

Abstract
Purpose
Lung squamous cell carcinoma (LSCC) currently lacks effective targeted therapies. Previous studies reported overexpression of HEDGEHOG (HH)-GLI signaling components in LSCC. However, they addressed neither the tumor heterogeneity nor the requirement for HH-GLI signaling. Here, we investigated the role of HH-GLI signaling in LSCC, and studied the therapeutic potential of HH-GLI suppression.
Experimental Design
Gene expression datasets of two independent LSCC patient cohorts were analyzed to study the activation of HH-GLI signaling. Four human LSCC cell lines were examined for HH-GLI signaling components. Cell proliferation and apoptosis were assayed in these cells after blocking the HH-GLI pathway by lentiviral-shRNA knockdown or small molecule inhibitors. Xenografts in immunodeficient mice were used to determine the in vivo efficacy of GLI inhibitor GANT61.
Results
In both cohorts, activation of HH-GLI signaling was significantly associated with the classical subtype of LSCC. In cell lines, genetic knockdown of SMO produced minor effects on cell survival, while GLI2 knockdown significantly reduced proliferation and induced extensive apoptosis. Consistently, the SMO inhibitor GDC-0449 resulted in limited cytotoxicity in LSCC cells, whereas the GLI inhibitor GANT61 was very effective. Importantly, GANT61 demonstrated specific in vivo anti-tumor activity in xenograft models of GLI-positive cell lines.
Conclusion
Our studies demonstrate an important role for GLI2 in LSCC, and suggest GLI inhibition as a novel and potent strategy to treat a subset of LSCC patients.
Keywords: Squamous cell lung cancer, HEDGEHOG, GLI
Introduction
Lung cancer is the leading cause of cancer-related death worldwide. Squamous cell carcinoma (LSCC) comprises ∼30% of non-small cell lung cancers and is associated strongly with smoking. Although oncogenic alterations have been gradually described in LSCC, druggable targets have not been identified, resulting in lack of effective targeted therapies (1). Analysis of gene expression of resected tumors has classified LSCC into four mRNA expression subtypes, defined as classical, primitive, secretory and basal (2). The oncogenic drivers and potential cells of origin of tumors within these subtypes are likely different (2, 3). However, the practical application of these expression subtypes in clinical care has not been determined.
Recently, growing evidence has suggested aberrant activation of the HEDGEHOG (HH) signaling pathway in lung cancer, which plays a critical role during lung development (4, 5). Canonical HH signaling is initiated by the binding of HH ligands (SONIC, INDIAN and DESERT HEDGEHOG) to a 12-transmembrane receptor PATCHED (PTCH), which relieves the catalytic inhibition of SMOOTHENED (SMO), a 7-transmembrane G-protein coupled receptor (GPCR)-like signal transducer. SMO de-repression triggers a series of intracellular events, resulting in the activation of downstream target genes through the zinc-finger transcription factors GLI1, GLI2 and GLI3 (6). GLI activation is complex with regulation at both the transcriptional and post-translational levels (7). GLI2 appears to be the primary activator of HH signaling in cancer, with GLI1 as a transcriptional target of GLI2 (8-10). GLI2 and GLI1 also induce transcription of overlapping and distinct sets of downstream targets (11). Several components of the HH pathway (PTCH, GLI1, GLI2, HHIP) are GLI transcriptional targets that induce positive or negative feedbacks (7). GLI targets mediate various cellular responses, notably enhanced cell proliferation and survival by upregulating D-type cyclins and anti-apoptotic proteins (12-14). Finally, phosphatidylinositol 3-kinase (PI3K)/AKT and RAS-MEK signals have been described as non-canonical HH activators in cancer (15-17).
Dysregulation of the HH pathway has been implicated in a variety of cancers (18). Although the involvement of HH signaling in small-cell lung cancer is well established (4, 5), the role of this pathway in non-small cell lung cancer remains poorly understood. However, multiple lines of evidence point to the potential importance of HH signaling in LSCC. Immunohistochemical studies in patient specimens reported the overexpression of HH signaling components (19, 20). Microarray analysis identified hyperactive HH signaling in LSCC (21). HH-GLI signaling has also been implicated in squamous cancer of other organs (22-24) . Despite these studies, very little is known regarding the specific role of HH signaling in regulating cellular survival and proliferation in LSCC.
Targeted inhibitors of the HH pathway have become available recently. Because of its accessibility on the membrane and its importance in regulation of the pathway, SMO has been the primary focus in the development of small molecule inhibitors of the HH pathway. GDC-0449 (vismodegib; Genentech) is an orally administered agent that selectively suppresses SMO activity and was the first SMO inhibitor to progress to clinical trials. It has produced promising antitumor responses in patients with advanced basal cell carcinoma and medulloblastoma (25, 26), but resistance has been reported (27, 28). The resistance to SMO inhibitors highlights the therapeutic need to target downstream effectors to maintain robust on-target responses. In a cell-based screen of GLI-mediated transcription (29), the small molecule GANT61 was identified as a specific inhibitor of GLI1 and GLI2. It suppresses the DNA binding capacity of GLIs and inhibits GLI-mediated transcription. GANT61 reduces proliferation and induces apoptosis in a GLI-specific fashion in prostate cancer (29), colon carcinoma (30, 31), oral squamous cell carcinoma (23), pancreatic cancer (32), neuroblastoma (33), and chronic lymphocytic leukemia (34). In this study, we investigate the role of HH-GLI signaling in LSCC and assess the clinical feasibility of using GDC-0449 or GANT61 to treat LSCC.
Materials and Methods
RNAseq and microarray analysis
RSEM values (35) for 178 tumor samples from The Cancer Genome Atlas (TCGA) LSCC study (3) were converted to expression measurements by replacing values equal to zero with the smallest non-zero value, taking a log2 transformation. After median centering by gene, heatmaps of the expression values from TCGA (3) and microarray data from 56 LSCC samples collected at the University of North Carolina (2) (UNC cohort) were produced with R 2.15.1 (36) and the gplots package. Hypotheses were subsequently tested: One-sided Wilcoxon Rank Sum tests were used to test the null hypothesis that the mean expression levels of PTCH1, GLI1, GLI2, GLI3, and SUFU are the same in the classical subtype as all other subtypes combined. For PTCH1, GLI1, and GLI2, the alternative hypothesis was that the expression levels are higher in the classical subtype, whereas for GLI3 and SUFU the alternative hypothesis was that the expression levels are lower in the classical subtype. A Bonferroni adjustment was applied to correct for multiple comparisons.
