

Abstract

The lysine methyltransferase SETD8 is the only known methyltransferase that catalyzes monomethylation of histone H4 lysine 20 (H4K20). Monomethylation of H4K20 has been implicated in regulating diverse biological processes including the DNA damage response. In addition to H4K20, SETD8 monomethylates non-histone substrates including proliferating cell nuclear antigen (PCNA) and promotes carcinogenesis by deregulating PCNA expression. However, selective inhibitors of SETD8 are scarce. The only known selective inhibitor of SETD8 to date is nahuoic acid A, a marine natural product, which is competitive with the cofactor. Here, we report the discovery of the first substrate-competitive inhibitor of SETD8, UNC0379 (1). This small-molecule inhibitor is active in multiple biochemical assays. Its affinity to SETD8 was confirmed by ITC (isothermal titration calorimetry) and SPR (surface plasmon resonance) studies. Importantly, compound 1 is selective for SETD8 over 15 other methyltransferases. We also describe structure–activity relationships (SAR) of this series.
Introduction
Protein lysine methyltransferases (PKMTs, also known as histone lysine methyltransferases (HKMTs)) catalyze the transfer of the methyl group from the cofactor S-adenosyl-l-methionine (SAM) to lysine residues of histone and non-histone substrates, leading to lysine mono-, di-, and/or trimethylation.1−3 Histone lysine methylation has been increasingly recognized as a major epigenetic gene regulation mechanism in eukaryotic cells.1−6 Therefore, PKMTs as a class of potential drug targets have received considerable attention from the medicinal chemistry and chemical biology community. With the exception of DOT1L, PKMTs contain an evolutionarily conserved SET (Su(var), E(z), and Trithorax) domain.7 This catalytic domain consists of a substrate binding groove and a cofactor binding site.8 A number of selective small-molecule inhibitors of PKMTs including G9a/GLP, EZH2, SMYD2, DOT1L, and SETD2 have been discovered.9−35 These inhibitors are competitive with either the peptide substrate or the cofactor SAM. In addition, a number of selective inhibitors of protein arginine methyltransferases (PRMTs), another class of protein methyltransferases, have been reported.36−38 Some of these inhibitors have subsequently been utilized as valuable tools for investigating the role of the corresponding methyltransferases in various human diseases.16,19,20,28,29,33,39−42
SETD8 (also known as SET8, PR-SET7, or KMT5A (lysine methyltransferase 5A)) is the sole methyltransferase that catalyzes monomethylation of histone H4 lysine 20 (H4K20).43−45 SETD8 and H4K20me (H4K20 monomethylation) have been implicated in regulating a diverse set of biological processes including the DNA damage response, DNA replication, and mitotic condensation.45 A recent study has shown that (1) SETD8 is physically associated with TWIST, a master regulator of EMT (epithelial–mesenchymal transition), (2) SETD8 and TWIST are functionally interdependent on promoting EMT, (3) SETD8 acts on the promoters of the TWIST target genes such as N-cadherin via exertion of its H4K20 monomethylation activity, and (4) SETD8 expression is positively correlated with metastasis and the expression of TWIST and N-cadherin in breast cancer cells.46 In addition to H4K20, SETD8 methylates many non-histone substrates including the tumor suppressor p53 and proliferating cell nuclear antigen (PCNA).47,48 The monomethylation of p53 at lysine 382 (p53K382me1) catalyzed by SETD8 suppresses p53-mediated transcription activation of highly responsive target genes.47 SETD8 and PCNA are coexpressed in lung cancer tissues.48 The monomethylation of PCNA at lysine 248 (PCNAK248me1) catalyzed by SETD8 stabilizes PCNA protein, enhances the interaction between PCNA and the flap endonuclease FEN1, and promotes the proliferation of cancer cells.48
However, selective inhibitors of SETD8 are scarce. To date, nahuoic acid A, a marine natural product, is the only known selective inhibitor of SETD8 (Figure 1).25 This inhibitor is competitive with the cofactor SAM and noncompetitive with the peptide substrate. Here we report the discovery of UNC0379 (1), the first substrate-competitive inhibitor of SETD8. Compound 1 is a synthetic small-molecule inhibitor that displays inhibitory activity in multiple biochemical assays and is selective for SETD8 over 15 other methyltransferases. The binding affinity of compound 1 to SETD8 was determined using biophysical assays such as ITC (isothermal titration calorimetry) and SPR (surface plasmon resonance) and is largely consistent with its potency in biochemical assays. We describe hit identification, analogue synthesis, structure–activity relationship (SAR) findings, and comprehensive characterization of compound 1 in a number of biochemical and biophysical assays including mechanism of action and selectivity studies.
Figure 1.

Structure of the known SETD8 inhibitor nahuoic acid A.25
Results and Discussion
Discovery of Compound 1 as a SETD8 Inhibitor
We previously reported that 2,4-diaminoquinazolines are selective, substrate-competitive inhibitors of the lysine methyltransferases G9a and GLP.10,12−14,30 To identify a substrate-competitive inhibitor of SETD8, we cross-screened our quinazoline-based inhibitor set, which consists of >150 compounds, against SETD8. From this study, we discovered compound 1 as an inhibitor of SETD8 (Figure 2). Interestingly, compound 1 was originally prepared for targeting L3MBTL1, a methyllysine reader protein,49 but showed no appreciable activity for L3MBTL1. On the other hand, compound 1 displayed inhibitory activity with an IC50 of 7.3 ± 1.0 μM (n = 2) in a radioactive biochemical assay that measures the transfer of the tritiated methyl group from 3H-SAM to a peptide substrate catalyzed by SETD8 (Figure 2). The inhibitory activity of compound 1 was confirmed in an orthogonal biochemical assay, microfluidic capillary electrophoresis (MCE) assay. This SETD8 MCE assay was developed analogously to the previously reported G9a MCE assay.50 Compound 1 exhibited an IC50 of 9.0 μM in the SETD8 MCE assay.
Figure 2.

Compound 1 was identified as an inhibitor of SETD8 by cross-screening a quinazoline-based inhibitor set. (A) Structure of compound 1. (B) Concentration–response curve of compound 1 in the SETD8 radioactive methyl transfer assay.
Analogue Synthesis
To determine SAR for this promising hit, we designed and synthesized a number of analogues that contain various 2- and 4-substituents at the quinazoline core. We synthesized compounds 1–24 from commercially available 2,4-dichloro-6,7-dimethoxyquinazoline and corresponding amines in good yields (Scheme 1 and Tables 1 and 2). Using the methods developed previously,10 we displaced the 4-chloro group with the first set of amines at room temperature and the 2-chloro group with the second set of amines under microwave heating conditions to yield the desired 2,4-diamino-6,7-dimethoxyquinazolines.
Scheme 1. Typical Synthesis of 2,4-Diamino-6,7-dimethoxyquinazolines.
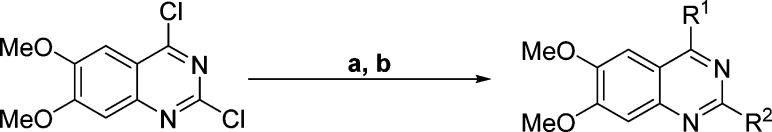
(a) R1 amines, THF, N,N-diisopropylethylamine, room temperature; (b) R2 amines, n-BuOH, DIPEA, microwave, 160 °C.
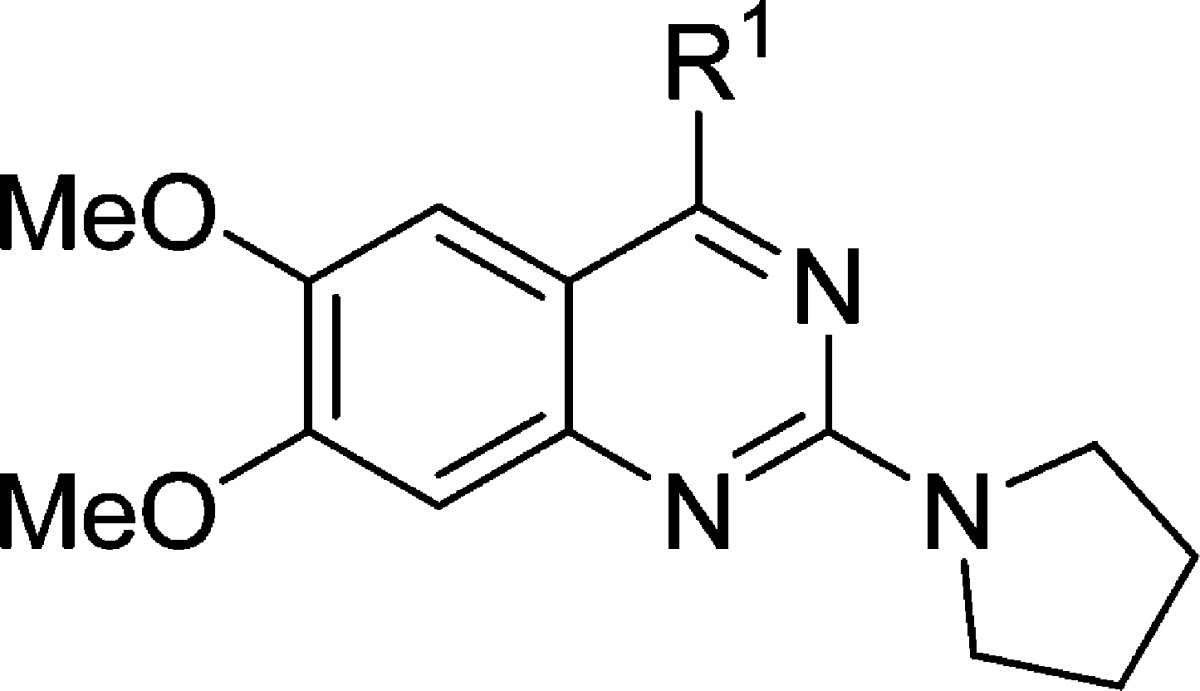
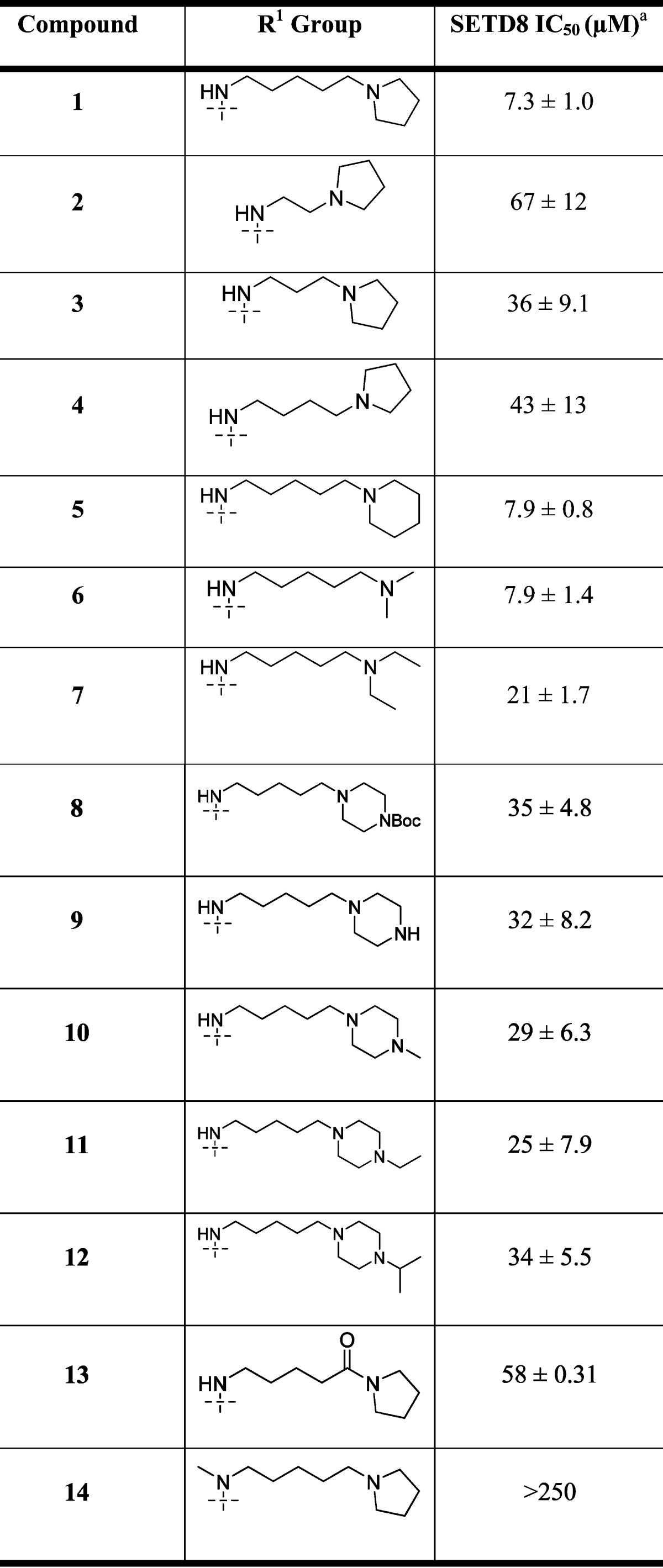
Table 1. SAR of the 4-Amino Group.
