

Abstract
Multidomain protein kinases, central controllers of signal transduction, use regulatory domains to modulate catalytic activity in a complex cellular environment. Additionally, these domains regulate noncatalytic functions, including cellular localization and protein–protein interactions. Src-family kinases (SFKs) are promising therapeutic targets for a number of diseases and are an excellent model for studying the regulation of multidomain kinases. Here, we demonstrate that the regulatory domains of the SFKs Src and Hck are divergently affected by ligands that stabilize two distinct inactive ATP-binding site conformations. Conformation-selective, ATP-competitive inhibitors differentially modulate the ability of the SH3 and SH2 domains of Src and Hck to engage in intermolecular interactions and the ability of the kinase–inhibitor complex to undergo post-translational modification by effector enzymes. This surprising divergence in regulatory domain behavior by two classes of inhibitors that each stabilize inactive ATP-binding site conformations is found to occur through perturbation or stabilization of the αC helix. These studies provide insight into how conformation-selective, ATP-competitive inhibitors can be designed to modulate domain interactions and post-translational modifications distal to the ATP-binding site of kinases.
Protein kinases are critical mediators of cellular signaling through the propagation of phosphorylation cascades. For all kinases, a highly conserved bilobal domain containing an ATP-binding cleft is responsible for phosphotransfer activity.1,2 In order to transmit signals with fidelity in the complex milieu of the cell, tight regulation of catalysis is required. This regulation is often achieved via fusion of the catalytic domain to regulatory or targeting domains.3 These domains can allosterically regulate the activity of the kinase domain through intramolecular engagement and suppression of the catalytic domain.4,5 Regulatory domains are not only important for modulating catalytic activity but also serve roles in other functions, including localization, DNA binding, and protein–protein interactions.6 Often, these domains facilitate functions that are independent of kinase catalytic activity in the cell.
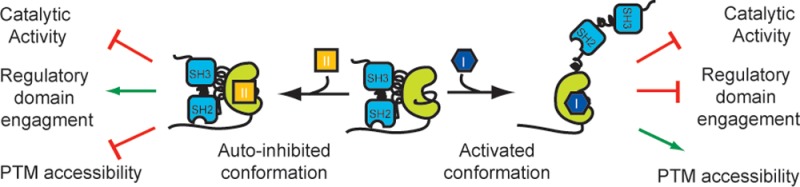
Src-family kinases (SFKs) are prototypical nonreceptor multidomain protein kinases consisting of regulatory SH2 and SH3 domains, a tyrosine kinase catalytic domain, and an N-terminal unique region. SFKs are involved in the regulation of important cellular processes including cell metabolism, proliferation, and differentiation.7−9 Additionally, SFKs have prominent roles in invasion and tumor progression, angiogenesis, and metastasis, making them a promising target for cancer therapy.10−12 More fundamentally, SFKs are a well-studied model for understanding how regulatory domains affect kinase catalysis.13,14 SFK activity is allosterically suppressed by two intramolecular binding events: the SH2 domain’s interaction with phospho-Tyr527 in the C-terminal tail and the SH3 domain’s interaction with a proline-containing linker (SH2-kinase linker) that connects the SH2 domain with the catalytic domain.14−16 Release of these interactions through dephosphorylation of pTyr527 or direct disruption of the intramolecular SH2 and SH3 regulatory domain interactions leads to activation of the catalytic domain (Figure 1A). Full activation is achieved by phosphorylation of Tyr416 in the activation loop.15
Figure 1.

Regulation of SFK catalytic activity and ATP-binding site conformational accessibility. (A) SFK activity is allosterically modulated by engagement of the SH2 and SH3 regulatory domains (PDB: 2SRC). Release of these interactions through dephosphorylation of pTyr527 or intermolecular displacement of the regulatory domains leads to increased catalytic activity (PDB: 1Y57). Phosphorylation of Tyr416 on the activation loop fully activates the catalytic domain. (B) ATP-binding site conformations in which SFKs have been observed. Left: The active ATP-binding site conformation of SFKs, where all conserved catalytic residues are aligned for facilitating phosphate transfer. Center: An inactive SFK ATP-binding site conformation, where the αC helix is rotated out of the active site, displacing a catalytically important glutamic acid. This inactive conformation is often referred to as the αC helix-out or Src/CDK-like inactive conformation. Right: The DFG-out inactive conformation, where flipping of a conserved tripeptide motif (DFG motif) at the base of the activation loop results in the displacement of an aspartate residue that is important for catalysis.
Just as SFK regulatory domains undergo large conformational changes, their ATP-binding sites are also highly dynamic. The ATP-binding site of Src has been structurally characterized in three distinct conformations: one active and two inactive (DFG-out and αC helix-out) forms (Figure 1). In the active conformation, all key catalytic residues are optimally positioned for catalysis, and two conserved networks of hydrophobic “spines” are aligned.1,17−19 Both inactive ATP-binding site conformations are characterized by displacement of at least one conserved catalytic residue from the active site and disruption of the regulatory hydrophobic spine. The DFG-out inactive conformation involves flipping of the conserved Asp-Phe-Gly (DFG) motif at the base of the activation loop, which results in the displacement of the catalytic aspartic acid residue and removal of the phenylalanine side chain from the regulatory hydrophobic spine. Kinases in the αC helix-out inactive conformation possess a disrupted salt bridge between the catalytic lysine (Lys295) and a conserved glutamic acid (Glu310) that is located on helix αC (Figure 1B). Importantly, conformation-selective inhibitors that stabilize each of the three conformations described above have been identified for the SFKs.
While the allosteric coupling between the regulatory and catalytic domains of SFKs has been extensively investigated, there are many aspects of their inter-relationship that are still not well understood. For example, the ATP-binding sites of Src and Hck constructs with fully engaged regulatory domains are in the αC helix-out inactive conformation in crystal structures,14,20 but it is unclear whether alternative inactive forms, such as DFG-out, are also accessible to autoinhibited SFKs. Furthermore, the full complement of regulatory interactions that are controlled by the conformation of SFK ATP-binding sites and the effects on SFK post-translational modification are still not known. Here, we biochemically profile how stabilizing the ATP-binding sites of the SFKs Src and Hck in two distinct inactive ATP-binding site conformations (αC helix-out and DFG-out) affect their regulatory interactions and their ability to undergo post-translational modification. Surprisingly, these studies show that different inactive ATP-binding site conformations lead to divergent SH2 and SH3 regulatory domain behaviors. Furthermore, we find that different classes of conformation-selective, ATP-competitive inhibitors also divergently modulate the ability of kinases and phosphatases to act upon inhibitor-bound SFKs as substrates. These new results give mechanistic insights into the regulation of SFKs and suggest strategies for the design of ATP-competitive inhibitors that are able to modulate protein–protein interactions and the post-translational modification of multidomain tyrosine kinases.
Results and Discussion
The SH3 Domains of Src and Hck Are Divergently Affected by Ligands That Stabilize Different Inactive ATP-Binding Site Conformations
To fully characterize how distinct inactive ATP-binding site conformations affect the regulatory domains of SFKs, we assembled a panel of conformation-selective inhibitors that stabilize both the αC helix-out and DFG-out forms (Figure 2A). Inhibitors 2 and 3 contain extended hydrophobic substituents at the C-3 positions of their pyrazolopyrimidine scaffolds that are incompatible with the active conformation of helix αC. Both inhibitors have been shown to stabilize the αC helix-out inactive conformation of tyrosine kinases in crystal structures.21,224 and 5 are ligands that stabilize the DFG-out inactive conformation of protein kinases.23 Both ligands contain a 3-trifluoromethylbenzamide group that can be accommodated only in the ATP-binding sites of kinases with “flipped” DFG motifs. Furthermore, both inhibitors contain an amide linker between the substituent that occupies the adenine pocket and the 3-trifluoromethylbenzamide group, which provides a hydrogen bond donor/acceptor interaction that is observed in almost all ligands that stabilize the DFG-out form. Alternative pyrazolopyrimidine-based inhibitors that stabilize the DFG-out conformation, but contain a urea linker, have been reported. However, crystal structures have shown that the ATP-binding site of Src can adopt either the DFG-out (PDB: 3EL8) or αC helix-out (PDB: 3QLF) conformation when bound to these ligands, most likely due to the conformational flexibility of the urea linkage. The more conformationally rigid amide linkages of 4 and 5 are most likely compatible only with the DFG-out conformation. PP2 (1), which does not contain any functionalities that favor a particular ATP-binding site conformation, was used as a control for all studies.