Two-sided Wilcoxon Rank Sum tests were used to test the null hypothesis that GLI1 and GLI2 expression values were equal in the TCGA and UNC cohorts. Spearman correlation coefficients were computed based on the uncentered expression values of GLI1, GLI2, TP63, PIK3CA, and SOX2 in both cohorts. The resulting unadjusted p values were used to assess the significance of these associations (Supplementary Table 2).
Gene expression data from 20 LSCC cell lines was obtained from the Cancer Cell Line Encyclopedia (CCLE) (37). After median centering the expression values by gene, the centroid classifier from (2) was used to predict expression subtypes for each line by finding the nearest centroid using a distance metric equal to one minus the Pearson correlation coefficient. Gene expression heatmaps were then produced using R2.15.1 (36) and the gplots package.
Cell culture and reagents
NCI-H520, NCI-H2170, NCI-H226 and SK-MES-1 cells were obtained from ATCC. Cell lines were routinely verified by morphology and growth characteristics, and verified biannually to be mycoplasma-free. NCI-H520, NCI-H2170 and NCI-H226 cells were maintained in the RPMI 1640 medium containing 10% FBS. SK-MES-1 cells were maintained in MEM medium containing 10% FBS, 0.1 mM non-essential amino acids, 1.0 mM sodium pyruvate. Antibodies: SHH, PTCH, SMO (Santa Cruz); GLI2, GAPDH (Abcam); GLI1 (Novus Biologicals); Cleaved Caspase-3, Cleaved PARP (Cell Signaling Technology); CCND1 (BD Biosciences). Compounds: GANT61 (Sigma) and GDC-0449 (Chemietek).
Lentiviral production and transduction
Lentiviral shRNA clones (Sigma Mission RNAi) targeting SMO, GLI2 and the non-targeting control (SHC002) were purchased from Sigma-Aldrich. 293T cells were plated in 10-cm plates 24 hours prior to transfection in DMEM medium containing 10% FBS without antibiotics. 5 μg shRNA plasmid, 4 μg psPAX2 and 1 μg pCI-VSVG packaging vectors (Addgene) were co-transfected into 293T cells using Lipofectamine 2000 Reagent (Invitrogen). Viral supernatants were collected, centrifuged and filtered with 0.45 μm PES Sterile Syringe Filter. Target cells were plated and incubated at 37°C, 5% CO2 overnight, and changed to medium containing lentivirus and 8 μg/mL polybrene. Control plates were incubated with medium containing 8 μg/mL polybrene. Cells were changed to fresh culture medium 24 hours after infection. Puromycin selection (5 μg/ml) was started 48 hours post infection and continued for 4∼5 days until no viable cells were observed in control plates. Once decreased expression of the targeted gene was confirmed, cells were used for subsequent experiments. Stable expression of non-targeting control, SMO or GLI2 shRNAs was ensured by culturing cells in the presence of puromycin.
The shRNA sequences:
SMO sh1 (5′- CCGGCCTGATGGACACAGAACTCATCTCGAGATGAGTTCTG TGTCCATCAGGTTTTT-3′)
SMO sh2 (5′- CCGGCATCTTTGTCATCGTGTACTACTCGAGTAGTACACGA TGACAAAGATGTTTTT-3′)
SMO sh3 (5′- CCGGGTGGAGAAGATCAACCTGTTTCTCGAGAAACAGGTTG ATCTTCTCCACTTTTT-3′)
GLI2 sh1 (5′- CCGGCCAACGAGAAACCCTACATCTCTCGAGAGATGTAGGGT TTCTCGTTGGTTTTTG-3′)
GLI2 sh2 (5′-CCGGCACTCAAGGATTCCTGCTCATCTCGAGATGAGCAGGAA TCCTTGAGTGTTTTTG-3′)
GLI2 sh3 (5′-CCGGGCTCTACTACTACGGCCAGATCTCGAGATCTGGCCGTA GTA GTAGAGCTTTTTG-3′)
Assessment of cell viability and caspase 3/7 activity
Cell viability and caspase 3/7 activity were determined by using ApoLive-Glo Multiplex Assay (Promega) according to the manufacturer's instructions. Briefly, cells were seeded in 96-well clear-bottom white plates at a density of 10,000 cells per well and incubated with complete medium overnight at 37°C, 5% CO2. The following day, cells were changed into 0.5% FBS-containing medium with either DMSO control or drugs at designated concentrations (0.1% final DMSO concentration) as triplicates and treated for 96 hours. At the end of treatment, viability reagent was added into all wells and gently mixed. After 1.5-hour incubation at 37°C, fluorescence was measured at the wavelength set 355EX/ 520EM by a FLUOstar Omega Microplate reader. Later, Caspase-Glo® 3/7 Reagent was added to all wells and gently mixed. Luminescence was measured after 1-hour incubation at room temperature. The reading of blank control was subtracted from readings of other wells as the background in the data analysis.
RNA isolation and quantitative PCR
Total RNA was isolated using the Qiagen RNeasy Mini Kit, treated with DNase I (Invitrogen) and converted to cDNA using iScript cDNA Synthesis Kit (BIO-RAD). Real-time PCR was performed using TaqMan Gene Expression Master Mix on an Eppendorf Mastercycler, and raw data were analyzed by Realplex software. TaqMan probes for SHH, PTCH1, SMO, GLI1, GLI2, HHIP and GAPDH were purchased from Applied Biosystems.
Western analysis
Total cellular lysates were prepared by using RIPA buffer (Sigma) with protease inhibitor cocktail (Sigma) and PhosSTOP (Roche). Protein concentrations were determined by Micro BCA Protein Assay Kit (Thermo Scientific). Proteins were separated on the NuPAGE 4-12% Bis-Tris Gel (Life Technologies) and transferred using Invitrolon PVDF Filter Paper Sandwich. Membranes were blocked with 5% nonfat dry milk or 5% BSA in 0.1%TBST for 1 hour in room temperature, then incubated with primary antibody overnight at 4°C. They were subsequently washed with 0.1%TBST and incubated with the secondary antibody for 1 hour at room temperature. Western Lightning- ECL (PerkinElmer) was used to develop the membranes.
