

Abstract
Hexavalent chromium (Cr(VI)) is a widespread environmental contaminant and a known human carcinogen, generally causing bronchial cancer. Recent studies have shown that the particulate forms of Cr(VI) are the potent carcinogens. Particulate Cr(VI) is known to induce a spectrum of DNA damage such as DNA single strand breaks, Cr-DNA adducts, DNA-protein crosslinks and chromosomal aberrations. However, particulate Cr(VI)-induced DNA double strand breaks (DSBs) have not been reported. Thus, the aim of this study was to determine if particulate Cr(VI)-induces DSBs in human bronchial cells. Using the single cell gel electrophoresis assay (comet assay), showed that lead chromate-induced concentration dependent increases in DSBs with 0.1, 0.5, 1 and 5 μg/cm2 lead chromate inducing a 20, 50, 67 and 109% relative increase in the tail integrated intensity ratio, respectively. Sodium chromate at concentrations of 1, 2.5 and 5 μM induced 38, 78 and 107% relative increase in the tail integrated intensity ratio, respectively. We also show that genotoxic concentrations of lead chromate activate the ataxia telangiectasia mutated (ATM) protein, which is thought to play a central role in the early stages of DSB detection and controls cellular responses to this damage. The H2A.X protein becomes rapidly phosphorylated on residue serine 139 in cells when DSBs are introduced into the DNA by ionizing radiation. By using immunofluorescence, we found that lead chromate-induced concentration-dependent increases in phosphorylated H2A.X (r-H2A.X) foci formation with 0.1, 0.5, 1, 5 and 10 μg/cm2 lead chromate inducing a relative increase in the number of cells with r-H2A.X foci formation of 43, 51, 115 and 129%, respectively.
Keywords: Hexavalent chromium, DNA double strand breaks, ATM, Smc1, Lead chromate
1. Introduction
Exposure to hexavalent chromium [Cr(VI)] has been known for more than a century to be associated with induction of cancer in humans, especially bronchial carcinoma, causing an 18–80-fold increased risk of lung cancer [1–4]. However, all Cr(VI) compounds are not equally potent as carcinogens. In particular, the water-insoluble (particulate) Cr(VI) salts are more potent carcinogens than the water soluble ones [2–5]. For example, only the particulate compounds are consistently tumorigenic in experimental animals [2–4] and only the particulates induce neoplastic transformation of C3H10T1/2 mouse embryo cells [5]. Further data from pathological studies of workers with Cr(VI)-induced lung cancers indicate that Cr(VI)-induced tumors originate in bronchial cells at the site of bifurcations where Cr(VI) particles are most likely to impact and persist. These observations strongly support the conclusion that particulate Cr(VI) compounds are the carcinogenic compounds [6,7]. However, the carcinogenic mechanisms of particulate Cr(VI) are poorly understood and the full spectrum of particulate Cr(VI)-induced genotoxic damage has not been established.
Most studies using particulate Cr(VI) have focused on lead chromate (LC) as a model compound for the particulate salts [8,9]. Studies in human lung cells showed that LC particles partially dissolve outside the cell releasing chromate and Pb ions, which enter the cell [10,11]. The internalized Cr ions then cause DNA damage including adducts, single strand breaks and chromosomal aberrations, cytotoxicity and growth inhibitory effects [10–17]. The internalized Pb ions have no apparent effect [10,16–17]. However, the full spectrum of damage induced by particulate Cr(VI) has not been determined as its ability to induce DNA double strand breaks (DSBs) has not been investigated. DSBs are of particular concern because they are most likely to induce genomic and chromosomal instability, which are hallmarks of Cr(VI)-induced tumors [18]. DSBs are particularly dangerous because, in addition to the potential for mutations to develop at their site of occurrence, incorrect DSB repair can lead to chromosomal translocations that generate chromosome instability [19].
Only two studies have considered soluble Cr(VI)-induced DSBs [20,21]. Each reported positive results; however, both studies considered only very acute exposures (0.5–3 h) with high concentrations and neither determined if the DSBs occur in human lung cells. Specifically, Wakeman et al. [21] treated HeLa cells with potassium chromate for 30 min and Ha et al. [20] treated primary human skin fibroblasts (HSF) for 1–3 h. Particulate chromate requires some time to dissolve and cannot achieve these levels in such a short period of time [8,10,16]. It remains to be seen if chromate induces DSBs at the lower and more chronic exposures that occur with particulate compounds and if these effects occur in human lung cells. Accordingly, the purpose of this study was to improve our current understanding of particulate Cr(VI) and study the induction and response to particulate chromate-induced DSBs in human lung cells.