Figure 2.

Conformation-selective inhibitors divergently modulate the SH3 domain accessibility of Src and Hck. (A) Panel of inhibitors that were used in this study. 2 and 3 stabilize the αC helix-out inactive conformation. 4 and 5 stabilize the DFG-out inactive conformation. (B) SH3 domain accessibility pull-down assay. Src or Hck was incubated with an immobilized SH3 domain ligand in the presence of a saturating amount of an inhibitor (1, 2, 4, or 5). After incubation, beads were washed, and retained SFKs were eluted with SDS. Retained SFKs were quantified by immunoblotting. (C) Quantification of the pull-down experiments performed with SrcY527F and HckY527F in the presence of 1, 2, 4, or 5 (mean ± SEM, n = 3). † = Previously reported and shown for comparison.21 (D) FRET assay to measure intermolecular SH3 domain accessibility. Hck was incubated with conformation-selective inhibitors in the presence of variable concentrations of a fluorophore-labeled SH3 peptide ligand (Peptide-FL). SH3 domain accessibility was determined by measuring FRET between the donor His-Tb, which is bound to the N-terminal His6 tag of Hck, and Peptide-FL. (E) Binding of fluorophore-labeled SH3 peptide ligand (Peptide-FL) in the presence of conformation-selective inhibitors (mean ± SEM, n = 3).
The effect of stabilizing different inactive ATP-binding site conformations on SFK regulatory domain interactions was first probed using SH3 domain pull-down experiments (Figure 2B). In this assay, unphosphorylated SFK constructs in the presence of a saturating amount of an ATP-competitive inhibitor are incubated with resin containing an immobilized SH3 peptide ligand. The beads are then washed before eluting bound kinase with detergent, and the amount of retained SFK is determined using western blot analysis (Figure 2B). In the presence of control compound 1, 11% of total Src and 2% of total Hck were retained on beads (Figure 2C). Because we have previously demonstrated that stabilizing the ATP-binding sites of Src and Hck in the αC helix-out inactive conformation with ligands 2 and 3 blocks the ability of their SH3 domains to engage in intermolecular interactions (Figure 2C), we were curious as to whether ligands 4 and 5, which stabilize the DFG-out conformation, would lead to the same effect. Surprisingly and in stark contrast to αC helix-out stabilizing inhibitors, 4 and 5 lead to a dramatic increase in SH3 domain accessibility for both Src and Hck (Figure 2C), with 74% of Src–4 and 38% of Hck–4 retained by the immobilized SH3 domain ligand. A similar increase in intermolecular SH3 domain engagement is observed when Src and Hck are bound to 5. Because ligands that stabilize the αC helix-out conformation lead to reduced SH3 domain accessibility by strengthening the intramolecular interaction between the SH3 domain and the SH2 linker, it is likely that the increased accessibility observed in the presence of DFG-out stabilizing inhibitors is due to destabilization of this same interaction. Thus, ligands 4 and 5 stabilize an inactive ATP-binding site conformation while stabilizing an active, extended global kinase conformation. Although this unexpected observation is novel for the SFKs, it has recently been observed that the DFG-out inhibitor, imatinib, stabilizes an open conformation of the kinase Abl, indicating that this phenomenon is not unique to SFKs and that it, in fact, may be general to many multidomain kinases.24
Next, a fluorescence resonance energy transfer (FRET)-based assay was employed to further characterize how conformation-selective inhibitors affect SH3 domain accessibility. To determine the ability of Hck’s SH3 domain to intermolecularly engage a ligand, FRET between a terbium-conjugated α-His6 antibody (Tb-His), which binds to the N-terminal His6 tag of Hck, and an Oregon Green-derivatized SH3 peptide ligand (Peptide-FL) was measured (Figure 2D). Relative differences in SH3 domain accessibility were determined by measuring FRET between Tb-His and variable concentrations of Peptide-FL in the presence of a saturating amount of conformation-selective inhibitors. Consistent with the lack of ATP-binding site conformational preference of ligand 1, similar FRET was observed for the Hck–1 complex and apo Hck at all Peptide-FL concentrations tested (Figure 2E). Similar to the pull-down assays, the SH3 domain of Hck is almost completely inaccessible when this kinase is bound to αC helix-out stabilizing inhibitor 2. However, ligand 4, which stabilizes the DFG-out inactive conformation, markedly increased the ability of Hck to bind Peptide-FL. Thus, the specific inactive ATP-binding site conformation that is stabilized, DFG-out or αC helix-out, leads to a dramatic difference in the SH3 domain accessibility of Hck.
Conformation-Selective Ligands Divergently Modulate SFK SH2 Domain Accessibility
The SH3 and SH2 regulatory domains of SFKs are believed to act in a “snap-lock” fashion, where intramolecular binding of one domain causes concerted engagement of the second domain.25−27 Therefore, increased intramolecular interactions between the SH3 domain and SH2 linker of SFKs should likely result in increased engagement of their SH2 domain with the C-terminal regulatory tail, reducing the capacity of the SH2 domain to partake in intermolecular binding events. To determine whether ATP-competitive ligands that decrease SH3 domain accessibility also reduce the ability of SFK SH2 domains to engage in intermolecular interactions, a series of SH2 pull down experiments was performed with an immobilized SH2 domain phospho-peptide ligand in the presence of saturating amounts of inhibitors (Figure 3A). Src–1 and Hck–1 complexes were used as references in the pull-down assay. A notable decrease in Hck SH2 domain accessibility was observed in the presence of inhibitors 2 or 3. Although the observed effect for inhibitor-bound Src was relatively small, 2% and 17% of Hck–2 and Hck–3 were retained by the immobilized SH2 ligand relative to that of Hck–1, respectively (Figure 3B). No change in retention was observed when Src was bound to DFG-out stabilizing ligands 4 or 5. However, a small increase in SH2 domain accessibility was observed for Hck–4 and Hck–5 relative to that of Hck–1. Therefore, stabilization of an αC helix-out ATP-binding site conformation also modulates the SH2 regulatory domain of SFKs. However, the magnitude of this effect is different for Src and Hck. The binding affinities of the SH2 domains of Src and Hck for the pYEEI phosphopeptide have been shown to be very similar.28 Therefore, a combination of differences in the SH2-linker sequence, residues linking the SH2 and SH3 domains, and residues comprising the C-terminal tail may account for these variations in coupled regulatory domain engagement.29−32 Further study is needed to dissect the exact sequence determinants of these regulatory differences.
Figure 3.
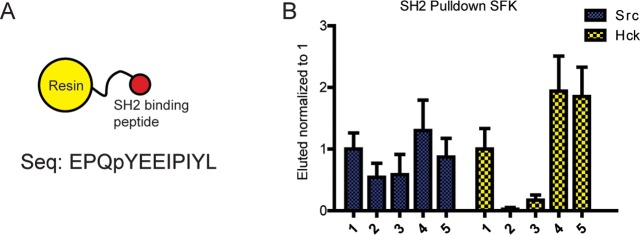
Conformation-selective inhibitors modulate SH2 domain accessibility. (A) SH2-binding peptide conjugated to resin. (B) Quantification of the SH2 pull-down assays performed with Src and Hck in the presence of various inhibitors (mean ± SEM, n = 3). The amount of kinase eluted from the beads is normalized to that of SFK–1.