IC50 determination experiments were performed in duplicate.
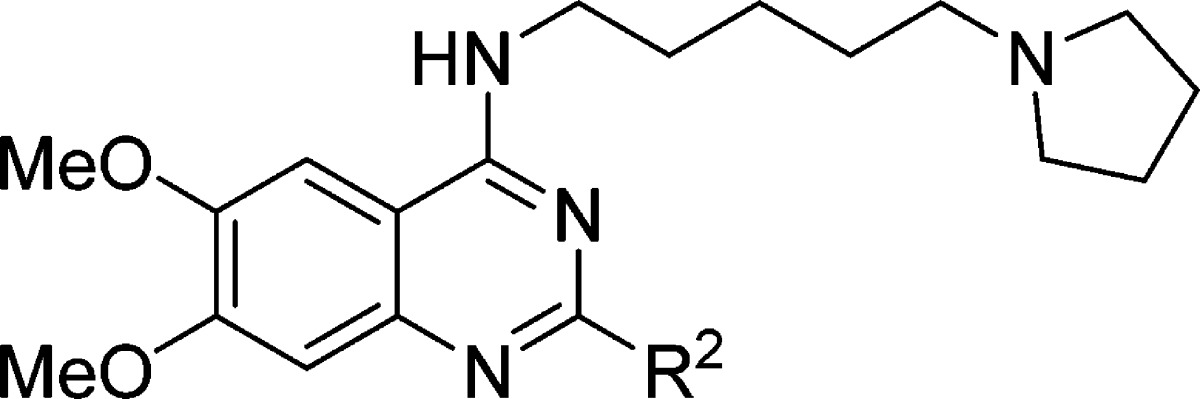
Table 2. SAR of the 2-Amino Group.
IC50 determination experiments were performed in duplicate.
SAR in SETD8 Biochemical Assay
The synthesized compounds were then evaluated in the SETD8 radioactive methyl transfer assay. IC50 values of these compounds in this biochemical assay are summarized in Tables 1 and 2.
We first explored the 4-amino group of the quinazoline scaffold (Table 1). Reducing the length of the alkyl linker between the two amino groups resulted in the decrease of the potency (compound 1 versus compounds 2–4). The distal amino group can be changed from the pyrrolidinyl to piperidinyl or dimethylamino without any potency loss (compound 1 versus compounds 5 and 6). Interestingly, the diethylamino analogue (compound 7) was somewhat less potent than compounds 1, 5, and 6. We also attempted to replace the piperidinyl group with a piperazinyl or N-substituted piperazinyl group (compound 1 versus compounds 8–12). However, these structural modifications led to a potency loss, suggesting that an additional basic amino group (compounds 9–12) or a large substituent (compound 8) is detrimental to inhibiting SETD8. In addition, we found that the basicity of the pyrrolidinyl group in compound 1 was an important contributor to its SETD8 inhibitory activity, as the corresponding amide analogue (compound 13) was about 8-fold less potent than compound 1. We also explored whether the NH group in compound 1 was potentially engaged in a hydrogen bond interaction and found that the N-methyl analogue (compound 14) was drastically less potent than compound 1, suggesting that the hydrogen of the secondary amine likely serves as a hydrogen bond donor.
We next explored the 2-amino group of the quinazoline scaffold (Table 2). The replacement of the pyrrolidinyl group with either the piperidinyl or azepanyl group resulted in a significant loss of the potency (compound 1 versus compounds 15 and 16), suggesting a large group is disfavored. On the other hand, the dimethylamino group (compound 17) did not lead to any significant potency loss. The introduction of an additional basic amino group (compound 15 versus the unsubstituted piperazinyl analogue 18 and the N-methylpiperazinyl analogue 19) resulted in a complete loss of the potency. We also explored other amino groups such as N-methyl-N-cyclopentylamine (compound 20) and N-methyl-N-cyclohexylamine (compound 21). Compared with compound 1, compounds 20 and 21 were about 4-fold less potent. Interestingly, removing the methyl group from either compound 20 or 21 led to the complete loss of inhibitory activity (compounds 22 and 23), suggesting that tertiary amino groups are preferred compared with secondary amino groups. In addition, the chloro analogue (compound 24) was inactive. Taken together, these results suggest that SETD8 inhibitory activity is very sensitive to the 2-substituent.
ITC and SPR Studies
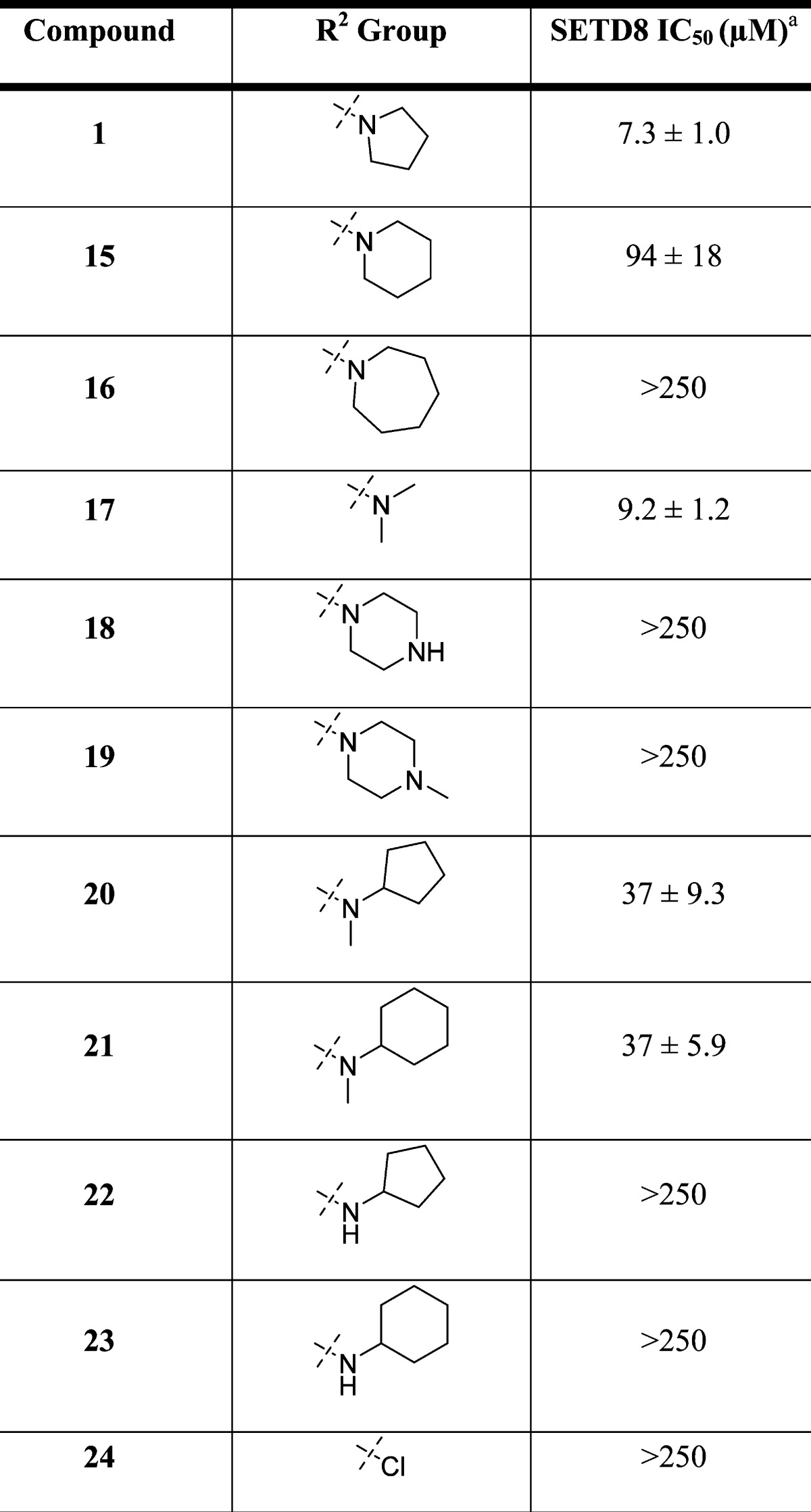
We next characterized compound 1 in biophysical assays. In ITC studies, compound 1 bound SETD8 with a KD of 18.3 ± 3.2 μM (n = 3) (Figure 3). In SPR studies, compound 1 behaved as a classic reversible inhibitor with a fast on rate (ka = 2.18 ± 0.36 × 104 1/Ms) and a fast off rate (kd = 7.82 ± 0.87 × 10–1 1/s) (Figure 4). The KD of compound 1 was determined to be 36.0 ± 2.3 μM (n = 3). The binding affinity of compound 1 to SETD8 determined by ITC and SPR is largely consistent with its potency in the biochemical assays.
Figure 3.
Compound 1 binds SETD8 with a KD of 18.3 ± 3.2 μM (n = 3) in ITC studies.
Figure 4.

Compound 1 exhibits rapid on and off rates in SPR studies.
MOA Studies
We next studied the MOA (mechanism of action) of the SETD8 inhibition by compound 1 via varying concentrations of the H4 peptide substrate or the cofactor SAM. As illustrated in Figure 5A, IC50 values of compound 1 increased linearly with H4 peptide concentrations. On the other hand, IC50 values of compound 1 remained constant in the presence of increasing concentrations of SAM (Figure 5B). These results indicate that compound 1 is competitive with the peptide substrate and noncompetitive with the cofactor SAM.
Figure 5.

MOA studies of compound 1. (A) Compound 1 is competitive with the peptide substrate, as its IC50 values increased linearly with H4 peptide concentrations. (B) Compound 1 is noncompetitive with the cofactor SAM, as its IC50 values remained constant in the presence of increasing concentrations of SAM.
Peptide Displacement Assay
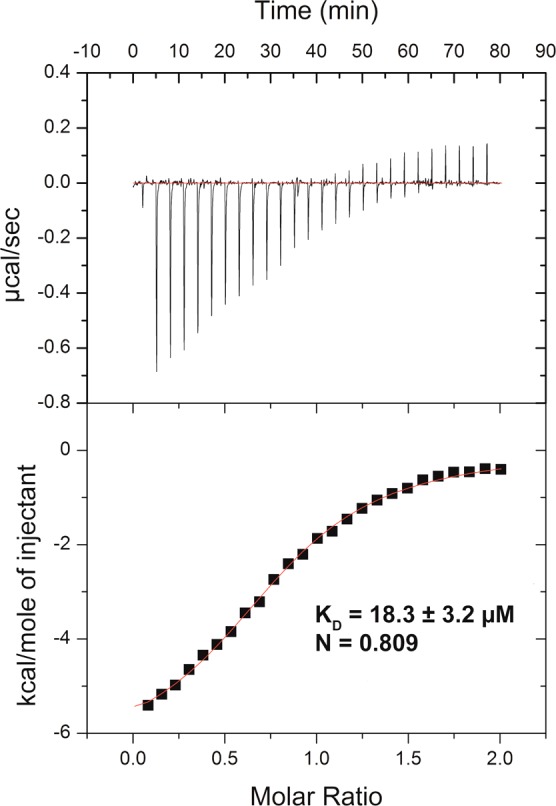
To confirm the findings from the MOA studies, we tested compound 1 and an inactive control (compound 14) in a peptide displacement assay using fluorescence polarization (FP), which measures effects of inhibitors on displacing the H4K20me (1–24) peptide with N-terminus labeled by fluorescein isothiocyanate (FITC). As illustrated in Figure 6, compound 1 effectively displaced the FITC-labeled peptide binding to SETD8 with an IC50 of 37.7 ± 7.2 μM (n = 2). On the other hand, our negative control (compound 14) did not displace the peptide in this FP assay. These results further support that compound 1 is competitive with the peptide substrate.
