

Abstract
Microalgae are considered a promising source for various high value products, such as carotenoids, ω-3 and ω-6 polyunsaturated fatty acids (PUFA). The unicellular green alga Lobosphaera (Parietochloris) incisa is an outstanding candidate for the efficient phototrophic production of arachidonic acid (AA), an essential ω-6 PUFA for infant brain development and a widely used ingredient in the baby formula industry. Although phototrophic production of such algal products has not yet been established, estimated costs are considered to be 2–5 times higher than competing heterotrophic production costs. This alga accumulates unprecedented amounts of AA within triacylglycerols and the molecular pathway of AA biosynthesis in L. incisa has been previously elucidated. Thus, progress in transformation and metabolic engineering of this high value alga could be exploited for increasing the efficient production of AA at competitive prices. We describe here the first successful transformation of L. incisa using the ble gene as a selection marker, under the control of the endogenous RBCS promoter. Furthermore, we have succeeded in the functional complementation of the L. incisa mutant strain P127, containing a mutated, inactive version of the delta-5 (Δ5) fatty acid desaturase gene. A copy of the functional Δ5 desaturase gene, linked to the ble selection marker, was transformed into the P127 mutant. The resulting transformants selected for zeocine resistant, had AA biosynthesis partially restored, indicating the functional complementation of the mutant strain with the wild-type gene. The results of this study present a platform for the successful genetic engineering of L. incisa and its long-chain PUFA metabolism.
Introduction
The green freshwater microalga Lobosphaera incisa (Reisigl) comb. nov. was isolated from snow water patches in the alpine environment of Mt. Tateyama, Japan [1]. It belongs to the Trebouxiophyceae, a class of Chlorophyte algae. Lobosphaera incisa was initially assigned to the genus Parietochloris rather than to Myrmecia [1] but was recently reclassified as belonging to Lobosphaera based on zoospore morphology and 18S rDNA analysis [2]. In contrast to most algae whose storage lipids triacylglycerols (TAGs) are mainly composed of saturated and monounsaturated fatty acids (FA), the main fatty acid in TAGs of L. incisa is arachidonic acid (AA, 20∶4 n-6). When cultivated under nitrogen starvation, the total FA content of the alga is over 35% of dry weight. AA constitutes about 60% of total FAs (TFA), and over 90% of cell AA is deposited in TAGs, making L. incisa the richest plant source of the pharmaceutically and nutraceutically valuable AA and a target organism of high biotechnological interest [3] [4].
The ability to store AA within reserve lipids, TAGs, was thought to be related to the need to maintain a buffering capacity for the long-chain polyunsaturated FA (LC-PUFA) pool under conditions of temperature change or growth recovery when rapid LC-PUFA incorporation into the cellular membranes is required for adjusting membrane fluidity [5] [6]. The conversion of dihomo-γ-linolenic acid (DGLA, 20∶3 n-6) to AA is mediated by the enzyme Δ5 desaturase (DES5, GenBank: GU390533). Based on its chlorotic phenotype at 15°C, we have been able to isolate P127, a L. incisa mutant strain in DES5 [7] [8]. The chemically induced non-sense mutation caused a nucleotide alteration in the DES5 gene, leading to the appearance of stop codon and the complete loss of AA biosynthesis. P127 accumulates DGLA, instead of AA, in TAGs and in polar lipids [8] [9].
In order to improve the biotechnological potential of L. incisa, we developed a stable nuclear transformation system for its metabolic engineering. Our initial attempts to transform this alga used the Streptomyces hindustanus ble gene as a selection marker connected to heterologous promoters, such as popular for transformation of Chlamydomonas reinhardtii tandem HSP70A/RBCS2 promoter [10], or the viral CaMV 35S. Using such constructs, transformation both by biolistic delivery and electroporation failed. Recent studies in microalgae engineering indicate that the best transformation results are achieved by using cloned endogenous promoters and regulatory untranslated regions (UTRs), fused to suitable selection markers. In C. reinhardtii, the PSAD, RBCS2 and HSP70A promoters have been widely used to drive nuclear gene expression [11]–[13]. Other endogenous promoters have been used to drive the expression of the ble gene, which encodes a protein that confers resistance to the antibiotic zeocin in many algal species. For example, the fcp promoter was successfully used for transformation of Phaeodactylum tricornutum [14] and likewise the bidirectional VCP2 promoter for transformation of Nannochloropsis sp. [15].
Here we describe the development and characterization of a transformation vector for the genetic engineering of L. incisa, based on the endogenous Ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO) small subunit (RBCS) promoter for expressing the ble gene as a selection marker. In order to demonstrate the successful metabolic engineering of L. incisa, we also transformed the P127 mutant, using the ble marker linked to the wild-type genomic DES5 and demonstrate successful restoration of AA biosynthesis.
Materials and Methods
Algal strains and culture conditions
We used the original isolate of L. incisa [1], maintained in the Microlagal Biotechnology Laboratory, Ben Gurion University, Israel, and now deposited at the Culture Collection of Algae at Göttingen University under accession no. SAG 2468. Axenic cultures were cultivated mixotrophically in LB-Miller Broth medium (BD, Franklin Lakes, NJ), either in liquid or on solid agar (Difco) medium. For C. reinhardtii, we used the cell-wall-deficient expression strain UVM4 [16], kindly provided by R. Bock, cultivated mixotrophically in a Tris-Acetate-Phosphate (TAP) medium [17]. All strains were cultivated in 250-mL Erlenmeyer flasks in an incubator shaker at a speed of 170 rpm, under an air/CO2 atmosphere (99∶1, v/v), at 25°C and an illumination of 100 µmol photons m−2 s−1 PAR.