Modulation of SFK Phosphorylation by ATP-Binding Site Occupancy
To the best of our knowledge, the effects that ATP-binding site occupation has on SFK regulatory post-translational processing have not been determined. Given that ATP-competitive inhibitors can differentially modulate the accessibility of SFK regulatory domains, we were interested to determine whether ATP-binding site occupancy can also influence the activating and deactivating regulatory phosphorylation of these enzymes. It has recently been reported that the binding of ATP-competitive inhibitors can increase the level of activation loop phosphorylation of some kinases, such as Akt, RAF, and Jak2.33−36 It has also been reported that when mitogen-activated protein kinases (MAPKs) are bound to inhibitors that stabilize the DFG-out inactive conformation their activation loops cannot be phosphorylated by upstream activators.37,38 Interestingly, this inhibitory effect is observed only when MAPKs are in the DFG-out inactive conformation. To determine whether conformation-selective inhibitors can likewise differentially influence the autoinhibitory phosphorylation site (Tyr527) of SFKs in a similar manner, a series of phosphorylation assays was performed with Src and Hck.
Although SFK C-terminal tail phosphorylation (Tyr527) can occur through trans-autophosphorylation, the major pathway for this post-translational modification is through the action of C-terminal Src kinase (Csk).39,40 Csk is a highly specific tyrosine kinase known to selectively phosphorylate a single tail tyrosine in SFKs.41,42 This phosphorylation event leads to increased intramolecular engagement between the C-terminal tail and the SH2 domain, resulting in SFK autoinhibition. In order to investigate the effects of ATP-binding site occupancy on this inhibitory modification, an assay that allows uninhibited Csk to act upon an inhibitor-bound SFK as a substrate was needed. Because of the similarities of their ATP-binding sites, many SFK inhibitors also potently inhibit Csk as well.1 Therefore, we developed a chemical genetic method that uses a drug-resistant Csk construct (CskDR). CskDR contains a T266 M gatekeeper mutation that provides resistance to many ATP-competitive inhibitors (Supporting Information Table S1). Src and HckY416F, which lacks the activation loop phosphorylation site, were used as inhibitor-bound substrates of CskDR (Figure 4A).
Figure 4.

SFK inhibitors can modulate C-terminal tail phosphorylation. (A) Experimental schematic of SFK Tyr527 phosphorylation. Src or HckY416F was incubated with CskDR in the presence of inhibitor. The concentration of each inhibitor is such that >85% of Src or HckY416F is bound to inhibitor and CskDR is inhibited by <13%. ATP was added to the mixture to initiate CskDR-mediated phosphorylation of the SFK–inhibitor complex. After 1 h of incubation, pTyr527 levels were determined by performing SDS-PAGE, followed by immunoblotting with an α-phospho-527 Src antibody for Src or α-pTyr antibody for HckY416F. All values are normalized to that of the SFK–1 complex. (B) Quantification of Src and HckY416F pTyr527 levels in the presence of inhibitors 1, 2, or 5. All values are normalized to that of the SFK–1 complex (mean ± SEM, n = 3). (C) Chemical genetic method for generating covalent SFK–inhibitor complexes to measure SFK Tyr527 phosphorylation. Covalent SFK–ligand complexes were generated by incubating SFKS245C with ligand 6 or 7. The resulting SFK–ligand substrate was purified and then incubated with Csk and γ32P-ATP. Phosphorylation levels were measured at 0.5, 1, and 1.5 h. (D) Structures of electrophile-containing analogues. (E) Quantification of SrcS245C pTyr527 levels in the presence of inhibitors 6 or 7 (mean ± SEM, n = 3). (F) Quantification of Hck245C pTyr527 levels in the presence of inhibitors 6 or 7 (mean ± SEM, n = 3).
Src or HckY416F was incubated with CskDR and ATP in the presence of a concentration of inhibitor that leads to minimal inhibition of CskDR (<13%) and >85% inhibitor-bound Src or HckY416F (see Supporting Information Table S2). Control activity assays were performed with a peptide substrate to verify that CskDR activity was the same at all inhibitor concentrations used in the phospho-Tyr527 assays (data not shown).42,43 Compound 1 was again used as a reference compound for the level of phosphorylation that is observed when an SFK construct is bound to an inhibitor that has minimal ATP-binding site conformational preference. The C-terminal tails of Src–1 and HckY416F–1 were phosphorylated by CskDR 3- and 5.5-fold more than that of Src–2 and Hck–2, respectively. Therefore, the C-terminal tails of SFKs are less accessible to Csk phosphorylation when their ATP-binding sites are occupied by inhibitors stabilizing the αC helix-out inactive conformation (Figure 4B). This correlates very well with our earlier observation that ATP-binding site occupation by this class of inhibitors results in decreased intermolecular accessibility of the SH2 domain, presumably through engagement of the C-terminal tail. Conversely, when Src or HckY416F was bound to ligands that stabilize the DFG-out conformation, an increase in C-terminal tail phosphorylation occurred. Src–5 and Hck–5 were phosphorylated 1.9- and 1.4-fold more than that of the SFK–1 substrate. Again, these results are consistent with the SH2 pull-down assay results shown in Figure 3, where ligands that stabilize the DFG-out conformation lead to an enhancement in the accessibility of SH2 domains to intermolecular binding events.
To provide a more quantitative comparison of how ATP-binding site occupancy affects C-terminal tail phosphorylation, we developed another chemical genetic method that allows formation of a covalent inhibitor–SFK complex to be used as a substrate for Csk. In this system, >99% of each SFK is bound to inhibitor without any resultant partial inhibition of Csk. To facilitate the formation of covalent inhibitor–SFK complexes, a cysteine was engineered into the active site of Src and Hck (S245C) at a position that is rare among nonreceptor kinases (Figure 4C; found only in the TEC family kinases BTK and ITK).44 Introduction of this mutation sensitizes the SFK constructs to pyrazolopyrimidine inhibitors containing an acryloyl functional group projecting from the N-1 position of the pyrazolopyrimidine ring (Figure 4D). Indeed, αC helix-in ligand 6 and αC helix-out ligand 7 inhibit SFKS245C constructs more potently than could be measured by activity assays, whereas Ki values measured for wild-type SFKs were at least 50-fold higher (Supporting Information Table S3).
Src– and Hck–ligand substrate complexes were generated by incubation of SFKS245C with 6 or 7. Excess small molecule was removed through Ni-affinity resin binding followed by desalting. Finally, unlabeled SFK was removed via incubation with resin displaying a potent kinase inhibitor, dasatinib, providing a single population of covalently labeled SFK. Activity assays were conducted to verify that all kinase activity was blocked (Supporting Information Table S4). We then verified that covalent modification of SFKs with 6 or 7 stabilized an αC helix-in or -out ATP-binding site conformation, respectively, by conducting an SH3 pull-down experiment. Indeed, similar to our studies with noncovalent analogues, the SH3 domain of the SrcS245C–6 complex was 3-fold more accessible than when SrcS245C was covalently modified by 7 (Supporting Information Figure S1). Next, we measured the ability of covalent SFK–ligand complexes to be phosphorylated by Csk. The covalent inhibitor–SFK complex substrate incubated with Csk and γ32P-ATP, and pTyr527 levels were determined at 0.5, 1, and 1.5 h (Figure 4C). No phosphorylation was observed when Csk or SFK substrate was absent from the reaction. Indeed, similar to the previous experiment, at 1.5 h the Src–6 complex was phosphorylated 3-fold more than the Src–7 substrate (Figure 4E). Likewise, Hck–6 was phosphorylated by Csk 5-fold more than Hck–7 (Figure 4E). Unfortunately a covalent ligand to stabilize a DFG-out conformation has not yet been developed, but these results corroborate our findings that stabilizing the ATP-binding site in an αC helix-out inactive conformation shields the C-terminal tail of SFKs from Csk phosphorylation.