2. Materials and methods
2.1. Chemicals and reagents
Lead chromate, sodium chromate, demecolchicine, D-ascorbic acid and potassium chloride (KCl) were purchased from Sigma (St. Louis, MO). Giemsa stain was purchased from Biomedical Specialties Inc. (Santa Monica, CA). Crystal violet, methanol and acetone were purchased from J.T. Baker (Phillips-burg, NJ). Dulbecco’s minimal essential medium and Ham’s F-12 medium (D-MEM/F-12) were purchased from Mediatech Inc. (Herndon, VA). Cosmic calf serum (CCS) was purchased from Hyclone, (Logan, UT). Gurr’s buffer, trypsin/EDTA, sodium pyruvate, penicillin/streptomycin and L-glutamine were purchased from Invitrogen Corporation (Grand Island, NY). Tissue culture dishes, flasks and plasticware were purchased from Corning Inc. (Acton, MA).
2.2. Cells and cell culture
Human lung fibroblasts, WTHBF-6 cells, were used in these studies. This cell line and the importance of studying Cr(VI) effects in fibroblasts have previously been described [10,11,13–15]. Briefly, WTHBF-6 cells, a clonal cell line derived from primary human bronchial fibroblasts (PHBF) with reconstituted telomerase activity, exhibit a diploid karyotype, normal growth parameters and an extended lifespan (currently greater than 700 population doublings). These cells have genotoxic and cytotoxic responses to metals that are the same as their parent primary human lung fibroblasts [15]. WTHBF-6 cells were cultured in a 50:50 mix of Dulbecco’s minimal essential medium and Ham’s F-12 medium plus 15% cosmic calf serum, 1% L-glutamine, 0.1 mM sodium pyruvate and 1% penicillin/streptomycin. All cells were maintained in a 37 °C, humidified incubator with 5% CO2.
2.3. Chromium preparations
Suspensions of lead chromate particles were prepared and administered as previously described [10] and were used as a model compound to study particulate chromium. The sizes of lead chromate particles ranged from 0.4 to 58.4 μm with a mean value of 2.7 μm, with less than 5% of the particles larger than 10 μm, and are described in more detail elsewhere [10]. Sodium chromate was used as a model for the soluble form of Cr(VI) and was administered as previously described [13].
2.4. Cytotoxicity assays
Cytotoxicity was determined by a clonogenic assay measuring the reduction in plating efficiency in treatment groups relative to controls as previously described [13]. There were four dishes per treatment group and each experiment was repeated at least three times.
2.5. Clastogenicity
Clastogenicity was determined by measuring the amount of chromosomal damage in treatment groups and controls exactly as previously described [13]. One hundred metaphases per data point were analyzed in each experiment. Each experiment was repeated at least three times.
2.6. Neutral comet assay
DSBs were measured using a gel electrophoresis assay (comet assay) adapted from the initial protocol of Singh et al. [22]. Briefly, 10 μl of cell suspension (~10 ×103 cells) was mixed with 75 μl of 0.5% low-melting agarose (Amresco, Solon, OH) and spread on MGE slides (Erie Scientific Inc, Portsmouth, NH), pre-coated with 100 μl 0.5% low-melting agarose. The gel was allowed to solidify under a coverslip on ice before a third layer containing 120 μl of low-melting agarose was placed on top and placed on ice to solidify. The cells were then lysed by immersing the slides for 2 h in a freshly prepared lysis solution (2.5 M NaCl, 100 mM EDTA (tetra sodium EDTA), 10 mM Tris, pH 10) containing additional 1% Triton X-100 at 4 °C. After cell lysis, the slides were immersed in lysis solution with 10 ug/ml of RNase (Sigma, St. Louis, MO) at 37 °C for 2 h followed by immersion in lysis solution with 1 mg/ml proteinase-K (Amresco, Solon, OH) overnight at 37 °C. Next, slides were dipped in cold deionized water and placed in a horizontal gel electrophoresis apparatus containing freshly prepared electrophoresis buffer (300 mM sodium acetate, 100 mM Tris, pH 9.0) for 20 min to allow equilibration of microgels. Electrophoresis was then carried out at 20 V, 140 mA (1 V/cm) for 10 min at 4 °C. All the steps described above were conducted under a reduced light level to prevent spurious DNA damage. Slides were neutralized by immersion in freshly prepared 1 M ammonium acetate in ethanol for 15 min and next in freshly prepared 1 mg/ml spermine (Sigma, St. Louis, MO) in 70% ethanol for 15 min. Slides were air-dried. One slide at a time was pre-stained with 50 μl of 5% sucrose in 10 mM NaH2PO4 and stained with 50 μl of 0.25 mM YOYO-1 in 5% sucrose and 5% DMSO. Images were captured using an Olympus fluorescence microscope equipped with a SensiCam camera. Images were analyzed using VisCOMET software (Inpuls, German). Analysis measures included the tail integrated intensity ratio (described in detail in 22) and tail length. Two slides were prepared for each concentration treatment with 50 cells analyzed per slide for a total of 100 nuclei per concentration.