DNA isolation
Genomic DNA (gDNA) was extracted from algal cells following the modified cetyltrimethyl ammonium bromide (CTAB) based protocol. CTAB buffer (2% CTAB, 1.4M NaCl, 0.2% β-mercaptanol, 20 mM EDTA, 100 mM Tris-HCl, pH 8.0) preheated to 60°C was added to cell pellets ground in liquid nitrogen. After incubation at 60°C in the water bath for 30 minutes, samples were extracted twice by chloroform-isoamyl alcohol (24∶1). Cold isopropanol was added to the upper aqueous phase, and the DNA was precipitated by centrifugation. The pellet was washed in 70% ethanol and resuspended in PCR grade water. The concentration and purity of the DNA was determined by Nano-Drop (Thermo-Scientific, Logan, UT).
Isolation of RNA and cDNA synthesis
The cells from 10 mL of culture with the chlorophyll content of 30 mg L-1 were harvested by centrifugation at 4000 rpm for 5 min, washed with double distilled sterilized water, flash-frozen in liquid N, and stored at −80°C until further use. Total RNA was isolated following the SV Total RNA Isolation system protocol (Promega, Madison, WI). cDNA was prepared from 1 µg of total RNA-template with the Verso cDNA kit (Thermo Fisher Scientific, Epson, UK).
Cloning procedures
PCR products were generated using the AccuPower PCR PreMix (Bioneer, Alameda, CA) and a TProfessional Thermocycler (Biometra, Goettingen, Germany). All PCR reactions were made on standard conditions. The sequences of all primers used are given in Table 1. PCR products were purified from agarose gel using the AccuPrep Gel Purification Kit (Bioneer). The In-Fusion HD Cloning Kit (Clontech, Mountain View, CA) was used for the directional cloning of DNA fragments into target vectors, according to the manufacturer's instructions.
Table 1. Primers used in this study.
| Primer | Sequence |
| RbcS1kbF | 5′-GAGTCACGACGGACTGTAGGA-3′ |
| RbcSR | 5′-TGCTTGGTAGCACAGTTCAGA-3′ |
| D5DF | 5′-AGTCTTGTACTCCTTGCCCTCCT-3′ |
| D5DR | 5′-GCTCTGTAAATCCATCCATCGTC-3′ |
| RbcEmptyF | 5′-TAAGCAACGAATGGCCTAG-3′ |
| RbcEmptyR | 5′-CATCTCGACTTGTTTAGTGTTG-3′ |
| BleF | 5′-ACACTAAACAAGTCGAGATGGCCAAGCTGACCAGCGCCGT-3′ |
| BleR | 5′-CCTAGGCCATTCGTTGCTTAGTCCTGCTCCTCGGCCACGAA-3′ |
| RbcS450F | 5′-GTTGATAGGCAAGACCCAACA-3′ |
| RbcS200F | 5′-GTTGCTTCACTTTCGCTTGAC-3′ |
| RT_LiDes5 F | 5′-TAAGTGCCAGGGCTGTGCTAGA-3′ |
| RT_LiDes5 R | 5′-GAACTGACCCTCCTCTGTGTCCT-3′ |
| RT_LiAct F | 5′-CGTCCAGCTCCACGATTGAGAAGA-3′ |
| RT_LiAct R | 5′-ATGGAGTTGAAGGCGGTCTCGT-3′ |
| LiDes5 F3 | 5′-GCTGTAACAGAGGGCGCTG-3′ |
Cloning of RBCS and DES5 genes
The RBCS gene, including 1 kb upstream of the predicted start codon and 430 bp downstream of the stop codon, was amplified using primers that annealed to the 5′ (RbcS1kbF) and 3′ (RbcSR) ends of the gene sequence. The 6048 bp genomic loci of L. incisa containing DES5 gene, including 1.8 kb upstream of the predicted start codon and 1 kb downstream of the stop codon, was amplified using primers that annealed to the 5′ (D5DF) and 3′ (D5DR) ends of the gene sequence. The amplification was carried out by PCR from 5 ng of total L. incisa gDNA as described above. The RBCS gene was cloned into the pGEM-T easy vector (Promega, Madison, WI) to create pLiRbcS plasmid. The DES5 gene was cloned into the pJET1.2 vector (Thermo-Scientific) to create pJET-LiD5Des plasmid.
Transformation vector design
The CDS of the ble selection marker was amplified from the plasmid pGenD-Ble [11] with the primers BleF and BleR. Using the In-Fusion protocol, it was cloned into pLiRbcS in replacement of the RBCS CDS, yielding the initial transformation vector pRbcS1kb. For the following cloning step, pRbcS1kb was used as a template for the amplification of expression cassettes with different promoter lengths. The forward primers RbcS450F, RbcS200F and BleF were used, in conjunction with the reverse primer RbcSR, to amplify the expression cassette with promoter lengths of 450 bp and 200 bp and without promoter, respectively. The PCR products were cloned into the pGEM-T easy vector to obtain three additional transformation vectors: pRbcS450, pRbcS200 and pRbcS0 (Fig. 1). Later, the pRbcS450 selection cassette was subcloned into pJET-LiD5Des by flanking NotI restriction sites to create the pD5Dselect cassette (Fig. 2). Plasmid midipreps of the constructs were prepared using the Fast Ion Midiprep Kit (RBC Bioscience, New Taipei City, Taiwan). The concentrations of the plasmids were determined by spectrophotometric analysis, and 1 µg/µL aliquots of DNA were prepared for later transformations.
Figure 1. Schematics of the LiRbcS transformation constructs and corresponding average number of resultant colonies per plate.
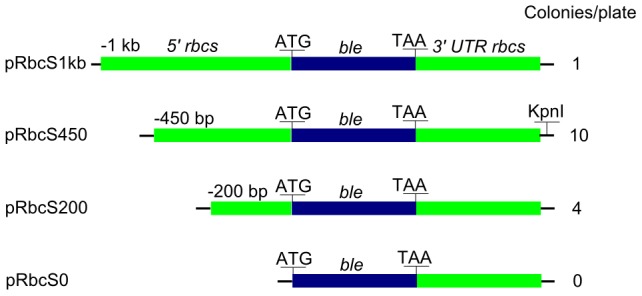
The number of colonies is the average of 12 different transformation events. The blue boxes represent the ble coding sequence, and the green boxes represent the LiRbcS promoter and terminating sequence. KpnI restriction site used in Southern blot hybridization is marked on the construct that was used for L. incisa transformation.
