min–1 M–1) of the T172R/G173Q/L196C/I301C mutant has a ∼150% improvement from that (kcat/KM = 9.2 × 108 min–1 M–1) of the T172R/G173Q mutant under the current clinical development.
Based on the encouraging kinetic data, the purified protein of the T172R/G173Q/L196C/I301C mutant was tested for the thermal stability at 37 °C. For this purpose, the enzyme was incubated at 37 °C, and the catalytic activity of the incubated enzyme against cocaine was assayed at different time points. As seen in Figure 3, the enzyme showed a relatively faster decrease of the activity (around 20%) during the first a few days, compared to the last 90 days. The relatively faster decrease of the activity during the first a few days is likely due to the possibility that certain percentage of the mutant protein molecules had not yet formed the expected cross-subunit disulfide bonds before the thermal stability test. Although the cross-subunit disulfide bonds were expected to form spontaneously (as no extra oxidation reagent was employed in this study to facilitate the disulfide bond formation), a small percentage of the new mutant (T172R/G173Q/L196C/I301C) molecules did not really form the cross-subunit disulfide bonds. Those T172R/G173Q/L196C/I301C mutant molecules without the cross-subunit disulfide bonds could lose the activity more rapidly, like the T172R/G173Q mutant which has an in vitro half-life of ∼6 h at 37 °C.11 However, after the first few days, the enzyme activity decreased very slowly. Within the last 90 days, the enzyme activity decreased for only ∼15%. Overall, the enzyme still retained more than 60% of the enzyme activity after incubation at 37 °C for 100 days, indicating that the in vitro half-life of the T172R/G173Q/L196C/I301C mutant at 37 °C should be longer than 100 days.
Figure 3.

Plot of the remaining enzyme activity of the T172R/G173Q/L196C/I301C CocE against cocaine versus the time of the enzyme incubation at 37 °C. The catalytic activity of the incubated enzyme was assayed after 0, 2, 9, 12, 31, and 100 days.
Confirmation of the Cross-Subunit Disulfide Bonds
With the encouraging data about the significant improvement in both the catalytic activity and thermal stability at 37 °C, we have determined the crystal structure of the T172R/G173Q/L196C/I301C mutant in order to directly confirm the formation of the cross-subunit disulfide bonds between L196C and I301C.
The CocE mutant crystallized with one monomer in the asymmetric unit, but a symmetry related molecule forms an extensive interface, burying over 1900 Å2 of solvent-accessible surface area as calculated by the PISA server.30 The dimer formed by these two molecules (Figure 4A and B) corresponds to previously reported structures of unlinked (PDB 3PUH) and covalently linked (PDB 3PUI) CocE dimers.26 Thus, the expected homodimer was formed in the crystals. Initial difference maps using the rigidly placed (without refinement) CocE mutant structure with glycine residues substituted at residue positions 196 and 301 showed strong positive Fo-Fc difference electron density for the cysteine side chains at the dimer interface (Figure 4C) consistent with disulfide bond formation. Subsequent refinement of the model with cysteine residues introduced at these positions and no disulfide restraints gave well-defined weighted 2Fo-Fc density and excellent geometry consistent with disulfide bond formation across the subunits (Figure 4D, see Figure 4A and B). The sulfur-to-sulfur distance is 2.0 Å and the dihedral angle is 94°. In summary, the crystal structure of the enzyme unambiguously confirms the presence of the engineered intersubunit disulfide bonds. Since the dimer axis corresponds to a crystallographic 2-fold axis (space group P6522), the structure was also refined in a lower symmetry group (P65), which places a disulfide-linked dimer in the asymmetric unit and does not therefore impose symmetry on the model. This refinement resulted in geometry for the two disulfide bonds that is nearly identical to that present in the dimer on the crystallographic 2-fold axis (Supporting Information Figure S3), eliminating the possibility of any artifact from this placement.
Figure 4.
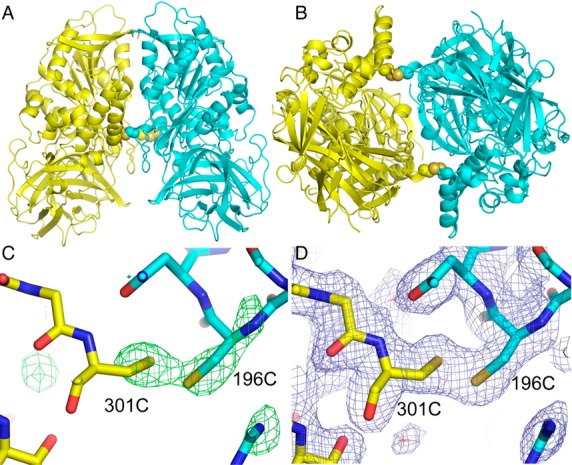
Crystal structure of the CocE mutant dimer. (A) Ribbons representation of the homodimeric molecule generated by applying 2-fold crystallographic symmetry. The side chains of the cysteine residues forming intersubunit disulfide bonds are shown in a space filling representation. (B) View of the dimer rotated 90° about a horizontal axis. (C) Fo-Fc electron density (green, 3.5 sigma contour) calculated with the rigidly placed (before refinement) molecular replacement model having residues 196 and 301 altered to glycines. The final refined model in a stick representation is superimposed on the map. (D) Final SIGMAA-weighted 2Fo-Fc electron density map (blue, 1.0 sigma cutoff) in the region of the disulfide bond with the final model shown in a stick representation.
