

Abstract
Long interspersed element-1 (L1) is an autonomous retroelement that is active in the human genome. The proposed mechanism of insertion for L1 suggests that cleavage of both strands of genomic DNA is required. We demonstrate that L1 expression leads to a high level of double-strand break (DSB) formation in DNA using immunolocalization of γ-H2AX foci and the COMET assay. Similar to its role in mediating DSB repair in response to radiation, ATM is required for L1-induced γ-H2AX foci and for L1 retrotransposition. This is the first characterization of a DNA repair response from expression of a non-long terminal repeat (non-LTR) retrotransposon in mammalian cells as well as the first demonstration that a host DNA repair gene is required for successful integration. Notably, the number of L1-induced DSBs is greater than the predicted numbers of successful insertions, suggesting a significant degree of inefficiency during the integration process. This result suggests that the endonuclease activity of endogenously expressed L1 elements could contribute to DSB formation in germ-line and somatic tissues.
Keywords: LINE, retrotransposition, genetic instability, L1
Introduction
Long interspersed element-1 (L1) is an active, autonomous family of retroelements that comprises 17% of the human genome.1 Another 11% of the human genome is composed of Alu and SVA elements that require L1 for mobilization in trans.2–4 L1 utilizes an APE-like endonuclease to insert into the target DNA.5 On the basis of in vitro analysis of the R2 element from the silkworm Bombyx mori (R2Bm), the current model for non-long terminal repeat (non-LTR) element insertion is target-primed reverse transcription (TPRT) (Figure 1).6,7 A variation of this model that incorporates the frequent creation of target-site duplications (TSDs) also likely explains the majority of human genomic and de novo L1 insertions.8–11 In these models of TPRT, cleavage of both strands of genomic DNA is required, which implies that an intermediate equivalent to a DSB is present during the integration process.
Figure 1.

Schematic of the L1 TPRT reaction. The top drawing shows a genomic site with a typical L1 endonuclease cleavage site (5'TTTTAA) that is exposed in the nucleus to the L1 RNP, where the ORF2 cleaves the DNA at the A+T-rich consensus sequence. In the second drawing, the T-rich region primes reverse transcription on the L1 mRNA poly(A) tail. The third drawing highlights the reverse transcriptase activity (formation of the blue cDNA) of the ORF2 and the need for a nick to occur on the second strand, made by an unknown source. The segment between the two nicks is highlighted in gray to illustrate the eventual formation of flanking direct repeats by the duplication of these segments. The fourth panel illustrates the possibility that the second-strand synthesis is primed by microhomology-mediated priming, which results in synthesis of a second copy of the gray segment (dotted arrow). Replication from the gray arrow completes synthesis across the 2nd strand of the cDNA creating a new L1 insert and completing synthesis across the other side of the direct repeat.
Recent studies have begun to recognize the interplay between mobile elements and DNA repair.12–14 As with retroviruses and transposons, which have well-characterized insertion mechanisms, host DNA repair enzymes are likely to both contribute to, and suppress, L1 retrotransposition by recognizing and processing the retrotransposon-initiated nicks or breaks. In contrast to transposons and retroviruses, with well-established in vitro models of the insertion process, molecular dissection of TPRT intermediates has been demonstrated only partially.6,7,15 Several other non-LTR retrotransposon systems have demonstrated a relationship between these elements, genetic instability, and host repair enzymes. Group II introns in yeast mitochondria induce DSBs in DNA16 and display hallmarks of DSB repair processing after successful integration,17,18 although the genetic requirements have not been demonstrated in that system. The L1.LtrB intron from the Lactococcus lactis bacterium has been shown in molecular assays to utilize TPRT as a subpathway for insertion,19,20 and it has been demonstrated recently to require host (Escherichia coli) repair proteins for later steps in integration.21 Other than L1 and R2Bm, the only other model non-LTR retrotransposon system in higher eukaryotes is the Drosophila I-factor, the controlling element in I-R hybrid dysgenesis.22–24 Chromosomal instability and elevated recombination (both hallmarks of DSB processing) have both been demonstrated in I-R crosses.25–27
L1 has dramatically impacted the human and mouse genomes through insertional mutagenesis,1 implying that L1 elements are expressed in germ-line tissues. Insertional mutagenesis is ongoing in the human population as evidenced by de novo and very recent disease-causing insertions.28,29 In the mouse male germ-line (prospermatogonia), L1 promoters are repressed by the action of the methyltransferase-related Dnmt3L protein.30,31 While the endogenous amount of L1 expression does not cause infertility in normal individuals, spermatocytes from dnmt3L knockout mice show greatly increased L1 expression. Correlated with this increased expression are aberrant chromosomal structures in meiosis that have been suggested to be due to elevated activity of endogenous retrotransposons and subsequent genetic instability. Though expression of L1 is very low in most normal somatic cells, expression of L1 is elevated in many cancer cells.32–34 Cancer cells generally show chromosome rearrangements indicative of DSB repair, and DSB repair responses in pre-oncogenic tissues adjacent to cancer cells suggests these lesions contribute to initial tumorigenesis.35,36 Given the correlations between DSB-related genetic instability, L1 expression, and the predictions of the TPRT model, we sought to characterize the ability of L1 to induce DSBs using immunolocalization of γ-H2AX foci and single-cell gel electrophoresis (COMET) analysis. To further establish the relevance of a DSB intermediate during L1 integration, we tested whether a DSB repair protein, ATM, is important for L1 retrotransposition.
Results
L1 expression induces γ-H2AX foci and fragmented DNA
DSB intermediates, predicted by the TPRT model for L1 insertions (Figure 1), were visualized indirectly by immunofluorescence after expression of a transiently transfected human L1 element, L1.3, which is known to be capable of retrotransposition in HeLa cells. Histone H2AX is phosphorylated in response to ionizing radiation (γ-H2AX) and is detectable as foci in response to DSBs.37,38 Several quantitative experiments have shown a 1:1 proportion of γ-H2AX foci to the number of IR-induced DSBs,39,40 and γ-H2AX is associated with DSB ends.41 γ-H2AX foci were present in L1.3-transfected HeLa cells in high numbers and high focus intensity 24 h after transfection (Figure 2(a)). The frequency of focus-positive cells correlated with transfection efficiency as measured using parallel transfection of EGFP expression constructs, approximately 60–70% (data not shown). The number of foci per focus-positive cell was generally within a range of 20–200 (mean of 68), with the majority demonstrating approximately the same number of foci as cells irradiated with 6 Gy of ionizing radiation (mean of 86). Although NeoR colonies from tagged L1.3 have been reported to occur at a rate as high as 1/20 cells in tissue culture assays,42 the L1 insertion frequency varies with different L1 vectors and transfection protocols. With our procedures, about 1/1000 cells produce L1-insertion colonies. However, an estimate of insertions based on integrated gene expression is bound to underestimate insertion frequencies of corresponding untagged vectors. We estimate that as many as nine out of ten integrants may be truncated sufficiently to not express a functional neomycin-resistance gene. Chromatin effects on the insertion location (possibly two out of three may be silent) will also underestimate the number of retrotransposition events. Untagged L1.3 produces tenfold more full-length mRNA than the corresponding tagged vector.43 Taking a relatively high estimate of L1 neoR colony formation as 1/100 cells under our conditions, there are likely to be as many as three inserts per cell from an untagged L1.3 (1/100×10×3×10). Therefore, it is conservative to estimate that tenfold more L1-induced DSBs are produced than insertions, although a 100-fold difference is within reason (our γ-H2AX counts slightly underestimate the expected by about twoto threefold39,40). However, it is also likely that these measurements on fixed cells underestimate the total number of L1-induced breaks, because an optimal assessment of DSB formation from a transfected and expressed element would integrate the frequency over time.