Modulation of SFK Dephosphorylation by ATP-Binding Site Occupancy
SFK activity is regulated through both phosphorylation and dephosphorylation. Given that ATP-competitive inhibitors can differentially modulate the phosphorylation of SFKs on Tyr527, we were interested to determine whether ATP-binding site occupancy can also influence dephosphorylation of this site. Intracellular SFK dephosphorylation is mediated by several protein tyrosine phosphatases.45 In order to probe the effects of stabilizing the ATP-binding sites of Src and Hck in the DFG-out or αC helix-out conformations on the modification of pTyr527, an assay that looks at the dephosphorylation of inhibitor-bound SFK complexes by tyrosine phosphatase PTP1b was developed. This phosphatase has been shown to regulate SFK phosphorylation in cells.46 In order to monitor dephosphorylation, SFKs were first radiolabeled at Tyr527 using γ32P-ATP. Following labeling, kinase activity was halted by the addition of inhibitors at a concentration that results in >98% SFK–inhibitor complex. No increase in phosphorylation was observed after inhibitor addition. PTP1b was added to the preformed SFK–inhibitor complex, and dephosphorylation was monitored by loss of 32P-labeled SFK over time (Figure 5A).
Figure 5.
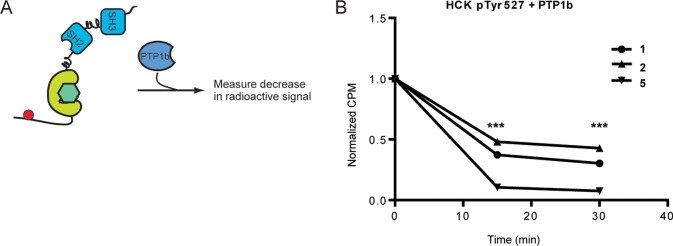
ATP-competitive inhibitors regulate the dephosphorylation of pTyr527. (A) Experimental schematic of SFK dephosphorylation. Hck was radiolabeled by Csk phosphorylation ((32P)pTyr527) with γ32P-ATP. Phosphorylation was then blocked by the addition of inhibitor. PTP1b was then added to the 32P-labeled SFK-inhibitor complex. Loss of radioactive signal was measured over time. (B) 32P-labeled pTyr527 HckY416F was incubated with PTP1b in the presence of 1, 2, or 5, and the decrease in phosphorylated Hck was measured at 15 and 30 min (mean ± SEM, n = 3). *** p < 0.001.
To investigate the dephosphorylation of pTyr527, 32P-labeled HckY416F was generated by incubating HckY416F with Csk and γ32P-ATP. Using a construct that lacks Tyr416 ensured that all radioactive signal was due to Csk phosphorylation of Tyr527 on Hck. Phosphorylation was stopped, and SFK–inhibitor complexes were formed by the addition of 1, 2, or 5. PTP1b was then added, and loss of radioactive signal was monitored over time (Figure 5B). Compound 1 was used as a reference compound for the level of dephosphorylation that is observed when an HckY416F construct is bound to an inhibitor that has minimal ATP-binding site conformational preference. Binding of inhibitor 2 shielded the C-terminal tail of the HckY416F–2 complex from dephosphorylation relative to that of 1. Shielding of pTyr527 from dephosphorylation in the Hck–2 complex agrees with the decreased rate of Tyr527 phosphorylation by Csk in this same complex (Figure 4B,E). 2’s occupation of the ATP-binding site of Hck strengthens the intramolecular interaction between the SH2 domain and the C-terminal tail, blocking not only phosphorylation but also dephosphorylation. In contrast, HckY416F–5 is dephosphorylated much more rapidly than the HckY416F–1 complex. Thus, an inhibitor that stabilizes the DFG-out inactive conformation greatly increases the accessibility of pTyr527 to phosphatases. These results further highlight how different inactive ATP-binding site conformations can lead to divergent regulatory domain behaviors.
Molecular Determinants of ATP-Binding Site Conformation Selectivity
Stabilizing the ATP-binding sites of SFKs in an αC helix-out inactive conformation induces a closed global conformation, which results in decreased accessibility to intermolecular binding events and reduced susceptibility to post-translational modifications. However, inhibitors that stabilize an alternative inactive ATP-binding site alignment, the DFG-out conformation, result in an extended and open global kinase conformation that is reminiscent of fully activated SFKs. In this conformation, SFKs are free to engage intermolecular binding partners and are highly susceptible to phosphorylating and dephosphorylating enzymes. Molecular dynamics studies have suggested that the DFG-out inactive conformation plays a role in the catalytic cycle of tyrosine kinases.47 Therefore, it is not surprising that DFG-out stabilizing ligands do not act entirely like inhibitors that are selective for the αC helix-out form. However, we were quite surprised that these two inactive forms have opposite effects on the regulatory interactions of SFKs. Inhibitors that stabilize the DFG-out and αC helix-out conformations make a number of different interactions with the ATP-binding sites of protein kinases that may account for their differing behaviors (Figure 6A). Therefore, we undertook a systematic analysis of each inhibitor class in order to understand the source of this divergence.
Figure 6.

Binding preferences of conformation-selective, ATP-competitive inhibitors. (A) Ligands that stabilize the DFG-out and αC helix-out conformations make a number of different interactions with the ATP-binding sites of kinases. Left: Src bound to 3 with the ATP-binding site in an inactive, αC helix-out conformation (PDB: 4DGG). The αC helix is rotated out of the active site, disrupting the interaction between Lys295 and Glu310. Right: Abl bound to 5 with the active site in an inactive, DFG-out conformation (PDB: 3OXZ). The DFG motif is flipped out of the active site, but Glu310 in the αC helix maintains a salt bridge with the catalytic lysine (the distance between the NH of 5 and the Glu310 side chain is 3.0 Å). The gatekeeper (Thr338), catalytic lysine (Lys295), and catalytic glutamic acid (Glu310) residues are shown in light blue (Src numbering). (B) Analogues of 3 that were tested in activity assays against Srcact, Hckact, SrcSH2eng, and HckSH2eng constructs. Quantitative comparison of the fold differences in Ki values between activated SFKs (SFKact) and their respective autoinhibited constructs (SFKSH2eng) allow systematic analysis of ATP-binding site conformational preference. Raw data for all assays are shown in Supporting Information Table S5.
As a preliminary screen of these interactions, the bidirectional nature of the interplay between the ATP-binding sites and regulatory domains of SFKs was exploited.21 Just as ATP-binding site conformation can affect regulatory domain engagement, conversely, the engagement of regulatory domains can influence ATP-binding site conformation. Therefore, the relative affinities of inhibitors for SFKs with different regulatory domain interactions is a reflection of the importance of active site features and inhibitor substituents toward specific ATP-binding site interactions. Autoinhibited SFK constructs (SFKSH2eng) that stabilize an αC helix-out ATP-binding site were generated by introducing mutations that strengthen intramolecular regulatory interactions between the C-terminal tail and the SH2 domain.48−51 Fully activated constructs (SFKact) that stabilize an αC helix-in ATP-binding site were generated via autophosphorylation of SFKY527F on Tyr416.15,52 As predicted, Srcact and Hckact are substantially more catalytically active than their autoinhibited counterparts, SrcSH2eng and HckSH2eng (Supporting Information Figure S2). The Ki values for various ligands against the SFKSH2eng and SFKact constructs were determined, and the Ki ratios were analyzed.