Figure 2. Schematic diagram showing the construction strategy of pD5Dselect.
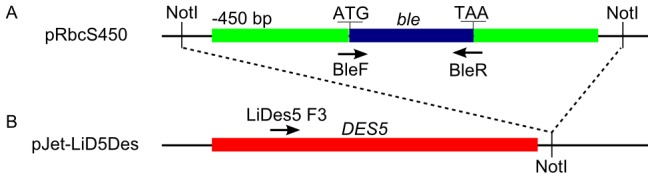
The pRbcS450 plasmid (A) contains the selection cassette, flanked by NotI restriction sites. The pJET-LiD5Des plasmid (B) contains the functional DES5 gene with its flanking regions. Following NotI digestion, the selection cassette was subcloned into pJET-LiD5Des. Primers used in analysis of P127-transformant are marked.
Transformation of C. reinhardtii
Cells of C. reinhardtii UVM4 in the log phase were transformed according to the PEG-glass beads method [18]. Prior to use, the vectors were linearized by ScaI. Cells were poured onto selective TAP agar plates (20 µg mL−1 zeocin) and incubated at 25°C under a light intensity of 20 µM photons m−2 s−1. Transformants appeared after 2–3 weeks of incubation.
Transformation of L. incisa by electroporation
Electroporation was performed using a Bio-Rad Gene PulserXcell as described by [19]. L. incisa was grown in 100 mL LB to a chlorophyll content of 30 µg mL−1. Then 20 mL of cell culture in 50 mL conical centrifuge tube were placed on ice, sonicated for 30 seconds at 50% power using a Digital Sonifier Cell Disruptor (Branson Ultrasonics Corp., Danbury, CT), centrifuged. The cell pellet was washed twice with sterile water by sequential centrifugation at 3000 rpm at ambient temperature. Prior to transformation by electroporation, the sample was observed under light microscope to ensure sufficient cell clusters disintegration. The pellet was resuspended in 0.4 mL of sterile water followed by addition of 10 µg of linearized plasmid DNA, and then transferred into a 2 mm electroporation cuvette for electroporation [one 500 V pulse (at 2500 V/cm), 50 mF capacity and infinitive shunting resistance]. Transformed cells were transferred to Erlenmeyer flasks containing 20 mL of LB medium and incubated for 24 h in the dark, followed by 24 h under light intensity of 20 µM photons m−2 s−1 in ambient air condition with moderate agitation. Afterwards, transformed cells were plated onto selective LB-agar plates, containing 10 µg mL−1 zeocin at 25°C under a light intensity of 20 µM photons m−2 s−1.
Southern blot analysis
Southern blot hybridization was performed using the North2South Chemiluminescent Hybridization and Detection Kit (Thermo Scientific) according to the manufacturer's instructions. Briefly, L. incisa genomic DNA (5 µg) was digested with KpnI endonuclease (NEB), desalinated by EtOH precipitation and separated by electrophoresis on a 0.7% agarose (Seakem, Lonza) gel. After depurination with 250 mM HCl, incubation in denaturation solution (0.5 M NaOH, 1.5 M NaCl) and neutralization with 0.5 M Tris-HCl pH 7.5, 1.5 M NaCl, the gel was equilibrated with 20× SSC (saline-sodium citrate buffer, 3 M sodium chloride and 300 mM trisodium citrate, pH 7.0). After overnight capillary transfer of digested and separated gDNA from the gel to a Magnagraph nylon membrane (GE Water & Process Technologies, USA) in 20× SSC buffer, the membrane was rinsed with 2× SSC and baked for 2 h at 80°C. The membrane was blocked, hybridized and washed, and bands were detected according to the kit manual. Chemiluminescent detection was performed using a Microchemi camera (Bioimaging System, Israel). The probe was synthesized by PCR using primers BleF and BleR (Table 1) from pRbcS450 plasmid with a GoTaq (Promega) PCR mix using the plasmid DNA as the template and biotinylated dUTP (Thermo Scientific) added to a final concentration of 50 nM. Efficiency of probe-labeling was verified by electrophoresis on an agarose gel and comparing molecular weights of labeled and unlabeled PCR products; the amount of probe was quantified using a ND-1000 spectrophotometer (NanoDrop).
Quantitative real-time PCR (qRT-PCR)
qRT-PCR analysis was carried out in optical 96-well plates using the CFX96 Touch Real-Time PCR Detection System (Bio-Rad, Hercules, CA). qRT-PCR primer pairs (Table 1) were designed for LiDES5 and the housekeeping gene LiAct (actin) and the reactions were performed using iTaq Universal SYBR Green Supermix (Bio-Rad). PCR cycling conditions consisted of an initial polymerase activation step at 95°C for 30 s followed by 40 cycles at 95°C for 5 s and 60°C for 30 s, and a final melting step at 65–95°C. Results were analyzed using the 2−ΔΔCt method, a function of the CFX Manager Software v3.0, using the relative expression value of the housekeeping gene as the calibrator. The experiment was performed twice, with two biological replicates and three technical replicates for each sample.
Fatty acid analysis
The direct transmethylation of algal biomass was performed by incubating freeze-dried biomass in dry methanol containing 2% (v/v) H2SO4 at 80°C for 1.5 h under argon atmosphere with continuous stirring as previously described [20]. Heptadecanoic acid (C17∶0) (Fluka, Buchs, Switzerland) was added as an internal standard. FAME were quantified on a Trace GC Ultra (Thermo, Milan, Italy) equipped with a flame ionization detector (FID) and a programmed temperature vaporizing (PTV) injector. The detector temperature was fixed at 280°C, and helium was used as a carrier gas. The PTV injector was programmed to increase the temperature from 40°C at time of injection to 300°C at time of sample transfer. Separation was achieved on fused silica capillary columns (SUPELCOWAX 10, Sigma-Aldrich, 30 m×0.32 mm). FAME were identified by co-chromatography with authentic standards (Sigma-Aldrich).