Figure 2.

L1.3 expression creates DNA DSBs. (a) L1 expression induces the formation of γ-H2AX foci. HeLa cells mock-transfected (control), transfected with the empty expression vector pCEP4 (vector), L1.3 (L1), or L1.3 EN– (L1 EN–) over-expressed from pCEP4, were fixed 24 h later, and stained with anti-γ-H2AX antibodies. Untransfected HeLa cells were also treated with 5 Gy of ionizing radiation (IR) and fixed 15 min later as a positive control for focus formation. (b) A time-course on the repair of γ-H2AX from L1 transfected cells in (a) is shown. γ-H2AX immunolocalization in (c) mouse NIH 3T3 cells and in (d) MCF7 cells 24 h after transfection. In addition to expression constructs from (a), an RT mutant, as well as an ORF2 over-expression vector were included in different panels.
Mutations in the endonuclease domain of the L1 ORF2 resulted in essentially complete loss of the γ-H2AX foci in HeLa cells (Figure 2(a)), consistent with the inability of the endonuclease mutant of L1 to support retrotransposition.44 This result argues that the endonuclease of L1 is required for this DSB formation, although it may not make the breaks on both strands (see Discussion). The L1-induced foci are largely repaired by 48 h post transfection, as evidenced by the decreasing percentage of cells with γ-H2AX foci and the decreasing number of γ-H2AX foci per cell (Figure 2(b)). The uniform disappearance of L1-induced breaks by 48 h suggests that cells may alter how they respond to L1 endonuclease activity over time or that cell-cycle synchrony affects L1 activity or localization. We do find that there are still significant levels of L1 RNA expressed 48 h after transfection (Victoria Perepelitsa-Belancio and P.L.D., unpublished results). Thus, cell synchrony due to transfection or DNA damage responses may be affecting L1 RNP localization and a second round of DSBs is not observed. Although, another possibility is that DNA repair proteins have been upregulated and they can now effectively limit new DSB formation.
We also characterized γ-H2AX foci after L1.3 expression in mouse 3T3 cells. At 24 h post L1.3 transfection, 3T3 cells have high numbers of distinct foci as observed for HeLa cells (Figure 2(c)). A few foci are observable in the L1.3 EN–transfected 3T3 cells relative to the empty vector. The ORF2-encoded reverse transcriptase (RT) is required for L1 retrotransposition.44,45 L1.3 RT– produced fewer foci than observed with wild-type L1.3, suggesting that high levels of focus formation may depend on the presence of the first L1 cDNA strand. This dependence could be due to a stabilization of break intermediates or that ORF2 requires cDNA synthesis to create the DSB. Detailed molecular analysis of the R2Bm reaction in vitro has shown that first and second strand cleavages occur separately, with an intermediate step requiring RNA, which supports the idea that DSB formation would require cDNA synthesis.46 The exact function of ORF1 in retrotransposition is unknown, but it has been proposed to act as a nucleic acid chaperone that could be involved directly in stabilizing integration intermediates.47 Mutations in ORF1 render L1 incompetent for retrotransposition.44,47 Over-expression of ORF2 alone resulted in the formation of only modest numbers of foci 24 h post transfection (Figure 2(c)). Thus, ORF2 expression alone is sufficient for generating DSBs, but it is probably facilitated by the presence of ORF1.
One possible scenario in which L1 could cause a large number of DSBs independent of insertion would be if apoptosis were induced leading to DNA fragmentation and foci formation.48 L1 insertions including a neomycin-resistance cassette have been suggested to invoke apoptosis in p53+ cells,49 and, therefore, one might consider that γ-H2AX foci could be created in response to the apoptosis-programmed cleavage of genomic DNA.48 The fact that γ-H2AX foci are dependent on the L1 endonuclease argues that the primary mechanism by which this would occur in our experiments is through a DNA damage-induced apoptosis and not ORF1 or ORF2 over-expression or trans activation per se. DNA damage-dependent apoptosis is dependent on p53.50,51 HeLa cells form L1 colonies robustly and do not have detectable levels of p53 protein,52 so an induction of foci due to L1-induced apoptosis is unlikely in that cell line. Further support for this comes from a fluorescence activated cell sorting (FACS) analysis of annexin V (Supplemental Figure 1) and TUNEL (data not shown) staining of HeLa cells following L1 transfection, which show no significant increase in apoptosis at either 24 or 48 h. In addition, we tested L1-induced foci formation in MCF7. MCF7 cells undergo a markedly delayed apoptotic response and do not readily form γ-H2AX foci in response to apoptosis.48 The observation that MCF7 cells show the same induction of γ-H2AX foci following wild-type L1 transfections suggests that the response is not likely due to apoptosis (Figure 2(d)).
To confirm the presence of fragmented DNA in cells after L1.3 expression, the neutral COMET assay was used.53 Transfection with L1.3 produced pronounced tail-moments at 24 h post transfection in the majority of cells (Figure 3). Similar to results with γ-H2AX foci, there were no tails at 48 h (data not shown). Thus, γ-H2AX foci likely represent true DSB formation directly induced by L1.
Figure 3.
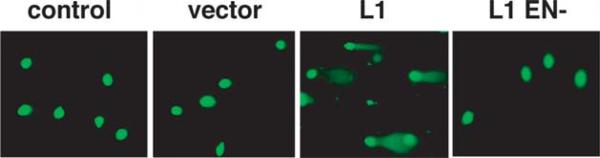
COMET assays on L1 transfected cells. HeLa cells were transfected either as a mock (control), or with vector, L1 or L1 EN– and then subjected to the neutral COMET assay for the periods of time shown.