Figure 6.
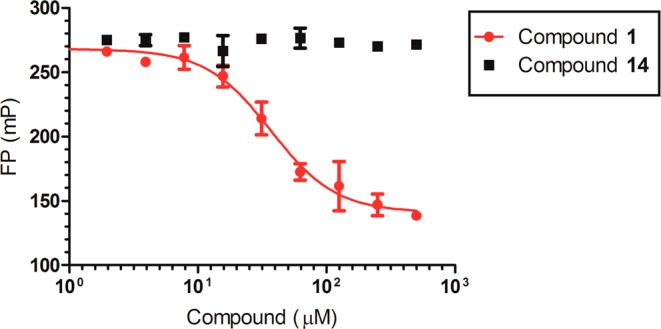
Peptide displacement assay. Compound 1 effectively displaced the FITC-labeled H4K20me (1–14) peptide, while an inactive control (compound 14) did not.
Selectivity
We next determined the selectivity of inhibitor 1 for SETD8 over 15 other methyltransferases (Figure 7). With the exception of PRC2 (polycomb repressive complex 2), the IC50 values of compound 1 for 14 other methyltransferases including G9a and GLP were all above 100 μM. For PRC2, compound 1 was active only at the two highest concentrations (50 and 100 μM) with an estimated IC50 value greater than 50 μM. Therefore, inhibitor 1 is a selective inhibitor of SETD8.
Figure 7.

Selectivity of inhibitor 1 versus 15 other methyltransferases.
Conclusions
We discovered the first selective, substrate competitive inhibitor of SETD8 by cross-screening our quinazoline-based epigenetic library. Our SAR studies of this series reveal that while the 4-amino moiety can be modified without a significant loss of potency, modifications to the 2-amino moiety are not well tolerated. We characterized compound 1 in a battery of biochemical, biophysical, MOA, and selectivity assays and found that the SETD8 inhibitory activity of this compound in biochemical assays was largely consistent with its binding affinity to SETD8 determined by ITC and SPR. Our MOA studies indicate that this inhibitor is competitive with the peptide substrate and noncompetitive with the cofactor SAM. This MOA was further supported by our finding that compound 1 effectively displaced the FITC-labeled H4 peptide in a FP assay. Importantly, this inhibitor is selective for SETD8 over 15 other methyltransferases including G9a and GLP, the two PKMTs that are known for being potently inhibited by quinazolines. These results provide the first evidence that 2,4-diaminoquinazolines can be modified to yield selective, substrate competitive inhibitors of PKMTs other than G9a and GLP.
Experimental Section
Chemistry General Procedures
HPLC spectra for all compounds were acquired using an Agilent 6110 series system with UV detector set to 254 nm. Samples were injected (5 μL) onto an Agilent Eclipse Plus 4.6 mm × 50 mm, 1.8 μm C18 column at room temperature. A linear gradient from 10% to 100% B (MeOH + 0.1% acetic acid) in 5.0 min was followed by pumping 100% B for another 2 min with A being H2O + 0.1% acetic acid. The flow rate was 1.0 mL/min. Mass spectra data were acquired in positive ion mode using an Agilent 6110 single quadrupole mass spectrometer with an electrospray ionization (ESI) source. Nuclear magnetic resonance (NMR) spectra were recorded at Varian Mercury spectrometer with 400 MHz for proton (1H NMR) and 100 MHz for carbon (13C NMR). Chemical shifts are reported in ppm (δ). Preparative HPLC was performed on Agilent Prep 1200 series with UV detector set to 254 nm. Samples were injected onto a Phenomenex Luna 75 mm × 30 mm, 5 μm C18 column at room temperature. The flow rate was 30 mL/min. A linear gradient was used with 10% (or 50%) of MeOH (A) in 0.1% TFA in H2O (B) to 100% of MeOH (A). HPLC was used to establish the purity of target compounds. All compounds had >95% purity using the HPLC methods described above. High-resolution (positive ion) mass spectrometry (HRMS) for compound 1 was performed using a Thermo LTqFT mass spectrometer under FT control at 100 000 resolution.
6,7-Dimethoxy-2-(pyrrolidin-1-yl)-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (1)
To a solution of 2-chloro-6,7-dimethoxy-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (24, 84 mg, 0.17 mmol) and n-butanol (1.5 mL) were added pyrrolidine (commercially available, 56 μL, 0.68 mmol) and N,N-diisopropylethylamine (89 μL, 0.51 mmol). The resulting solution was stirred inside a microwave at 160 °C for 30 min. After the mixture was cooled, TLC indicated the completion of the reaction. After removal of the solvent by rotary evaporation, the residue was redissolved in CH2Cl2 and washed with brine. The organic layer was dried, concentrated, and purified by HPLC to give the title compound as a TFA salt, white solid (70 mg, yield 64%). 1H NMR (400 MHz, MeOH-d4) δ 7.56 (s, 1H), 7.10 (s, 1H), 3.95 (s, 3H), 3.92 (s, 3H), 3.78–3.57 (m, 8H), 3.22–3.16 (m, 2H), 3.10–3.01 (m, 2H), 2.19–1.97 (m, 8H), 1.86–1.73 (m, 4H), 1.55–1.47 (m, 2H); 13C NMR (100 MHz, MeOH-d4) δ 160.40, 157.17, 151.52, 148.80, 136.95, 105.04, 103.62, 99.62, 56.93, 56.79, 56.11, 55.08 (four carbons), 42.48, 29.31, 26.87, 25.20, 23.95 (four carbons). HPLC purity: >95%; tR = 3.32 min. MS (ESI): 414 [M + H]+. HRMS calcd for C23H35N5O2 + H, 414.2869; found, 414.2862 [M + H]+.
6,7-Dimethoxy-2-(pyrrolidin-1-yl)-N-(2-(pyrrolidin-1-yl)ethyl)quinazolin-4-amine (2)
2-Chloro-6,7-dimethoxy-N-(2-(pyrrolidin-1-yl)ethyl)quinazolin-4-amine was prepared according to the procedure for making 24 from 2,4-dichloro-6,7-dimethoxylquinazoline, 1-(2-aminoethyl)pyrrolidine (commercially available), N,N-diisopropylethylamine, and THF. To a solution of 2-chloro-6,7-dimethoxy-N-(2-(pyrrolidin-1-yl)ethyl)quinazolin-4-amine (72 mg, 0.13 mmol) and isopropanol (0.7 mL) were added pyrrolidine (21 μL, 0.26 mmol) and HCl in dioxane (4.0 M, 63 μL, 0.26 mmol). The resulting solution was stirred inside a microwave at 160 °C for 20 min. After the mixture was cooled, TLC indicated the completion of the reaction. After removal of the solvent by rotary evaporation, the residue was redissolved in CH2Cl2, washed with brine. The organic layer was dried, concentrated, and purified by HPLC to give the title compound 2 as a TFA salt, white solid (56 mg, yield 73%). 1H NMR (400 MHz, MeOH-d4) δ 7.50 (s, 1H), 7.12 (s, 1H), 4.06 (t, J = 6.0 Hz, 2H), 3.96 (s, 3H), 3.92 (s, 3H), 3.82–3.71 (m, 4H), 3.68–3.56 (m, 4H), 3.22–3.10 (m, 2H), 2.22–1.99 (m, 8H). HPLC purity: >95%; tR = 2.88 min. MS (ESI): 372 [M + H]+.
6,7-Dimethoxy-2-(pyrrolidin-1-yl)-N-(3-(pyrrolidin-1-yl)propyl)quinazolin-4-amine (3)
2-Chloro-6,7-dimethoxy-N-(3-(pyrrolidin-1-yl)propyl)quinazolin-4-amine was prepared according to the procedure for making 24 from 2,4-dichloro-6,7-dimethoxylquinazoline, 1-(3-aminopropyl)pyrrolidine (commercially available), N,N-diisopropylethylamine, and THF. Compound 3 was prepared according to the procedure for making 2 from 2-chloro-6,7-dimethoxy-N-(3-(pyrrolidin-1-yl)propyl)quinazolin-4-amine (66 mg, 0.19 mmol), pyrrolidine (30 μL, 0.37 mmol), HCl in dioxane (4.0 M, 93 μL, 0.37 mmol), and isopropanol (1.0 mL). The title compound 3 was obtained as a TFA salt, brown solid (45 mg, yield 40%). 1H NMR (400 MHz, MeOH-d4) δ 7.53 (s, 1H), 7.10 (s, 1H), 3.95 (s, 3H), 3.91 (s, 3H), 3.83–3.60 (m, 8H), 3.35–3.28 (m, 2H), 3.13–3.02 (m, 2H), 2.25–2.00 (m, 10H); 13C NMR (100 MHz, MeOH-d4) δ 160.68, 157.34, 151.49, 148.83, 137.08, 105.03, 103.57, 99.57, 56.91, 56.82, 55.11 (four carbons), 53.95, 39.75, 26.51, 23.96 (four carbons). HPLC purity: >95%; tR = 3.24 min. MS (ESI): 386 [M + H]+.
6,7-Dimethoxy-2-(pyrrolidin-1-yl)-N-(4-(pyrrolidin-1-yl)butyl)quinazolin-4-amine (4)
2-Chloro-6,7-dimethoxy-N-(4-(pyrrolidin-1-yl)butyl)quinazolin-4-amine was prepared according to the procedure for making 24 from 2,4-dichloro-6,7-dimethoxylquinazoline, 1-pyrrolidinebutanamine (commercially available), N,N-diisopropylethylamine, and THF. Compound 4 was prepared according to the procedure for making 1 from 2-chloro-6,7-dimethoxy-N-(4-(pyrrolidin-1-yl)butyl)quinazolin-4-amine (109 mg, 0.30 mmol), pyrrolidine (99 μL, 1.2 mmol), N,N-diisopropylethylamine (105 μL, 0.60 mmol), and n-butanol (1.0 mL). The title compound 4 was obtained as a TFA salt, white solid (144 mg, yield 77%). 1H NMR (400 MHz, MeOH-d4) δ 7.54 (s, 1H), 7.09 (s, 1H), 3.94 (s, 3H), 3.91 (s, 3H), 3.79–3.54 (m, 8H), 3.28–3.19 (m, 2H), 3.12–3.01 (m, 2H), 2.19–1.97 (m, 8H), 1.89–1.79 (m, 4H). HPLC purity: >95%; tR = 3.02 min. MS (ESI): 400 [M + H]+.