2.7. Western blot analysis
Cells were lysed in 50 mM Tris (pH 8.0), 200 mM NaCl, 1% Igepal CA-630 supplemented with 5 ug/ml aprotinin, 5 ug/ml leupeptin, 1 mM NaF, 20 mM β-glycerophosphate, 1 mM sodium vanadate, 1 mM dithiothreitol and 1 mM phenylmethylsulfonyl fluoride. Cell lysates were clarified by microcentrifugation, and the supernatant fractions were saved prior to protein concentration determination. Equal samples were resolved by 3–8% SDS–PAGE for ATM and 4–15% gradient SDS–PAGE gels for SMC1 and then transferred to nitrocellulose membranes. The membranes were probed with anti-phospho-ATM antibody (Rockland, Gilbertsville, PA) overnight for ATM and 2 h with polyclonal SMC1 antibody (Bethyl Laboratories) or polyclonal phospho-specific SMC1 Ser966 antibody (Bethyl Laboratories). For ATM the membranes were then probed with secondary antibody Alexa Fluor 680 antibody (Molecular Probes, Eugene, OR) and IRDye 800 antibody (Rockland, Gilbertsville, PA). The secondary antibody was visualized by LI-COR Odyssey Infrared Imaging System (LI-COR Bioscience, Lincoln, Nebraska). Equal protein loading was confirmed by immunblotting with an antibody to constitutively expressed β-actin (Abcam, Cambridge, MA). For SMC1 the membranes were then probed with HRP-conjugated secondary antibody for 1 h and the blots visualized by ECL western blotting detection reagents (Amersham Biosciences, Piscataway, NJ).
2.8. Immunofluorescence for r-H2A.X foci formation
Gamma-H2A.X foci were also used to detect the presence of chromate-induced DSBs. Cells were grown on chamber slides. After treatment with Cr(VI) for 24 h, the cells were fixed in 4% paraformaldehyde for 10 min, permeablilized with 0.2% Triton X-100 for 5 min and blocked with 1% BSA and 5% horse serum (Jackson Immunolaboratories, West Grove, PA) for 1 h. Cells were then incubated with anti-r-H2A.X antibody (Cell Signaling, Beverly, MA) at 4 °C overnight and incubated with a FITC AlexaFluor 484-conjugated goat anti-rabbit Ig G second antibody for 1 h. Nuclei were counterstained with propidium iodide. The slides were mounted and viewed with an Olympus laser scanning confocal microscope (LSCM) using a 60× objective. Images of the same antibody staining were obtained using the same LCSM parameters (brightness, contrast, pinhall, etc.).
2.9. Apoptosis detection by flow cytometry
Cells were seeded at 2 ×10 5/60 mm dishes and treated with 0, 0.1, 0.5, 1 and 5 μg/cm2 lead chromate or 0, 0.5, 1, 2.5 and 5 μM sodium chromate for 24 h and then washed with PBS. Cells were harvested with trypsin and the cell pellets were resuspended in cell medium at 1 ×106 cells/ml. Cells were incubated with Annexin V-biotin for 15 min at room temperature. After adding conjugated streptavidin and propidium iodide, cells were analyzed on a BD FACS Calibur flow cytometer immediately and the data analyzed with Cel-lQuest V. 4.0 software (BD Bioscience).
2.10. Cell cycle analysis
Subconfluent cell monolayers were treated for 24 h with lead chromate or sodium chromate and analyzed for cell cycle effects. Ice cold nuclear isolation media (330 μl) was added to each suspension to disrupt cell membranes and stain nuclei. Fluorescing nuclei were analyzed on NPE Systems cell cycle analyzer. Twenty thousand events were recorded for each suspension producing histograms identifying G0/G1, S and G2/M phases. Histograms were analyzed using Modfit LT 5.0 software (Verity Software House, Topsham, ME).
2.11. Determination of intracellular Cr Ion levels
Cells were prepared for determination of intracellular Cr levels as previously described (10). A Perkin-Elmer Optima 2000 ICP-AES, equipped with a gem cone low flow nebulizer, was used to determine the intracellular levels of Cr. Solutions were introduced to the nebulizer using a peristaltic pump operating at 2 ml/min. Samples of extracellular and intracellular fluids were diluted 4–5× in 0.16 M aqueous HNO3 prior to analysis. Cr was determined using emission wavelength at 267.716 with a minimum detection limit of 2 ppb. Y was used as an internal standard for Cr determinations.
2.12. Statistics
P values were calculated based on t-test for determining the statistical significance of the difference in means at 99 or 95% confidence intervals. No adjustment was made for multiple comparisons.
3. Results
3.1. Cytotoxicity and clastogenicity of lead chromate particles
The aim was to determine if lead chromate induces DSBs and to determine the initial cellular response to that damage. Thus, the intent was to select concentrations of lead chromate that induced DNA damage, but that were not completely cytotoxic. We selected concentrations over a range of cytotoxicity from low to high toxicity in WTHBF-6 cells (Table 1). Concentrations of 0.1, 0.5, 1 and 5 μg/cm2 induced 85, 60, 36 and 3% relative survival, respectively. This cytotoxicity assay measures the long-term survival of these cells and not the amount of death in the first 24 h of exposure. To account for the potential confounding role of apoptosis, the amount of apoptosis that occurred at these concentrations in the first 24 h was determined and found that it was minimal (Table 1).