Results
Cloning and analysis of the L. incisa RBCS and DES5 genes
The RBCS and DES5 genes, their putative promoters and 3′-flanking flanking regions were identified in a draft assembly of the L. incisa genome generated in the framework of the EU FP7 GIAVAP project (www.GIAVAP.eu). The sequences were submitted to Genbank, accession numbers KJ633120 and KJ633121 for RBCS and DES5 genes respectively. The RBCS gene contains two introns within the 570 bp coding sequence, while DES5 contains seven introns within the 1356 bp of CDS. Genes and their upstream and downstream sequences were cloned into plasmid vectors.
Characterization of L. incisa RBCS promoter and UTRs in C. reinhardtii
The RBCS based transformation vectors (Fig. 1) were used to transform C. reinhardtii cells. After 2–3 weeks on zeocin selection plates, resistant colonies appeared. For all transformants, the presence of the ble gene was confirmed by PCR (not shown). Each construct was used for 12 different transformation events. The highest number of colonies was recovered with the pRbcS450 construct with an average of 10 colonies per plate. Four colonies per plate, on average, were recovered using pRbcS200, but none ever with pRbcS0, indicating that the RBCS promoter was indeed necessary to drive ble expression. Constructs with the 1 kb promoter sequence resulted in a very low transformation efficiency, suggesting that the −1000 to −450 region of the L. incisa RBCS promoter contains elements counteracting transformation or gene expression in C. reinhardtii.
Genetic transformation of L. incisa
L. incisa is usually cultivated and maintained in the photoautotrophic mineral medium BG11 [21]. In this medium, L. incisa divides through the multiple fission of mother cells, producing 8–16 daughter cells after hatching. After release, daughter cells primarily remain attached to each other, giving rise to large cell clusters (palmelloids) (Fig. 3a). We assume that our previous failures in transformation attempts were due to difficulties in delivering plasmid-DNA into a single cell. Indeed, while cells remain in clusters, many dividing cells are shielded by sister-cells that might prevent plasmid penetration. Moreover, we assume that even if one cell from a cluster obtains and inserts into its genome a copy of plasmid DNA, the chances for survival of the single transformed cell during selection would also be low, because of the proximity of dying cells, which might release toxic and cell-death signaling compounds. Hence, our first aim for achieving a successful L. incisa transformation was the production of a culture enriched in single cells.
Figure 3. The effect of growth media and sonication on the morphology of L. incisa cells.
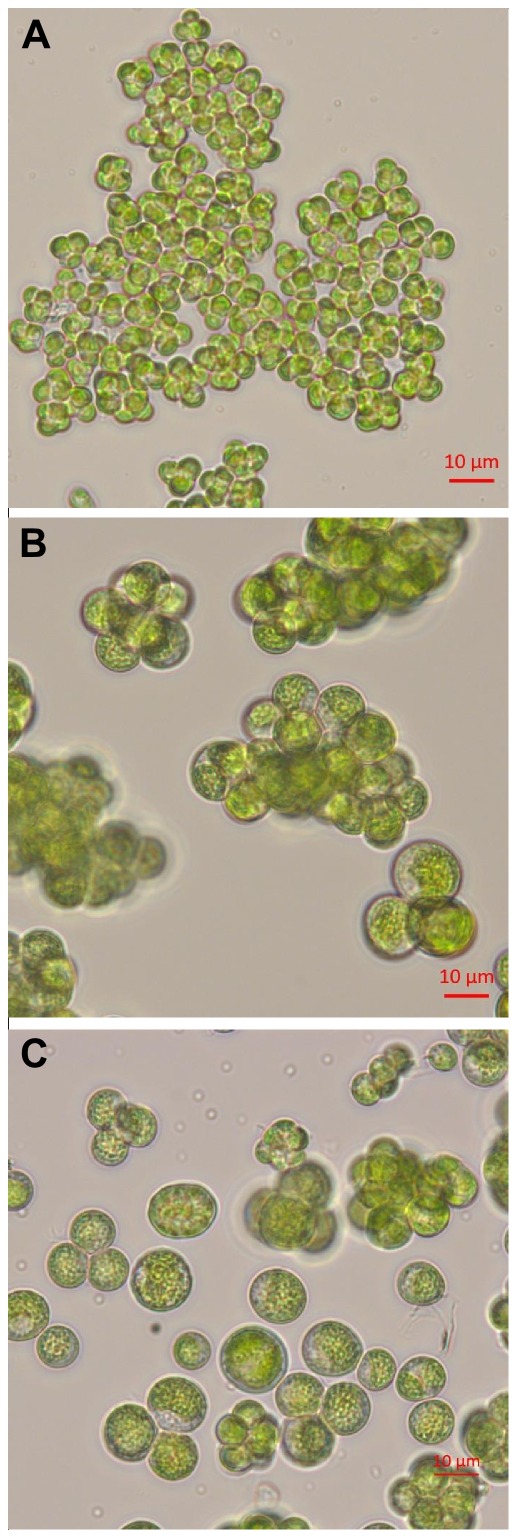
(A) cells grown in a mineral BG11 medium, (B) cells grown in an LB medium, (C) cells grown in an LB medium, after sonication.