6,7-Dimethoxy-N-(5-(piperidin-1-yl)pentyl)-2-(pyrrolidin-1-yl)quinazolin-4-amine (5)
2-Chloro-6,7-dimethoxy-N-(5-(piperidin-1-yl)pentyl)quinazolin-4-amine was prepared according to the procedure for making 24 from 2,4-dichloro-6,7-dimethoxylquinazoline, 1-piperidinepentanamine (commercially available), N,N-diisopropylethylamine, and THF. Compound 5 was prepared according to the procedure for making 2 from 2-chloro-6,7-dimethoxy-N-(5-(piperidin-1-yl)pentyl)quinazolin-4-amine (79 mg, 0.13 mmol), pyrrolidine (21 μL, 0.26 mmol), HCl in dioxane (4.0 M, 63 μL, 0.26 mmol), and isopropanol (0.7 mL). The title compound 5 was obtained as a TFA salt, brown solid (58 mg, yield 68%). 1H NMR (400 MHz, MeOH-d4) δ 7.55 (s, 1H), 7.09 (s, 1H), 3.95 (s, 3H), 3.91 (s, 3H), 3.79–3.57 (m, 6H), 3.56–3.47 (m, 2H), 3.12–3.05 (m, 2H), 2.90 (td, J = 12.5, 2.7 Hz, 2H), 2.22–2.01 (m, 4H), 1.97–1.89 (m, 2H), 1.88–1.70 (m, 7H), 1.58–1.43 (m, 3H); 13C NMR (100 MHz, MeOH-d4) δ 160.40, 157.17, 151.53, 148.81, 136.96, 105.05, 103.62, 99.62, 58.12, 56.93, 56.79, 54.27 (four carbons), 42.50, 29.32, 25.29, 24.92, 24.24 (four carbons), 22.72. HPLC purity: >95%; tR = 3.26 min. MS (ESI): 428 [M + H]+.
N1-(6,7-Dimethoxy-2-(pyrrolidin-1-yl)quinazolin-4-yl)-N5,N5-dimethylpentane-1,5-diamine (6)
N1-(2-Chloro-6,7-dimethoxyquinazolin-4-yl)-N5,N5-dimethylpentane-1,5-diamine was prepared according to the procedure for making 24 from 2,4-dichloro-6,7-dimethoxylquinazoline, 5-(dimethylamino)amylamine (commercially available), N,N-diisopropylethylamine, and THF. Compound 6 was prepared according to the procedure for making 2 from N1-(2-chloro-6,7-dimethoxyquinazolin-4-yl)-N5,N5-dimethylpentane-1,5-diamine (70 mg, 0.15 mmol), pyrrolidine (21 μL, 0.30 mmol), HCl in dioxane (4.0 M, 75 μL, 0.30 mmol), and isopropanol (1.0 mL). The title compound 6 was obtained as a TFA salt, white solid (57 mg, yield 62%). 1H NMR (400 MHz, MeOH-d4) δ 7.55 (s, 1H), 7.09 (s, 1H), 3.95 (s, 3H), 3.91 (s, 3H), 3.78–3.54 (m, 6H), 3.16–3.09 (m, 2H), 2.87 (s, 6H), 2.22–1.98 (m, 4H), 1.89–1.72 (m, 4H), 1.58–1.39 (m, 2H); 13C NMR (100 MHz, MeOH-d4) δ 160.41, 157.19, 151.53, 148.82, 136.96, 105.05, 103.62, 99.62, 58.87, 56.93, 56.79, 43.37 (four carbons), 42.48, 29.32 (two carbons), 25.47 (two carbons), 25.06. HPLC purity: >95%; tR = 3.02 min. MS (ESI): 388 [M + H]+.
N1-(6,7-Dimethoxy-2-(pyrrolidin-1-yl)quinazolin-4-yl)-N5,N5-diethylpentane-1,5-diamine (7)
N1-(2-Chloro-6,7-dimethoxyquinazolin-4-yl)-N5,N5-diethylpentane-1,5-diamine was prepared according to the procedure for making 24 from 2,4-dichloro-6,7-dimethoxylquinazoline, 5-(diethylamino)pentylamine (commercially available), N,N-diisopropylethylamine, and THF. Compound 7 was prepared according to the procedure for making 1 from N1-(2-chloro-6,7-dimethoxyquinazolin-4-yl)-N5,N5-diethylpentane-1,5-diamine (62 mg, 0.16 mmol), pyrrolidine (54 μL, 0.64 mmol), N,N-diisopropylethylamine (113 μL, 0.64 mmol), and n-butanol (0.2 mL). The title compound 7 was obtained as a TFA salt, amber solid (79 mg, yield 76%). 1H NMR (400 MHz, MeOH-d4) δ 7.53 (s, 1H), 7.07 (s, 1H), 3.93 (s, 3H), 3.90 (s, 3H), 3.77–3.54 (m, 6H), 3.22 (q, J = 7.4 Hz, 4H), 3.16–3.09 (m, 2H), 2.21–2.01 (m, 4H), 1.87–1.72 (m, 4H), 1.57–1.45 (m, 2H), 1.30 (t, J = 7.3 Hz, 6H). HPLC purity: >95%; tR = 3.54 min. MS (ESI): 416 [M + H]+.
tert-Butyl 4-(5-((6,7-Dimethoxy-2-(pyrrolidin-1-yl)quinazolin-4-yl)amino)pentyl)piperazine-1-carboxylate (8)
tert-Butyl 4-(5-((2-chloro-6,7-dimethoxyquinazolin-4-yl)amino)pentyl)piperazine-1-carboxylate was prepared according to the procedure for making 24 from 2,4-dichloro-6,7-dimethoxylquinazoline, tert-butyl 4-(5-aminopentyl)piperazine-1-carboxylate (commercially available), N,N-diisopropylethylamine, and THF. Compound 8 was prepared according to the procedure for making 1 from tert-butyl 4-(5-((2-chloro-6,7-dimethoxyquinazolin-4-yl)amino)pentyl)piperazine-1-carboxylate (200 mg, 0.40 mmol), pyrrolidine (135 μL, 1.6 mmol), N,N-diisopropylethylamine (280 μL, 1.6 mmol), and n-butanol (0.5 mL). The title compound 8 was obtained as a TFA salt, yellow solid (180 mg, yield 60%). 1H NMR (400 MHz, MeOH-d4) δ 7.54 (s, 1H), 7.09 (s, 1H), 4.33–4.08 (m, 2H), 3.95 (s, 3H), 3.91 (s, 3H), 3.77–3.50 (m, 8H), 3.29–3.12 (m, 4H), 3.10–2.90 (m, 2H), 2.21–2.01 (m, 4H), 1.87–1.77 (m, 4H), 1.55–1.49 (m, 2H), 1.47 (s, 9H). HPLC purity: >95%; tR = 3.98 min. MS (ESI): 529 [M + H]+.
6,7-Dimethoxy-N-(5-(piperazin-1-yl)pentyl)-2-(pyrrolidin-1-yl)quinazolin-4-amine (9)
To the solution of tert-butyl 4-(5-((6,7-dimethoxy-2-(pyrrolidin-1-yl)quinazolin-4-yl)amino)pentyl)piperazine-1-carboxylate (8) in MeOH and THF was added TFA at room temperature and stirred overnight at 50 °C. LC–MS indicated the completion of the reaction. After removal of the solvent by rotary evaporation, the residue was purified by HPLC to give 14 mg of the title compound 9 as a TFA salt, white solid (14 mg, yield 80%). 1H NMR (400 MHz, MeOH-d4) δ 7.56 (s, 1H), 7.11 (s, 1H), 3.96 (s, 3H), 3.92 (s, 3H), 3.82–3.49 (m, 14H), 3.23–3.17 (m, 2H), 2.21–2.02 (m, 4H), 1.87–1.78 (m, 4H), 1.56–1.47 (m, 2H). HPLC purity: 95%; tR = 3.50 min. MS (ESI): 429 [M + H]+.
6,7-Dimethoxy-N-(5-(4-methylpiperazin-1-yl)pentyl)-2-(pyrrolidin-1-yl)quinazolin-4-amine (10)
To the solution of 6,7-dimethoxy-N-(5-(piperazin-1-yl)pentyl)-2-(pyrrolidin-1-yl)quinazolin-4-amine (9, 80 mg, 0.12 mmol) in MeOH (1.0 mL) were added formaldehyde (commercially available, 39 μL, 1.4 mmol), acetic acid (80 μL, 1.4 mmol), and sodium cyanoborohydride (44 mg, 0.7 mmol) at 0 °C. The resulting mixture was warmed to room temperature and stirred overnight. LC–MS indicated the completion of the reaction. After removal of the solvent by rotary evaporation, the residue was purified by HPLC to give the title compound 10 as a TFA salt, gray solid (25 mg, yield 31%). 1H NMR (400 MHz, MeOH-d4) δ 7.56 (s, 1H), 7.10 (s, 1H), 3.96 (s, 3H), 3.92 (s, 3H), 3.81–3.53 (m, 14H), 3.26–3.19 (m, 2H), 2.97 (s, 3H), 2.22–2.02 (m, 4H), 1.88–1.77 (m, 4H), 1.56–1.46 (m, 2H). HPLC purity: >95%; tR = 3.59 min. MS (ESI): 443 [M + H]+.
N-(5-(4-Ethylpiperazin-1-yl)pentyl)-6,7-dimethoxy-2-(pyrrolidin-1-yl)quinazolin-4-amine (11)
Compound 11 was prepared according to the procedure for making 10 from 6,7-dimethoxy-N-(5-(piperazin-1-yl)pentyl)-2-(pyrrolidin-1-yl)quinazolin-4-amine (9, 80 mg, 0.12 mmol), acetaldehyde (commercially available, 56 μL, 1.4 mmol), acetic acid (80 μL, 1.4 mmol) and sodium cyanoborohydride (44 mg, 0.7 mmol) and MeOH (1.0 mL). The title compound 11 was obtained as a TFA salt, gray solid (74 mg, yield 90%). 1H NMR (400 MHz, MeOH-d4) δ 7.55 (s, 1H), 7.10 (s, 1H), 3.95 (s, 3H), 3.92 (s, 3H), 3.81–3.55 (m, 14H), 3.35–3.26 (m, 2H), 3.26–3.20 (m, 2H), 2.18–2.02 (m, 4H), 1.88–1.78 (m, 4H), 1.56–1.48 (m, 2H), 1.37 (t, J = 7.3 Hz, 3H). HPLC purity: >95%; tR = 3.60 min. MS (ESI): 457 [M + H]+.
N-(5-(4-Isopropylpiperazin-1-yl)pentyl)-6,7-dimethoxy-2-(pyrrolidin-1-yl)quinazolin-4-amine (12)
Compound 12 was prepared according to the procedure for making 10 from 6,7-dimethoxy-N-(5-(piperazin-1-yl)pentyl)-2-(pyrrolidin-1-yl)quinazolin-4-amine (9, 80 mg, 0.12 mmol), acetone (commercially available, 103 μL, 1.4 mmol), acetic acid (80 μL, 1.4 mmol) and sodium cyanoborohydride (44 mg, 0.7 mmol) and MeOH (1.0 mL). The title compound 12 was obtained as a TFA salt, brown solid (61 mg, yield 73%). 1H NMR (400 MHz, MeOH-d4) δ 7.56 (s, 1H), 7.10 (s, 1H), 3.96 (s, 3H), 3.92 (s, 3H), 3.82–3.48 (m, 15H), 3.24–3.18 (m, 2H), 2.20–2.03 (m, 4H), 1.88–1.78 (m, 4H), 1.56–1.47 (m, 2H), 1.40 (s, 3H), 1.38 (s, 3H). HPLC purity: >95%; tR = 3.61 min. MS (ESI): 471 [M + H]+.
5-((6,7-Dimethoxy-2-(pyrrolidin-1-yl)quinazolin-4-yl)amino)-1-(pyrrolidin-1-yl)pentan-1-one (13)
5-((tert-Butoxycarbonyl)amino)pentanoic acid (commercially available, 0.93 g, 4.3 mmol) and pyrrolidine (238 μL, 2.9 mmol) were dissolved in 23 mL of DMF. To this solution were added N,N-diisopropylethylamine (2.5 mL, 14.5 mmol) and HATU (2.18 g, 5.8 mmol). The resulting solution was stirred at 50 °C for 3 h. After the mixture was cooled, TLC indicated the completion of the reaction. After removal of the solvent by rotary evaporation, the residue was redissolved in CH2Cl2, washed with brine. The organic layer was dried, concentrated, and purified by ISCO to give tert-butyl (5-oxo-5-(pyrrolidin-1-yl)pentyl)carbamate. To the solution of tert-butyl (5-oxo-5-(pyrrolidin-1-yl)pentyl)carbamate in MeOH and THF was added TFA at room temperature and stirred overnight at room temperature. LC–MS indicated the completion of the reaction. After removal of the solvent by rotary evaporation, the product 5-amino-1-(pyrrolidin-1-yl)pentan-1-one obtained was used for the next step without further purification.