Xenograft and tumor treatment
106 NCI-H520 cells, 106 NCI-H2170 cells or 5×106 NCI-H226 cells were suspended in a total volume of 100 μl of a 1:1 mixture of RPMI 1640 medium: Matrigel (BD Biosciences). Cells were injected subcutaneously in the right posterior flank of 6∼8 week C.129S7 (B6)-Rag1tm1Mom/ J (Rag1-/-) mice. Tumors were grown until they reached a median size of ≈250 mm3 (NCI-H520), ≈230 mm3 (NCI-H2170), and ≈150 mm3 (NCI-H226). Animals were randomly divided into groups and treated with solvent only (corn oil: ethanol, 4:1) or GANT61 in solvent (50 mg/kg). Treatments were given every other day for 20 days by intraperitoneal injection. Tumor volumes were calculated by the formula 0.52×length×(width)2. At the end of treatment, tumors were removed, weighed and processed for subsequent analysis. All animal experiments were approved by and conformed to the policies and regulations of Institutional Animal Care and Use Committees at Duke University.
Results
Activation of HH-GLI signaling is associated with the classical subtype of human LSCC
To ascertain whether HH signaling is upregulated in a particular subset of LSCC patients, the RNA expression data of 178 patient samples from the Cancer Genome Atlas (TCGA) LSCC study (3) was queried. As Figure 1A demonstrates, the expression of HH target genes (PTCH1, GLI1, GLI2) was significantly higher, while expression of negative regulators (GLI3, SUFU) was substantially lower in the classical subtype in comparison to the other subtypes. One-sided Wilcoxon Rank Sum Test confirmed these observations (Supplementary Table 1) even after applying a Bonferroni adjustment for multiple comparisons. Similar expression patterns were seen in an independent cohort of 56 LSCC samples collected at the University of North Carolina (2) (UNC cohort) (Supplementary Figure S1A). In both cohorts, GLI2 mRNA level was significantly higher than GLI1 (Figure 1B, Supplementary Figure S1B). Samples with high GLI2 expression were mainly found in the classical subtype although occasionally in other subtypes. When taking the 75th percentile of all GLI2 expression values in a given cohort as the threshold for high GLI2, 55% (TCGA cohort) and 52% (UNC cohort) of all classical subtype samples exhibited high GLI2 expression (Figure 1C, Supplementary Figure S1C).
Figure 1. HH pathway components are overexpressed in the classical subtype of LSCC in the TCGA cohort.
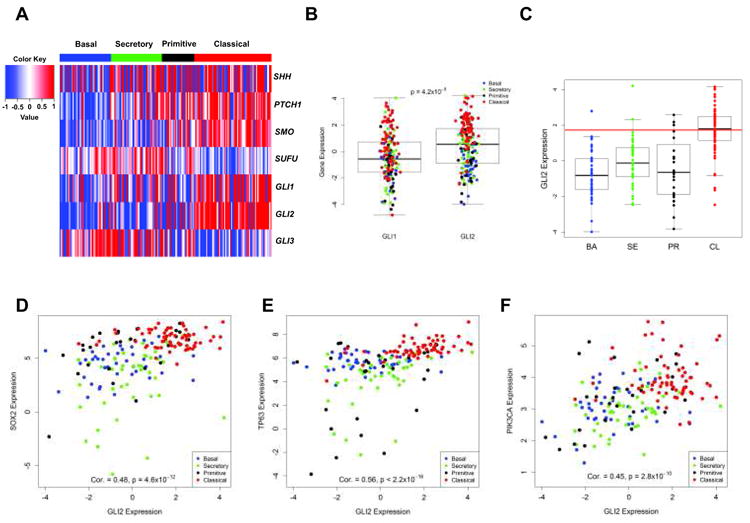
A. Heatmap of HH signaling components. Tumor samples are displayed as columns, grouped by gene expression subtypes. Selected HH genes are rows. Displayed genes of HH pathway except SHH, showed highly significant association with gene expression subtype (p<0.001) (Supplementary Table 1). B. GLI2 expression is significantly higher than GLI1 (p=4.2×10-5). C. Gene expression values were plotted according to subtypes, and the 75th percentile of all GLI2 expression is shown as a horizontal red line. D∼F. Scatterplots of gene expression measurements between GLI2 and SOX2 (D), TP63 (E) and PIK3CA (F). Spearman correlation coefficients and corresponding p values were provided in Supplementary Table 2.
Strong positive correlations between GLI2 and the prominent markers for the classical subtype (SOX2, TP63 and PIK3CA) on chromosome 3q were observed in both cohorts (Figure 1D∼F, Supplementary Figure S1D∼F). However, GLI1 was only associated with classical chr3q genes in the TCGA cohort (Supplementary Figure S1G∼I), suggesting that GLI2 is highly likely to be the major signaling transducer in LSCC. Spearman correlation coefficients between GLI2/ GLI1 and the classical subtype markers with corresponding p values were provided in Supplementary Table 2.
HH-GLI signaling components are expressed in human LSCC cell lines
The difficulty of growing human LSCC cells in vitro limits available primary cancer cells. Therefore, we chose the four most widely used human LSCC cell lines to analyze active HH signaling: NCI-H520 and NCI-H2170, derived from primary tumors; and NCI-H226 and SK-MES-1, derived from metastatic pleural effusions. By real-time PCR (Figure 2A) and Western blots (Figure 2B), high levels of SHH were detected only in NCI-H520, whereas PTCH1 and SMO were expressed universally across all four lines. Neither GLI1 nor GLI2 was detected in NCI-H2170. In the remaining three lines, GLI1 was expressed at a low level, whereas high levels of GLI2 were consistently detected at both the mRNA and protein level. To ascertain whether these cell lines represent different LSCC subtypes by mRNA expression, gene expression profiles from CCLE (37) were analyzed. NCI-H520 and NCI-H2170 were predicted to be classical subtype, and NCI-H226 and SK-MES-1 as secretory subtype. Gene expression of the subtypes between the cell lines and patient tumors is consistent over the validation gene set (Supplementary Figure S2).