To determine the importance of specific interactions toward stabilizing the αC helix-out conformation and inducing a closed SFK regulatory state, analogues of inhibitor 3 were tested (Figure 6B). Consistent with its effects on global kinase conformation, 3 is highly selective for autoinhibited SFKs over their fully activated forms. Ligand 8, which also contains a 6-benzyloxy-2-naphthyl substituent but is missing the 3-chloro group of 3, is also selective for autoinhibited Src and Hck, although to a lesser degree. However, an analogue of 3 that lacks a hydrophobic substituent at the 6-position of the naphthyl ring, ligand 9, is selective for activated SFKs. Thus, the presence of a hydrophobic substituent to fill the pocket created by movement of the αC helix is necessary to stabilize this inactive conformation.
Ligands 4 and 5 are only sterically compatible with the ATP-binding sites of protein kinases that possess a flipped DFG motif (the DFG-out conformation). Unlike the αC helix-out conformation, the alignment of the αC helix in the DFG-out conformation is minimally perturbed from an active alignment. In fact, almost all ligands that stabilize the DFG-out ATP-binding site alignment in SFKs form a hydrogen bond with Glu310 in the αC helix that is possible only when this structural motif is in an active conformation (Figure 6A, right). Therefore, we predicted that the unexpected abilities of inhibitors that stabilize the DFG-out inactive conformation to induce an active global SFK conformation may be due to the interactions that these ligands make with the αC helix. Unfortunately, because of their highly potent inhibition of Src and Hck, we were unable to determine the selectivity of inhibitor 4 or 5 toward activated or autoinhibited SFK constructs. However, on the basis of their propensity to induce an open and active SFK global conformation, these inhibitors are most likely selective for activated SFKs over their autoinhibited forms.
Although not a DFG-out stabilizing ligand, as noted above, 9 is selective for activated SFKs. We hypothesized that this selectivity is likely due to hydrogen bonding with the αC helix. The 6-hydroxyl group of the naphthyl substituent should be able to hydrogen bond with Glu310 in an αC helix-in conformation, which is reflected by its pronounced selectivity toward activated SFKs. In fact, a pyrazolopyrimidine-based ligand, 10, whose azaindole C-3 substituent contains a hydrogen-bond donor, has been crystallized bound to Src, revealing an active ATP-binding site conformation with hydrogen bonding to Glu310 of the αC helix (Figure 7A).
Figure 7.

Biochemical profiling of ligands predicted to stabilize an active, αC helix-in ATP-binding site conformation. (A) Src bound to 8, with the ATP-binding site in an active conformation (PDB: 3EN4). The hydrogen bond between 8 and Glu310 is shown (the distance between NH and the Glu310 side chain is 2.7 Å). The gatekeeper threonine (Thr338), catalytic lysine (Lys295), and catalytic glutamic acid (Glu310) residues are shown in light blue (Src numbering). (B) Chemical structures of the ATP-competitive SFK inhibitors that can potentially hydrogen bond with the αC helix. Quantitative comparison of the fold differences in Ki values between an activated SFK (Srcact and Hckact) and their respective SH2 domain-engaged construct (SrcSH2eng and HckSH2eng). † = The absolute fold difference in Ki value could not be determined because inhibitor affinity is less than the enzyme concentration used in the assay. (C) Quantification of the SH3 pull-down assays performed with Src and Hck in the presence of various inhibitors (mean ± SEM, n = 3).
To further probe the importance of hydrogen bonding with Glu310 in the αC helix, we assembled a small panel of pyrazolopyrimidine-based ligands that contain substituents at the C-3 position that should be capable of hydrogen bonding (Figure 7A,B). Because of the small size of their C-3 substituents, these ligands cannot stabilize the αC helix-out or DFG-out conformation but should interact with an αC helix-in ATP-binding site conformation. Consistent with this notion, each of these ligands was also selective for activated SFKs over the autoinhibited constructs. Furthermore, ligand 1, which is structurally very similar to 11 and 12 but lacks a hydrogen-bond donor, shows little selectivity for either SFK construct, highlighting the importance of hydrogen bonding for activated SFK preference.
On the basis of the bidirectional relationship observed between the ATP-binding sites and regulatory domains of SFKs, inhibitors that are selective for activated SFK constructs should promote a more open global SFK conformation. Furthermore, if αC helix alignment is the molecular switch regulating the divergent outcomes observed between the two inactive conformation-stabilizing ligand classes, then active conformation-selective, αC helix-in ligands should behave similarly to that of DFG-out ligands. To test this, biochemical profiling of Src and Hck in complex with an active conformation-selective ligand was performed. Ligand 11 was chosen as a representative member of this class of inhibitors because of its structural similarity to 1. In SH3 pull-down experiments, DFG-out inhibitors dramatically increased SH3 accessibility. For active-conformation selective inhibitors, SH3 domain accessibility was increased 2- and 3.2-fold for Src and Hck, respectively, relative to that of 1 (Figure 7C). SFKs bound to active-conformation selective inhibitors do, in fact, behave similarly to DFG-out inhibitors and not like αC helix-out inhibitors. These data indicate that a ligand’s ability to control the conformation of the αC helix either through hydrogen bonding or steric perturbation is the major determinant of its influence on noncatalytic SFK regulation. This helix acts as a switch regulating both global kinase conformation and ability to undergo post-translational modification.
Conclusions
In this study, we have determined that two distinct inactive ATP-binding site conformations can differentially modulate the engagement or disengagement of the SH2 and SH3 regulatory domains of SFKs through perturbation or stabilization of the αC helix. Additionally, these conformations can regulate the accessibility of the C-terminal tail towards post-translational modification. Concisely, SFKs can be divergently modulated through the use of inhibitors that stabilize an αC helix-in or -out conformation.
These results have direct implications on the pharmacological inhibition of multidomain kinases. Differential modulation of noncatalytic roles, post-translational modification, and catalytic activity through stabilization of discrete ATP-binding sites with particular inhibitor classes allows for greater control of aberrant signaling enzymes. Recent studies have shown that kinase inhibition in different ATP-binding site conformations can greatly alter downstream signaling.33−36,53,54 Better understanding of how ATP-binding site stabilization affects intermolecular interactions is critical to predicting off-target effects of kinase inhibition. The potential to develop kinase inhibitors that selectively or cooperatively inactivate catalytic and noncatalytic functions should be of great use in both dissecting signaling pathways and in designing future therapeutic inhibitors of aberrant signaling networks.
Methods
Protein Expression
The Src-family kinases, Src (residues 84–533) and Hck (residues 84–531), were expressed with YopH and GroEL and purified as previously described.21,55 Csk and CskDR plasmids were transformed into BL21 (DE3) competent cells and purified by GST resin following the manufacturer’s protocol.
Synthetic Methods
All compounds were synthesized as previously described.56−60
Pull-Down Assay To Determine SH3 Domain Accessibility
Formation of the Kinase–Inhibitor Complex
The kinase of interest (100 nM) and mammalian lysate (0.2 mg mL–1) were diluted in immobilization buffer (50 mM Tris, 100 mM NaCl, and 1 mM DTT, pH 7). The inhibitor of interest (5 μM) was added to this kinase dilution. The mixture was allowed to incubate for 30 min before loading on the resin.