L1 expression is toxic and induces G2/M cell-cycle arrest
The high levels of DSBs observed would be expected to induce significant levels of cell death. Previous studies have suggested that p53-positive cells were relatively incompetent for L1 retro-transposition, potentially due to toxicity.49 Other investigators have had difficulties with expression of ORF2 that was abrogated by endonuclease-defective mutants (EN–).54 To test potential toxicity from expression of L1.3, we co-transfected a neoR expression vector along with either one of the L1 expression constructs or with the empty backbone vector (pCEP4A) into HeLa cells. Cotransfection with L1.3 reduced the colony yield by 60% compared to vector (Figure 4(a)). L1 EN– showed no significant toxicity, while L1.3 RT– showed toxicity levels similar to that of the wild-type L1.3. Thus, the primary toxic lesion is dependent on the L1 endonuclease and not the synthesis of cDNA intermediates. This result suggests that L1 RT– makes sufficient DSBs to kill cells, which supports the idea that L1 cDNA is not required for the formation of L1-induced DSBs. Non-homologous end-joining (NHEJ) repairs flush DSB ends,55–57 which could occur rapidly in the absence of cDNA containing intermediates and thus a lower level of DSBs is observed at the characterized time-point. L1-induced DSBs appear to be less toxic than those from IR.58 L1 expression after transfection could result in asynchronous endonuclease activity, thus producing an effectively lower rate of DSB formation compared to high dose-rate IR. Alternatively, the enzyme-induced breaks from L1 are likely to be repaired more easily than the variable ends created by IR.
Figure 4.

L1.3 expression induces toxicity and cell cycle arrest. (a) HeLa cells were cotransfected with neoR expression vector and the indicated L1-expression construct or empty vector. Colony formation was assayed after two weeks under neomycin selection. The chart shows the average ±S.E. of four independent experiments. Values are relative to the empty vector control. (b) G2/M arrest induced by L1 expression. HeLa cells were transfected with the noted L1 expression construct or empty vector. At 24 h after transfection, the mitotic index of cells was analyzed using phosphorylated histone 3 staining, shown as % H3-P positive G2 cells in the whole FSC/SSC gated population. The chart shows the mean ± S.E. of three observations of cells from parallel transfections.
A secondary response to high levels of DSBs includes cell-cycle arrest. To determine whether L1-induced DSBs caused cell-cycle arrest, the mitotic index of transfected cells was determined.59 L1.3 expression reduced the number of cells entering mitosis, whereas neither empty vector nor EN– induced checkpoint responses in HeLa cells (Figure 4(b)). Typically, IR produces a much stronger cell-cycle arrest.59 However, in the case of L1, the arrest is probably limited because of the asynchrony of the DSBs inherent to transfections.
ATM is required for formation of γ-H2AX foci from L1 expression
The ATM protein is a central regulator of the cellular response to DSBs and contains PI3-like protein kinase activity.60,61 ATM kinase activity is required to activate cell-cycle arrest, but it also phosphorylates several proteins known to be involved in the repair of DSBs. Consistent with the important role of ATM to repair DSBs, ATM-deficient cells demonstrate increased sensitivity to ionizing radiation.58,62–64 IR-induced γ-H2AX foci are dependent on ATM,65 although some redundancy with the repair-associated protein kinases ATR66 and DNAPKos67 is evident also in some cells in response to DNA damage. To determine whether L1-induced g-H2AX foci are also dependent on ATM, we over-expressed L1.3 in isogenic ATMC+/p=n- cell lines, YZ-5 (ATMC) and EBS-7 (ATM–),68 and analyzed focus formation. L1.3 expression induced γ-H2AX foci in ATMC but not in ATMK cells with exposure times normalized to observation of IR-induced foci in ATM+ (Figure 5). L1 EN– failed to induce a punctate staining pattern in the ATM+ cells consistent with results in the other cell lines.
Figure 5.

L1-induced γ-H2AX foci require ATM. EBS7 (AT–) and YZ-5 (cATM) cells were also analyzed as in Figure 1(a): 10 Gy of IR was used as a positive control.
To characterize the potential requirement for ATM for productive L1 retrotransposition, L1 retrotransposition was analyzed in YZ-5 (ATM+) and EBS-7 (ATM–). We utilized a highly efficient L1-retro-transposition vector, synL1_optORF1_neo (the sequence is available upon request) we developed recently. As shown in Table 1, synL1_optORF1_neo produced colonies efficiently in ATM+ cells. No colonies were detected in ATM– cells. Control transfections withthe neoR expression vector showed similar numbers of colonies between these two matched cell lines, although the EBS-7 neoR colonies were somewhat less distinct (Supplementary Figure 2). Given that the reduction inneoR integration colonies in the absence of ATM was reduced only twofold compared to ATM+, the absolute defect in L1 retrotransposition is not likely to be due to a difference in colony formation or transfection competence between the two cell lines. Notably, a modified toxicity assay shows no L1-induced toxicity in the ATM– cell line (data not shown), suggesting that ATM is specifically required for retrotransposition and not the survival of cells after L1 expression. Transient inhibition of ATM with kinase inhibitors or a dominant negative plasmid in HeLa cells (data not shown) also demonstrate the involvement of ATM in promoting retrotransposition.
Table 1.
L1 retrotransposition in ATM+/– cells
| Cell line | ATM | Test | No. L1 coloniesa (±SD) | No. neoR plasmid coloniesb (±SD) |
|---|---|---|---|---|
| YZ-5 | + | 1 | 62 (6) | 207 (38) |
| EBS7 | – | 1 | 0 | 133 (10) |
| YZ-5 | + | 2 | 24 (10) | 115 (12) |
| EBS7 | – | 2 | 0 | 91 (4.7) |
Optimized human L1 with selection cassette synwt-opt ORF1-neo.
Neomycin-resistance expression plasmid pIRES-EGFP.