Table 1.
Cytotoxicity and clastogenicity of lead chromatea
| Treatment (μg/cm2 LC) | Relative survival (% of control) | Percent of metaphases with damageb,c | Total damage in 100 metaphasesb,c | Percent of apoptosis |
|---|---|---|---|---|
| 0d | 100 | 2 ±0.4 | 2 ±0.4 | 2.9 ±0.5 |
| 0.1 | 83 | 9 ±1.1e | 9 ±1.2e | 3.3 ±0.2 |
| 0.5 | 66 | 23 ±2.9e | 27 ±4.2e | 4.1 ±0.7 |
| 1.0 | 56 | 34 ±1.9e | 42 ±3.0e | 4.2 ±0.2 |
| 5.0 | 8 | NMf | NM | 6.8 ±0.7 |
Average of three experiments.
±Standard error of the mean.
Average of five experiments.
0: vehicle control (acetone).
Significantly (p < 0.001) different compared to vehicle control.
NM: no metaphases.
Table 1 also shows that lead chromate was clastogenic in a concentration-dependent manner. Concentrations of 0.1, 0.5 and 1.0 μg/cm2 damaged 9, 23 and 34% of metaphases, respectively. There was also a concentration-dependent increase in the number of metaphases with multiple aberrations as these concentrations induced 9, 27 and 42 aberrations per 100 metaphases, respectively. Metaphases were not observed at concentrations of 5 and 10 μg/cm2, which indicated that complete cell cycle arrest had occurred. Accordingly we focused our efforts on a concentration range of 0.1–5 μg/cm2.
3.2. Lead chromate particles induce DNA double strand breaks
We next took two approaches to determine if lead chromate-induced DNA DSBs. Breaks were measured directly using a single cell microgel electrophoresis assay also known as the ‘comet’ assay and indirectly by measuring r-H2A.X foci formation. The comet assay showed that lead chromate did induce concentration-dependent increases in DSBs with 0.1, 0.5, 1 and 5 μg/cm2 lead chromate inducing a 20, 50, 67 and 109% relative increase in the tail integrated intensity ratio (Fig. 1). These results were confirmed by measuring r-H2A.X foci formation, and showed that concentrations of 0.1, 0.5, 1 and 5 μg/cm2 lead chromate induced a 43, 51, 115 and 129% relative increase in the number of cells with r-H2A.X foci (Fig. 2). H2A.X is phosphorylated by ATM in response to DNA damage resulting in the formation of discrete repair foci at the sites of DNA DSBs [23,24]. The presence of DNA DSBs and r-H2A.X foci suggest that ATM is activated by lead chromate.
Fig. 1.

Lead chromate induces DNA DSBs in human lung cells. (A) Photomicrographs of human lung fibroblasts following lead chromate treatment and neutral microgel eletrophoresis. (I) Control; (II) 0.1 μg/cm2 LC; (III) 5 μg/cm2 LC. Magnification: 400×, dye: YOYO-1. (B–E) Quantitation of DNA DSBs in human lung fibroblasts following lead chromate treatment and neutral microgel electrophoreses. Data represent a minimum of three experiments. Error bars = standard error of the mean. (*) Significantly (p < 0.01) different when compared to vehicle control.
Fig. 2.

Lead chromate induces r-H2A.X foci formation in human lung cells. (A) Photomicrographs of r-H2A.X foci (green) in WTHBF-6 cells treated with lead chromate. Nuclei were stained with propidium iodide (red). Cells were analyzed for r-H2A.X foci formation by immunofluorescence with anti-r-H2A.X antibody. Hydrogen peroxide was used as the positive control. (B and C) Quantitation of r-H2A.X foci in human lung fibroblasts following lead chromate treatment. Data represent a minimum of three experiments. Error bars = standard error of the mean. (*) Significantly (p < 0.05) different compared to vehicle control.
3.3. ATM activation phosphorylation
To investigate the potential role of ATM in lead chromate-induced DSBs, ATM phosphorylation after lead chromate exposure of WTHBF-6 cells was measured. The results demonstrated an increase in phosphorylation of Ser-1981 on ATM after lead chromate exposure, indicating that ATM is indeed activated (Fig. 3).
Fig. 3.
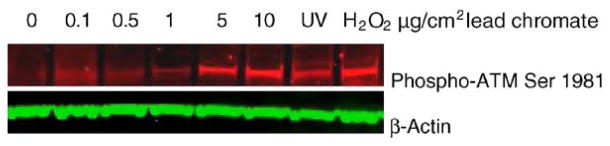
ATM undergoes inter-molecular autophosphorylation at Ser1981 in human lung cells after lead chromate exposure. Protein extracts were analyzed for ATM phosphorylation by Western blotting with anti-phospho-ATM antibody (red). Equal protein loading was confirmed by immunblotting with anti β-actin antibody (green). Hydrogen peroxide was used as the positive control.