Pull-Down
Forty microliters of a 50% slurry of SNAP-Capture pull-down resin (NEB) was placed in a microcentrifuge tube. The resin was washed (2×, 10 bed volumes) with immobilization buffer. A SNAP tag–polyproline peptide fusion (VSLARRPLPPLP) (10 μM) was loaded onto the resin at a final volume of 100 μL in buffer. The resin was shaken at rt for 90 min with agitation by a pipet every 15 min. After polyproline peptide immobilization, the resin was washed (2×, 10 bed volumes), and 100 μL of the kinase–inhibitor complex was loaded. The resin was allowed to shake at rt for 1 h. After incubation with the kinase–inhibitor complex, the flow through was collected, and the resin was washed (4×, 10 bed volumes). To elute the retained kinase, 100 μL of 1× SDS loading buffer was added, and the beads were boiled at 90 °C for 10 min. All samples were separated by SDS-PAGE and visualized by western blotting using a His6-specific antibody (at a 1:5000 dilution (abm, HIS.H8)). The scanned blots were quantified with LI-COR Odyssey software to determine the percentage of kinase retained on the resin on the basis of the loaded and eluted fractions (mean ± SEM, n = 2).
Pull-Down Assay To Determine SH2 Domain Accessibility
Formation of the Kinase–Inhibitor Complex
Kinase inhibitor complexes were prepared as described above.
Pull-Down
Pull-down experiments were performed as described above using a resin conjugated to an SH2 affinity peptide (sequence: EPQpYEEIPIYL).
FRET Assay
SFK (25 nM) was incubated with inhibitor (10 μM) and titrated with Peptide-FL in buffer (final volume = 30 μL, 50 mM HEPES, pH 7.5,10 mM MgCl2, 2.5 mM EGTA, and 100 mM NaCl). After 1 h, His-Tb was added, and FRET signal was monitored on a plate reader.
Phosphorylation of pTyr527 Assay with CskDR
Src or HckY416F (25 nM) was incubated with inhibitor in the presence of CskDR (25 nM) with BSA (0.1 mg mL–1) in activation buffer. Following 0.5 h incubation, ATP (1 μM) was added. After 1 h, the reaction was quenched by the addition of 3× loading dye. All samples were separated by SDS-PAGE and visualized by western blotting using pTyr527 antibody for Src (1:2000 P-Src (Tyr527) Cell Signaling) or a P-tyrosine antibody for Hck (1:2000 (P Tyr-100) Cell Signaling) and a His6-specific antibody (at a 1:5000 dilution (abm, HIS.H8). The scanned blots were quantified with LI-COR Odyssey software to determine the phosphorylation levels of the SFK pTyr527 treated with inhibitors relative to that of 1 (mean ± SEM, n = 3). For inhibitor concentrations, see Supporting Information Table S2.
Preparation of Covalent SFK–Inhibitor Substrate Complex
SFK (2 μM) was incubated with inhibitor (100 μM) in buffer (50 mM Tris, 100 mM NaCl, and 0.5 mM DTT, pH 7) overnight at 4 °C. SFK–inhibitor complexes were bound to Ni-NTA resin to wash away excess inhibitor (ThermoScientific). The kinase was then eluted and desalted using a Zeba spin column following the manufacturer’s protocol. The kinase was then incubated with 10 μL of dasatinib-conjugated resin for 1 h at rt to remove any unmodified kinase. The supernatant was then used in the Csk Tyr527 phosphorylation assay.
Phosphorylation of pTyr527 Assay with Covalent SFK–Inhibitor Substrates
Covalent SFK-inhibitor substrate (25 nM) was incubated with Csk (50 nM) and BSA (0.1 mg mL–1) in activation buffer. Following the addition of γ32P-ATP (0.2 μCi/well), reactions were incubated at rt for 0.5 h and then spotted on a nitrocellulose membrane. Membranes were washed with 0.5% phosphoric acid (3×, 10 min each wash) and dried, and the radioactivity was determined by phosphorimaging with a GE Typhoon FLA 9000 phosphor scanner. The scanned membranes were quantified with ImageQuant. Data was plotted using GraphPad Prism software.
Dephosphorylation of pTyr527 Assay
HckY416F (250 nM) was phosphorylated at Tyr527 by Csk (100 nM) in activation buffer containing BSA (0.25 mg mL–1) and radiolabeled ATP. After 1 h, inhibitor (5 μM) was added, and the mixture was incubated for 30 min. Following incubation, 10 μL of PTP1b (500 nM) and DTT (15 mM) was added to 20 μL of the hot reaction mixture. Reactions were spotted at 0, 15, and 30 min onto a nitrocellulose membrane. Membranes were washed with 0.5% phosphoric acid (3×, 10 min each wash) and dried, and the radioactivity was determined by phosphorimaging with a GE Typhoon FLA 9000 phosphor scanner. The scanned membranes were quantified with ImageQuant. Data was plotted using GraphPad Prism software.
Activity Assay
Inhibitors (initial concentration = 10 μM, 3-fold serial dilutions, 10 data points) were assayed in triplicate against Src ([SrcSH2eng] = 20 nM and [Srcact] = 0.5 nM) or Hck ([HckSH2eng] = 10 nM and [Hckact] = 0.5 nM) in assay buffer containing 75 mM HEPES, pH 7.5, 15 mM MgCl2, 3.75 mM EGTA, 150 mM NaCl, 0.2 mg mL–1 BSA, γ32P-ATP (0.2 μCi/well), and an optimized Src peptide substrate of the sequence Ac-EIYGEFKKK-OH (final concentration = 100 μM). The final volume of each assay well was 30 μL. The enzymatic reaction was run at rt for 2 h and then terminated by spotting 4.6 μL of the reaction mixture onto a phosphocellulose membrane. Membranes were washed with 0.5% phosphoric acid (3×, 10 min each wash) and dried, and the radioactivity was determined by phosphorimaging with a GE Typhoon FLA 9000 phosphor scanner. The scanned membranes were quantified with ImageQuant and converted to percent inhibition. Data was analyzed using GraphPad Prism software, and IC50 values were determined using nonlinear regression analysis.
Acknowledgments
This work was supported by the NIH (R01GM086858 to D.J.M. and F32CA174346 to S.E.L.) and by the Alfred P. Sloan and Camille and Henry Dreyfus Foundations (to D.J.M.).