Discussion
Mechanistic implications of L1 DSBs
L1 elements are known to be responsible for a wide range of diseases through insertional muta-genesis.1,28 The formation of γ-H2AX foci (Figures 2 and 5) and COMET assays (Figure 3) demonstrate that expression of the human L1 mobile element creates DSBs as predicted by the TPRT model. The requirement for the endonuclease activity of ORF2 to produce these effects suggests a direct role for ORF2. However, whether the second nick (Figure 1) takes advantage of a separate enzyme for creating DSBs, is created by ORF2, or even whether the L1 ribonucleoprotein complex “homes in” to already existing nicks, remains to be determined. These events could even take advantage of lagging strand nicks created during S phase, as observed for the RmInt1 group II intron.69
The reason for the excess of DSBs in the L1 retrotransposition process is not understood at this time. The DSB intermediates in the retrotransposition process may be repaired efficiently most of the time and therefore do not result in an insertion event (see Figure 1). Alternatively, it may be that the ORF2 expressed from L1 elements can create DNA nicks independent of the retrotransposition process. The presence of moderate levels of breaks when ORF2 is overexpressed (Figure 2) suggests that the presence of ORF2 alone is insufficient for efficient DSB activity. However, these studies are complicated by our poor understanding regarding cytoplasmic/nuclear transport of L1 RNA and proteins. ORF2 made by the expression vector may not be transported back to the nucleus in the way that might occur if it is expressed as part of the full L1 RNA if ORF1 is required for efficient localization of the L1 ribonucleoprotein (RNP) complex with genomic DNA prior to cleavage.70 However, at this point similar arguments can be made for most of the point mutants made and expressed from the authentic L1 mRNA. Another relevant concern would be that over-expression of L1 leads to a situation in which the DSB:insertion ratio is skewed, perhaps by titration of an important repair factor or ORF2 activity unrelated to integration. These possibilities are difficult to reconcile until a quantitative assay of L1 integration intermediates can be developed without the use of selectable marker cassettes.
Host DNA repair proteins may act to either counteract, or promote, L1 integration (Figure 1). Activation of H2AX and the disappearance of DSBs in a time-course (Figures 2 and 3), suggests that L1 induces a DNA repair response. ATM is required both for L1-induced γ-H2AX foci (Figure 5) and L1 retrotransposition (Table 1). This apparent interaction between L1 and the DSB-repair activity is consistent with previous evidence that enzymes involved in DSB repair in CHO cells influence retrotransposition with endonuclease-deficient L1 elements.71
This is the first evidence that the non-LTR class of retrotransposons depend on a cellular DNA repair enzyme for standard TPRT mobilization in eukaryotic cells. Recently, the action of host DNA repair enzymes has been demonstrated in E. coli for group II intron retrohoming.21 This last report and our results demonstrate that non-LTR retrotransposons encode protein machinery that initiates insertion into DNA but largely requires host factors for many of the subsequent steps. A requirement for host DNA repair functions for mobilization has been demonstrated recently for other classes of mobile DNA. The DNA transposon Sleeping Beauty has been shown to require NHEJ and ATM for high levels of transposition.13 The HIV-1 retrovirus has been shown to depend on ATM (and potentially ATR and DNAPK) for integration.72–74 The common requirement of these different insertional elements on host DSB DNA repair proteins is likely due to the required modification of double-stranded DNA for insertion. Because non-LTR insertion proceeds through a different integration mechanism in which cDNA synthesis precedes endonuclease activity, this dependence represents a novel understanding of the L1 life cycle.
We consider two primary distinct possibilities for the dependence of L1 specifically on ATM. One possibility is that ATM is required for a processing step involving DSB-like intermediates. ATM is important for homologous recombination repair of DSBs,75,76 and for a subset of NHEJ repair.77 Computational analyses of de novo L1 integrations and genomic L1 integrations have shown that most integrations proceed through a microhomology process for priming L1 DNA synthesis.9 Therefore, the requirement for ATM in L1 integration could be at the step of the microhomology-promoted synthesis of L1 DNA. It is unclear whether ATM is required specifically for this microhomology-driven pathway but breaks due to Sleeping Beauty excision in ATM– cells that show a decrease in micro-homology-repaired breaks.13 The genetic requirement for a DSB repair protein by L1 underscores the likelihood that DSBs represent a normal L1 integration intermediate. The other possibility for the ATM dependence is that ATM phosphorylates either the ORF1 or ORF2 proteins encoded by L1, and that this phosphorylation is required for either endonucleolytic cleavage or some other pre or post-integration step of L1 retrotransposition.
Although most full-length L1 elements in the human genome are defective, our data suggest that more may be capable of causing genomic damage than the limited number capable of active retro-transposition.78 Many elements with ORF1 or RT-inactivating mutations may still be capable of expressing a functional endonuclease that produces genomic DSBs. This could be particularly true of older L1 subfamilies, which are no longer retrotranspositionally active. Nonetheless, full levels of DSB activity per element are likely to still require a largely intact ORF1 for both RNP formation/ localization and interactions with the genome.
Cellular implications of L1 DSBs
An important implication of L1-induced DSBs is that they could be a major contributor to chromosomal damage, including translocations, deletions and duplications. One expectation for L1-induced translocations would be breakpoint sequences consistent with L1 endonuclease cleavage sites (AT-rich). Another common feature of chromosomal deletions and translocation breakpoints is proximity to Alu or L1 elements.79 Alu-Alu recombination is a prevalent form of chromosome rearrangement in the germ-line and in cancer cells.80 The common occurrence of repeat element recombination has generally been interpreted to be a result of ectopic recombination between repeated sequences due to the high prevalence and homology of these sequences. Alu elements are often flanked by consensus L1 endonuclease cleavage sites caused by retention of the original site of insertion and formation of A-rich 3' tails (Figure 1), and it has been proposed that re-use of these sites may facilitate instability around Alu elements.81,82 Thus, the L1 endonuclease may be responsible for insertion of the dispersed sites of homology, as well as creating the DSBs adjacent to them that facilitate the instability. Our data suggest that L1 endonuclease function may promote many “retrotransposition silent” chromosomal rearrangements. In light of the recent characterization of L1 expression in some normal somatic tissues, vascular endothelium,83 steroidigenic tissues,84,85 and neuronal precursors,86 L1 may represent an important endogenous source of DSB-related genetic instability. The ATM requirement for L1 integration could have potential consequences for human health in ATM-compromised individuals. If L1-induced breaks are processed by alternative repair pathways, this could account for the genetic instability phenotype of AT cells showing translocation or deletion products. Although increased genetic instability in ATM-deficient lymphoid cells is predominantly due to misrepair of RAG1/2-induced lesions,87 genetic instability effects might be enhanced in non-lymphoid tissues that have high levels of L1 expression.
The role of genetic instability in diverse pheno-types like aging, fertility, and cancer has highlighted the importance of understanding how cells respond to endogenous sources of DNA damage. This work demonstrates that the L1 integration process produces DSBs and requires a DSB-repair protein. Thus, further understanding the extent of DSB damage from expression of these endogenous elements and how intermediates are processed is potentially important for human health.
Materials and Methods
Tissue culture
HeLa cells (ATCC Manassas, VA) were cultured in EMEM plus non-essential amino acids and sodium pyruvate supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco/Invitrogen Carlsbad, CA). YZ-5 and EBS-768 cells were maintained in the same medium but supplemented with hygromycinB at 120 μg/ml to maintain the cATM expression cassette or the corresponding empty vector. The 3T3 and MCF7 (ATCC) cells were cultured in DMEM plus non-essential amino acids and sodium pyruvate supplemented with 10% FBS.