Structure–activity correlation
Superposition between the X-ray crystal structures of E172-173 (representing the T172R/G173Q mutant) and E196-301 (representing the T172R/G173Q/L196C/I301C mutant) revealed a RMSD value of 0.299 Å, indicating the high similarity of the two protein structures. However, the superposition also revealed a slight shift of two α-helices (H2 and H3 in domain II) in E196-301 compared to that in E172-173 (see Figure S1 of Supporting Information). The shift created a slightly enlarged active-site cavity in E196-301, which could possibly favor the catalytic reaction process. A slightly larger active site could better accommodate such a large substrate like cocaine and, thus, make the enzyme more active against cocaine; the similar type of structure–activity correlation was noted for our previously designed mutants of human butyrylcholinesterase (BChE).31 The BChE mutants with a slightly larger active site have a significantly improved catalytic efficiency against cocaine without changing substrate specificity.31,32
To further understand why E196-301 has an improved catalytic activity against cocaine compared to E172-173, we needed to perform MD simulations on the transition state (TS1) for the initial reaction step of (−)-cocaine hydrolysis catalyzed by E172-173 and E196-301. Previous computational studies33 on the catalytic reaction mechanism for wild-type CocE-catalyzed hydrolysis of (−)-cocaine revealed that the CocE-catalyzed cocaine hydrolysis is initialized by the nucleophilic attack on the carbonyl carbon of (−)-cocaine benzoyl ester by the hydroxyl oxygen of Ser117, and that the transition state is stabilized by hydrogen bonding of the carbonyl oxygen of cocaine benzoyl ester with the hydroxyl group of Y44 side chain and the NH group of the Y118 backbone.33 The (−)-cocaine hydrolysis catalyzed by E172-173 or E196-301 is expected to follow the same catalytic reaction mechanism, as the residues #196 and #301 are all far away from the active site. Based on the established mechanistic understanding, the stronger the hydrogen bonding of the carbonyl oxygen of the substrate with Y44 side chain and Y118 backbone, the more active the enzyme.
Our general strategy and protocol for performing MD simulation on a transition state of enzymatic reaction using the classical force field have been described in detail elsewhere.34,35 Based on the protocol,34 the lengths of the transition bonds (i.e., the covalent bonds that gradually form or break during the reaction step associated with the transition state) in the transition state are restrained according to the previous QM/MM reaction-coordinate calculations, assuming that the these bond lengths do not significantly change after the mutations. The transition-state modeling in the present study was based on our QM/MM-optimized TS1 structure33 for CocE-catalyzed hydrolysis of (−)-cocaine. It is reasonable to assume that the transition bond lengths in the TS1 structure will not significantly change after the T172R/G173Q or T172R/G173Q/L196C/I301C mutations. So, we carried out the MD simulations on the TS1 structures corresponding to E172-173 and E196-301, with the transition bond lengths restrained.
Depicted in Figure S2 of Supporting Information are the simulated time-dependent H···O distances (relevant to the hydrogen bonds) in E172-173 and E196-301 during the MD simulations for 50 ns. The detailed analysis of the key H···O distances between enzyme residues and cocaine is summarized in Table 3. In E172-173, the H···O distance between CocO and Y44HH was 3.02 Å in maximum, 1.44 Å in minimum, and 1.87 Å in average, while the H···O distance between CocO and Y114H was 3.14 Å in maximum, 1.61 Å in minimum, and 2.16 Å in average. In E196-301, the H···O distance between Y44HH and CocO was 3.17 Å in maximum, 1.44 Å in minimum, and 1.80 Å in average, while the H···O distance between CocO and Y114H was 2.89 Å in maximum, 1.61 Å in minimum, and 2.13 Å in average. According to these simulated H···O distances, the H···O distances of the two hydrogen bonds in E196-301 are all shorter than the corresponding ones in E172–173, suggesting that cocaine has the stronger hydrogen bonding with E196-301 compared to that with E172-173 in the TS1 structure. The enhanced hydrogen bonding helps to stabilize the transition-state (TS1) structure during the catalytic reaction process and, thus, lower the energy barrier, which explains the improved catalytic activity of the new mutant.