In the course of our studies on L. incisa, we have determined that this alga can grow well mixotrophically in LB medium. We have observed that in these cultures, L. incisa seems to form smaller clusters that further disassemble into single cells, even under the weight of a microscopic cover-glass (Fig. 3b). We assumed that medium enrichment with the organic carbon and nitrogen sources, such as sugars, amino acids and peptides, that are abundant in LB medium, might have exerted a substantial impact on the cell cycle and/or cell wall composition, resulting in the reduced attachment of cells and clusters formation. We have hypothesized that the single-celled form of the culture might be produced from cells growing in the LB medium after gentle mechanical treatment. Thus, in order to develop a stable transformation system, we identified sonication with precise power and time settings as the most suitable treatment for preparing high quality single-cell-enriched cultures for transformation (Fig. 3c). Since counting of individual cells was difficult even after ultrasonication, due to some remaining clusters, we estimated transformation efficiency not per number of cells in transformation reaction but per standard reaction of 20 mL containing 30 µg mL−1 chlorophyll and 10 µg of linear plasmid DNA. The transformation efficiency in optimized conditions was approximately 10 clones per 10 µg of plasmid DNA for wild-type L. incisa and a magnitude lower for the P127 mutant strain.
Confirmation of stable DNA integration to the L. incisa genome
Zeocin resistant colonies were transferred to new selective media, and the integration of the transgene was confirmed by PCR and Southern blot analysis. The PCR analysis was carried out on five zeocin-resistant clones, obtained after transformation with plasmid pRbcS450. The expected 415 bp ble fragment was detected in all five clones but not in non-transformed cells (Fig. 4A). Southern blot analysis was performed to confirm the stable integration of the transgene (ble), as well as to estimate the number of copies of the transgene inserted into the genome of transgenic clones. The gDNAs of the five transformants and wild-type were digested with KpnI restriction enzyme and analyzed by hybridization with a ble-specific probe (Fig. 4B). Since pRbcS450 plasmid was digested only once with KpnI restriction enzyme at the 3′ flanking region of ble gene-marker, we expected that size of hybridized bands would be unique for each integration event since a second 5′ KpnI restriction site should be randomly located at the 5′ chromosome flanking region. According to the hybridization pattern, two out of the five lines exhibited two hybridization bands (400-2 and 600-2), while two lines (400-1 and 600-1) exhibited only one. Clone 400-3 did not exhibit any hybridization signal, probably due to poor DNA quality. No bands were observed in the wild-type non-transformed cells, and a single sharp band was observed in the digested plasmid DNA used as a positive control.
Figure 4. Molecular analyses of L. incisa clones transformed with a pRbcS450 construct.
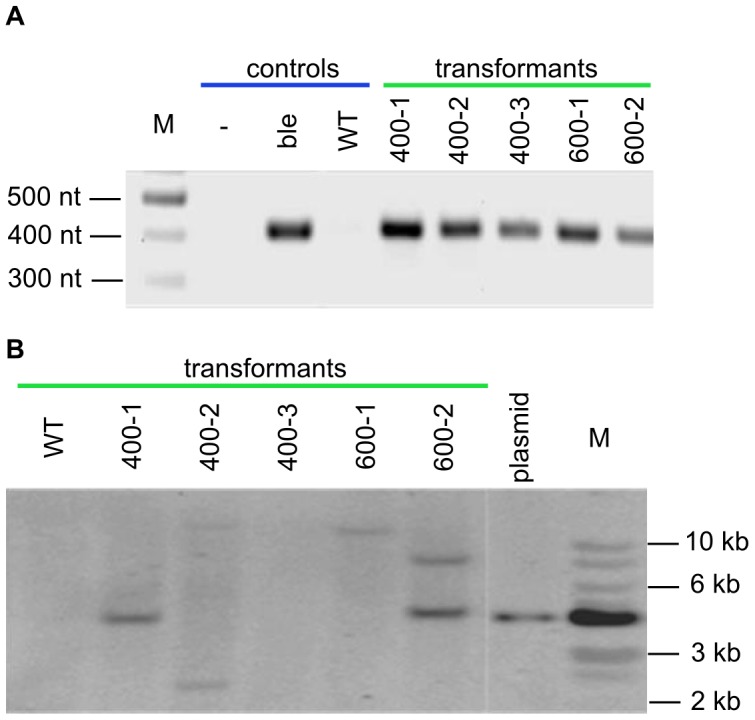
(A) PCR analysis: gDNA was amplified with BleF and BleR primers yielding a 415-bp fragment. Lanes: (M) DNA ladder; (-) no template, (Ble) ble plasmid control, (wt) negative control (non-transformed cells); five transformed clones. (B) Southern blot analysis: gDNA isolated from both transgenic and non-transgenic cells, as well as plasmid DNA (pRbcS450), were digested with KpnI restriction enzyme. The blot was hybridized with a probe derived from a 415-bp amplified fragment of the ble gene. Lanes: (M) 1 Kb ladder; (plasmid) positive control; five transformed clones; (wt) negative control (non-transformed cells).
Transformation stability was assessed by growing the resistant clones on non-selective plates for three months and then, for an additional month, on selective plates (Fig. 5). All clones retained their antibiotic resistance.
Figure 5. Transformation stability analysis.

(A) five resistant clones and a wild-type control were grown on non-selective plates for three months. The cell-lines were then restreaked on a selective plate. (B) all clones retained their antibiotic resistance. The wild-type did not grow under selection (20 µg mL−1 zeocin).
Complementation of the mutant P127 with functional DES5
Having established the transformation platform, we attempted to restore AA biosynthesis in the previously isolated des5 mutant strain P127 [9] by metabolic engineering. The linearized plasmid pD5Dselect harboring the genomic version of DES5 was introduced into P127 cells by electroporation, as described above. Notably, the transformation efficiency, even with pRbcS450 plasmid, of the P127 mutant appeared to be substantially lower than that of the wild-type. Zeocin resistant colonies appeared after four weeks. Insertion of the pD5Dselect cassette containing wt-DES5 gene linked to ble gene marker was confirmed by PCR amplification from gDNA isolated from P127-transformants. We used BleF and BleR primers for ble gene amplification and LiDes5 F3 and BleR primers (Table 1) for conformation of a linkage DES5 and ble genes in the genome of P127-transformants (Fig. 6C).
Figure 6. Analysis of L. incisa P127 clones transformed with a pD5Dselect construct.