Plasmids
The L1.3 expression construct without a selectable cassette was JM101/L1.3 CEP no tag. The L1.3 RT– construct is the same but with a D702A mutation in ORF2. Both were gifts (John moran, University of Michigan Medical School, Ann Arbor, MI). To construct L1 EN– no tag, pJM102/L1.3/H230A (a gift from John Moran)71 was digested with SapI and Bst11071 to isolate the appropriate fragment and cloned into the same digest of JM101/L1.3 CEP no tag.
The NeoR expression plasmid used for toxicity is pIRES2-EGFP (BD Biosciences Clontech, Palo Alto, CA).
The ORF2 expression construct includes a codon-optimized version of the L1-RP ORF2 with a canonical Kozak consensus sequence. This sequence was constructed synthetically by Blue Heron Biotechnology, Inc (Bothell, WA) and subcloned into the pBudCE4.1 (Invitrogen) under control of the CMV promoter.
SynL1_optORF1_neo is described elsewhere.88
Transfection
For COMET and FACS analyses, 8.0×105–1.0×106 HeLa cells were seeded in 60 mm dishes. Cells were transfected the next day with 4 mg of L1 expression construct and 3 μl of Lipofectamine/dish for 4 h. Transfection was stopped by aspiration and addition of growth medium. For immunofluorescence, the same conditions were used but cells were seeded in 4-chamber Falcon culture slides. For γ-H2AX immunostaining in ATMC/– cells, 200,000 cells were seeded on 22 mm× 22 mm glass coverslips in six-well plates and transfected the next day. Samples (2 μg) of pCEP4A and L1 expression constructs were transfected using 12 μl of Lipofectamine and 2 μl of PLUS per six-well plate. For colony plating assays, HeLa cells were seeded in six-well plates at 100,000 cells/well and transfected the following day. Samples (2 μg) of pCEP4A and L1 expression constructs were cotransfected with 0.15 mg of pIRES2-EGFP using 12 μl of Lipofectamine and 2 μl of PLUS per six-well plate. Selection medium (400 μg/ml Geneticin, Invitrogen) was added the following day and refreshed every three or four days for two weeks. For L1 and neoR assays with YZ-5 and EBS-7, 500,000 cells were seeded and transfected with 6 μl of Lipofectamine, 1 μg of synL1_optORF1_neo or pIRES2-EGFP per T75. G418 medium was added two days post-transfection and maintained for 14 days. For colony counting, cells were fixed and stained for 30 min with crystal violet (0.2% (w/ v) crystal violet in 5% (v/v) acetic acid and 2.5% (v/v) isopropanol). Colony counts for experiments in T75s utilized the ColCount from Oxford Optronix.
Single-cell gel electrophoresis (COMET assay)
The COMET assay is based on the method outlined by Singh et al.53 75 μl of 1% (w/v) low melting point agarose diluted in 2H2O was heated to 37 °8C, ~104 control or transfected cells were added and this material was applied to the agarose-coated slide with a third layer of agarose overlay. The slides were placed in lysis solution (10 mM Tris (pH 10.0), 2.5 M NaCl, 100 mM EDTA, 1% (v/v) Triton X-100, 10% (v/v) Me2SO) at 4 °C for 1 h. The slides were then transferred to an electrophoresis apparatus containing a neutral solution (300 mM NaOH, 1 mM EDTA (pH 8 after addition of HCl)). The slides remained in this solution for 1 h to promote DNA unwinding and were subsequently subjected to electric current (300 mA) for 1 h. The slides were removed, washed three times for 5 min in neutralizing buffer (0.4 M Tris–HCl, pH 7.5) at room temperature, and stained in a 1:10,000 dilution of Sybr Gold (Molecular Probes). Cells were photographed using a Leitz microscope equipped with epi-fluorescence optics and a SPOT CCD camera.
Immunofluorescence microscopy
HeLa, MCF7, and 3T3 cells were transfected with empty vector, L1, or L1 EN–. Following transfection, cells were harvested and then fixed in 2% (v/v) paraformaldehyde diluted in PBS for 15 min, washed three times in PBS, permeabilized for 5 min on ice in PBS, 0.2% Triton X-100, and blocked in PBS, 1% (w/v) BSA for 30 min at room temperature. The slides were incubated with 1/100 diluted anti-γ-H2AX (Trevigen, Gaithersburg, MD) for 1 h at room temperature, and subsequently incubated with FITC-conjugated goat anti-rabbit secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h at room temperature. Cells were washed in PBS and mounted using Vectashield mounting medium containing 4',6-diamidino-2-phenylindole (DAPI) to counterstain nuclei (Vector Laboratories, Burlingame, CA). For ATMC/K cells, fixation and staining were performed as above with the exception that primary antibodies were incubated overnight at 4 °C. Anti-γ-H2AX was used at 1/4000 dilution. Anti-rabbit secondary conjugated to Alexa 488 (Molecular Probes, Eugene, OR) was used at 1/4000 dilution for 2 h at room temperature. Images were captured with a Leitz epifluorescence microscope equipped with a SPOT CCD camera.
Supplementary Material
Acknowledgements
We thank Melanie Palmisano for technical help with DNA preparation and tissue culture. We thank Deininger laboratory members, Jeremy Stark, Erik Flemington, and Charles Hemenway for comments on the manuscript. S.L.G. was supported by postdoctoral fellowship PF-01-077-01-LIB from the American Cancer Society and from the Brown Foundation through the Tulane Cancer Center. The P.L.D. laboratory is supported by grants from the USPHS (grant R01GM45668), the National Science Foundation (grant EPS-0346411) and the State of Louisiana Board of Regents Support Fund. The B.X. laboratory is supported by grants P20RR020152-01 and R03ES013301-01 from the NIH and by a grant from the Department of Defense (DAM 17-03-1-0709). S.L.G. specifically thanks the American Society of Human Genetics for post-Katrina funding as well as the Berry College Department of Communications (Rome, GA) and the Doug Bishop Laboratory (University of Chicago) for computer support during the writing of the manuscript.