The major signaling role of ATM after DNA DSBs detection is to signal the cells to stop dividing, particularly causing S-phase arrest (19). We investigated the effects of lead chromate on the cell cycle and confirmed that these concentrations did indeed induce S-phase arrest (Fig. 4A). Specifically, 0.5, 1 and 5 μg/cm2 induced a concentration-dependent decrease in the cells in G1 and an increase in the cells in S-phase. ATM mediated S-phase arrest has been shown to be mediated by SMC1 (21) and consistent with this arrest, lead chromate-induced concentration-dependent increases in SMC1 (Fig. 4B).
Fig. 4.

Lead chromate induces cell cycle arrest in human lung cells. (A) Cell cycle analysis of cells treated with lead chromate for 24 h as described in Section 2. Data represent a minimum of three experiments. Error bars = standard error of the mean. (*) Statistically different from control (p < 0.01). (#) Statistically different from control (p < 0.015). (B) Smc1 is phosphorylated at Ser 966 after lead chromate treatment. Smc1 phosphorylation was assessed by western blotting analyses using the phosphoserine-specific antibodies (top) or an anti-Smc1 antibody (bottom panel).
3.4. Comparison of lead chromate and sodium chromate
Previous studies have indicated that the physico-chemical mechanism for the cytotoxic and clastogenic effects of lead chromate is mediated by soluble chromate ions [10]. We investigated whether this was also true for DNA DSBs. Soluble chromate ions (from sodium chromate) also induced DNA DSBs after a 24 h exposure (Fig. 5A). Specifically, treatments with concentrations of 1, 2.5 and 5 μM resulted in a 38, 78 and 107% relative increase in the tail integrated intensity ratio respectively as detected with the comet assay. Similarly, soluble chromate ions induced concentration-dependent increases in r-H2A.X foci (Fig. 5B). Concentrations of 1, 2.5 and 5 μM sodium chromate induced 56, 128 and 104% relative increases in cells with r-H2A.X foci respectively. Comparing the damage measured after both lead chromate and sodium chromate exposure based on the amount of soluble intracellular chromium ions that they produce indicates that both were the same (Fig. 6A and B). We investigated the cellular response to soluble chromate ions and found that it matched that of lead chromate with ATM induction, S-phase arrest and SMC1 activation (Fig. 7A, B and C). Thus, altogether, these data confirm that lead chromate-induced DNA DSBs in human lung cells are mediated by soluble chromate ions.
Fig. 5.

Sodium chromate induces DNA DSBs in human lung cells. (A–D) Quantitation of DNA DSBs in human lung fibroblasts following sodium chromate treatment and neutral microgel electrophoreses. Tail integrated intensity ratio is the product measurements of breadth, position and total intensity divided by the sum of all intensity values. (E–F) Quantitation of r-H2A.X foci in human lung fibroblasts following sodium chromate treatment. For all panels, data represent a minimum of three experiments. Error bars = standard error of the mean. (*) Significantly (p < 0.01) different compared to vehicle control. (#) Significantly (p < 0.05) different compared to vehicle control.
Fig. 6.

Lead chromate and sodium chromate induce the same level of DNA DSBs in human cells when compared by intracellular chromium ion levels. (A) Quantitation of DNA DSBs in human lung fibroblasts following lead chromate or sodium chromate treatment and neutral microgel electrophoreses based on measured intracellular chromium ion levels. Tail integrated intensity ratio is the product measurements of breadth, position and total intensity divided by the sum of all intensity values. Data represent a minimum of three experiments. Error bars = standard error of the mean. (B) Quantitation of r-H2A.X foci in human lung fibroblasts following lead chromate or sodium chromate treatment based on measured intracellular chromium ion levels. Data represent a minimum of three experiments. Error bars = standard error of the mean.
Fig. 7.

Sodium chromate induces similar cell cycle effects as lead chromate in human lung cells. (A) ATM undergoes inter-molecular autophosphorylation at Ser1981 in human lung cells after sodium chromate exposure. Protein extracts were analyzed for ATM phosphorylation by Western blotting with anti-phospho-ATM antibody (red). Equal protein loading was confirmed by immunblotting with anti β-actin antibody (green). Hydrogen peroxide was used as the positive control. (B) Cell cycle analysis of cells treated with lead chromate for 24 h as described in Section 2. Sodium chromate at the highest concentration causes cells to accumulate in S-phase. Data represent a minimum of three experiments. Error bars = standard error of the mean. (*) Statistically different from control (p = 0.005). (C) Smc1 is phosphorylated at Ser 966 after sodium chromate treatment. Smc1 phosphorylation was assessed by immunoblot analyses using the phosphoserine-specific antibodies (top) or an anti-Smc1 antibody (bottom panel).