Figure 2. HH signaling components are expressed in human LSCC cell lines.
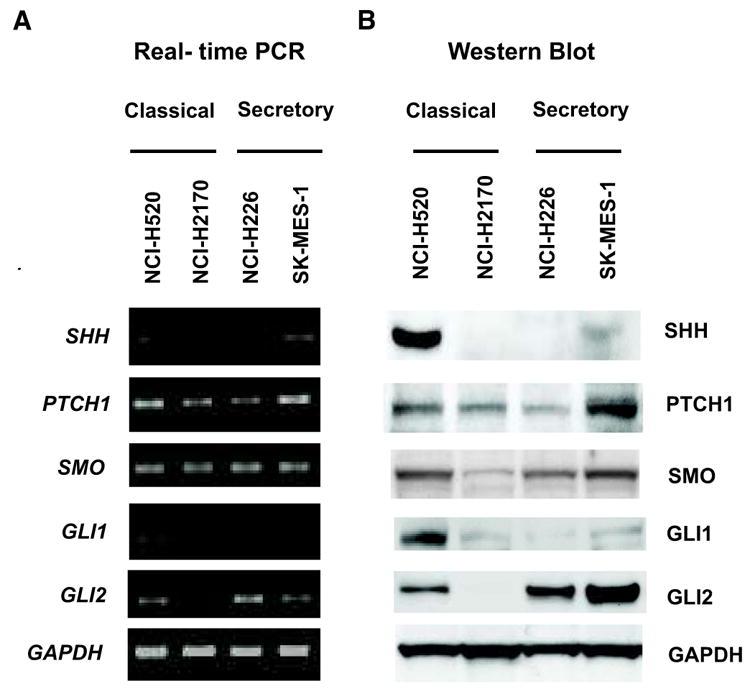
A. Real-time PCR for indicated genes, with GAPDH as the quantitative control. B, Western Blot for the SHH-GLI pathway components, with GAPDH as the loading control. Classical subtype: NCI-H520 and NCI-H2170; Secretory subtype: NCI-H226 and SK-MES-1.
shRNA knockdown of SMO produces minor effects on LSCC survival
The universal expression and the ability to target SMO with multiple available inhibitors prompted us to investigate the importance of SMO in LSCC cells. Lentiviral mediated expression of independent SMO shRNA constructs successfully reduced the SMO mRNA level by 70∼90% in four cell lines (Figure 3A). However, only minor effects on cell viability and apoptosis were observed in these cells (Figure 3B, C). SMO knockdown caused a moderate decrease of PTCH1 mRNA in NCI-H520 and NCI-H226 (Figure 3D), but no significant reduction of HHIP mRNA in any of four lines (Figure 3E). These data suggest a minimal role for SMO in regulating LSCC survival via the canonical HH pathway. Interestingly, loss of SMO did not reduce GLI2 mRNA level in three GLI-positive cell lines. Instead, we noted a slight increase of GLI2 mRNA (Figure 3F), which may be caused by compensatory upregulation of GLI2 by other SMO-independent mechanisms.
Figure 3. SMO plays a minor role while GLI2 is required for LSCC survival in vitro.
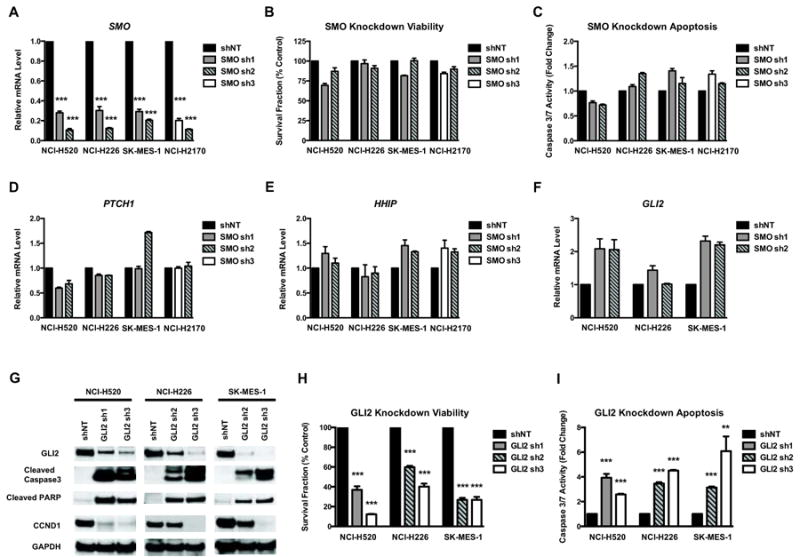
A. Change of GAPDH-normalized SMO mRNA level by real-time PCR following lentiviral shRNA knockdown. Non-targeting shRNA control (shNT) or 2 independent shRNAs (SMO sh1, 2, 3) targeting SMO were used in each cell line. B∼C. Measurements of viability (B) and apoptosis (C) in cells after SMO knockdown. Data were normalized to shNT control and represent the mean ± SD of 3 independent experiments. D∼F. Real-time PCR showing the GAPDH-normalized mRNA level of PTCH1 (D), HHIP (E) and GLI2 (F) after SMO knockdown. G. Western blots showing GLI2 knockdown by independent shRNAs (GLI2 sh1, 2, 3). GLI2 shRNAs significantly reduced GLI2 protein levels, in comparison to the shNT control. GLI2 knockdown also caused induction of cleaved caspase-3, cleaved PARP and reduction of CCND1. GAPDH was used as the loading control. A representative Western blot from 3 independent experiments is shown. Knockdown of GLI2 in LSCC cells significantly reduced proliferation (H) and induced apoptosis (I). Data were normalized to shNT control and represent the mean ± SD of 3 independent experiments. (Two-tailed t test, **: p<0.01; ***: p<0.001)
Targeting GLI2 with shRNAs inhibits LSCC cell growth and induces extensive apoptosis
Since GLI1 is hardly detectable and high level of GLI2 is consistently expressed in three cell lines and across human LSCC tumors, we focused on GLI2. Independent lentiviral-based GLI2 shRNAs achieved satisfactory knockdown of GLI2 protein in all three GLI2-positive lines (Figure 3G). Knockdown of GLI2 reduced the protein level of the GLI target CCND1 (Figure 3G), corresponding to a strong inhibition of cell proliferation and survival (Figure 3H). Loss of GLI2 also induced extensive apoptosis, demonstrated by elevated caspase 3/7 activity (Figure 3I), and the detection of cleaved caspase-3 and cleaved PARP (Figure 3G). These data suggest an important role of GLI2 in regulating LSCC cell survival, raising the possibility that GLI2 is a therapeutic target in human LSCC.