5-((2-Chloro-6,7-dimethoxyquinazolin-4-yl)amino)-1-(pyrrolidin-1-yl)pentan-1-one was prepared according to the procedure for making 24 from 2,4-dichloro-6,7-dimethoxylquinazoline, 5-amino-1-(pyrrolidin-1-yl)pentan-1-one, N,N-diisopropylethylamine, and THF. Compound 13 was prepared according to the procedure for making 1 from 5-((2-chloro-6,7-dimethoxyquinazolin-4-yl)amino)-1-(pyrrolidin-1-yl)pentan-1-one (87 mg, 0.22 mmol), pyrrolidine (74 μL, 0.88 mmol), N,N-diisopropylethylamine (154 μL, 0.88 mmol), and n-butanol (0.25 mL). The title compound 13 was obtained as a TFA salt, brown solid (16 mg, yield 13%). 1H NMR (400 MHz, MeOH-d4) δ 7.57 (s, 1H), 7.09 (s, 1H), 3.96 (s, 3H), 3.93 (s, 3H), 3.80–3.59 (m, 6H), 3.48 (t, J = 6.8 Hz, 2H), 3.40 (t, J = 6.9 Hz, 2H), 2.41 (t, J = 7.0 Hz, 2H), 2.20–2.03 (m, 4H), 2.01–1.93 (m, 2H), 1.91–1.84 (m, 2H), 1.83–1.71 (m, 4H). HPLC purity: >95%; tR = 4.58 min. MS (ESI): 428 [M + H]+.
6,7-Dimethoxy-N-methyl-2-(pyrrolidin-1-yl)-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (14)
2-Chloro-6,7-dimethoxy-N-methyl-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine was prepared according to the procedure for making 24 from 2,4-dichloro-6,7-dimethoxylquinazoline, N-methyl-5-(pyrrolidin-1-yl)pentan-1-amine (synthesized according to the precedures preported previously),51N,N-diisopropylethylamine, and THF. Compound 14 was prepared according to the procedure for making 2 from 2-chloro-6,7-dimethoxy-N-methyl-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (50 mg, 0.13 mmol), pyrrolidine (21 μL, 0.26 mmol), HCl in dioxane (4.0 M, 65 μL, 0.26 mmol), and isopropanol (0.7 mL). The title compound 14 was obtained as a TFA salt, white solid (25 mg, yield 30%). 1H NMR (400 MHz, MeOH-d4) δ 7.47 (s, 1H), 7.17 (s, 1H), 3.97 (s, 3H), 3.91 (s, 3H), 3.90–3.85 (m, 2H), 3.75–3.60 (m, 6H), 3.57 (s, 3H), 3.24–3.17 (m, 2H), 3.11–3.01 (m, 2H), 2.22–2.06 (m, 6H), 2.05–1.97 (m, 2H), 1.96–1.87 (m, 2H), 1.86–1.77 (m, 2H), 1.55–1.45 (m, 2H); 13C NMR (100 MHz, MeOH-d4) δ 162.30, 157.06, 150.22, 147.38, 139.69, 109.50, 104.02, 99.83, 56.97, 56.79, 56.04 (two carbons), 55.09 (two carbons), 54.45 (two carbons), 41.10, 27.43, 26.91 (two carbons), 25.08 (two carbons), 23.97 (two carbons). HPLC purity: >95%; tR = 3.28 min. MS (ESI): 428 [M + H]+.
6,7-Dimethoxy-2-(piperidin-1-yl)-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (15)
Compound 15 was prepared according to the procedure for making 2 from 2-chloro-6,7-dimethoxy-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (24, 70 mg, 0.19 mmol), piperidine (commercially available, 37 μL, 0.38 mmol), HCl in dioxane (4.0 M, 95 μL, 0.38 mmol), and isopropanol (1.0 mL). The title compound 15 was obtained as a TFA salt, light yellow solid (101 mg, yield 83%). 1H NMR (400 MHz, MeOH-d4) δ 7.54 (s, 1H), 7.09 (s, 1H), 3.94 (s, 3H), 3.91 (s, 3H), 3.88–3.82 (m, 4H), 3.69–3.61 (m, 4H), 3.22–3.16 (m, 2H), 3.10–3.01 (m, 2H), 2.20–2.08 (m, 2H), 2.07–1.96 (m, 2H), 1.85–1.69 (m, 10H), 1.55–1.46 (m, 2H); 13C NMR (100 MHz, MeOH-d4) δ 160.55, 157.21, 152.43, 149.01, 137.00, 104.87, 103.58, 99.79, 56.90, 56.81, 56.08, 55.06 (two carbons), 47.16 (two carbons), 42.56, 29.29, 26.87, 26.69 (two carbons), 25.19, 25.13, 23.95 (two carbons). HPLC purity: >95%; tR = 3.22 min. MS (ESI): 428 [M + H]+.
2-(Azepan-1-yl)-6,7-dimethoxy-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (16)
Compound 16 was prepared according to the procedure for making 2 from 2-chloro-6,7-dimethoxy-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (24, 70 mg, 0.19 mmol), hexamethyleneimine (commercially available, 42 μL, 0.38 mmol), HCl in dioxane (4.0 M, 95 μL, 0.38 mmol), and isopropanol (1.0 mL). The title compound 16 was obtained as a TFA salt, light yellow solid (35 mg, yield 28%). 1H NMR (400 MHz, MeOH-d4) δ 7.56 (s, 1H), 7.18 (s, 1H), 3.95 (s, 3H), 3.92 (s, 3H), 3.90–3.70 (m, 4H), 3.70–3.61 (m, 4H), 3.22–3.16 (m, 2H), 3.10–3.01 (m, 2H), 2.19–2.08 (m, 2H), 2.07–1.97 (m, 2H), 1.95–1.86 (m, 4H), 1.85–1.75 (m, 4H), 1.69–1.61 (m, 4H), 1.56–1.47 (m, 2H); 13C NMR (100 MHz, MeOH-d4) δ 160.40, 157.23, 152.91, 148.99, 137.10, 104.89, 103.68, 99.85, 56.91, 56.80, 56.10, 55.09 (two carbons), 49.16 (two carbons), 42.59, 29.47, 28.58 (three carbons), 27.85, 26.91, 25.23, 23.95 (two carbons). HPLC purity: >95%; tR = 3.67 min. MS (ESI): 442 [M + H]+.
6,7-Dimethoxy-N2,N2-dimethyl-N4-(5-(pyrrolidin-1-yl)pentyl)quinazoline-2,4-diamine (17)
To a solution of Pd(OAc)2 (2 mg, 0.01 mmol), (+)-BINAP (6 mg, 0.01 mmol), and THF (0.5 mL) were added 2-chloro-6,7-dimethoxy-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (24, 46 mg, 0.10 mmol), dimethylamine hydrochloride (commercially available, 10 mg, 0.12 mmol), and Cs2CO3 (73 mg, 0.24 mmol). The resulting solution was stirred inside a microwave at 140 °C for 30 min. After the mixture was cooled, TLC indicated the completion of the reaction. After removal of the solvent by rotary evaporation, the residue was redissolved in CH2Cl2, washed with brine. The organic layer was dried, concentrated, and purified by HPLC to give 7.0 mg of the title compound 17 as a TFA salt, white solid (yield 13%). 1H NMR (400 MHz, MeOH-d4) δ 7.57 (s, 1H), 7.15 (s, 1H), 3.97 (s, 3H), 3.92 (s, 3H), 3.71 (t, J = 7.1 Hz, 2H), 3.68–3.60 (m, 2H), 3.31 (s, 6H), 3.23–3.16 (m, 2H), 3.10–3.01 (m, 2H), 2.21–2.09 (m, 2H), 2.07–1.96 (m, 2H), 1.87–1.75 (m, 4H), 1.56–1.48 (m, 2H); 13C NMR (100 MHz, MeOH-d4) δ 160.39, 157.35, 153.70, 149.10, 137.07, 104.92, 103.55, 99.79, 56.93, 56.82, 56.13, 55.14 (two carbons), 42.57 (two carbons), 37.96, 29.36, 26.92, 25.23, 23.96 (two carbons). HPLC purity: >95%; tR = 3.04 min. MS (ESI): 388 [M + H]+.
6,7-Dimethoxy-2-(piperazin-1-yl)-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (18)
Boc-protected compound 18 was prepared according to the procedure for making 1 from 2-chloro-6,7-dimethoxy-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (24, 56 mg, 0.11 mmol), 1-Boc-piperazine (commercially available, 82 mg, 0.44 mmol), N,N-diisopropylethylamine (57 μL, 0.33 mmol), and n-butanol (0.7 mL). To the resulting mixture was added TFA, and the mixture was stirred overnight at room temperature. LC–MS indicated the completion of the reaction. After removal of the solvent by rotary evaporation, the residue was purified by HPLC to give 40 mg of the title compound 18 as a TFA salt, white solid (yield 55%, two steps). 1H NMR (400 MHz, MeOH-d4) δ 7.62 (s, 1H), 7.16 (s, 1H), 4.22–4.14 (m, 4H), 3.96 (s, 3H), 3.93 (s, 3H), 3.71 (t, J = 7.0 Hz, 2H), 3.69–3.61 (m, 2H), 3.48–3.38 (m, 4H), 3.23–3.16 (m, 2H), 3.11–3.01 (m, 2H), 2.20–2.09 (m, 2H), 2.07–1.96 (m, 2H), 1.86–1.73 (m, 4H), 1.57–1.48 (m, 2H). HPLC purity: >95%; tR = 1.76 min. MS (ESI): 429 [M + H]+.
6,7-Dimethoxy-2-(4-methylpiperazin-1-yl)-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (19)
Compound 19 was prepared according to the procedure for making 1 from 2-chloro-6,7-dimethoxy-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (24, 56 mg, 0.11 mmol), 1-methylpiperazine (commercially available, 49 μL, 0.44 mmol), N,N-diisopropylethylamine (57 μL, 0.33 mmol), and n-butanol (0.7 mL). The title compound 19 was obtained as a TFA salt, gray solid (54 mg, yield 73%). 1H NMR (400 MHz, MeOH-d4) δ 7.62 (s, 1H), 7.16 (s, 1H), 3.96 (s, 3H), 3.93 (s, 3H), 3.90–3.24 (m, 12H), 3.22–3.16 (m, 2H), 3.10–3.02 (m, 2H), 2.99 (s, 3H), 2.19–2.10 (m, 2H), 2.07–1.96 (m, 2H), 1.86–1.75 (m, 4H), 1.56–1.47 (m, 2H). HPLC purity: >95%; tR = 0.55 min. MS (ESI): 443 [M + H]+.
N2-Cyclopentyl-6,7-dimethoxy-N2-methyl-N4-(5-(pyrrolidin-1-yl)pentyl)quinazoline-2,4-diamine (20)
Compound 20 was prepared according to the procedure for making 1 from 2-chloro-6,7-dimethoxy-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (24, 56 mg, 0.11 mmol), N-methylcyclopentanamine (commercially available, 44 mg, 0.44 mmol), N,N-diisopropylethylamine (57 μL, 0.33 mmol), and n-butanol (0.7 mL). The title compound 20 was obtained as a TFA salt, yellow solid (40 mg, yield 54%). 1H NMR (400 MHz, MeOH-d4) δ 7.57 (s, 1H), 7.18 (s, 1H), 5.15–5.01 (m, 1H), 3.96 (s, 3H), 3.92 (s, 3H), 3.72–3.61 (m, 4H), 3.23–3.16 (m, 2H), 3.14 (s, 3H), 3.10–3.01 (m, 2H), 2.19–2.10 (m, 2H), 2.05–1.93 (m, 4H), 1.89–1.68 (m, 10H), 1.56–1.47 (m, 2H); 13C NMR (100 MHz, MeOH-d4) δ 160.35, 157.29, 153.54, 149.07, 137.09, 104.90, 103.75, 99.90, 58.92, 56.92, 56.80, 56.10, 55.10 (three carbons), 42.62, 30.41, 29.65 (two carbons), 29.39, 26.93, 25.28, 25.25, 23.95 (two carbons). HPLC purity: >95%; tR = 3.83 min. MS (ESI): 442 [M + H]+.