Abbreviations used
- LINE
long interspersed element
- DSB
double-strand break
- L1
LINE-1 retroelement
- ORF
open reading frame
- NeoR
neomycin resistance
- FOV
fields of view
- TPRT
target-primed reverse transcription
- EN
endonuclease
- RT
reverse transcriptase
- RNP
ribonucleic/protein complex
- TSD
target site duplication
- IR
ionizing radiation
Footnotes
Supplementary Data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j. jmb.2006.01.089
References
- 1.Ostertag EM, Kazazian HH., Jr Biology of mammalian L1 retrotransposons. Annu. Rev. Genet. 2001;35:501–538. doi: 10.1146/annurev.genet.35.102401.091032. [DOI] [PubMed] [Google Scholar]
- 2.Dewannieux M, Esnault C, Heidmann T. LINE-mediated retrotransposition of marked Alu sequences. Nature Genet. 2003;35:41–48. doi: 10.1038/ng1223. [DOI] [PubMed] [Google Scholar]
- 3.Ostertag EM, Goodier JL, Zhang Y, Kazazian HH., Jr SVA elements are nonautonomous retrotransposons that cause disease in humans. Am. J. Hum. Genet. 2003;73:1444–1451. doi: 10.1086/380207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.International Human Genome Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 5.Feng Q, Moran JV, Kazazian HH, Jr, Boeke JD. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. 1996;87:905–916. doi: 10.1016/s0092-8674(00)81997-2. [DOI] [PubMed] [Google Scholar]
- 6.Luan DD, Korman MH, Jakubczak JL, Eickbush TH. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell. 1993;72:595–605. doi: 10.1016/0092-8674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- 7.Luan DD, Eickbush TH. RNA template requirements for target DNA-primed reverse transcription by the R2 retrotransposable element. Mol. Cell. Biol. 1995;15:3882–3891. doi: 10.1128/mcb.15.7.3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jurka J. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc. Natl Acad. Sci. USA. 1997;94:1872–1877. doi: 10.1073/pnas.94.5.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zingler N, Willhoeft U, Brose HP, Schoder V, Jahns T, Hanschmann KM, et al. Analysis of 5′ junctions of human LINE-1 and Alu retrotransposons suggests an alternative model for 5′-end attachment requiring microhomology-mediated end-joining. Genome Res. 2005;15:780–789. doi: 10.1101/gr.3421505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ostertag EM, Kazazian HH., Jr Twin priming: a proposed mechanism for the creation of inversions in L1 retrotransposition. Genome Res. 2001;11:2059–2065. doi: 10.1101/gr.205701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilbert N, Lutz-Prigge S, Moran JV. Genomic deletions created upon LINE-1 retrotransposition. Cell. 2002;110:315–325. doi: 10.1016/s0092-8674(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 12.Skalka AM, Katz RA. Retroviral DNA integration and the DNA damage response. Cell Death Differ. 2005;12(Suppl 1):971–978. doi: 10.1038/sj.cdd.4401573. [DOI] [PubMed] [Google Scholar]
- 13.Izsvak Z, Stuwe EE, Fiedler D, Katzer A, Jeggo PA, Ivics Z. Healing the wounds inflicted by sleeping beauty transposition by double-strand break repair in mammalian somatic cells. Mol. Cell. 2004;13:279–290. doi: 10.1016/s1097-2765(03)00524-0. [DOI] [PubMed] [Google Scholar]
- 14.Yant SR, Kay MA. Nonhomologous-end-joining factors regulate DNA repair fidelity during Sleeping Beauty element transposition in mammalian cells. Mol. Cell. Biol. 2003;23:8505–8518. doi: 10.1128/MCB.23.23.8505-8518.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cost GJ, Feng Q, Jacquier A, Boeke JD. Human L1 element target-primed reverse transcription in vitro. EMBO J. 2002;21:5899–5910. doi: 10.1093/emboj/cdf592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zimmerly S, Guo H, Perlman PS, Lambowitz AM. Group II intron mobility occurs by target DNA-primed reverse transcription. Cell. 1995;82:545–554. doi: 10.1016/0092-8674(95)90027-6. [DOI] [PubMed] [Google Scholar]
- 17.Eskes R, Liu L, Ma H, Chao MY, Dickson L, Lambowitz AM, Perlman PS. Multiple homing pathways used by yeast mitochondrial group II introns. Mol. Cell. Biol. 2000;20:8432–8446. doi: 10.1128/mcb.20.22.8432-8446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moran JV, Zimmerly S, Eskes R, Kennell JC, Lambowitz AM, Butow RA, Perlman PS. Mobile group II introns of yeast mitochondrial DNA are novel site-specific retroelements. Mol. Cell. Biol. 1995;15:2828–2838. doi: 10.1128/mcb.15.5.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cousineau B, Lawrence S, Smith D, Belfort M. Retrotransposition of a bacterial group II intron. Nature. 2000;404:1018–1021. doi: 10.1038/35010029. [DOI] [PubMed] [Google Scholar]
- 20.Ichiyanagi K, Beauregard A, Lawrence S, Smith D, Cousineau B, Belfort M. Retrotransposition of the Ll.LtrB group II intron proceeds predominantly via reverse splicing into DNA targets. Mol. Microbiol. 2002;46:1259–1272. doi: 10.1046/j.1365-2958.2002.03226.x. [DOI] [PubMed] [Google Scholar]
- 21.Smith D, Zhong J, Matsuura M, Lambowitz AM, Belfort M. Recruitment of host functions suggests a repair pathway for late steps in group II intron retrohoming. Genes Dev. 2005;19:2477–2487. doi: 10.1101/gad.1345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pelisson A, Finnegan DJ, Bucheton A. Evidence for retrotransposition of the I factor, a LINE element of Drosophila melanogaster. Proc. Natl Acad. Sci. USA. 1991;88:4907–4910. doi: 10.1073/pnas.88.11.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fawcett DH, Lister CK, Kellett E, Finnegan DJ. Transposable elements controlling I-R hybrid dysgenesis in D. melanogaster are similar to mammalian LINEs. Cell. 1986;47:1007–1015. doi: 10.1016/0092-8674(86)90815-9. [DOI] [PubMed] [Google Scholar]
- 24.Chambeyron S, Bucheton A. I elements in Drosophila: in vivo retrotransposition and regulation. Cytogenet. Genome Res. 2005;110:215–222. doi: 10.1159/000084955. [DOI] [PubMed] [Google Scholar]
- 25.Chaboissier MC, Lemeunier F, Bucheton A. IR hybrid dysgenesis increases the frequency of recombination in Drosophila melanogaster. Genet. Res. 1995;65:167–174. doi: 10.1017/s0016672300033255. [DOI] [PubMed] [Google Scholar]