Table 3. Summary of the MD-Simulated Key Distances (in Å) between the Hydrogen Atoms of Key Residue and the Carbonyl Oxygen of (−)-Cocaine Benzoyl Ester in the Rate-Determining Transition-State Structures of CocE.
| distances
(Å) |
||||
|---|---|---|---|---|
| hydrogen bond | max. | min. | avg | |
| Y44HH-CocOb | E172-173a | 3.02 | 1.44 | 1.87 |
| E196-301a | 3.17 | 1.44 | 1.80 | |
| Y118H-CocOc | E172-173a | 3.14 | 1.61 | 2.16 |
| E196-301a | 2.89 | 1.61 | 2.13 | |
E172-173 represents T172R/G173Q CocE, and E196-301 refers to T172R/G173Q/L196C/I301C CocE.
Y44HH–CocO represents the distance between the hydroxyl hydrogen (denoted as HH) of the Y44 side chain and the carbonyl oxygen (denoted as CocO) of (−)-cocaine benzoyl ester.
Y118H–CocO refers to the distance between the hydrogen (H) of the Y118 backbone and the carbonyl oxygen (CocO) of (−)-cocaine benzoyl ester.
In Vivo Protection of Mice against Cocaine-Induced Lethality
For development of an effective cocaine abuse treatment using a cocaine-metabolizing enzyme, it is highly desired to have a long residence time of the enzyme in the body. To have a long residence time in the body, the enzyme must be thermostable at 37 °C for a sufficiently long time. So, it is a necessary condition, but not a sufficient condition, for an enzyme having a long residence time in the body to have a long in vitro half-life of the enzyme at 37 °C. An enzyme may be eliminated rapidly from the body, even if it is very thermostable at 37 °C. In this consideration, PEGylation is a popularly used strategy to prevent the possible rapid elimination of a protein from the body. Hence, we further engineered E196-301 through the PEGylation modification. Our activity assays confirmed that the PEGylated E196-301 completely maintained its activity against cocaine as that of E196-301.
To test the ability of E196–301 (unPEGylated, unless specified otherwise) and the PEGylated E196-301 in protecting mice (n = 5) from a lethal dose of cocaine, E196–301 or the PEGylated E16-301 was administered i.v. (at a single dose of 30 mg/kg) 1 min before the first i.p. administration of 180 mg/kg cocaine (LD100). Depicted in Figure 5 are the data for the in vivo protection of mice provided by E196–301 and the PEGylated E196–301 against the cocaine-induced lethality. As seen in Figure 5, E196–301 protected the mice from death after the first injection of 180 mg/kg cocaine, but lost the efficacy at the second injection of 180 mg/kg cocaine 24 h later. The PEGylated E196–301 was able to fully protect the mice (n = 5) for at least 72 h from the acute toxicity of a lethal dose of cocaine (180 mg/kg, LD100): no mouse died after the fourth cocaine challenge at 72 h, three mice died after the fifth cocaine challenge at 96 h, and the remaining two mice died after the final (sixth) cocaine challenge at 120 h (see Figure 5). According to the data depicted in Figure 5, the PEGylated E196-301 can protect the three mice (60%) with the protection time (tp) being between 72 and 96 h: 72 h < tp < 96 h, or we have tp = 84 ± 12 h. For the remaining two mice (40%), 96 h < tp < 120 h, or we have tp = 108 ± 12 h according to the data in Figure 5. Overall, the PEGylated E196–301 can protect the mice with an average protection time (denoted as <tp> for convenience) of ∼94 h, i.e. we have <tp> = ∼94 h. This is the longest in vivo protection of mice from a lethal dose of cocaine (180 mg/kg, LD100) demonstrated so far within all in vivo studies using an exogenous enzyme.
Figure 5.

In vivo effectiveness of E196-301 (black squares) and the PEGylated E196-301 (red triangles) in the protection of mice from cocaine-induced lethality. A single dose (30 mg/kg) of E196-301 (PEGylated or unPEGylated) was administered (i.v.) 1 min before the first i.p. administration of 180 mg/kg cocaine (n = 5). The mice were challenged daily with 180 mg/kg cocaine until no mouse survived. E196–301 refers to the T172R/G173Q/L196C/I301C mutant of CocE.
It should be pointed out that the in vivo studies described above are a simplified animal model (using a high dose of enzyme and high doses of cocaine for convenience of animal behavior observation) to show the long residence time of the PEGylated E196-301. As discussed earlier in this report, the long residence time of the enzyme is crucial for an effective enzyme-based cocaine addiction treatment. For practical cocaine addiction treatment using a cocaine-metabolizing enzyme in humans, the cocaine doses are expected to be much lower (a typical cocaine addiction dose is ∼1 mg/kg which is much lower than the lethal dose of 180 mg/kg used in our animal model) and, correspondingly, the required dose of the enzyme for effective cocaine metabolism may be significantly lower than 30 mg/kg.