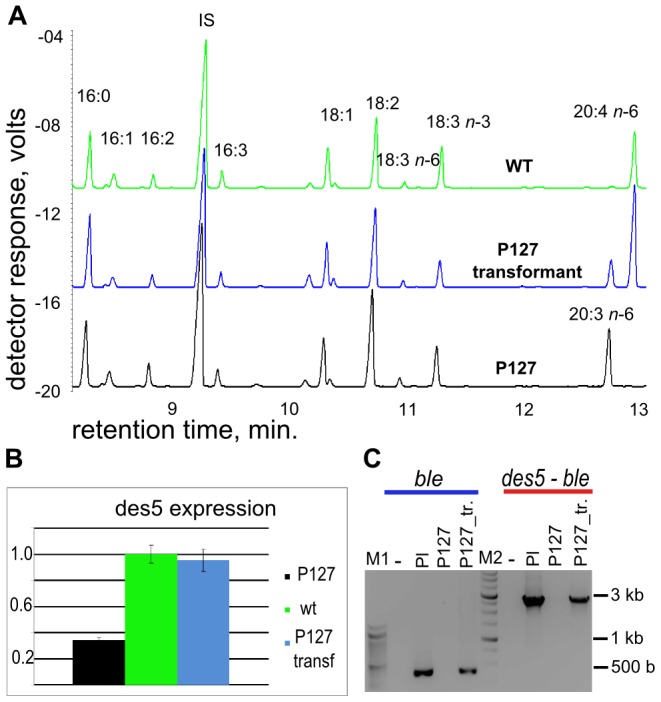
(A) GC-FID analysis of the fatty acid profiles of P127 mutant, P127-transformant and wild type cells, (B) qRT-PCR analysis of expression of DES5 gene in P127 mutant, P127-transformant and wild type cells. (C) PCR analysis of gDNA isolated from P127 mutant, P127-transformant and wild type cells with primers LiDes5 F3 and BleR, demonstrating transgene integration in conjunction with the ble gene.
The P127 mutant strain, the P127-transformant and the wild type strain were cultivated in mBG-11 liquid medium and harvested for FA analysis. A gas chromatography (GC) analysis of FA composition, following the growth in the complete mBG-11 nutrient medium with daily dilution for 4 days to keep the logarithmic growth, revealed a drastic decrease in the proportion of DGLA in the fatty acid profile (from ca. 16% to 6.3% of TFA) with a concomitant rise in that of AA in the P127-transformant as compared to the untransformed P127 strain. The representative chromatograms are shown on Fig. 6A. Notably, the AA proportion in the FA profile of the recombinant strain (25% of TFA) reached and even surpassed the AA proportion in the WT (16% of TFA). Furthermore, as determined by qPCR, while the level of DES5 transcript expression was lower in the mutant than in the WT, pD5Dselect integration led to a similar level of expression in the WT and in the P127-transformant (Fig. 6B). However, the P127-transformants that featured relatively high proportion of DGLA simultaneously with the restored AA biosynthesis, appeared to display an altered slow-growth phenotype. The impaired growth of the P127-transformant may be responsible for its higher AA percentage compared to the WT because growth inhibition in this alga is associated with the production of storage lipids that are rich in LC-PUFA. At present, we do not have a plausible explanation for this phenomenon; future studies will shed more light on the effect of altered LC-PUFA content on physiological response of L. incisa to cultivation conditions.
Discussion
Microalgae have a high potential for the production of biofuels and high value products, such as PUFA and carotenoids [22]. However, microalgal production processes require significant improvements at various levels to compete successfully with products currently used in food, cosmetics, aquaculture and agriculture that are prepared synthetically or extracted from other natural feedstock. Significant progress in strain development and sustainable cultivation technologies are required to reduce the currently high production costs for algal biomass that is produced phototrophically. To date, a few green microalgae species such as Chlamydomonas, Dunaliella or Chlorella, and heterokont microalgae such as Phaeodactylum or Nannochloropsis have been successfully transformed [23]. Beyond mere transformation, adequate tools for the actual expression of unselected transgenes are required: they have been efficiently demonstrated so far in only a few algal species. Here, we demonstrate the ability: i) to genetically transform L. incisa and ii) to use this transformation platform for the metabolic engineering of its FA composition. L. incisa is highly attractive alga species, that can accumulate over 20% of its dry-weight (% of DW) as AA under nitrogen starvation [3] [4], but its industrial use is hampered by relatively slow growth and sensitivity to environmental stress, such as combined stress caused by high light and nitrogen depletion. The technology presented here can thus assist in modulating the LC-PUFA composition of this alga through sequential metabolic engineering of FA desaturation level and carbon chain-length. Dramatic increases in the C22 ω-3 LC-PUFA by iterative engineering of the biosynthesis pathway have been achieved in P. tricornutum [24]. The developed tools for L. incisa pave the way to further engineering efforts to improve growth performance and/or lipid productivity, as exemplified in several microalgae [24]–[26]. Further progress could come, for example, from downregulation of lipid catabolism, which in Thalassiosira pseudonana improved lipid yield without affecting growth performance [26], and enhancement of photosynthesis by modulating inorganic carbon assimilation (Spalding MH. personal communication). An expected doubling of productivity would allow the use of this phototrophic organism for competitive PUFA production in a rapidly growing market [22].
The transformation of novel non-model microalgae species is a multi-dimensional puzzle with several unknowns. The successful transfer of the DNA into the algal cell without causing lethal damage, the sufficient expression of a suitable selection marker and the efficient recovery of the transformed cells are all essential for achieving adequate transformation efficiency. This had been achieve in several representatives of heterokont algae [27] [28]. Green algae appear to be more difficult to transform, and until recently, C. reinhardtii remained the only green algal species accessible to metabolic engineering. Recently many protocols for transformation of other green algae species have been presented [27]–[35], but efficient metabolic engineering remains to be confirmed.