- 26.Dimitri P, Arca B, Berghella L, Mei E. High genetic instability of heterochromatin after transposition of the LINE-like I factor in Drosophila melanogaster. Proc. Natl Acad. Sci. USA. 1997;94:8052–8057. doi: 10.1073/pnas.94.15.8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hiraizumi Y. Heterochromatic recombination in germ cells of Drosophila melanogaster females. Genetics. 1981;98:105–114. doi: 10.1093/genetics/98.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen JM, Stenson PD, Cooper DN, Ferec C. A systematic analysis of LINE-1 endonuclease-dependent retrotranspositional events causing human genetic disease. Hum. Genet. 2005;117:411–427. doi: 10.1007/s00439-005-1321-0. [DOI] [PubMed] [Google Scholar]
- 29.Kazazian HH, Jr, Wong C, Youssoufian H, Scott AF, Phillips DG, Antonarakis SE. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature. 1988;332:164–166. doi: 10.1038/332164a0. [DOI] [PubMed] [Google Scholar]
- 30.Bourćhis D, Bestor TH. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature. 2004;431:96–99. doi: 10.1038/nature02886. [DOI] [PubMed] [Google Scholar]
- 31.Webster KE, O'bryan MK, Fletcher S, Crewther PE, Aapola U, Craig J, et al. Meiotic and epigenetic defects in Dnmt3L-knockout mouse spermatogenesis. Proc. Natl Acad. Sci. USA. 2005;102:4068–4073. doi: 10.1073/pnas.0500702102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asch HL, Eliacin E, Fanning TG, Connolly JL, Bratthauer G, Asch BB. Comparative expression of the LINE-1 p40 protein in human breast carcinomas and normal breast tissues. Oncol. Res. 1996;8:239–247. [PubMed] [Google Scholar]
- 33.Bratthauer GL, Fanning TG. Active LINE-1 retrotransposons in human testicular cancer. Oncogene. 1992;7:507–510. [PubMed] [Google Scholar]
- 34.Nangia-Makker P, Sarvis R, Visscher DW, Bailey-Penrod J, Raz A, Sarkar FH. Galectin-3 and L1 retrotransposons in human breast carcinomas. Breast Cancer Res. Treat. 1998;49:171–183. doi: 10.1023/a:1005913810250. [DOI] [PubMed] [Google Scholar]
- 35.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 36.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 37.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 38.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rothkamm K, Lobrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc. Natl Acad. Sci. USA. 2003;100:5057–5062. doi: 10.1073/pnas.0830918100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sedelnikova OA, Rogakou EP, Panyutin IG, Bonner WM. Quantitative detection of (125)IdU-induced DNA double-strand breaks with gamma-H2AX antibody. Radiat. Res. 2002;158:486–492. doi: 10.1667/0033-7587(2002)158[0486:qdoiid]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 41.d'Adda d. F., Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 42.Wei W, Morrish TA, Alisch RS, Moran JV. A transient assay reveals that cultured human cells can accommodate multiple LINE-1 retrotransposition events. Anal. Biochem. 2000;284:435–438. doi: 10.1006/abio.2000.4675. [DOI] [PubMed] [Google Scholar]
- 43.Perepelitsa-Belancio V, Deininger P. RNA truncation by premature polyadenylation attenuates human mobile element activity. Nature Genet. 2003;35:363–366. doi: 10.1038/ng1269. [DOI] [PubMed] [Google Scholar]
- 44.Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH., Jr High frequency retrotransposition in cultured mammalian cells. Cell. 1996;87:917–927. doi: 10.1016/s0092-8674(00)81998-4. [DOI] [PubMed] [Google Scholar]
- 45.Mathias SL, Scott AF, Kazazian HH, Jr, Boeke JD, Gabriel A. Reverse transcriptase encoded by a human transposable element. Science. 1991;254:1808–1810. doi: 10.1126/science.1722352. [DOI] [PubMed] [Google Scholar]
- 46.Christensen SM, Eickbush TH. R2 target-primed reverse transcription: ordered cleavage and polymerization steps by protein subunits asymmetrically bound to the target DNA. Mol. Cell. Biol. 2005;25:6617–6628. doi: 10.1128/MCB.25.15.6617-6628.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin SL, Cruceanu M, Branciforte D, Wai-Lun LP, Kwok SC, Hodges RS, Williams MC. LINE-1 retrotransposition requires the nucleic acid chaperone activity of the ORF1 protein. J. Mol. Biol. 2005;348:549–561. doi: 10.1016/j.jmb.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 48.Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem. 2000;275:9390–9395. doi: 10.1074/jbc.275.13.9390. [DOI] [PubMed] [Google Scholar]
- 49.Haoudi A, Semmes OJ, Mason JM, Cannon RE. Retrotransposition-competent human LINE-1 induces apoptosis in cancer cells with intact p53. J. Biomed. Biotechnol. 2004;2004:185–194. doi: 10.1155/S1110724304403131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 51.Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 52.Bohnke A, Westphal F, Schmidt A, El Awady RA, Dahm-Daphi J. Role of p53 mutations, protein function and DNA damage for the radiosensitivity of human tumour cells. Int. J. Radiat. Biol. 2004;80:53–63. doi: 10.1080/09553000310001642902. [DOI] [PubMed] [Google Scholar]
- 53.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Expt. Cell Res. 1988;175:184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 54.Goodier JL, Ostertag EM, Engleka KA, Seleme MC, Kazazian HH., Jr potential role for the nucleolus in L1 retrotransposition. Hum. Mol. Genet. 2004;13:1041–1048. doi: 10.1093/hmg/ddh118. [DOI] [PubMed] [Google Scholar]
- 55.Kuhne C, Tjornhammar ML, Pongor S, Banks L, Simoncsits A. Repair of a minimal DNA double-strand break by NHEJ requires DNA-PKcs and is controlled by the ATM/ATR checkpoint. Nucl. Acids Res. 2003;31:7227–7237. doi: 10.1093/nar/gkg937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin Y, Lukacsovich T, Waldman AS. Multiple pathways for repair of DNA double-strand breaks in mammalian chromosomes. Mol. Cell. Biol. 1999;19:8353–8360. doi: 10.1128/mcb.19.12.8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guirouilh-Barbat J, Huck S, Bertrand P, Pirzio L, Desmaze C, Sabatier L, Lopez BS. Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol. Cell. 2004;14:611–623. doi: 10.1016/j.molcel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 58.Elkind MM. Repair processes in radiation biology. Radiat. Res. 1984;100:425–449. [PubMed] [Google Scholar]
- 59.Xu B, Kastan MB. Analyzing cell cycle checkpoints after ionizing radiation. Methods Mol. Biol. 2004;281:283–292. doi: 10.1385/1-59259-811-0:283. [DOI] [PubMed] [Google Scholar]