Concluding Remarks
Molecular dynamics simulation and subsequent structural analysis on the dimer structure of a therapeutic cocaine–metabolizing enzyme, i.e. E172–173 (which is the T172R/G173Q mutant of CocE), led us to predict that the extra L196C/I301C mutations on E172–173 can produce cross-subunit disulfide bonds in the dimer. The formation of cross-subunit disulfide bonds were expected to stabilize the dimer structure and also improve the catalytic activity against cocaine. Following the computational prediction, our in vitro experimental studies have demonstrated that the computationally designed new CocE mutant (T172R/G173Q/L196C/I301C), i.e. E196–301, indeed has a significantly improved catalytic efficiency against cocaine and a considerably extended in vitro half-life (>100 days) at 37 °C. The predicted cross-subunit disulfide bonds in the E196–301 dimer structure were confirmed by X-ray diffraction. In addition, in vivo studies in mice demonstrated that the PEGylated E196–301 can fully protect mice from a lethal dose of cocaine (180 mg/kg, LD100) for at least 3 days, with the average protection time being ∼94 h. All of the data suggest that the currently designed enzyme E196–301 with improved thermal stability and catalytic activity against cocaine is more valuable than the existing therapeutic enzyme E172–173 which is under clinical trial phase II for cocaine overdose treatment. The encouraging outcomes of this study also suggest that the structure-and-mechanism-based computational design and integrated computational-experimental approach are promising for rational protein drug design. The general computational protein design strategy and approach to simultaneously improve both the protein stability and function may also be valuable for engineering other proteins.
Material and Methods
Computational Methods Used for the Mutant Design
For the molecular dynamics (MD) simulations on the CocE dimer structures, the starting structure of the E172–173 dimer was the X-ray crystal structure (deposited in the Protein Data Bank) at 2.0 Å resolution (PDB ID: 3I2F),26 and the starting structure of the E196–301 dimer was the X-ray crystal structure determined in the present study. In order to simulate the TS1 structures for the enzymatic hydrolysis of cocaine, the transition bond lengths in the TS1 structure were restrained as those in our previously QM/MM-optimized TS1 structure33 for CocE-catalyzed hydrolysis of (−)-cocaine. A transition bond in the TS1 structure refers to a covalent bond which gradually forms or breaks in the transition state (TS1) during the first step of the chemical reaction process. According to our previously reported QM/MM reaction-coordinate calculations33 on CocE-catalyzed hydrolysis of (−)-cocaine, there are three transition bonds in the TS1 structure: (1) the internuclear distance (1.93 Å) between the carbonyl carbon of (−)-cocaine benzoyl ester and the hydroxyl oxygen (Oγ) of Ser117 side chain; (2) the internuclear distance (1.38 Å) between the hydroxyl oxygen (Oγ) and hydroxyl hydrogen (Hγ) of Ser117 side chain; (3) the internuclear distance (1.19 Å) between the hydroxyl hydrogen (Hγ) of Ser117 side chain and the nitrogen (Nε) atom of His287 side chain. These three transition bond lengths were used in all of our MD simulations on the TS1 structures. All of the mutations (T172R/G173Q and L196C/I301C) examined in the present study were made on the amino acid residues (#172, #173, #196, and #301) that are far away from the active site. So, these mutations are not expected to dramatically change the catalytic mechanism or significantly affect the transition bond lengths. The similar approximation was used in previously reported computational modeling studies34,36 on other esterases, leading to successful design and discovery of new mutants with a significantly improved catalytic activity.
The general procedure for carrying out the MD simulations in the present study was similar to that used in our previously reported computational studies.34,37−39 Briefly, all molecular mechanics-based energy minimization and MD simulations were carried out by using the AMBER 9 program package. The Amber force field (ff03) was used to establish the potentials of protein.40 For each system, counterions (Na+) were used to neutralize the system and, then, the neutralized system was immersed in an orthorhombic box of TIP3P water molecules41 with a minimum solute-wall distance of 10 Å. The whole system was carefully equilibrated and fully energy-minimized. After that, the system was gradually heated in the NPT ensemble from 10 to 300 K over 60 ps. Then, a 50 ns MD simulation was performed under the normally adopted temperature (300 K). During the MD simulation, the Particle Mesh Ewald (PME) method was employed to deal with the long-range electrostatic interactions.42 The SHAKE procedure was applied to constrain the lengths of all covalent bonds involving hydrogen atoms,43 with a time step of 2.0 fs. The atomic coordinates were saved every 1 ps for subsequent sampling and analysis.