4. Discussion
Particulate chromate compounds target fibroblasts and epithelial cells at bronchial bifurcation sites in human lungs [7,25]. These particulates are potent carcinogens because they persist in the lungs for extended periods of time and induce genotoxic damage [26–29]. However, the full spectrum of damage induced by these compounds has not been determined. Specifically, the ability of particulate chromates to induce DNA DSBs had not been investigated. DSBs are one of the most dangerous types of DNA damage as even a single break leading to cell death and their repair is potentially highly promutagenic [19]. This is the first study to report that particulate chromate induces DSBs. We found that H2A.X is phosphorylated and forms repair foci in response to these breaks. In addition the cell-cycle apparatus is signaled through the phosphorylation of the checkpoint kinase ATM, which in turn activates SMC1 and induces S-phase arrest. This damage response is consistent with the pathways found for ionizing radiation-induced DNA DSB.
We also found that the induction of and response to particulate Cr(VI)-induced DSBs is mediated by soluble chromate ions. These observations are consistent with our previous studies showing that in the first 24 h of exposure, soluble chromate ions are responsible for the chromosome damage, cytotoxicity, growth inhibition and cell cycle arresting effects of lead chromate (10–17). They are the first data to extend this Cr ion-mediated effect to a cell signaling pathway, and they extend our understanding of the physico-chemical mechanism of particulate Cr(VI) in human lung cells to include DSBs.
A previous report indicated that soluble chromate can induce DSB and activate ATM [20]. However, this study was done after acute (3 h) exposures in skin cells. Similarly, a second study showed that soluble chromate-induced DSB and activated both ATM and SMC1 after very acute (30 min) exposures in HeLa cells [21]. Our data concerning soluble chromate are consistent with these earlier studies and extend the knowledge of soluble chromate’s effects to include longer exposures (24 h) in actual target cells (bronchial cells).
The ultimate cause of chromate-induced DSBs is uncertain. Ha et al. [20] showed data indicating that the breaks did not occur in G1, but rather most likely occurred in S-phase, though the study did not account for possible breaks in G2/M. This suggests, and indeed the authors argue, that the breaks may be related to stalled replication forks, possibly caused by DNA–DNA crosslinks and thus are formed indirectly as a result of crosslink repair. However, lead chromate has not been shown to induce DNA–DNA crosslinks and so it is uncertain if this mechanism applies to particulate chromate. In addition, it is still possible that a portion of the breaks may have occurred in G2/M and thus some portion of the breaks may reflect direct strand breakage, perhaps as a consequence of exposure to one of the highly reactive metabolites such as pentavalent chromium or free radical species, produced during the intracellular reduction of Cr(VI).
Our data do indicate that these are DSBs induced either through misrepair of crosslinks or direct strand damage and not indirectly as the result of apoptosis. Specifically, we find only background levels of apoptosis occurring at the concentrations we studied. Moreover, when DNA is fragmented during apoptosis, such fragmentation is in the size of nucleosome oligomers, and studies have shown that such small pieces of DNA are not generally detected by the comet assay [30]. In addition, several studies showed that the comet assay is a valid indicator of genotoxic damage and that the comet images are not the result of apoptotic nuclear fragmentation [31,32].
Our data expand the overall hypothesis for particulate chromate-induced carcinogenesis in human lung cells. We extended the hypothesis (Fig. 8) to indicate that particles dissolve outside lung cells to release chromate and Pb ions, which are taken up into the cell [9,10]. Once inside the cell, chromate ions are reduced to trivalent chromium complexes and several possible and potentially reactive intermediates [33]. Next, either trivalent chromium, a reactive metabolic intermediate, or some combination of these species induces DSB [this report], DNA adducts, single strand breaks and chromosomal aberrations [10–15], cell cycle arrest and growth inhibition [16] and cell death [10,11,13]. In response to DSBs, ATM is auto-phosphorylated and transduces signals to SMC1 and possibly H2A.X leading to cell cycle arrest [11], growth inhibition [11], repair and apoptosis (12, Fig. 8). Unrepaired or mis-repaired DSBs may also be involved in the generation of chromosomal lesions by chromate. Ultimately, a mutated or chromosomally unstable cell emerges from these events and progresses to tumorigenesis. Future research is aimed at exploring the relationship of Cr(VI)-induced DSBs to chromosomal instability.
Fig. 8.

Proposed mechanism of particulate Cr(VI) genotoxicity. In response to DNA DSBs induced by Cr ions released from the extracellular dissolution of lead chromate, ATM kinases are activated. ATM, through the intermediacy of H2A.X, phosphorylates SMC1 on Ser 966 which leading to cell cycle arrest, growth inhibition, repair and apoptosis.
Acknowledgments
We thank Shawn Holt and Lynne Elmore for their expertise and assistance in creating the hTERT immortalized cell line; Dan Swett for technical assistance with the apoptosis assay; Jonathan Moreland for technical assistance with the cell cycle arrest assay; and Geron Corporation for the use of the hTERT materials. This work was supported by NIEHS grant ES10838 (J.P.W.), the Maine Center for Toxicology and Environmental Health at the University of Southern Maine, NIEHS grant ES013301 (B.X), NIOSH student training grant (T.P.W.) and Cancer Association of Greater New Orleans (CAGNO).