Based on our experience with transformation of L. incisa, we think that success depends on consideration of all of the following issues:
A suitable combination of promoter and selection marker must be established. Our initial attempts with plasmid pSP124 designed for C. reinhardtii transformation [35] were unsuccessful in L. incisa. Selecting the endogenous L. incisa RBCS promoter and testing it in C. reinhardtii allowed us to determine its optimal length for driving transformation, which proved effective in L. incisa. Our results open the door to further improvement. For example, one could try to introduce sequence elements that might insulate the promoter from negative effects from the surrounding context, as was achieved with the HSP70A-RBCS2 combination in C. reinhardtii [36]. Introduction of an intron might also improve transformation yield, as shown for C. reinhardtii [35]. Other resistance markers also need to be tested.
The clumping behavior of L. incisa represented a major challenge in developing adequate transformation and gene transfer technologies. Finally, the cultivation of the strain in a rich organic medium, accompanied by mild mechanical treatment, was successful in producing cultures composed of predominantly single cells, adequate for efficient transformation.
Electroporation, at carefully optimized conditions, was found to be the most effective method of gene transfer, allowing the recovery of transformed cells at an adequate frequency. But we had no success with particle gun bombardment. Electroporation has the significant advantage of requiring only one plating step following transformation, when using antibiotics selection, whereas particle gun bombardment requires replating on selective medium. In Haematococcus pluvialis, we estimate that only five percent of cells survive the plating steps required during the bombardment, recovery and replating procedures even before the selection for transformants becomes effective (S. Boussiba, unpublished results). Usefulness of bombardment may still be explored with non-lethal antibiotics like spectinomycin [37], or complementation approaches, where no replating is necessary.
The adequate expression of heterologous marker genes is an additional and final obstacle for achieving satisfactory transformation efficiency, mostly due to different codon usage in different organisms. The codon usage of the ble gene is quite similar to that in L. incisa. However, it is generally accepted that the highest expression rate of transgenes in recombinant microalgae is achieved with constructs containing endogenous selection markers as compared to the heterologous genes. This is why the development of selection markers, based on endogenous genes, could be an important part of an efficient “molecular toolbox” and an essential step in the future.
In summary, we have managed to demonstrate the successful stable transformation of L. incisa utilizing a partial set of possible optimization steps available. Moreover, the usage of the transformation method described above will lead to the development of a complete molecular toolbox, including endogenous markers and sets of constitutive and inducible promoters. We have furthermore demonstrated successful metabolic engineering in this alga by restoring AA biosynthesis to the mutant P127, via transformation of the wild-type DES5 gene with its own promoter linked to the ble selection marker. The approach developed in this work may now permit the engineering of the LC-PUFA metabolism in L. incisa by expression of homologous and heterologous desaturases and elongases for the production of high value ω-3 LC-PUFA of commercial interest.
Acknowledgments
The authors would like to thank Prof. Zvi Cohen and Prof. Dina Raveh (BGU) for their valuable suggestions and recommendations during the research.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. L. incisa strain used in this work is deposited at the Culture Collection of Algae at Göttingen University under accession no. SAG 2468.
Funding Statement
This research was financially supported by the European Commission's Seventh Framework Program for Research and Technology Development (FP7), project GIAVAP, Grant No. 266401. BZ, OG and YK acknowledge support from the Kreitman School of Advanced Graduate Studies at Ben-Gurion University. OV acknowledges support from the French state "Initiative d'Excellence" program (Grant "DYNAMO", ANR-11-LABX-0011-01). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Watanabe S, Hirabayahi S, Boussiba S, Vonshak A, Richmond A (1996) Parietochloris incisa comb. nov. (Trebouxiophyceae, Chlorophyta). Phycological Res 44: 107–108. [Google Scholar]
- 2. Karsten U, Friedl T, Schumann R, Hoyer K, Lembcke S (2005) Mycosporine-like amino acids and phylogenies in green algae: Prasiola and its relatives from the Trebouxiophyceae (Chlorophyta). J Phycol 41: 557–566. [Google Scholar]
- 3. Bigogno C, Khozin-Goldberg I, Cohen Z (2002) Accumulation of arachidonic acid and triacylglycerols in the microalga Parietochloris incisa (Chlorophyceae). Phytochemistry 60: 135–143. [DOI] [PubMed] [Google Scholar]
- 4. Khozin-Goldberg I, Bigogno C, Shrestha P, Cohen Z (2002) Nitrogen starvation induces the accumulation of arachidonic acid in the freshwater green alga Parietochloris incisa (Trebouxiophyceae). J. Phycol 38: 991–994. [Google Scholar]
- 5. Bigogno C, Khozin-Goldberg I, Boussiba S, Vonshak A, Cohen Z (2002) Lipid and fatty acid composition of the green alga Parietochloris incisa . Phytochemistry 60: 497–503. [DOI] [PubMed] [Google Scholar]
- 6. Khozin-Goldberg I, Shrestha P, Cohen Z (2005) Mobilization of arachidonyl moieties from triacylglycerols into chloroplastic lipids following recovery from nitrogen starvation of the microalga Parietochloris incisa . Biochim Biophys Acta 1738: 63–71. [DOI] [PubMed] [Google Scholar]
- 7.Cohen Z, Khozin Goldberg I, Boussiba S, Vonshak A (2009) Over-production of dihomo gamma linolenic acid by a mutant strain of Parietochloris incisa. WO/2009/022323
- 8.Cohen Z, Khozin-Goldberg I (2010) Searching for PUFA-rich microalgae. In Cohen Z, Ratledge C (editors.) Single Cell Oils, 2-nd edition, American Oil Chemists' Society, Champaign IL, pp.201–224.