Site-Directed Mutagenesis
Point mutations were generated using the QuikChange method.44 Further mutations required to produce a new CocE mutant cDNA were generated from the cDNA corresponding to the E172–173 in the pET-22b (+) bacterial expression vector. All mutants were sequenced in both directions over the entire coding region. Using plasmid DNA as template and primers with specific base-pair alterations, mutations were made by polymerase chain reaction with Pfu DNA polymerase for replication fidelity. The PCR product was treated with DpnI endonuclease to digest the parental DNA template. The digested product was transformed into Escherichia coli, amplified, and purified. The DNA sequences of the mutants were confirmed by DNA sequencing.
Protein Expression and Purification
The CocE mutants were expressed in Escherichia coli BL-21 (DE3) cells grown at 37 °C. Protein expression was induced with 1 mM isopropyl-β-thiogalactopyranoside (Sigma-Aldrich) for ∼15 h at 18 °C. Cells were pelleted, resuspended in 50 mM Tris-HCl, pH 8.0, 150 mM NaCl buffer with protease inhibitor cocktail (Sigma) and lysed using a French press (Thermo Fisher Scientific). The 6His-tagged enzymes were then enriched using HisPur cobalt resin (Thermo Fisher Scientific). The eluted fractions were concentrated by using an Amicon Ultra-50K centrifuge (Millipore, Billerica, MA). The enzyme concentrations were determined using a CB-Protein Assay kit (from CALBIOCHEM) with bovine serum albumin as a standard.
Enzyme Activity Assays
To measure (−)-cocaine and benzoic acid, the product of (−)-cocaine hydrolysis catalyzed by BChE, sensitive radiometric assays were used based on toluene extraction of [3H](−)-cocaine labeled on its benzene ring.45 In brief, to initiate the enzymatic reaction, 100 nCi of [3H](−)-cocaine was mixed with the solution of the purified enzyme. The enzymatic reactions proceeded at 37 °C with varying concentrations of (−)-cocaine. The reactions were stopped by adding 200 μL of 0.1 M HCl, which neutralized the liberated benzoic acid whereas ensuring a positive charge on the residual (−)-cocaine. [3H]Benzoic acid was extracted by 1 mL of toluene and measured by scintillation counting. Finally, the measured (−)-cocaine concentration-dependent radiometric data were analyzed by using the standard Michaelis–Menten kinetics with Prism 5 (GraphPad Software Inc., San Diego, CA).
To determine the in vitro half-life of the enzyme at 37 °C, the enzyme was diluted to 200 μg/mL, stored in sealed glass tubes and incubated at 37 °C. The tubes were sealed to avoid the possible vaporization-associated change in the volume. One tube will be taken out of the incubation cabinet at various time points (0, 2, 9, 12, 31, and 100 days) and assayed for the catalytic activity against (−)-cocaine as mentioned above. The percentage of remaining activity was plotted against the incubation time.
Crystallization and Structure Determination
Crystals of the designed new mutant of CocE were grown by hanging drop vapor diffusion, screening for conditions against the JCSG Core Suite (Qiagen). At a protein concentration of 10 mg mL–1 and 1:1 well-to-protein ratio, several screen conditions gave spindle-shaped or fusiform crystals. The most ordered crystals grew against wells containing 0.1 M phosphate-citrate (pH 4.2), 1.6 M sodium dihydrogen phosphate, and 0.4 M dipotassium hydrogen phosphate (JCSG IV # 94). The largest crystals were ∼0.2 mm in the longest dimension.
Crystals were mounted in Mylar loops (LithoLoops, Molecular Dimensions) and flash frozen46 in liquid nitrogen after passing for a few seconds through a solution containing the well solutes plus 20% glycerol. X-ray data were collected at beamline 22ID (SER-CAT sector) at the Advanced Photon Source, Argonne National Laboratory at a temperature of 110 K. Data were reduced with the program HKL200047 and all aspects of structure determination and refinement were carried out in the Phenix suite.48 Initial phasing was done by molecular replacement (Phaser(49) module) using the structure of unliganded cocaine esterase25 (PDB code 3I2J). Subsequent model refinement and addition of ordered solvent was carried out using the autobuild and refinement modules of Phenix with manual rebuilding in Coot,50 which was also used to introduce the sequence changes into the model (T172R, G173Q, L196C, and I301C). Data reduction and model parameters are given in Table S1 of the Supporting Information.
PEGylation
Purified enzyme was conjugated with maleimide-linked branched poly(ethylene glycol) (PEG) with molecular weight of 40 kDa (JenKem Technology, Allen, TX) overnight in PBS buffer, pH 7.4 at the PEG to enzyme molar ratio of 20. The PEGylated protein was purified by using the same HisPur cobalt resin mentioned above.