References
- 1.Newman DA. A case of adeno-carcinoma of the left inferior turbinated body, and perforation of the nasal septum, in the person of a worker in chrome pigments. Glasgow Med J. 1981;33:469–470. [Google Scholar]
- 2.IARC. Monographs on the Evaluation of Carcinogenic Risks to Humans: Chromium, Nickel and Welding. Vol. 49. International Agency for Research on Cancer; Lyons, France: 1990. [PMC free article] [PubMed] [Google Scholar]
- 3.Léonard A, Lauwerys RR. Carcinogenicity and mutagenicity of chromium. Mutat Res. 1980;76:227–239. doi: 10.1016/0165-1110(80)90018-4. [DOI] [PubMed] [Google Scholar]
- 4.Levy LS, Vanitt S. Carcinogenicity and mutagenicity of chromium compounds: the association between bronchial metaplasia and neoplasia. Carcinogenesis. 1986;7:831–835. doi: 10.1093/carcin/7.5.831. [DOI] [PubMed] [Google Scholar]
- 5.Patierno SR, Banh D, Landolph JR. Transformation of C3H/10T1/2 mouse embryo cells by insoluble lead chromate but not soluble calcium chromate: relationship to mutagenesis and internalization of lead chromate particles. Cancer Res. 1988;47:3815–3823. [PubMed] [Google Scholar]
- 6.Ishikawa Y, Nakagawa K, Satoh Y, Kitagawa T, Sugano H, Hirano T, Tsuchiya E. Characteristics of chromate workers’ cancers, chromium lung deposition and precancerous bronchial lesions: an autopsy study. Br J Cancer. 1994;70:160–166. doi: 10.1038/bjc.1994.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishikawa Y, Nakagawa K, Satoh Y, Kitagawa T, Sugano H, Hirano T, Tsuchiya E. “Hot spots” of chromium accumulation at bifurcations of chromate workers’ bronchi. Cancer Res. 1994;54:2342–2346. [PubMed] [Google Scholar]
- 8.Wise JP, Orenstein JM, Patierno SR. Inhibition of lead chromate clastogenesis by ascorbate: relationship to particle dissolution and uptake. Carcinogenesis. 1993;14:429–434. doi: 10.1093/carcin/14.3.429. [DOI] [PubMed] [Google Scholar]
- 9.Wise JP, Sr, Stearns DM, Wetterhahn KE, Patierno SR. Cell-enhanced dissolution of carcinogenic lead chromate particles: the role of individual dissolution products in clastogenesis. Carcinogenesis. 1994;15:2249–2254. doi: 10.1093/carcin/15.10.2249. [DOI] [PubMed] [Google Scholar]
- 10.Xie H, Holmes AL, Wise SS, Gordon N, Wise JP., Sr Lead chromate-induced chromosome damage requires extracellular dissolution to liberate chromium ions but does not require particle internalization or intracellular dissolution. Chem Res Toxicol. 2004;17:1362–1367. doi: 10.1021/tx0498509. [DOI] [PubMed] [Google Scholar]
- 11.Wise SS, Holmes AL, Ketterer ME, Hartsock WJ, Fomchenko E, Katsifis S, Thompson WD, Wise JP., Sr Chromium is the proximate clastogenic species for lead chromate-induced clastogenicity in human bronchial cells. Mutat Res. 2004;560:79–89. doi: 10.1016/j.mrgentox.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 12.Singh J, Pritchard DE, Carlisle DL, Mclean JA, Montaser A, Orenstein JM, Patierno SR. Internalization of carcinogenic lead chromate particles by cultured normal human lung epithelial cells: formation of intracellular lead-inclusion bodies and induction of apoptosis. Toxicol Appl Pharmol. 1999;161:240–248. doi: 10.1006/taap.1999.8816. [DOI] [PubMed] [Google Scholar]
- 13.Wise JP, Sr, Wise SS, Little JE. The cytotoxicity and genotoxicity of particulate and soluble hexavalent chromium in human lung cells. Mutat Res. 2002;517:221–229. doi: 10.1016/s1383-5718(02)00071-2. [DOI] [PubMed] [Google Scholar]
- 14.Wise SS, Schuler JHC, Katsifis SP, Wise JP., Sr Barium chromate is cytotoxic and genotoxic to human lung cells. Environ Mol Mutagen. 2003;42:274–278. doi: 10.1002/em.10203. [DOI] [PubMed] [Google Scholar]
- 15.Wise SS, Elmore LW, Holt SE, Little JE, Bryant BH, Wise JP., Sr Telomerase-mediated lifespan extension of human bronchial cells does not affect hexavalent chromium-induced cytotoxicity or genotoxicity. Mol Cell Biochem. 2004;255:103–111. doi: 10.1023/b:mcbi.0000007266.82705.d9. [DOI] [PubMed] [Google Scholar]