N2-Cyclohexyl-6,7-dimethoxy-N2-methyl-N4-(5-(pyrrolidin-1-yl)pentyl)quinazoline-2,4-diamine (21)
Compound 21 was prepared according to the procedure for making 1 from 2-chloro-6,7-dimethoxy-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (24, 56 mg, 0.11 mmol), N-methylcyclohexylamine (commercially available, 44 mg, 0.44 mmol), N,N-diisopropylethylamine (57 μL, 0.33 mmol), and n-butanol (0.7 mL). The title compound 21 was obtained as a TFA salt, brown solid (47 mg, yield 62%). 1H NMR (400 MHz, MeOH-d4) δ 7.56 (s, 1H), 7.18 (s, 1H), 4.65–4.49 (m, 1H), 3.95 (s, 3H), 3.92 (s, 3H), 3.71–3.61 (m, 4H), 3.23–3.16 (m, 2H), 3.14 (s, 3H), 3.10–3.01 (m, 2H), 2.21–2.08 (m, 2H), 2.07–1.98 (m, 2H), 1.97–1.90 (m, 2H), 1.86–1.75 (m, 6H), 1.71–1.60 (m, 2H), 1.56–1.42 (m, 4H), 1.40–1.17 (m, 2H); 13C NMR (100 MHz, MeOH-d4) δ 160.35, 157.24, 153.14, 149.04, 137.14, 104.89, 103.79, 99.93, 57.59, 56.91, 56.80, 56.08, 55.09 (three carbons), 42.68, 30.85 (two carbons), 30.09, 29.40, 26.93 (two carbons), 26.38, 25.34, 23.95 (two carbons). HPLC purity: >95%; tR = 3.54 min. MS (ESI): 456 [M + H]+.
N2-Cyclopentyl-6,7-dimethoxy-N4-(5-(pyrrolidin-1-yl)pentyl)quinazoline-2,4-diamine (22)
Compound 22 was prepared according to the procedure for making 1 from 2-chloro-6,7-dimethoxy-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (24, 56 mg, 0.11 mmol), cyclopentylamine (commercially available, 44 μL, 0.44 mmol), N,N-diisopropylethylamine (57 μL, 0.33 mmol), and n-butanol (0.7 mL). The title compound 22 was obtained as a TFA salt, light yellow solid (41 mg, yield 57%). 1H NMR (400 MHz, MeOH-d4) δ 7.52 (s, 1H), 6.95 (s, 1H), 4.41–4.28 (m, 1H), 3.95 (s, 3H), 3.90 (s, 3H), 3.73–3.60 (m, 4H), 3.23–3.16 (m, 2H), 3.10–2.99 (m, 2H), 2.20–1.98 (m, 6H), 1.88–1.75 (m, 6H), 1.73–1.60 (m, 4H), 1.56–1.46 (m, 2H). HPLC purity: >95%; tR = 3.58 min. MS (ESI): 428 [M + H]+.
N2-Cyclohexyl-6,7-dimethoxy-N4-(5-(pyrrolidin-1-yl)pentyl)quinazoline-2,4-diamine (23)
Compound 23 was prepared according to the procedure for making 1 from 2-chloro-6,7-dimethoxy-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (24, 56 mg, 0.11 mmol), N-cyclohexylamine (commercially available, 50 μL, 0.44 mmol), N,N-diisopropylethylamine (57 μL, 0.33 mmol), and n-butanol (0.7 mL). The title compound 23 was obtained as a TFA salt, brown semisolid (45 mg, yield 61%). 1H NMR (400 MHz, MeOH-d4) δ 7.50 (s, 1H), 6.91 (s, 1H), 4.00–3.85 (m, 1H), 3.94 (s, 3H), 3.89 (s, 3H), 3.70–3.61 (m, 4H), 3.23–3.15 (m, 2H), 3.10–3.01 (m, 2H), 2.19–2.10 (m, 2H), 2.06–1.99 (m, 3H), 1.88–1.75 (m, 7H), 1.54–1.28 (m, 8H). HPLC purity: >95%; tR = 3.69 min. MS (ESI): 442 [M + H]+.
2-Chloro-6,7-dimethoxy-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine (24)
To the solution of 2,4-dichloro-6,7-dimethoxyquinazoline (commercially available, 1.00 g, 3.86 mmol) in THF (9.5 mL) was added 5-(pyrrolidin-1-yl)pentan-1-amine (commercially available, 1.33 g, 8.49 mmol), followed by the addition of N,N-diisopropylethylamine (612 μL, 3.51 mmol). And the resulting mixture was stirred at room temperature for 6 h until TLC showed that the starting material had disappeared. Water was added to the reaction mixture, and the resulting solution was extracted with ethyl acetate. The organic layer was washed with brine, dried, and concentrated to give the crude product, which was purified on ISCO using 24 g silica gel column, eluting with hexane–ethyl acetate to give 754 mg of the title compound 24 as a yellow semisolid (yield 52%). 1H NMR (400 MHz, MeOH-d4) δ 7.61 (s, 1H), 6.99 (s, 1H), 3.98 (s, 3H), 3.96 (s, 3H), 3.73 (t, J = 7.2 Hz, 2H), 3.69–3.61 (m, 2H), 3.24–3.16 (m, 2H), 3.11–3.01 (m, 2H), 2.20–2.09 (m, 2H), 2.07–1.96 (m, 2H), 1.87–1.76 (m, 4H), 1.57–1.47 (m, 2H); 13C NMR (100 MHz, MeOH-d4) δ 161.38, 157.90, 152.68, 151.65, 141.15, 107.16, 103.64, 102.16, 57.02, 56.99, 56.08, 55.12 (two carbons), 42.73, 29.27, 26.64, 24.82, 23.95 (two carbons). HPLC purity: >95%; tR = 3.60 min. MS (ESI): 379 [M + H]+.
Radioactive Assay (Also Known as Scintillation Proximity Assay)
SETD8 catalytic domain was expressed in E. coli and purified as reported.52 Radioactive assay was developed to screen our chemical library for small molecule inhibitors. Methylation (10 μL) reactions were carried out in a buffer containing 50 mM Tris-HCl (pH 8.0), 10 mM GSH, 0.1%Triton X-100, at room temperature using 50 nM SETD8, 1.5 μM tritium labeled SAM (catalog no. NET155V250UC, PerkinElmer), and 5 μM biotinylated H4 (1–24) peptide substrate (SGRGKGGKGLGKGGAKRHRKVLRDK-biotin) in 384-well plates in the presence of 50 μM compounds. The reactions were then quenched by addition of equal volume of 7.5 M guanidine hydrochloride after a 1 h incubation. Then 40 μL of buffer (20 mM Tris-HCl, pH 8.0) was added into the quenched samples, and all samples were then transferred into a streptavidin/scintillant-coated microplate (catalog no. SMP410, PerkinElmer). The amount of methylated peptide was quantified by tracing the radioactivity (counts per minute) as measured after 1 h using a TopCount plate reader (PerkinElmer). For IC50 values determination, the compounds were serially diluted 2-fold in DMSO for a total of 11 concentrations, beginning at 0.25 mM and tested in the same condition.
Microfluidic Capillary Electrophoresis Assay
Inhibition of SETD8 methyltransferase activity was analyzed by monitoring decrease in methylation of the fluorescein labeled peptide, TW21 (NH2-LGKGGAKRHRKVLRDNIQGITK(5Fam)-OH), essentially as described previously.50
The test compounds were solubilized in 100% DMSO to 10 mM and then plated in Greiner 384-well polypropylene plates using 3-fold dilution scheme over 10 points spanning the concentration range from 3 mM to 0.15 μM using TECAN Genesis liquid handler. Then 1 μL of the serial dilution was transferred into compound dilution plate using Nanoscreen MultiMek liquid handling robot. Prior to performing the assay the compounds were diluted 10-fold in 1× assay buffer (20 mM Tris, pH 8.0, 25 mM NaCl, 2 mM DTT, and 0.05% Tween-20), and an amount of 2.5 μL of the resulting dilution was transferred into assay plate by Nanoscreen MultiMek liquid handling robot. To this plate 20 μL of 50 nM SETD8 and 2 μM TW21 peptide cocktail in 1× assay buffer were added using multidrop liquid dispenser. Following a 10 min incubation of the compounds with the enzyme/peptide mix, the reaction was initiated by adding 2.5 μL of 150 μM SAM in 1× buffer. For 100% inhibition controls 1× buffer was added instead of SAM. The reaction was allowed to proceed at room temperature for 120 min, and then the reaction was terminated by adding 35 μL of 0.08 ng/μL Endo-LysC protease solution. Following an additional 1 h incubation the plate was read on a Caliper Life Sciences EZ reader II using upstream voltage −500 V, downstream voltage −1800 V, and pressure of −1.5 psi. The IC50 values were determined using Screenable Solutions software.
Isothermal Titration Calorimetry
All ITC measurements were performed at 25 °C using an AutoITC200 microcalorimeter (MicroCal/GE Healthcare). The calorimeter cell (volume 200 μL) was loaded with SETD8 protein in the full salt dialysis buffer (150 mM NaCl) at a concentration of 100 μM. The syringe was loaded with compound 1 (dissolved in the same buffer) at a concentration of 1 mM. A typical injection protocol included a single 0.2 μL first injection followed by 26 1.5 μL injections of the compound into the calorimeter cell. The spacing between injections was kept at 180 s and the reference power at 8 μcal/s. A control experiment was performed by titrating compound 1 into buffer under identical settings to determine the heat signals that arose from compound dilution; these were subtracted from the heat signals of protein–compound interaction. The data were analyzed using Origin for ITC, version 7.0, software supplied by the manufacturer and fitted well to a one-site binding model.
Surface Plasmon Resonance
The interaction between compound 1 and protein SETD8 was further explored using a ProteOn XPR36 biosensor (Bio-Rad Laboratories, Inc.) at 25 °C. PBS buffer (phosphate buffered saline, pH 7.4) supplemented with 0.005% Tween-20 was used as the running buffer. A GLH (catalog no. 176-5013, Bio-Rad Laboratories, Inc.) sensor chip was first activated by flowing a mixture of 20 mM sulfo-NHS and 20 mM EDC over the chip surface for 5 min at a flow rate of 30 μL/min. SETD8 was then diluted to 20 and 10 μg/mL in 5 mM NaOAc, pH 5.0, and immobilized onto two ligand channels (30 μL/min for 2 min). The surface was then deactivated by flowing 1 M ethanolamine for 5 min at a flow rate 60 μL/min. A blank injection of the running buffer was made, after which four concentrations of the compound in the running buffer (100, 33.3, 11.1, and 3.7 μM) and a running buffer were injected simultaneously at a flow rate of 50 μL/min for 60 s. The sensorgrams obtained at the four concentrations of the compound were fit simultaneously after subtracting a ligand reference (inner spot) and a double compound reference (buffer) using a Langmuir model to obtain on (ka) and off (kd) rates. The kinetic KD was calculated based on the on and off rates for three replicates. An equilibrium analysis of the data was also performed to calculate the KD.
MOA Studies
IC50 values were determined for compound 1 at varying concentrations of SAM, 50 nM SETD8, 200 μM peptide H4 (1–24) or at varying concentrations of substrate H4 (1–24) peptide, 50 nM of SETD8, 250 μM SAM. The reaction mixtures were incubated 15 min at 23 °C. To stop the enzymatic reactions, an equal volume of 7.5 M guanidine hydrochloride was added and mixed. Then 10 μL of mixture was spotted onto a square of SAM2 biotin capture membrane (catalog no. V2861, Promega), allowed absorption for 5 min at room temperature, washed with 2 M of NaCl solution several times followed by two deionized water wash, and dried, and scintillation liquid was added and CPM (counts per minute) were read using the scintillation counter.