GLI inhibitor (GANT61) leads to significant growth inhibition and apoptosis, and demonstrates greater efficacy than the SMO inhibitor (GDC-0449)
To investigate the feasibility of pharmacologically targeting SMO or GLI proteins in LSCC, we studied the therapeutic potential of a clinically available SMO inhibitor, GDC-0449, and a GLI inhibitor, GANT61. To maintain physiologic relevance and minimize off-target toxicity, we assessed the efficacy of GDC-0449 and GANT61 in four LSCC cell lines at the concentrations of 2.5, 5 and 10 μmol/L. Cells were treated in triplicate with either DMSO control, GDC-0449 or GANT61 for 96 hours, and then assayed for viability and caspase 3/7 activation.
As Figure 4A and 4B demonstrate, GDC-0449 showed limited growth inhibition and apoptosis induction only in NCI-H520 and NCI-H226 cells at 10 μmol/L despite the universal expression of SMO. Consistently, GDC-0449 only caused modest reduction of PTCH1 mRNA in NCI-H226 cells (Figure 5A). Among three GLI-positive cell lines, GDC-0449 led to slight decrease of GLI2 mRNA in NCI-H226 and SK-MES-1 (Figure 5B), again suggesting a minimal role of SMO in mediating HH-GLI signaling in LSCC.
Figure 4. GANT61 is more effective than GDC-0449 in reducing cell survival and inducing apoptosis.

A∼B. 96-hour treatment of GDC-0449 generated limited growth inhibition (A) and apoptosis (B). Only moderate cytotoxicity was observed in NCI-H520 and NCI-H226 cells at 10 μmol/L. C∼D. 96-hour treatment of GANT61 significantly reduced cell survival (C) and induced extensive apoptosis (D) in all GLI-positive cell lines. GLI-negative NCI-H2170 was not affected by GANT61. Data were normalized to DMSO control and represent the mean ± SD of 3 independent experiments. (Two-tailed t test, *: p<0.05; **: p<0.01; ***: p<0.001)
Figure 5. GANT61 is more efficient than GDC-0449 in suppressing the expression of GLI target genes.
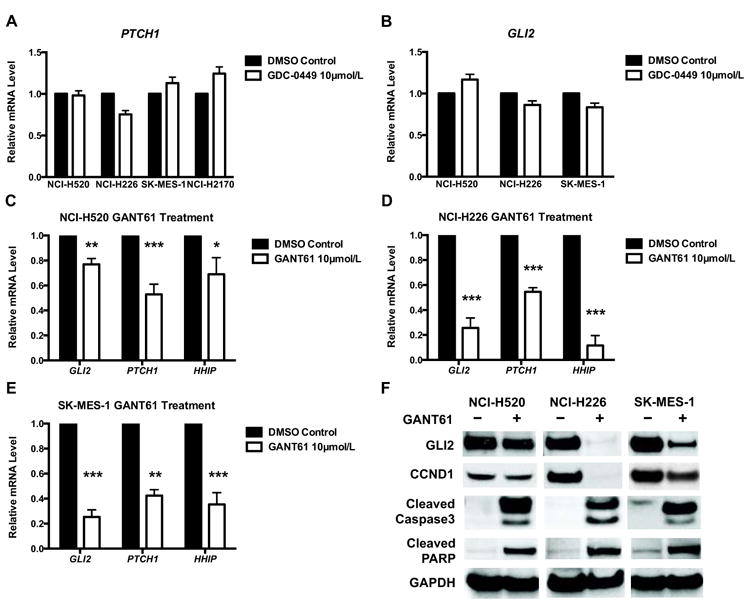
A∼B. Real-time PCR of GAPDH-normalized mRNA level of PTCH1 (A) and GLI2 (B) in cells treated with DMSO or 10 μmol/L GDC-0449 for 96 hours. C∼E. 96-hour exposure to 10 μmol/L GANT61 reduced mRNA levels of GLI targets. Gene expressions were normalized to endogenous GAPDH in each cell line. Data represent the mean ± SD of 3 independent experiments. (Two-tailed t test, *: p<0.05; **: p<0.01; ***: p<0.001) F. 96-hour treatment of 10 μmol/L GANT61 reduced protein levels of GLI2 and CCND1, and induced cleaved caspase-3 and cleaved PARP. GAPDH was used as the loading control. A representative Western blot from 3 independent experiments is shown.
In contrast, GANT61 demonstrated greater efficacy in a dose-dependent manner in all GLI-positive cells. The originally reported IC50 of GANT61 to reduce GLI-luciferase reporter activity is ≈5 μmol/L(29), and 5∼30 μmol/L is commonly used (29-31). In our studies, the IC50 of growth inhibition for three GLI-positive lines was ≈5 μmol/L (Figure 4C). Both NCI-H520 and NCI-H226 showed a 55% reduction at 5 μmol/L and 90% reduction at 10 μmol/L in cell survival. SK-MES-1 displayed approximate 40% and 60% decrease in viability at 5 and 10 μmol/L, respectively. As expected, GANT61 exhibited little cytotoxicity in GLI-negative NCI-H2170 cells. Consistently, increased apoptosis was seen in GLI-positive cell lines at corresponding GANT61 concentrations: NCI-H520 (1.8∼2.3 fold), NCI-H226 (2.8∼4 fold) and SK-MES-1 (2.4∼2.6 fold), but not in GLI-negative NCI-H2170 (Figure 4D).
Real-time PCR demonstrated significant reduction of HH downstream targets (GLI2, PTCH1 and HHIP) in NCI-H520 with a greater decrease in NCI-H226 and SK-MES-1 in comparison to DMSO control (Figure 5C∼E). Western analysis confirmed the reduction of GLI2 protein in GANT61-treated cells (Figure 5F). Cleaved caspase-3 and cleaved PARP were detected in cells receiving GANT61 (Figure 5F). The protein level of CCND1 was also decreased by GANT61 treatment, indicating impaired cell proliferation in addition to increased cell death (Figure 5F).