Supporting Information Available
Quantification of the pull-down experiments performed with SrcS245C in the presence of 6 or 7; catalytic activity of SFK constructs; Ki values for drug resistant constructs, covalent inhibitors, and SFKs; pY527 chemical genetic assay parameters; and activity of SFK treated with covalent inhibitors for the pY527 assay. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Taylor S. S.; Kornev A. P. (2011) Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem. Sci. 36, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard S. R.; Till J. H. (2000) Protein tyrosine kinase structure and function. Annu. Rev. Biochem. 69, 373–398. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya R. P.; Remenyi A.; Yeh B. J.; Lim W. A. (2006) Domains, motifs, and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annu. Rev. Biochem. 75, 655–680. [DOI] [PubMed] [Google Scholar]
- Kuriyan J.; Eisenberg D. (2007) The origin of protein interactions and allostery in colocalization. Nature 450, 983–990. [DOI] [PubMed] [Google Scholar]
- Pellicena P.; Kuriyan J. (2006) Protein–protein interactions in the allosteric regulation of protein kinases. Curr. Opin. Struct. Biol. 16, 702–709. [DOI] [PubMed] [Google Scholar]
- Rauch J.; Volinsky N.; Romano D.; Kolch W. (2011) The secret life of kinases: functions beyond catalysis. Cell Commun. Signaling 9, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame M. C. (2002) Src in cancer: deregulation and consequences for cell behaviour. Biochim. Biophys. Acta 1602, 114–130. [DOI] [PubMed] [Google Scholar]
- Chong Y. P.; Ia K. K.; Mulhern T. D.; Cheng H. C. (2005) Endogenous and synthetic inhibitors of the Src-family protein tyrosine kinases. Biochim. Biophys. Acta 1754, 210–220. [DOI] [PubMed] [Google Scholar]
- Parsons S. J.; Parsons J. T. (2004) Src family kinases, key regulators of signal transduction. Oncogene 23, 7906–7909. [DOI] [PubMed] [Google Scholar]
- Summy J. M.; Gallick G. E. (2003) Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 22, 337–358. [DOI] [PubMed] [Google Scholar]
- Sen B.; Johnson F. M. (2011) Regulation of SRC family kinases in human cancers. J. Signal Transduction 2011, 865819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarino M. (2010) Src signaling in cancer invasion. J. Cell. Physiol. 223, 14–26. [DOI] [PubMed] [Google Scholar]
- Jura N.; Zhang X.; Endres N. F.; Seeliger M. A.; Schindler T.; Kuriyan J. (2011) Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol. Cell 42, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W.; Doshi A.; Lei M.; Eck M. J.; Harrison S. C. (1999) Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol. Cell 3, 629–638. [DOI] [PubMed] [Google Scholar]
- Osusky M.; Taylor S. J.; Shalloway D. (1995) Autophosphorylation of purified c-Src at its primary negative regulation site. J. Biol. Chem. 270, 25729–25732. [DOI] [PubMed] [Google Scholar]
- Sicheri F.; Moarefi I.; Kuriyan J. (1997) Crystal structure of the Src family tyrosine kinase Hck. Nature 385, 602–609. [DOI] [PubMed] [Google Scholar]
- Johnson L. N.; Noble M. E.; Owen D. J. (1996) Active and inactive protein kinases: structural basis for regulation. Cell 85, 149–158. [DOI] [PubMed] [Google Scholar]
- Kornev A. P.; Taylor S. S.; Ten Eyck L. F. (2008) A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. U.S.A. 105, 14377–14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornev A. P.; Taylor S. S. (2010) Defining the conserved internal architecture of a protein kinase. Biochim. Biophys. Acta 1804, 440–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennequin L. F.; Allen J.; Breed J.; Curwen J.; Fennell M.; Green T. P.; Lambert-van der Brempt C.; Morgentin R.; Norman R. A.; Olivier A.; Otterbein L.; Ple P. A.; Warin N.; Costello G. (2006) N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J. Med. Chem. 49, 6465–6488. [DOI] [PubMed] [Google Scholar]
- Krishnamurty R.; Brigham J. L.; Leonard S. E.; Ranjitkar P.; Larson E. T.; Dale E. J.; Merritt E. A.; Maly D. J. (2013) Active site profiling reveals coupling between domains in SRC-family kinases. Nat. Chem. Biol. 9, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte D. J.; Liu Y. T.; Arduini R. M.; Hession C. A.; Miatkowski K.; Wildes C. P.; Cullen P. F.; Hong V.; Hopkins B. T.; Mertsching E.; Jenkins T. J.; Romanowski M. J.; Baker D. P.; Silvian L. F. (2010) Structures of human Bruton’s tyrosine kinase in active and inactive conformations suggest a mechanism of activation for TEC family kinases. Protein Sci. 19, 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T.; Commodore L.; Huang W. S.; Wang Y.; Thomas M.; Keats J.; Xu Q.; Rivera V. M.; Shakespeare W. C.; Clackson T.; Dalgarno D. C.; Zhu X. (2011) Structural mechanism of the pan-BCR-ABL inhibitor ponatinib (AP24534): lessons for overcoming kinase inhibitor resistance. Chem. Biol. Drug Des. 77, 1–11. [DOI] [PubMed] [Google Scholar]
- Skora L.; Mestan J.; Fabbro D.; Jahnke W.; Grzesiek S. (2013) NMR reveals the allosteric opening and closing of Abelson tyrosine kinase by ATP-site and myristoyl pocket inhibitors. Proc. Natl. Acad. Sci. U.S.A. 110, E4437–E4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young M. A.; Gonfloni S.; Superti-Furga G.; Roux B.; Kuriyan J. (2001) Dynamic coupling between the SH2 and SH3 domains of c-Src and Hck underlies their inactivation by C-terminal tyrosine phosphorylation. Cell 105, 115–126. [DOI] [PubMed] [Google Scholar]
- Ulmer T. S.; Werner J. M.; Campbell I. D. (2002) SH3–SH2 domain orientation in Src kinases: NMR studies of Fyn. Structure 10, 901–911. [DOI] [PubMed] [Google Scholar]
- Cowan-Jacob S. W.; Fendrich G.; Manley P. W.; Jahnke W.; Fabbro D.; Liebetanz J.; Meyer T. (2005) The crystal structure of a c-Src complex in an active conformation suggests possible steps in c-Src activation. Structure 13, 861–871. [DOI] [PubMed] [Google Scholar]
- Sicilia R. J.; Hibbs M. L.; Bello P. A.; Bjorge J. D.; Fujita D. J.; Stanley I. J.; Dunn A. R.; Cheng H. C. (1998) Common in vitro substrate specificity and differential Src homology 2 domain accessibility displayed by two members of the Src family of protein-tyrosine kinases, c-Src and Hck. J. Biol. Chem. 273, 16756–16763. [DOI] [PubMed] [Google Scholar]
- Gonfloni S.; Williams J. C.; Hattula K.; Weijland A.; Wierenga R. K.; Superti-Furga G. (1997) The role of the linker between the SH2 domain and catalytic domain in the regulation and function of Src. EMBO J. 16, 7261–7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs S. D.; Smithgall T. E. (1999) SH2-kinase linker mutations release Hck tyrosine kinase and transforming activities in Rat-2 fibroblasts. J. Biol. Chem. 274, 26579–26583. [DOI] [PubMed] [Google Scholar]
- LaFevre-Bernt M.; Sicheri F.; Pico A.; Porter M.; Kuriyan J.; Miller W. T. (1998) Intramolecular regulatory interactions in the Src family kinase Hck probed by mutagenesis of a conserved tryptophan residue. J. Biol. Chem. 273, 32129–32134. [DOI] [PubMed] [Google Scholar]
- Williams J. C.; Weijland A.; Gonfloni S.; Thompson A.; Courtneidge S. A.; Superti-Furga G.; Wierenga R. K. (1997) The 2.35 A crystal structure of the inactivated form of chicken Src: a dynamic molecule with multiple regulatory interactions. J. Mol. Biol. 274, 757–775. [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G.; Song K.; Yen I.; Brandhuber B. J.; Anderson D. J.; Alvarado R.; Ludlam M. J.; Stokoe D.; Gloor S. L.; Vigers G.; Morales T.; Aliagas I.; Liu B.; Sideris S.; Hoeflich K. P.; Jaiswal B. S.; Seshagiri S.; Koeppen H.; Belvin M.; Friedman L. S.; Malek S. (2010) RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464, 431–435. [DOI] [PubMed] [Google Scholar]