- 16.Wise SS, Holmes AL, Moreland JA, Xie H, Sandwick SJ, Stackpole MM, Fomchenko E, Teufack S, May AJ, Jr, Katsfis SP, Wise JP., Sr Human lung cell growth is not stimulated by lead ions after lead chromate-induced genotoxicity. Mol Cell Biochem. doi: 10.1007/s11010-005-8217-0. in press. [DOI] [PubMed] [Google Scholar]
- 17.Holmes AL, Wise SS, Xie H, Gordon N, Thompson WD, Wise JP., Sr Lead ions do not cause human lung cells to escape chromate-induced cytotoxicity. Toxicol Appl Pharmacol. 2005;203:167–176. doi: 10.1016/j.taap.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 18.Hirose T, Kondo K, Takahashi Y, Ishikura H, Fujino H, Tsuyuguchi M, Hashimoto M, Yokose T, Mukai K, Kodama T, Monden Y. Frequent microsatellite instability in lung cancer from chromate-exposed workers. Mol Carcinogen. 2002;33(3):172–180. doi: 10.1002/mc.10035. [DOI] [PubMed] [Google Scholar]
- 19.Jackson SP. Detecting, signalling and repairing DNA double-strand breaks. Biochem Soc Trans. 2001;29:655–661. doi: 10.1042/0300-5127:0290655. [DOI] [PubMed] [Google Scholar]
- 20.Ha L, Ceryak S, Patierno SR. Generation of S phase-dependent DNA double-strand breaks by Cr(VI) exposure: involvement of ATM in Cr(VI) induction of gamma-H2AX. Carcinogenesis. 2004;25(11):2265–2274. doi: 10.1093/carcin/bgh242. [DOI] [PubMed] [Google Scholar]
- 21.Wakeman TP, Kim WJ, Callens S, Chiu A, Brown KD, Xu B. The ATM-SMC1 pathway is essential for activation of the chromium(VI)-induced S-phase checkpoint. Mutat Res. 2004;554(1–2):241–251. doi: 10.1016/j.mrfmmm.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 22.Singh NP, Stephens RE. X-ray induced DNA double-strand breaks in human sperm. Mutagenesis. 1998;13:75–79. doi: 10.1093/mutage/13.1.75. [DOI] [PubMed] [Google Scholar]
- 23.Aten JA, Stap J, Krawczyk PM, van Oven CH, Hoebe RA, Essers J, Kanaar R. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science. 2004;303(5654):92–95. doi: 10.1126/science.1088845. [DOI] [PubMed] [Google Scholar]
- 24.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–915. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.IARC. IARC Monographs on the Evaluation of the Carcinogenic Risk to Humans: Chromium, Nickel and Welding. Vol. 49. International Agency for Research on Cancer; Lyon: 1990. pp. 1–648. [PMC free article] [PubMed] [Google Scholar]
- 26.Cohen MD, Kargacin B, Klein CB, Costa M. Mechanisms of chromium carcinogenicity and toxicity. CRC Crit Rev Toxicol. 1993;23:255–281. doi: 10.3109/10408449309105012. [DOI] [PubMed] [Google Scholar]
- 27.Luippold RS, Mundt KA, Austin RP, Liebig E, Panko J, Crump C, Crump K, Proctor D. Lung cancer mortality among chromate production workers. Occup Environ Med. 2003;60(6):451–457. doi: 10.1136/oem.60.6.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gambelunghe A, Piccinini R, Ambrogi M, Villarini M, Moretti M, Marchetti C, Abbritti G, Muzi G. Primary DNA damage in chrome-plating workers. Toxicology. 2003;188(2–3):187–195. doi: 10.1016/s0300-483x(03)00088-x. [DOI] [PubMed] [Google Scholar]
- 29.Borska L, Fiala Z, Smejkalova J, Tejral J. Health risk of occupational exposure in welding processes I. Genotoxic risk. Acta Med (Hradec Kralove) 2003;46(1):25–29. [PubMed] [Google Scholar]
- 30.Collins AR. The comet assay for DNA damage and repair. Mol Biotechnol. 2004;26:249–261. doi: 10.1385/MB:26:3:249. [DOI] [PubMed] [Google Scholar]
- 31.Rundell MS, Wagner ED, Plewa MJ. The comet assay: genotoxic damage or nuclear fragmentation? Environ Mol Mutagen. 2003;42:61–67. doi: 10.1002/em.10175. [DOI] [PubMed] [Google Scholar]
- 32.Roser S, Zobel B, Rechkemmer G. Contribution of apoptosis to response in the comet assay. Mutat Res. 2001;497:169–175. doi: 10.1016/s1383-5718(01)00255-8. [DOI] [PubMed] [Google Scholar]
- 33.De Flora S, Wetterhahn KE. Mechanisms of chromium metabolism and genotoxicity. Life Chem Rep. 1989;7:169–244. [Google Scholar]