In Vivo Studies
Male CD-1 mice (25–30 g) were purchased from Harlan (Indianapolis, IN) and were housed in groups of four mice per cage. All mice were allowed ad libitum access to food and water and were maintained on a 12 h light–dark cycle with lights on at 6:30 a.m. in a room kept at a temperature of 21–22 °C. Experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health. The experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Kentucky.
The purified enzyme was administrated intravenously (i.v., via tail vein) and (−)-cocaine HCl (obtained from National Institute on Drug Abuse, Bethesda, MD) was administered intraperitoneally, at a volume of ∼0.2 mL/mouse. Cocaine-induced toxicity was characterized by the occurrence of lethality. Lethality was defined as cessation of observed movement and respiration. A single dose (30 mg/kg) of E196–301 with or without the PEGylation was administered intravenously (i.v.) 1 min before the first intraperitoneal (i.p.) administration of 180 mg/kg cocaine (n = 5). Then, the mice were challenged again daily with 180 mg/kg cocaine (i.p.) until no mouse survived. Following cocaine administration, mice were immediately placed individually for observation. The presence or absence of lethality was recorded for 60 min following cocaine administration.
Acknowledgments
This work was supported by the National Institutes of Health (grants R01 DA035552, R01 DA032910, R01 DA013930, and R01 DA025100 to Zhan). X-ray diffraction data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. We thank the staff of SER-CAT for assistance in data collection. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No.W-31-109-Eng-38. Use of the Protein Core of the Center for Molecular Medicine was supported by NIH Grant No. P20GM103486 from the National Institute of General Medical Sciences. The authors also acknowledge the Computer Center at the University of Kentucky for supercomputing time on a Dell X-series Cluster with 384 nodes or 4768 processors.
Supporting Information Available
X-ray crystallography data collection and refinement statistics (Table S1). Backbone superposition between the X-ray crystal structures of E172-173 and E196-301 (Figure S1). Time-dependence of important H···O distances (relevant to hydrogen bonds) from the MD-simulated E172-173 and E196-301 structures (Figure S2). Intermonomer disulfide bonds in the CocE mutant (E196-301) dimer refined in space group P65 (Figure S3). This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The atomic coordinates and structure factors (code 4P08) for the crystal structure of the T172R/G173Q/L196C/I301C mutant of CocE have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- UNODC. (2010) World Drug Report 2010, United Nations Publication, Sales No. E.10.XI.13. [Google Scholar]
- Karila L.; Gorelick D.; Weinstein A.; Noble F.; Benyamina A.; Coscas S.; Blecha L.; Lowenstein W.; Martinot J. L.; Reynaud M.; Lépine J. P. (2008) New treatments for cocaine dependence: A focused review. Int. J. Neuropsychoph. 11, 425–438. [DOI] [PubMed] [Google Scholar]
- Xi Z.-X.; Gardner L. E. (2008) Hypothesis-driven medication discovery for the treatment of psychostimulant addiction. Curr. Drug Abuse Rev. 1, 303–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrera M. R. A.; Kaufmann G. F.; Mee J. M.; Meijler M. M.; Koob G. F.; Janda K. D. (2004) Treating cocaine addiction with viruses. Proc. Natl. Acad. Sci. U.S.A. 101, 10416–10421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamendulis L. M.; Brzezinski M. R.; Pindel E. V.; Bosron W. F.; Dean R. A. (1996) Metabolism of cocaine and heroin is catalyzed by the same human liver carboxylesterases. J. Pharmacol. Exp. Ther. 279, 713–717. [PubMed] [Google Scholar]
- Meijler M. M.; Kaufmann G. F.; Qi L.; Mee J. M.; Coyle A. R.; Moss J. A.; Wirsching P.; Matsushita M.; Janda K. D. (2005) Fluorescent cocaine probes: A tool for the selection and engineering of therapeutic antibodies. J. Am. Chem. Soc. 127, 2477–2484. [DOI] [PubMed] [Google Scholar]
- Zhan C.-G.; Deng S.-X.; Skiba J. G.; Hayes B. A.; Tschampel S. M.; Shields G. C.; Landry D. W. (2005) First-principle studies of intermolecular and intramolecular catalysis of protonated cocaine. J. Comput. Chem. 26, 980–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry D. W.; Zhao K.; Yang G. X.; Glickman M.; Georgiadis T. M. (1993) Antibody-catalyzed degradation of cocaine. Science 259, 1899–1901. [DOI] [PubMed] [Google Scholar]