- Poulikakos P. I.; Zhang C.; Bollag G.; Shokat K. M.; Rosen N. (2010) RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan T. O.; Zhang J.; Rodeck U.; Pascal J. M.; Armen R. S.; Spring M.; Dumitru C. D.; Myers V.; Li X.; Cheung J. Y.; Feldman A. M. (2011) Resistance of Akt kinases to dephosphorylation through ATP-dependent conformational plasticity. Proc. Natl. Acad. Sci. U.S.A. 108, E1120–E1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andraos R.; Qian Z.; Bonenfant D.; Rubert J.; Vangrevelinghe E.; Scheufler C.; Marque F.; Regnier C. H.; De Pover A.; Ryckelynck H.; Bhagwat N.; Koppikar P.; Goel A.; Wyder L.; Tavares G.; Baffert F.; Pissot-Soldermann C.; Manley P. W.; Gaul C.; Voshol H.; Levine R. L.; Sellers W. R.; Hofmann F.; Radimerski T. (2012) Modulation of activation-loop phosphorylation by JAK inhibitors is binding mode dependent. Cancer Discovery 2, 512–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hari S. B.; Merritt E. A.; Maly D. J. (2014) Conformation-selective ATP-competitive inhibitors control regulatory interactions and noncatalytic functions of mitogen-activated protein kinases. Chem. Biol. 21, 628–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan J. E.; Holdgate G. A.; Campbell D.; Timms D.; Gerhardt S.; Breed J.; Breeze A. L.; Bermingham A.; Pauptit R. A.; Norman R. A.; Embrey K. J.; Read J.; VanScyoc W. S.; Ward W. H. (2005) Prevention of MKK6-dependent activation by binding to p38alpha MAP kinase. Biochemistry 44, 16475–16490. [DOI] [PubMed] [Google Scholar]
- Murphy S. M.; Bergman M.; Morgan D. O. (1993) Suppression of c-Src activity by C-terminal Src kinase involves the c-Src SH2 and SH3 domains: analysis with Saccharomyces cerevisiae. Mol. Cell. Biol. 13, 5290–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M.; Nada S.; Yamanashi Y.; Yamamoto T.; Nakagawa H. (1991) CSK: a protein-tyrosine kinase involved in regulation of src family kinases. J. Biol. Chem. 266, 24249–24252. [PubMed] [Google Scholar]
- Cole P. A.; Shen K.; Qiao Y.; Wang D. (2003) Protein tyrosine kinases Src and Csk: a tail’s tale. Curr. Opin. Chem. Biol. 7, 580–585. [DOI] [PubMed] [Google Scholar]
- Sondhi D.; Xu W.; Songyang Z.; Eck M. J.; Cole P. A. (1998) Peptide and protein phosphorylation by protein tyrosine kinase Csk: insights into specificity and mechanism. Biochemistry 37, 165–172. [DOI] [PubMed] [Google Scholar]
- Hill Z. B.; Perera B. G.; Andrews S. S.; Maly D. J. (2012) Targeting diverse signaling interaction sites allows the rapid generation of bivalent kinase inhibitors. ACS Chem. Biol. 7, 487–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z.; Scheerens H.; Li S. J.; Schultz B. E.; Sprengeler P. A.; Burrill L. C.; Mendonca R. V.; Sweeney M. D.; Scott K. C.; Grothaus P. G.; Jeffery D. A.; Spoerke J. M.; Honigberg L. A.; Young P. R.; Dalrymple S. A.; Palmer J. T. (2007) Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem. 2, 58–61. [DOI] [PubMed] [Google Scholar]
- Roskoski R. Jr. (2005) Src kinase regulation by phosphorylation and dephosphorylation. Biochem. Biophys. Res. Commun. 331, 1–14. [DOI] [PubMed] [Google Scholar]
- Bjorge J. D.; Pang A.; Fujita D. J. (2000) Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J. Biol. Chem. 275, 41439–41446. [DOI] [PubMed] [Google Scholar]
- Shan Y.; Seeliger M. A.; Eastwood M. P.; Frank F.; Xu H.; Jensen M. O.; Dror R. O.; Kuriyan J.; Shaw D. E. (2009) A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc. Natl. Acad. Sci. U.S.A. 106, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarado J. J.; Betts L.; Moroco J. A.; Smithgall T. E.; Yeh J. I. (2010) Crystal structure of the Src family kinase Hck SH3–SH2 linker regulatory region supports an SH3-dominant activation mechanism. J. Biol. Chem. 285, 35455–35461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayrapetov M. K.; Wang Y. H.; Lin X.; Gu X.; Parang K.; Sun G. (2006) Conformational basis for SH2–Tyr(P)527 binding in Src inactivation. J. Biol. Chem. 281, 23776–23784. [DOI] [PubMed] [Google Scholar]
- Lerner E. C.; Smithgall T. E. (2002) SH3-dependent stimulation of Src-family kinase autophosphorylation without tail release from the SH2 domain in vivo. Nat. Struct. Biol. 9, 365–369. [DOI] [PubMed] [Google Scholar]
- Moarefi I.; LaFevre-Bernt M.; Sicheri F.; Huse M.; Lee C. H.; Kuriyan J.; Miller W. T. (1997) Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature 385, 650–653. [DOI] [PubMed] [Google Scholar]
- Smart J. E.; Oppermann H.; Czernilofsky A. P.; Purchio A. F.; Erikson R. L.; Bishop J. M. (1981) Characterization of sites for tyrosine phosphorylation in the transforming protein of Rous sarcoma virus (pp60v-src) and its normal cellular homologue (pp60c-src). Proc. Natl. Acad. Sci. U.S.A. 78, 6013–6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Perera B. G.; Hari S. B.; Bhhatarai B.; Backes B. J.; Seeliger M. A.; Schurer S. C.; Oakes S. A.; Papa F. R.; Maly D. J. (2012) Divergent allosteric control of the IRE1alpha endoribonuclease using kinase inhibitors. Nat. Chem. Biol. 8, 982–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppikar P.; Bhagwat N.; Kilpivaara O.; Manshouri T.; Adli M.; Hricik T.; Liu F.; Saunders L. M.; Mullally A.; Abdel-Wahab O.; Leung L.; Weinstein A.; Marubayashi S.; Goel A.; Gonen M.; Estrov Z.; Ebert B. L.; Chiosis G.; Nimer S. D.; Bernstein B. E.; Verstovsek S.; Levine R. L. (2012) Heterodimeric JAK–STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 489, 155–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeliger M. A.; Young M.; Henderson M. N.; Pellicena P.; King D. S.; Falick A. M.; Kuriyan J. (2005) High yield bacterial expression of active c-Abl and c-Src tyrosine kinases. Protein Sci. 14, 3135–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S. M.; Murphy R. C.; Geiger J. A.; DeRocher A. E.; Zhang Z.; Ojo K. K.; Larson E. T.; Perera B. G.; Dale E. J.; He P.; Reid M. C.; Fox A. M.; Mueller N. R.; Merritt E. A.; Fan E.; Parsons M.; Van Voorhis W. C.; Maly D. J. (2012) Development of Toxoplasma gondii calcium-dependent protein kinase 1 (TgCDPK1) inhibitors with potent anti-toxoplasma activity. J. Med. Chem. 55, 2416–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apsel B.; Blair J. A.; Gonzalez B.; Nazif T. M.; Feldman M. E.; Aizenstein B.; Hoffman R.; Williams R. L.; Shokat K. M.; Knight Z. A. (2008) Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat. Chem. Biol. 4, 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigham J. L.; Perera B. G.; Maly D. J. (2013) A hexylchloride-based catch-and-release system for chemical proteomic applications. ACS Chem. Biol. 8, 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapf C. W.; Gerstenberger B. S.; Xing L.; Limburg D. C.; Anderson D. R.; Caspers N.; Han S.; Aulabaugh A.; Kurumbail R.; Shakya S.; Li X.; Spaulding V.; Czerwinski R. M.; Seth N.; Medley Q. G. (2012) Covalent inhibitors of interleukin-2 inducible T cell kinase (Itk) with nanomolar potency in a whole-blood assay. J. Med. Chem. 55, 10047–10063. [DOI] [PubMed] [Google Scholar]
- Chen W., and Loury D. J. (2013) Preparation of pyrazolo[3,4-d]pyrimidine and pyrrolo[2,3-d]pyrimidine derivatives as Btk kinase inhibitors, p191, Patent US8377946 B1.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.