Peptide Displacement Assay
H4K20me (1–24) peptide (SGRGKGGKGLGKGGAKRHRKme1VLRD) with N-terminus labelled by FITC was ordered from Tufts University Core Services (Boston, MA). The sample was prepared in a buffer containing 50 mM Tris-HCl (pH 8.0), 10 mM GSH, 0.01% Triton X-100. 50 nM peptide was incubated with 20 μM SETD8 protein, 1 mM S-adenosyl-l-homocysteine (SAH), and different concentrations of compounds. The final volume was 10 μL with less than 5% DMSO. The FP measurement was performed in black, 384-well PCR plate (catalog no. 47744-828, VWR), and signal was read with a Synergy H4 microplate reader (BioTek). The polarization values in millipolarization units were measured at an excitation wavelength of 485 nm and an emission wavelength of 528 nm.
Selectivity Assay
Selectivity of compound 1 against a panel of methyltransferases was assessed as previously reported.24,36,53 In brief, the effect of compound 1 on activity of G9a, SETDB1, GLP, SUV39H2, SETD7, PRMT3, PRMT5-MEP50 complex, PRMT1, SUV420H1, SUV420H2, SMYD2, DNMT1, PRC2 complex, MLL1 complex, and DOT1L was assessed by monitoring the incorporation of tritium-labeled methyl group to lysine or arginine residues of peptide substrates using radioactive assay. Assays were performed in a 20 μL reaction mixture containing 3H-SAM (catalog no. NET155V250UC, Perkin Elmer) at substrate concentrations close to Km values for each enzyme. Compound concentrations from 100 nM to 100 μM were used in all selectivity assays. To stop the enzymatic reactions, 7.5 M guanidine hydrochloride was added, followed by 180 μL of buffer (20 mM Tris, pH 8.0), mixed, and then transferred to a 96-well FlashPlate (catalog no. SMP103, PerkinElmer). After mixing, the reaction mixtures in FlashPlates were incubated for 1 h and the CPM were measured using TopCount plate reader (PerkinElmer). The CPM in the absence of compound for each data set were defined as 100% activity. In the absence of the enzyme, the CPM in each data set were defined as background (0%). Data were plotted using SigmaPlot software. For DNMT1 the dsDNA substrate was prepared by annealing two complementary strands (biotintlated forward strand B-GAGCCCGTAAGCCCGTTCAGGTCG and reverse strand CGACCTGAACGGGCTTACGGGCTC), synthesized by Eurofins MWG Operon. For DOT1L, a filter-based assay was used. In this assay, 20 μL of reaction mixtures was incubated at room temperature for 1 h, 100 μL of 10% trichloroacetic acid (TCA) was added, mixed, and transferred to filter plates (catalog no. MSFBN6B10, Millipore). Plates were centrifuged at 2000 rpm (Allegra X-15R; Beckman Coulter, Inc.) for 2 min followed by two additional 10% TCA washes and one ethanol wash (180 μL) followed by centrifugation. Plates were dried, and 100 μL of MicroScint-O (catalog no. 6013611, PerkinElmer) was added to each well, centrifuged, and removed. Then 70 μL of MicroScint-O was added again, and CPM were measured using TopCount plate reader.
Acknowledgments
We thank Dr. Raymond C. Trievel at University of Michigan for providing the SETD8 vector, Jacob Stuckey for assisting with ITC studies, and Catherine Simpson for assisting with MCE assay studies. The research described here was supported by Grant R01GM103893 from the National Institute of General Medical Sciences of the National Institutes of Health, the University Cancer Research Fund from University of North Carolina at Chapel Hill, the V Foundation for Cancer Research, and the Structural Genomics Consortium, a registered charity (No. 1097737) that receives funds from the Canada Foundation for Innovation, Eli Lilly Canada, GlaxoSmithKline, the Ontario Ministry of Economic Development and Innovation, the Novartis Research Foundation, Pfizer, AbbVie, Takeda, Janssen, Boehringer Ingelheim, Bayer, and the Wellcome Trust.
Glossary
Abbreviations Used
- H4K20
histone H4 lysine 20
- H4K20me
histone H4 lysine 20 monomethylation
- PCNA
proliferating cell nuclear antigen
- MOA
mechanism of action
- ITC
isothermal titration calorimetry
- SPR
surface plasmon resonance
- SAR
structure–activity relationship
- PKMT
protein lysine methyltransferase
- HKMT
histone lysine methyltransferase
- SAM
S-adenosyl-l-methionine
- SET
Su(var), E(z), and Trithorax
- PRMT
protein arginine methyltransferase
- KMT5A
lysine methyltransferase 5A
- EMT
epithelial–mesenchymal transition
- p53K382me1
monomethylation of p53 at lysine 382
- PCNA
proliferating cell nuclear antigen
- PCNAK248me1
monomethylation of proliferating cell nuclear antigen at lysine 248
- MCE
microfluidic capillary electrophoresis
- FP
fluorescence polarization
- FITC
fluorescein isothiocyanate
- PRC2
polycomb repressive complex 2
Supporting Information Available
1H and 13C NMR spectra of compound 1. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∞ A.M. and W.Y. contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Copeland R. A.; Solomon M. E.; Richon V. M. Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discovery 2009, 8, 724–732. [DOI] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Bountra C.; Fish P. V.; Lee K.; Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discovery 2012, 11, 384–400. [DOI] [PubMed] [Google Scholar]
- Helin K.; Dhanak D. Chromatin proteins and modifications as drug targets. Nature 2013, 502, 480–488. [DOI] [PubMed] [Google Scholar]
- Bernstein B. E.; Meissner A.; Lander E. S. The mammalian epigenome. Cell 2007, 128, 669–681. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Martin C.; Zhang Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [DOI] [PubMed] [Google Scholar]
- Jenuwein T.; Laible G.; Dorn R.; Reuter G. SET domain proteins modulate chromatin domains in eu- and heterochromatin. Cell. Mol. Life Sci. 1998, 54, 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B.; Wilson J. R.; Gamblin S. J. SET domains and histone methylation. Curr. Opin. Struct. Biol. 2003, 13, 699–705. [DOI] [PubMed] [Google Scholar]
- Kubicek S.; O’Sullivan R. J.; August E. M.; Hickey E. R.; Zhang Q.; Teodoro M. L.; Rea S.; Mechtler K.; Kowalski J. A.; Homon C. A.; Kelly T. A.; Jenuwein T. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 2007, 25, 473–481. [DOI] [PubMed] [Google Scholar]
- Liu F.; Chen X.; Allali-Hassani A.; Quinn A. M.; Wasney G. A.; Dong A.; Barsyte D.; Kozieradzki I.; Senisterra G.; Chau I.; Siarheyeva A.; Kireev D. B.; Jadhav A.; Herold J. M.; Frye S. V.; Arrowsmith C. H.; Brown P. J.; Simeonov A.; Vedadi M.; Jin J. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a. J. Med. Chem. 2009, 52, 7950–7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y.; Ganesh T.; Horton J. R.; Spannhoff A.; Liu J.; Sun A.; Zhang X.; Bedford M. T.; Shinkai Y.; Snyder J. P.; Cheng X. Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases. J. Mol. Biol. 2010, 400, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Chen X.; Allali-Hassani A.; Quinn A. M.; Wigle T. J.; Wasney G. A.; Dong A.; Senisterra G.; Chau I.; Siarheyeva A.; Norris J. L.; Kireev D. B.; Jadhav A.; Herold J. M.; Janzen W. P.; Arrowsmith C. H.; Frye S. V.; Brown P. J.; Simeonov A.; Vedadi M.; Jin J. Protein lysine methyltransferase G9a inhibitors: design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines. J. Med. Chem. 2010, 53, 5844–5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedadi M.; Barsyte-Lovejoy D.; Liu F.; Rival-Gervier S.; Allali-Hassani A.; Labrie V.; Wigle T. J.; DiMaggio P. A.; Wasney G. A.; Siarheyeva A.; Dong A.; Tempel W.; Wang S.-C.; Chen X.; Chau I.; Mangano T.; Huang X.-P.; Simpson C. D.; Pattenden S. G.; Norris J. L.; Kireev D. B.; Tripathy A.; Edwards A.; Roth B. L.; Janzen W. P.; Garcia B. A.; Petronis A.; Ellis J.; Brown P. J.; Frye S. V.; Arrowsmith C. H.; Jin J. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 2011, 7, 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Barsyte-Lovejoy D.; Allali-Hassani A.; He Y.; Herold J. M.; Chen X.; Yates C. M.; Frye S. V.; Brown P. J.; Huang J.; Vedadi M.; Arrowsmith C. H.; Jin J. Optimization of cellular activity of G9a inhibitors 7-aminoalkoxy-quinazolines. J. Med. Chem. 2011, 54, 6139–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson A. D.; Larsen N. A.; Howard T.; Pollard H.; Green I.; Grande C.; Cheung T.; Garcia-Arenas R.; Cowen S.; Wu J.; Godin R.; Chen H.; Keen N. Structural basis of substrate methylation and inhibition of SMYD2. Structure 2011, 19, 1262–1273. [DOI] [PubMed] [Google Scholar]
- Daigle S. R.; Olhava E. J.; Therkelsen C. A.; Majer C. R.; Sneeringer C. J.; Song J.; Johnston L. D.; Scott M. P.; Smith J. J.; Xiao Y.; Jin L.; Kuntz K. W.; Chesworth R.; Moyer M. P.; Bernt K. M.; Tseng J. C.; Kung A. L.; Armstrong S. A.; Copeland R. A.; Richon V. M.; Pollock R. M. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y.; Chen P.; Diao J.; Cheng G.; Deng L.; Anglin J. L.; Prasad B. V. V; Song Y. Selective inhibitors of histone methyltransferase DOT1L: design, synthesis and crystallographic studies. J. Am. Chem. Soc. 2011, 13316746–16749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y.; Wang Q.; Paulk J.; Kubicek S.; Kemp M. M.; Adams D. J.; Shamji A. F.; Wagner B. K.; Schreiber S. L. A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem. Biol. 2012, 7, 1152–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson S. K.; Wigle T. J.; Warholic N. M.; Sneeringer C. J.; Allain C. J.; Klaus C. R.; Sacks J. D.; Raimondi A.; Majer C. R.; Song J.; Scott M. P.; Jin L.; Smith J. J.; Olhava E. J.; Chesworth R.; Moyer M. P.; Richon V. M.; Copeland R. A.; Keilhack H.; Pollock R. M.; Kuntz K. W. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 2012, 8, 890–896. [DOI] [PubMed] [Google Scholar]
- McCabe M. T.; Ott H. M.; Ganji G.; Korenchuk S.; Thompson C.; Van Aller G. S.; Liu Y.; Graves A. P.; Della Pietra A. III; Diaz E.; Lafrance L. V.; Mellinger M.; Duquenne C.; Tian X.; Kruger R. G.; McHugh C. F.; Brandt M.; Miller W. H.; Dhanak D.; Verma S. K.; Tummino P. J.; Creasy C. L. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [DOI] [PubMed] [Google Scholar]