Taken together, our results suggest that targeting HH signaling at the level of GLI proteins may be more effective than targeting either the ligand SHH or the receptor SMO in LSCC, potentially due to the existence of the ligand or receptor independent pathway activation.
GANT61 suppresses GLI-positive tumor progression in vivo
Currently, there are no available transgenic murine models that faithfully recapitulate human LSCC. Recently, patient-derived xenograft models of LSCC have shown promise, but have not yet achieved satisfactory progress. Therefore, we used a xenograft model of representative human LSCC cell lines to determine the efficacy of GANT61 in vivo. GLI-positive NCI-H520 or NCI-H226 and GLI-negative NCI-H2170 cancer cells were injected subcutaneously into the right flank of immune deficient Rag1-/- mice. Mice were randomly divided into two groups when tumors reached median size ≈250 mm3 for NCI-H520 (n=8 for each group), ≈150 mm3 for NCI-H226 (n=5 for each group) and ≈230 mm3 for NCI-H2170 (n=5 for each group). We began treatment with either solvent control or GANT61 at a previously described dose of 50 mg/kg (29) by intraperitoneal injection every other day. During a 20-day treatment period, suppression of tumor growth was observed in the groups receiving GANT61 (Figure 6A and 6B) for both NCI-H520 and NCI-H226. In contrast, no significant difference of tumor growth was found in the NCI-H2170 xenograft (Figure 6C). GANT61 led to a significant 40% reduction of tumor weight for both GLI-positive cell lines in comparison to solvent control, but had no effects on GLI-negative NCI-H2170 tumors (Figure 6D∼F), suggesting specific anti-tumor efficacy of GANT61. No adverse side effects, such as weight loss, ulcerations, or general illness of the animals, were observed. Real-time PCR analysis confirmed that GANT61 reduced the mRNA level of GLI target genes PTCH1 and HHIP in NCI-H520 and NCI-H226 xenografts, but not in NCI-H2170 tumors (Figure 6G∼I).
Figure 6. GANT61 treatment suppressed GLI-positive tumor progression in vivo.

A∼C, Growth of NCI-H520 (A, n=8), NCI-H226 (B, n=5), and NCI-H2170 (C, n=5) xenografts (mean ± SD). D∼F, Measurement of tumor weight at the end of treatment for NCI-H520 (D, n=8), NCI-H226 (E, n=5), and NCI-H2170 (F, n=5). Shown is the mean ± SD. G∼I, Quantification of PTCH1 and HHIP mRNA by real-time PCR in treated tumors for NCI-H520 (G, n=4), NCI-H226 (H, n=5), and NCI-H2170 (I, n=5). Values were normalized against GAPDH. Shown is the mean ± SD of independent tumors in each group. In A∼I, data were analyzed by two-tailed t test. (*: p<0.05; **: p<0.01; ***: p<0.001, NS: not significant.)
Discussion
Aberrant HH signaling has been implicated in a diverse spectrum of human cancers. Previous studies have reported hyperactive HH signaling in a subset of LSCC, but they failed to address the complexity and heterogeneity of the disease. Four distinct molecular subtypes, which have different survival outcomes, patient populations, and biological processes, were identified by gene expression-subtype signatures (2). In the recent study of TCGA (3), we found that the HH activation is associated with the classical subtype (∼36% of LSCC). A consistent pattern was observed in an independent UNC microarray dataset (2). This observation is consistent with previous immunohistochemical studies (19, 20), which showed high activation of HH signaling in approximately 27% of LSCC patients. Among all four subtypes, the classical subtype (2, 3) has the highest proportion of smokers and the heaviest smoking history, as well as the greatest overexpression of three known oncogenes on 3q26 amplicon: SOX2, TP63 and PIK3CA (2, 3).
While GLI2 was consistently highly expressed in the classical subtype, strong positive correlations between GLI2 and the three best-known markers of the classical subtype on chromosome 3q were observed, together suggesting a critical role of GLI2 in LSCC, as found in SCC in other organs (22-24). Interestingly, the expression of ligand SHH within the classical subtype varied markedly and was not significantly different between subtypes. The expression patterns of other two HH ligands, IHH and DHH, were similar to SHH (Supplementary Figure S1J). These data indicate the existence of ligand-independent GLI activation in the classical subtype. Genetic alterations, including loss of PTCH function, constitutively active SMO, and amplification of GLI1/GLI2 have been reported to activate downstream HH signaling independent of ligands in various cancers. Moreover, GLI function can be modulated in a SMO-independent manner by PI3K/AKT (15), RAS-MEK signaling (16, 17), which may contribute to the hyperactive HH-GLI signaling in LSCC in addition to canonical HH signaling.
Recent development of multiple SMO inhibitors for human administration prompted us to test the potential clinical application to treat LSCC patients by targeting SMO. However, shRNAs knockdown of SMO showed only minor effects on LSCC cell survival and apoptosis, with little effect on HH downstream target gene expression. Consistently, GDC-0449 produced limited cytotoxicity despite the universal expression of SMO, suggesting the existence of SMO-independent regulation of GLI signaling. The strong and unique expression pattern of GLI2 prompted the query whether it is necessary for LSCC tumor progression. Knockdown of GLI2 induced apoptosis and significant growth inhibition in GLI2-positive LSCC cells, demonstrating an essential role of GLI2 in regulating cell viability and death. Importantly, the GLI inhibitor, GANT61, effectively blocked GLI-mediated signal transduction and suppressed tumor growth both in vitro and in vivo. Recent studies in neuroblastoma (33) and chronic lymphocytic leukemia (34) also showed that GANT61 effectively suppressed tumor progression in cancers insensitive to SMO inhibition. Interestingly, amplification of GLI2 or CCND1 have been proposed as additional mechanisms responsible for resistance to SMO inhibitors (27), and GANT61 treatment significantly reduced expression of GLI2 and CCND1 in LSCC cells.