- Larsen N. A.; Turner J. M.; Stevens J.; Rosser S. J.; Basran A.; Lerner R. A.; Bruce N. C.; Wilson I. A. (2002) Crystal structure of a bacterial cocaine esterase. Nat. Struct. Mol. Biol. 9, 17–21. [DOI] [PubMed] [Google Scholar]
- Ko M.-C.; Bowen L. D.; Narasimhan D.; Berlin A. A.; Lukacs N. W.; Sunahara R. K.; Cooper Z. D.; Woods J. H. (2007) Cocaine esterase: Interactions with cocaine and immune responses in mice. J. Pharmacol. Exp. Ther. 320, 926–933. [DOI] [PubMed] [Google Scholar]
- Gao D.; Narasimhan D. L.; Macdonald J.; Ko M.-C.; Landry D. W.; Woods J. H.; Sunahara R. K.; Zhan C.-G. (2009) Thermostable variants of cocaine esterase for long-time protection against cocaine toxicity. Mol. Pharmacol. 75, 318–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sticke D. F.; Presta L. G.; Dill K. A.; Rose G. D. (1992) Hydrogen bonding in globular proteins. J. Mol. Biol. 226, 1143–1159. [DOI] [PubMed] [Google Scholar]
- Kabashima T.; Li Y.; Kanada N.; Ito K.; Yoshimoto T. (2001) Enhancement of the thermal stability of pyroglutamyl peptidase I by introduction of an intersubunit disulfide bond. Biochim. Biophys. Acta 1547, 214–220. [DOI] [PubMed] [Google Scholar]
- Shoichet B. K.; Baase W. A.; Kuroki R.; Matthews B. W. (1995) A relationship between protein stability and protein function. Proc. Natl. Acad. Sci. U.S.A. 92, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørk A.; Dalhus B.; Mantzilas D.; Eijsink V. G. H.; Sirevåg R. (2003) Stabilization of a tetrameric malate dehydrogenase by introduction of a disulfide bridge at the dimer–dimer interface. J. Mol. Biol. 334, 811–821. [DOI] [PubMed] [Google Scholar]
- Han Z.-l.; Han S.-y.; Zheng S.-p.; Lin Y. (2009) Enhancing thermostability of a Rhizomucor miehei lipase by engineering a disulfide bond and displaying on the yeast cell surface. Appl. Microbiol. Biotechnol. 85, 117–126. [DOI] [PubMed] [Google Scholar]
- Beadle B. M.; Shoichet B. K. (2002) Structural bases of stability–function tradeoffs in enzymes. J. Mol. Biol. 321, 285–296. [DOI] [PubMed] [Google Scholar]
- Bloom J. D.; Wilke C. O.; Arnold F. H.; Adami C. (2004) Stability and the evolvability of function in a model protein. Biophys. J. 86, 2758–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatani R. A.; Gonzalez A.; Shoichet B. K.; Brinen L. S.; Babbitt P. C. (2007) Stability for function trade-offs in the enolase superfamily “catalytic module”. Biochemistry 46, 6688–6695. [DOI] [PubMed] [Google Scholar]
- Tokuriki N.; Stricher F.; Serrano L.; Tawfik D. S. (2008) How protein stability and new functions trade off. PLoS Comput. Biol. 4, e1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas V. L.; McReynolds A. C.; Shoichetb B. K. (2010) Structural bases for stability-function tradeoffs in antibiotic resistance. J. Mol. Biol. 396, 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merski M.; Shoichet B. K. (2012) Engineering a model protein cavity to catalyze the Kemp elimination. Proc. Natl. Acad. Sci. U.S.A. 109, 16179–16183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brim R. L.; Nance M. R.; Youngstrom D. W.; Narasimhan D.; Zhan C.-G.; Tesmer J. J.; Sunahara R. K.; Woods J. H. (2010) A thermally stable form of bacterial cocaine esterase: A potential therapeutic agent for treatment of cocaine abuse. Mol. Pharmacol. 77, 593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins G. T.; Brim R. L.; Narasimhan D.; Ko M.-C.; Sunahara R. K.; Zhan C.-G.; Woods J. H. (2009) Cocaine esterase prevents cocaine-induced toxicity and the ongoing intravenous self-administration of cocaine in rats. J. Pharmacol. Exp. Ther. 331, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan D.; Nance M. R.; Gao D.; Ko M.-C.; Macdonald J.; Tamburi P.; Yoon D.; Landry D. M.; Woods J. H.; Zhan C.-G.; Tesmer J. J. G.; Sunahara R. K. (2010) Structural analysis of thermostabilizing mutations of cocaine esterase. Protein. Eng. Des. Sel. 23, 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan D.; Collins G. T.; Nance M. R.; Nichols J.; Edwald E.; Chan J.; Ko M.-C.; Woods J. H.; Tesmer J. J. G.; Sunahara R. K. (2011) Subunit stabilization and polyethylene glycolation of cocaine esterase improves in vivo residence time. Mol. Pharmacol. 