- Verma S. K.; Tian X.; LaFrance L. V.; Duquenne C.; Suarez D. P.; Newlander K. A.; Romeril S. P.; Burgess J. L.; Grant S. W.; Brackley J. A.; Graves A. P.; Scherzer D. A.; Shu A.; Thompson C.; Ott H. M.; Aller G. S. V.; Machutta C. A.; Diaz E.; Jiang Y.; Johnson N. W.; Knight S. D.; Kruger R. G.; McCabe M. T.; Dhanak D.; Tummino P. J.; Creasy C. L.; Miller W. H. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med. Chem. Lett. 2012, 3, 1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W.; Ibáñez G.; Wu H.; Blum G.; Zeng H.; Dong A.; Li F.; Hajian T.; Allali-Hassani A.; Amaya M. F.; Siarheyeva A.; Yu W.; Brown P. J.; Schapira M.; Vedadi M.; Min J.; Luo M. Sinefungin derivatives as inhibitors and structure probes of protein lysine methyltransferase SETD2. J. Am. Chem. Soc. 2012, 134, 18004–18014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W.; Chan H.; Teng L.; Li L.; Chuai S.; Zhang R.; Zeng J.; Li M.; Fan H.; Lin Y.; Gu J.; Ardayfio O.; Zhang J.-H.; Yan X.; Fang J.; Mi Y.; Zhang M.; Zhou T.; Feng G.; Chen Z.; Li G.; Yang T.; Zhao K.; Liu X.; Yu Z.; Lu C. X.; Atadja P.; Li E. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 21360–21365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W.; Chory E. J.; Wernimont A. K.; Tempel W.; Scopton A.; Federation A.; Marineau J. J.; Qi J.; Barsyte-Lovejoy D.; Yi J.; Marcellus R.; Iacob R. E.; Engen J. R.; Griffin C.; Aman A.; Wienholds E.; Li F.; Pineda J.; Estiu G.; Shatseva T.; Hajian T.; Al-awar R.; Dick J. E.; Vedadi M.; Brown P. J.; Arrowsmith C. H.; Bradner J. E.; Schapira M. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 2012, 3, 1288. [DOI] [PubMed] [Google Scholar]
- Williams D. E.; Dalisay D. S.; Li F.; Amphlett J.; Maneerat W.; Chavez M. A. G.; Wang Y. A.; Matainaho T.; Yu W.; Brown P. J.; Arrowsmith C. H.; Vedadi M.; Andersen R. J. Nahuoic acid A produced by a streptomyces sp. isolated from a marine sediment is a selective SAM-competitive inhibitor of the histone methyltransferase SETD8. Org. Lett. 2013, 15, 414–417. [DOI] [PubMed] [Google Scholar]
- Anglin J. L.; Deng L.; Yao Y.; Cai G.; Liu Z.; Jiang H.; Cheng G.; Chen P.; Dong S.; Song Y. Synthesis and structure–activity relationship investigation of adenosine-containing inhibitors of histone methyltransferase DOT1L. J. Med. Chem. 2012, 55, 8066–8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konze K. D.; Ma A.; Li F.; Barsyte-Lovejoy D.; Parton T.; MacNevin C. J.; Liu F.; Gao C.; Huang X. P.; Kuznetsova E.; Rougie M.; Jiang A.; Pattenden S. G.; Norris J. L.; James L. I.; Roth B. L.; Brown P. J.; Frye S. V.; Arrowsmith C. H.; Hahn K. M.; Wang G. G.; Vedadi M.; Jin J. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013, 8, 1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson S. K.; Warholic N. M.; Wigle T. J.; Klaus C. R.; Allain C. J.; Raimondi A.; Porter Scott M.; Chesworth R.; Moyer M. P.; Copeland R. A.; Richon V. M.; Pollock R. M.; Kuntz K. W.; Keilhack H. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 7922–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beguelin W.; Popovic R.; Teater M.; Jiang Y.; Bunting K. L.; Rosen M.; Shen H.; Yang S. N.; Wang L.; Ezponda T.; Martinez-Garcia E.; Zhang H.; Zheng Y.; Verma S. K.; McCabe M. T.; Ott H. M.; Van Aller G. S.; Kruger R. G.; Liu Y.; McHugh C. F.; Scott D. W.; Chung Y. R.; Kelleher N.; Shaknovich R.; Creasy C. L.; Gascoyne R. D.; Wong K. K.; Cerchietti L.; Levine R. L.; Abdel-Wahab O.; Licht J. D.; Elemento O.; Melnick A. M. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013, 23, 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Barsyte-Lovejoy D.; Li F.; Xiong Y.; Korboukh V.; Huang X. P.; Allali-Hassani A.; Janzen W. P.; Roth B. L.; Frye S. V.; Arrowsmith C. H.; Brown P. J.; Vedadi M.; Jin J. Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J. Med. Chem. 2013, 56, 8931–8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweis R. F.; Pliushchev M.; Brown P. J.; Guo J.; Li F. L.; Maag D.; Petros A. M.; Soni N. B.; Tse C.; Vedadi M.; Michaelides M. R.; Chiang G. G.; Pappano W. N. Discovery and development of potent and selective inhibitors of histone methyltransferase G9a. ACS Med. Chem. Lett. 2014, 5, 205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garapaty-Rao S.; Nasveschuk C.; Gagnon A.; Chan E. Y.; Sandy P.; Busby J.; Balasubramanian S.; Campbell R.; Zhao F.; Bergeron L.; Audia J. E.; Albrecht B. K.; Harmange J. C.; Cummings R.; Trojer P. Identification of EZH2 and EZH1 small molecule inhibitors with selective impact on diffuse large B cell lymphoma cell growth. Chem. Biol. 2013, 20, 1329–1339. [DOI] [PubMed] [Google Scholar]
- Daigle S. R.; Olhava E. J.; Therkelsen C. A.; Basavapathruni A.; Jin L.; Boriack-Sjodin P. A.; Allain C. J.; Klaus C. R.; Raimondi A.; Scott M. P.; Waters N. J.; Chesworth R.; Moyer M. P.; Copeland R. A.; Richon V. M.; Pollock R. M. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasveschuk C. G.; Gagnon A.; Garapaty-Rao S.; Balasubramanian S.; Campbell R.; Lee C.; Zhao F.; Bergeron L.; Cummings R.; Trojer P.; Audia J. E.; Albrecht B. K.; Harmange J.-C. P. Discovery and optimization of tetramethylpiperidinyl benzamides as inhibitors of EZH2. ACS Med. Chem. Lett. 2014, 5, 378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Korboukh I.; Jin J.; Huang J. Targeting protein lysine methylation and demethylation in cancers. Acta Biochim. Biophys. Sin. 2012, 44, 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siarheyeva A.; Senisterra G.; Allali-Hassani A.; Dong A.; Dobrovetsky E.; Wasney G. A.; Chau I.; Marcellus R.; Hajian T.; Liu F.; Korboukh I.; Smil D.; Bolshan Y.; Min J.; Wu H.; Zeng H.; Loppnau P.; Poda G.; Griffin C.; Aman A.; Brown P. J.; Jin J.; Al-awar R.; Arrowsmith C. H.; Schapira M.; Vedadi M. An allosteric inhibitor of protein arginine methyltransferase 3. Structure 2012, 20, 1425–1435. [DOI] [PubMed] [Google Scholar]
- Liu F.; Li F.; Ma A.; Dobrovetsky E.; Dong A.; Gao C.; Korboukh I.; Liu J.; Smil D.; Brown P. J.; Frye S. V.; Arrowsmith C. H.; Schapira M.; Vedadi M.; Jin J. Exploiting an allosteric binding site of PRMT3 yields potent and selective inhibitors. J. Med. Chem. 2013, 56, 2110–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost J. M.; Korboukh I.; Liu F.; Gao C.; Jin J. Targets in epigenetics: inhibiting the methyl writers of the histone code. Curr. Chem. Genomics 2011, 5, 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnertz B.; Pabst C.; Su L.; Miller M.; Liu F.; Yi L.; Zhang R.; Krosl J.; Yung E.; Kirschner J.; Rosten P.; Underhill T. M.; Jin J.; Hebert J.; Sauvageau G.; Humphries R. K.; Rossi F. M. The methyltransferase G9a regulates HoxA9-dependent transcription in AML. Genes Dev. 2014, 28, 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Skutt-Kakaria K.; Davison J.; Ou Y. L.; Choi E.; Malik P.; Loeb K.; Wood B.; Georges G.; Torok-Storb B.; Paddison P. J. G9a/GLP-dependent histone H3K9me2 patterning during human hematopoietic stem cell lineage commitment. Genes Dev. 2012, 26, 2499–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antignano F.; Burrows K.; Hughes M. R.; Han J. M.; Kron K. J.; Penrod N. M.; Oudhoff M. J.; Wang S. K.; Min P. H.; Gold M. J.; Chenery A. L.; Braam M. J.; Fung T. C.; Rossi F. M.; McNagny K. M.; Arrowsmith C. H.; Lupien M.; Levings M. K.; Zaph C. Methyltransferase G9A regulates T cell differentiation during murine intestinal inflammation. J. Clin. Invest. 2014, 124, 1945–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konze K. D.; Pattenden S. G.; Liu F.; Barsyte-Lovejoy D.; Li F.; Simon J. M.; Davis I. J.; Vedadi M.; Jin J. A chemical tool for in vitro and in vivo precipitation of lysine methyltransferase g9a. ChemMedChem 2014, 9, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka K.; Rice J. C.; Sarma K.; Erdjument-Bromage H.; Werner J.; Wang Y.; Chuikov S.; Valenzuela P.; Tempst P.; Steward R.; Lis J. T.; Allis C. D.; Reinberg D. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol. Cell 2002, 9, 1201–1213. [DOI] [PubMed] [Google Scholar]
- Fang J.; Feng Q.; Ketel C. S.; Wang H.; Cao R.; Xia L.; Erdjument-Bromage H.; Tempst P.; Simon J. A.; Zhang Y. Purification and functional characterization of SET8, a nucleosomal histone H4-lysine 20-specific methyltransferase. Curr. Biol. 2002, 12, 1086–1099. [DOI] [PubMed] [Google Scholar]
- Beck D. B.; Oda H.; Shen S. S.; Reinberg D. PR-Set7 and H4K20me1: at the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev. 2012, 26, 325–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F.; Sun L.; Li Q.; Han X.; Lei L.; Zhang H.; Shang Y. SET8 promotes epithelial–mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J. 2012, 31, 110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X.; Kachirskaia I.; Yamaguchi H.; West L. E.; Wen H.; Wang E. W.; Dutta S.; Appella E.; Gozani O. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell 2007, 27, 636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takawa M.; Cho H. S.; Hayami S.; Toyokawa G.; Kogure M.; Yamane Y.; Iwai Y.; Maejima K.; Ueda K.; Masuda A.; Dohmae N.; Field H. I.; Tsunoda T.; Kobayashi T.; Akasu T.; Sugiyama M.; Ohnuma S.; Atomi Y.; Ponder B. A.; Nakamura Y.; Hamamoto R. Histone lysine methyltransferase SETD8 promotes carcinogenesis by deregulating PCNA expression. Cancer Res. 2012, 72, 3217–3227. [DOI] [PubMed] [Google Scholar]
- Min J.; Allali-Hassani A.; Nady N.; Qi C.; Ouyang H.; Liu Y.; MacKenzie F.; Vedadi M.; Arrowsmith C. H. L3MBTL1 recognition of mono- and dimethylated histones. Nat. Struct. Mol. Biol. 2007, 14, 1229–1230. [DOI] [PubMed] [Google Scholar]
- Wigle T. J.; Provencher L. M.; Norris J. L.; Jin J.; Brown P. J.; Frye S. V.; Janzen W. P. Accessing protein methyltransferase and demethylase enzymology using microfluidic capillary electrophoresis. Chem. Biol. 2010, 17, 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. Y.; Seo H. N.; Lee M. J.; Park S. J.; Jeon J. Y.; Kang J. H.; Pae A. N.; Rhim H.; Lee J. Y. Synthesis and biological evaluation of novel T-type calcium channel blockers. Bioorg. Med. Chem. Lett. 2007, 17, 471–475. [DOI] [PubMed] [Google Scholar]
- Couture J. F.; Collazo E.; Brunzelle J. S.; Trievel R. C. Structural and functional analysis of SET8, a histone H4 Lys-20 methyltransferase. Genes Dev. 2005, 19, 1455–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senisterra G.; Wu H.; Allali-Hassani A.; Wasney G. A.; Barsyte-Lovejoy D.; Dombrovski L.; Dong A.; Nguyen K. T.; Smil D.; Bolshan Y.; Hajian T.; He H.; Seitova A.; Chau I.; Li F.; Poda G.; Couture J. F.; Brown P. J.; Al-Awar R.; Schapira M.; Arrowsmith C. H.; Vedadi M. Small-molecule inhibition of MLL activity by disruption of its interaction with WDR5. Biochem. J. 2013, 449, 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.