- 60.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nature Rev. Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 61.Kurz EU, Lees-Miller SP. DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair (Amst) 2004;3:889–900. doi: 10.1016/j.dnarep.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 62.Kuhne M, Riballo E, Rief N, Rothkamm K, Jeggo PA, Lobrich M. A double-strand break repair defect in ATM-deficient cells contributes to radiosensitivity. Cancer Res. 2004;64:500–508. doi: 10.1158/0008-5472.can-03-2384. [DOI] [PubMed] [Google Scholar]
- 63.Meyn MS. High spontaneous intrachromosomal recombination rates in ataxia-telangiectasia. Science. 1993;260:1327–1330. doi: 10.1126/science.8493577. [DOI] [PubMed] [Google Scholar]
- 64.Taylor AM, Harnden DG, Arlett CF, Harcourt SA, Lehmann AR, Stevens S, Bridges BA. Ataxia telangiectasia: a human mutation with abnormal radiation sensitivity. Nature. 1975;258:427–429. doi: 10.1038/258427a0. [DOI] [PubMed] [Google Scholar]
- 65.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 66.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001;276:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 67.Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–2396. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- 68.Ziv Y, Bar-Shira A, Pecker I, Russell P, Jorgensen TJ, Tsarfati I, Shiloh Y. Recombinant ATM protein complements the cellular A-T phenotype. Oncogene. 1997;15:159–167. doi: 10.1038/sj.onc.1201319. [DOI] [PubMed] [Google Scholar]
- 69.Martinez-Abarca F, Barrientos-Duran A, Fernandez-Lopez M, Toro N. The RmInt1 group II intron has two different retrohoming pathways for mobility using predominantly the nascent lagging strand at DNA replication forks for priming. Nucl. Acids Res. 2004;32:2880–2888. doi: 10.1093/nar/gkh616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kulpa DA, Moran JV. Ribonucleoprotein particle formation is necessary but not sufficient for LINE-1 retrotransposition. Hum. Mol. Genet. 2005;14:3237–3248. doi: 10.1093/hmg/ddi354. [DOI] [PubMed] [Google Scholar]
- 71.Morrish TA, Gilbert N, Myers JS, Vincent BJ, Stamato TD, Taccioli GE, et al. DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nature Genet. 2002;31:159–165. doi: 10.1038/ng898. [DOI] [PubMed] [Google Scholar]
- 72.Lau A, Swinbank KM, Ahmed PS, Taylor DL, Jackson SP, Smith GC, O'Connor MJ. Suppression of HIV-1 infection by a small molecule inhibitor of the ATM kinase. Nature Cell Biol. 2005;7:493–500. doi: 10.1038/ncb1250. [DOI] [PubMed] [Google Scholar]
- 73.Nunnari G, Argyris E, Fang J, Mehlman KE, Pomerantz RJ, Daniel R. Inhibition of HIV-1 replication by caffeine and caffeine-related methylxanthines. Virology. 2005;335:177–184. doi: 10.1016/j.virol.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 74.Daniel R, Katz RA, Skalka AM. A role for DNA-PK in retroviral DNA integration. Science. 1999;284:644–647. doi: 10.1126/science.284.5414.644. [DOI] [PubMed] [Google Scholar]
- 75.Morrison C, Sonoda E, Takao N, Shinohara A, Yamamoto K, Takeda S. The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J. 2000;19:463–471. doi: 10.1093/emboj/19.3.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Golding SE, Rosenberg E, Khalil A, McEwen A, Holmes M, Neill S, et al. Double strand break repair by homologous recombination is regulated by cell cycle-independent signaling via ATM in human glioma cells. J. Biol. Chem. 2004;279:15402–15410. doi: 10.1074/jbc.M314191200. [DOI] [PubMed] [Google Scholar]
- 77.Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol. Cell. 2004;16:715–724. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 78.Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH., Jr Hot L1s account for the bulk of retrotransposition in the human population. Proc. Natl Acad. Sci. USA. 2003;100:5280–5285. doi: 10.1073/pnas.0831042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abeysinghe SS, Chuzhanova N, Krawczak M, Ball EV, Cooper DN. Translocation and gross deletion breakpoints in human inherited disease and cancer I: nucleotide composition and recombination-associated motifs. Hum. Mutat. 2003;22:229–244. doi: 10.1002/humu.10254. [DOI] [PubMed] [Google Scholar]
- 80.Deininger PL, Batzer MA. Alu repeats and human disease. Mol. Genet. Metab. 1999;67:183–193. doi: 10.1006/mgme.1999.2864. [DOI] [PubMed] [Google Scholar]
- 81.Babcock M, Pavlicek A, Spiteri E, Kashork CD, Ioshikhes I, Shaffer LG, et al. Shuffling of genes within low-copy repeats on 22q11 (LCR22) by Alu-mediated recombination events during evolution. Genome Res. 2003;13:2519–2532. doi: 10.1101/gr.1549503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gentles AJ, Kohany O, Jurka J. Evolutionary diversity and potential recombinogenic role of integration targets of non-LTR retrotranspo-sons. Mol. Biol. Evol. 2005;22:1983–1991. doi: 10.1093/molbev/msi188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ergun S, Buschmann C, Heukeshoven J, Dammann K, Schnieders F, Lauke H, et al. Cell type-specific expression of LINE-1 open reading frames 1 and 2 in fetal and adult human tissues. J. Biol. Chem. 2004;279:27753–27763. doi: 10.1074/jbc.M312985200. [DOI] [PubMed] [Google Scholar]
- 84.Branciforte D, Martin SL. Developmental and cell type specificity of LINE-1 expression in mouse testis: implications for transposition. Mol. Cell. Biol. 1994;14:2584–2592. doi: 10.1128/mcb.14.4.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Trelogan SA, Martin SL. Tightly regulated, developmentally specific expression of the first open reading frame from LINE-1 during mouse embryogenesis. Proc. Natl Acad. Sci. USA. 1995;92:1520–1524. doi: 10.1073/pnas.92.5.1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH. Somatic mosaicism in neuronal precursor cells mediated by L1 retro-transposition. Nature. 2005;435:903–910. doi: 10.1038/nature03663. [DOI] [PubMed] [Google Scholar]
- 87.Revy P, Buck D, le Deist F, de Villartay JP. The repair of DNA damages/modifications during the maturation of the immune system: lessons from human primary immunodeficiency disorders and animal models. Advan. Immunol. 2005;87:237–295. doi: 10.1016/S0065-2776(05)87007-5. [DOI] [PubMed] [Google Scholar]
- 88.Gasior SL, Palmisano M, Deininger PL. Alu-linked hairpins efficiently mediate RNA interference with less toxicity than do H1-expressed short hairpin RNAs. Anal. Biochem. 2006;349:41–48. doi: 10.1016/j.ab.2005.11.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.