80, 1056–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins G. T.; Zaks M. E.; Cunningham A. R. St; Clair C.; Nichols J.; Narasimhan D.; Ko M.-C.; Sunahara R. K.; Woods J. H. (2011) Effects of a long-acting mutant bacterial cocaine esterase on acute cocaine toxicity in rats. Drug Alcohol Depend. 118, 158–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins G. T.; Narasimhan D.; Cunningham A. R.; Zaks M. E.; Nichols J.; Ko M.-C.; Sunahara R. K.; Woods J. H. (2012) Long-lasting effects of a PEGylated mutant cocaine esterase (CocE) on the reinforcing and discriminative stimulus effects of cocaine in rats. Neuropsychopharmacology 37, 1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins G. T.; Brim R. L.; Noon K. R.; Narasimhan D.; Lukacs N. W.; Sunahara R. K.; Woods J. H.; Ko M.-C. (2012) Repeated administration of a mutant cocaine esterase: Effects on plasma cocaine levels, cocaine-induced cardiovascular activity, and immune responses in rhesus monkeys. J. Pharmacol. Exp. Ther. 342, 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Yang W.; Pan Y.; Zheng F.; Cho H.; Tai H.-H.; Zhan C.-G. (2009) Free-energy perturbation simulation on transition states and redesign of butyrylcholinesterase. Biophys. J. 96, 1931–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou S.; Xue L.; Yang W.; Fang L.; Zheng F.; Zhan C.-G. (2013) Substrate selectivity of high-activity mutants of human butyrylcholinesterase. Org. Biomol. Chem. 11, 7477–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Hamza A.; Zhan C.-G. (2009) Fundamental reaction mechanism and free energy profile for (−)-cocaine hydrolysis catalyzed by cocaine esterase. J. Am. Chem. Soc. 131, 11964–11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y.; Gao D.; Yang W.; Cho H.; Yang G.; Tai H.-H.; Zhan C.-G. (2005) Computational redesign of human butyrylcholinesterase for anticocaine medication. Proc. Natl. Acad. Sci. U.S.A. 102, 16656–16661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F.; Yang W.; Xue L.; Hou S.; Liu J.; Zhan C.-G. (2010) Design of high-activity mutants of human butyrylcholinesterase against (−)-cocaine: structural and energetic factors affecting the catalytic efficiency. Biochemistry 49, 9113–9119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D.; Zhan C.-G. (2006) Modeling evolution of hydrogen bonding and stabilization of transition states in the process of cocaine hydrolysis catalyzed by human butyrylcholinesterase. Proteins 62, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza A.; Cho H.; Tai H. H.; Zhan C.-G. (2005) Molecular dynamics simulation of cocaine binding with human butyrylcholinesterase and its mutants. J. Phys. Chem. B 109, 4776–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y.; Gao D.; Yang W.; Cho H.; Zhan C.-G. (2007) Free energy perturbation (FEP) simulation on the transition states of cocaine hydrolysis catalyzed by human butyrylcholinesterase and its mutants. J. Am. Chem. Soc. 129, 13537–13543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F.; Yang W.; Ko M.-C.; Liu J.; Cho H.; Gao D.; Tong M.; Tai H.-H.; Woods J. H.; Zhan C.-G. (2008) Most efficient cocaine hydrolase designed by virtual screening of transition states. J. Am. Chem. Soc. 130, 12148–12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong D.; Chun W.; Shibasish C.; Mathew C. L.; Guoming X.; Wei Z.; Rong Y.; Piotr C.; Ray L.; Taisung L.; James C.; Junmei W.; Peter K. (2003) A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. [Google Scholar]
- Darden T.; York D.; Pedersen L. (1993) Particle mesh Ewald: An N [center-dot] log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341. [Google Scholar]
- Braman J.; Papworth C.; Greener A. (2000) Site-directed mutagenesis using double-stranded plasmid DNA templates. Nucleic Acid Protocols Handbook 835–844. [DOI] [PubMed] [Google Scholar]
- Sun H.; Shen M. L.; Pang Y.-P.; Lockridge O.; Brimijoin S. (2002) Cocaine metabolism accelerated by a re-engineered human butyrylcholinesterase. J. Pharmacol. Exp. Ther. 302, 710–716. [DOI] [PubMed] [Google Scholar]
- Rodgers D. W. (1997) Practical cryocrystallography, in Methods in Enzymology (Charles W., Carter, Ed.), pp 183–203, Academic Press, New York. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode, in Methods in Enzymology (Charles W., Carter, Ed.), pp 307–326, Academic Press, New York. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.