GANT61 is a relatively new member in the HH inhibitor family, since most known HH pathway antagonists focus on the transmembrane activator SMO. Other readily available agents that inhibit GLI2 are rare. Arsenic trioxide (ATO), which is FDA-approved treatment for acute promyelocytic leukemia, has recently been described as a potent HH inhibitor. ATO has been shown to inhibit HH signaling by inhibiting GLI2 ciliary accumulation and promoting its degradation (38), and inhibit tumor growth in cancers with known drug-resistant SMO mutations and in the context of GLI2 overexpression (39). Due to the likelihood that compounds suppressing HH pathway-dependent proliferation in one cell type may be inactive in others, the clinical relevance of ATO in LSCC treatment is currently under investigation.
A recently emerging idea in clinical treatment is to combine several anti-tumor agents that specifically target different signaling pathways. It has been reported that GLI function can be modulated in a SMO-independent manner by PI3K/AKT signaling (15). Pharmacologic inhibition of PI3K/AKT signaling reduced tumor growth in GDC-0449-resistant medulloblastoma (27). Several inhibitors of the PI3K pathway are undergoing clinical evaluation. PIK3CA copy number gains and loss of PTEN function are prevalent in the classical subtype of LSCC, where the activation of HH signaling was observed. This coexistence raises the possibility that a combined therapy may be more beneficial than a monotherapy to enhance efficiency and overcome drug resistance.
Treatment options for LSCC overall are disappointing. Different from standard-of-care chemotherapy or small molecule inhibition of kinase signaling cascades, we present a potential strategy to treat a subset of LSCC patients by targeting the GLI transcriptional network. Our studies also highlight the need for agents that suppress GLI effectors with high efficacy and selectivity.
Supplementary Material
Translational Relevance.
Targeted therapeutics for lung squamous cell carcinoma (LSCC) are currently lacking. In this study, we have analyzed molecular subtypes of LSCC and identified overexpression of HEDGEHOG family members in the classical subtype. In representative LSCC cell lines, genetic deletion of SMOOTHENED (SMO) produced minor effects on cell survival, while GLI2 knockdown greatly reduced cell viability and induced extensive apoptosis. Using both in vitro and in vivo approaches, we evaluated therapeutic efficacy of GDC-0449, a clinically-available SMO inhibitor as well as GANT61, a targeted GLI inhibitor. GANT61 was significantly more effective than GDC-0449 in reduction of proliferation and induction of apoptosis. We report SMO-independent regulation of GLI in LSCC, and present a potential strategy of targeting GLIs to treat a subset of LSCC patients.
Acknowledgments
We would like to acknowledge Jing Zhang and Lixia Luo for animal husbandry.
Supported by HHMI Early Career Grant, DOD Promising Young Investigator Grant, TSFRE Research Grant
References
- 1.Drilon A, Rekhtman N, Ladanyi M, Paik P. Squamous-cell carcinomas of the lung: emerging biology, controversies, and the promise of targeted therapy. The Lancet Oncology. 2012;13(10):e418–e26. doi: 10.1016/S1470-2045(12)70291-7. [DOI] [PubMed] [Google Scholar]
- 2.Wilkerson MD, Yin X, Hoadley KA, Liu Y, Hayward MC, Cabanski CR, et al. Lung Squamous Cell Carcinoma mRNA Expression Subtypes Are Reproducible, Clinically Important, and Correspond to Normal Cell Types. Clinical Cancer Research. 2010;16(19):4864–75. doi: 10.1158/1078-0432.CCR-10-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422(6929):313–7. doi: 10.1038/nature01493. [DOI] [PubMed] [Google Scholar]
- 5.Park KS, Martelotto LG, Peifer M, Sos ML, Karnezis AN, Mahjoub MR, et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat Med. 2011;17(11):1504–8. doi: 10.1038/nm.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lum L, Beachy PA. The Hedgehog Response Network: Sensors, Switches, and Routers. Science. 2004;304(5678):1755–9. doi: 10.1126/science.1098020. [DOI] [PubMed] [Google Scholar]
- 7.Ruiz i Altaba A, Mas C, Stecca B. The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends in Cell Biology. 2007;17(9):438–47. doi: 10.1016/j.tcb.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thiyagarajan S, Bhatia N, Reagan-Shaw S, Cozma D, Thomas-Tikhonenko A, Ahmad N, et al. Role of GLI2 Transcription Factor in Growth and Tumorigenicity of Prostate Cells. Cancer Research. 2007;67(22):10642–6. doi: 10.1158/0008-5472.CAN-07-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikram MS, Neill GW, Regl G, Eichberger T, Frischauf AM, Aberger F, et al. GLI2 Is Expressed in Normal Human Epidermis and BCC and Induces GLI1 Expression by Binding to its Promoter. J Investig Dermatol. 2004;122(6):1503–9. doi: 10.1111/j.0022-202X.2004.22612.x. [DOI] [PubMed] [Google Scholar]
- 10.Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development. 2002;129(20):4753–61. doi: 10.1242/dev.129.20.4753. [DOI] [PubMed] [Google Scholar]
- 11.Eichberger T, Sander V, Schnidar H, Regl G, Kasper M, Schmid C, et al. Overlapping and distinct transcriptional regulator properties of the GLI1 and GLI2 oncogenes. Genomics. 2006;87(5):616–32. doi: 10.1016/j.ygeno.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Duman-Scheel M, Weng L, Xin S, Du W. Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature. 2002;417(6886):299–304. doi: 10.1038/417299a. [DOI] [PubMed] [Google Scholar]
- 13.Bigelow RLH, Chari NS, Undén AB, Spurgers KB, Lee S, Roop DR, et al. Transcriptional Regulation of bcl-2 Mediated by the Sonic Hedgehog Signaling Pathway through gli-1. Journal of Biological Chemistry. 2004;279(2):1197–205. doi: 10.1074/jbc.M310589200. [DOI] [PubMed] [Google Scholar]
- 14.Regl G, Kasper M, Schnidar H, Eichberger T, Neill GW, Philpott MP, et al. Activation of the BCL2 Promoter in Response to Hedgehog/GLI Signal Transduction Is Predominantly Mediated by GLI2. Cancer Research. 2004;64(21):7724–31. doi: 10.1158/0008-5472.CAN-04-1085. [DOI] [PubMed] [Google Scholar]
- 15.Riobó NA, Lu K, Ai X, Haines GM, Emerson CP. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(12):4505–10. doi: 10.1073/pnas.0504337103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, et al. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proceedings of the National Academy of Sciences. 2007;104(14):5895–900. doi: 10.1073/pnas.0700776104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nolan-Stevaux O, Lau J, Truitt ML, Chu GC, Hebrok M, Fernández-Zapico ME, et al. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes & Development. 2009;23(1):24–36. doi: 10.1101/gad.1753809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katoh Y, Katoh M. Hedgehog Target Genes: Mechanisms of Carcinogenesis Induced by Aberrant Hedgehog Signaling Activation. Current Molecular Medicine. 2009;9(7):873–86. doi: 10.2174/156652409789105570. [DOI] [PubMed] [Google Scholar]
- 19.Gialmanidis IP, Bravou V, Amanetopoulou SG, Varakis J, Kourea H, Papadaki H. Overexpression of hedgehog pathway molecules and FOXM1 in non-small cell lung carcinomas. Lung Cancer. 2009;66(1):64–74. doi: 10.1016/j.lungcan.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 20.Raz G, Allen KE, Kingsley C, Cherni I, Arora S, Watanabe A, et al. Hedgehog signaling pathway molecules and ALDH1A1 expression in early-stage non-small cell lung cancer. Lung Cancer. 2012;76(2):191–6. doi: 10.1016/j.lungcan.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 21.Shi I, Sadraei NH, Duan ZH, Shi T, Shi I, Sadraei NH, et al. Aberrant Signaling Pathways in Squamous Cell Lung Carcinoma. Cancer Informatics. 2011;10:273–85. doi: 10.4137/CIN.S8283. 2923-CIN-Aberrant-Signaling-Pathways-in-Squamous-Cell-Lung-Carcinoma2.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Snijders AM, Schmidt BL, Fridlyand J, Dekker N, Pinkel D, Jordan RCK, et al. Rare amplicons implicate frequent deregulation of cell fate specification pathways in oral squamous cell carcinoma. Oncogene. 2005;24(26):4232–42. doi: 10.1038/sj.onc.1208601. [DOI] [PubMed] [Google Scholar]
- 23.Yan M, Wang L, Zuo H, Zhang Z, Chen W, Mao L, et al. HH/GLI signalling as a new therapeutic target for patients with oral squamous cell carcinoma. Oral Oncology. 2011;47(6):504–9. doi: 10.1016/j.oraloncology.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 24.Yang L, Wang LS, Chen XL, Gatalica Z, Qiu S, Liu Z, et al. Hedgehog signaling activation in the development of squamous cell carcinoma and adenocarcinoma of esophagus. Int J Biochem Mol Biol. 2012;3(1):46–57. Epub 2012 Feb 10th. [PMC free article] [PubMed] [Google Scholar]
- 25.Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, et al. Treatment of Medulloblastoma with Hedgehog Pathway Inhibitor GDC-0449. New England Journal of Medicine. 2009;361(12):1173–8. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, et al. Inhibition of the Hedgehog Pathway in Advanced Basal-Cell Carcinoma. New England Journal of Medicine. 2009;361(12):1164–72. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 27.Dijkgraaf GJP, Alicke B, Weinmann L, Januario T, West K, Modrusan Z, et al. Small Molecule Inhibition of GDC-0449 Refractory Smoothened Mutants and Downstream Mechanisms of Drug Resistance. Cancer Research. 2011;71(2):435–44. doi: 10.1158/0008-5472.CAN-10-2876. [DOI] [PubMed] [Google Scholar]
- 28.Yauch RL, Dijkgraaf GJP, Alicke B, Januario T, Ahn CP, Holcomb T, et al. Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science. 2009;326(5952):572–4. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lauth M, Bergström Å, Shimokawa T, Toftgård R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proceedings of the National Academy of Sciences. 2007;104(20):8455–60. doi: 10.1073/pnas.0609699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazumdar T, DeVecchio J, Shi T, Jones J, Agyeman A, Houghton JA. Hedgehog Signaling Drives Cellular Survival in Human Colon Carcinoma Cells. Cancer Research. 2011;71(3):1092–102. doi: 10.1158/0008-5472.CAN-10-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazumdar T, DeVecchio J, Agyeman A, Shi T, Houghton JA. Blocking Hedgehog Survival Signaling at the Level of the GLI Genes Induces DNA Damage and Extensive Cell Death in Human Colon Carcinoma Cells. Cancer Research. 2011;71(17):5904–14. doi: 10.1158/0008-5472.CAN-10-4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu J, Rodova M, Roy SK, Sharma J, Singh KP, Srivastava RK, et al. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer letters. 2013;330(1):22–32. doi: 10.1016/j.canlet.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wickström M, Dyberg C, Shimokawa T, Milosevic J, Baryawno N, Fuskevåg OM, et al. Targeting the hedgehog signal transduction pathway at the level of GLI inhibits neuroblastoma cell growth in vitro and in vivo. International Journal of Cancer. 2013;132(7):1516–24. doi: 10.1002/ijc.27820. [DOI] [PubMed] [Google Scholar]
- 34.Desch P, Asslaber D, Kern D, Schnidar H, Mangelberger D, Alinger B, et al. Inhibition of GLI, but not Smoothened, induces apoptosis in chronic lymphocytic leukemia cells. Oncogene. 2010;29(35):4885–95. doi: 10.1038/onc.2010.243. [DOI] [PubMed] [Google Scholar]
- 35.Li B, Dewey C. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12(1):323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.R Core Team. R Foundation for Statistical Computing V, Austria. 2012. R: A language and environment for statistical computing. URL http://www.R-project.org.
- 37.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–307. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim J, Lee JJ, Kim J, Gardner D, Beachy PA. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proceedings of the National Academy of Sciences. 2010;107(30):13432–7. doi: 10.1073/pnas.1006822107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim J, Aftab Blake T, Tang Jean Y, Kim D, Lee Alex H, Rezaee M, et al. Itraconazole and Arsenic Trioxide Inhibit Hedgehog Pathway Activation and Tumor Growth Associated with Acquired Resistance to Smoothened Antagonists. Cancer Cell. 2013;23(1):23–34. doi: 10.1016/j.ccr.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.