- 9. Iskandarov U, Khozin-Goldberg I, Cohen Z (2011) Selection of a DGLA-producing mutant of the microalga Parietochloris incisa: I. Identification of mutation site and expression of VLC-PUFA biosynthesis genes. Appl Microbiol Biotechnol 90: 249–256. [DOI] [PubMed] [Google Scholar]
- 10. Schroda M, Blocker D, Beck CF (2000) The HSP70A promoter as a tool for the improved expression of transgenes in Chlamydomonas. Plant J 21: 121–31. [DOI] [PubMed] [Google Scholar]
- 11. Fischer N, Rochaix JD (2001) The flanking regions of PsaD drive efficient gene expression in the nucleus of the green alga Chlamydomonas reinhardtii . Mol Genet Genomics 265: 888–894. [DOI] [PubMed] [Google Scholar]
- 12. Stevens DR, Rochaix JD, Purton S (1996) The bacterial phleomycin resistance gene ble as a dominant selectable marker in Chlamydomonas. Mol Gen Genet 251: 23–30. [DOI] [PubMed] [Google Scholar]
- 13. von Gromoff ED, Schroda M, Oster U, Beck CF (2006) Identification of a plastid response element that acts as an enhancer within the Chlamydomonas HSP 70A promoter. Nucleic Acids Res 34: 4767–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Apt K, Grossman A, Kroth-Pancic P (1996) Stable nuclear transformation of the diatom Phaeodactylum tricornutum . Mol Gen Gene 252: 572–557. [DOI] [PubMed] [Google Scholar]
- 15. Kilian O, Benemann CS, Niyogi KK, Vick B (2011) High-efficiency homologous recombination in the oil-producing alga Nannochloropsis sp. Proc Natl Acad Sci U S A 108: 21265–21269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Neupert J, Karcher D, Bock R (2009) Generation of Chlamydomonas strains that efficiently express nuclear transgenes. Plant J 57: 1140–1150. [DOI] [PubMed] [Google Scholar]
- 17.Harris EH (1989) The Chlamydomonas Sourcebook. San Diego: Academic Press.
- 18. Kindle KL (1990) High-frequency nuclear transformation of Chlamydomonas reinhardtii . Proc Natl Acad Sci U S A 87: 1228–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dower WJ, Miller JF, Ragsdale CW (1988) High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res 16: 6127–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pal D, Khozin-Goldberg I, Cohen Z, Boussiba S (2011) The effect of light, salinity, and nitrogen availability on lipid production by Nannochloropsis sp. Appl Microbiol Biotechnol 90: 1429–1441. [DOI] [PubMed] [Google Scholar]
- 21. Stanier RY, Kunisawa MM, Cohen-Bazir G (1971) Purification and properties of unicellular blue-green algae (order Chlorococcales). Bacteriol Rev 35: 171–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leu S, Boussiba S (2014) Advances in the production of high value products by microalgae. Ind Biotechnol (New Rochelle N Y) 10(3): 169–183. [Google Scholar]
- 23. Qin S, Lin H, Jiang P (2012) Advances in genetic engineering of marine algae. Biotechnol Adv 30: 1602–1613. [DOI] [PubMed] [Google Scholar]
- 24. Hamilton ML, Haslam RP, Napier JA, Sayanova O (2014) Metabolic engineering of Phaeodactylum tricornutum for the enhanced accumulation of omega-3 long chain polyunsaturated fatty acids. Metab Eng 22: 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mussgnug JH, Thomas-Hall S, Rupprecht J, Foo A, Klassen V, et al. (2007) Engineering photosynthetic light capture: impacts on improved solar energy to biomass conversion. Plant Biotechnol J 5: 802–814. [DOI] [PubMed] [Google Scholar]
- 26. Trentacoste EM, Shrestha RP, Smith SR, Glé C, Hartmann AC, et al. (2013) Metabolic engineering of lipid catabolism increases microalgal lipid accumulation without compromising growth. Proc Natl Acad Sci U S A 110: 19748–19753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang C, Hu H (2013) High-efficiency nuclear transformation of the diatom Phaeodactylum tricornutum by electroporation. Mar Genomics Available: http://dx.doi.org/10.1016/j.margen.2013.10.003 [DOI] [PubMed]
- 28. Vieler A, Wu G, Tsai CH, Bullard B, Cornish AJ, et al. (2012) Genome, functional gene annotation, and nuclear transformation of the heterokont oleaginous alga Nannochloropsis oceanica CCMP1779. PLoS Genet 8(11): e1003064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Talebi AF, Tohidfar M, Tabatabaei M, Bagheri A, Mohsenpor M, et al. (2013) Genetic manipulation, a feasible tool to enhance unique characteristic of Chlorella vulgaris as a feedstock for biodiesel production. Mol Biol Rep 40: 4421–4428. [DOI] [PubMed] [Google Scholar]
- 30. Lerche K, Hallmann A (2013) Stable nuclear transformation of Eudorina elegans . BMC Biotechnol 13: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guo SL, Zhao XQ, Tang Y, Wan C, Alam MA, et al. (2013) Establishment of an efficient genetic transformation system in Scenedesmus obliquus . J Biotechnol 163: 61–68. [DOI] [PubMed] [Google Scholar]
- 32. van Ooijen G, Knox K, Kis K, Bouget FY, Millar AJ (2012) Genomic transformation of the picoeukaryote Ostreococcus tauri . J Vis Exp (65): e4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Imamura S, Hagiwara D, Suzuki F, Kurano N, Harayama S (2012) Genetic transformation of Pseudochoricystis ellipsoidea, an aliphatic hydrocarbon-producing green alga. J Gen Appl Microbiol 58: 1–10. [DOI] [PubMed] [Google Scholar]
- 34. Ishida K (2007) Sexual pheromone induces diffusion of the pheromone-homologous polypeptide in the extracellular matrix of Volvox carteri . Eukaryot Cell 6: 2157–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lumbreras V, Stevens DR, Purton S (1998) Efficient foreign gene expression in Chlamydomonas reinhardtii mediated by an endogenous intron. Plant J 14: 441–447. [Google Scholar]
- 36. Schroda M1, Beck CF, Vallon O (2002) Sequence elements within an HSP70 promoter counteract transcriptional transgene silencing in Chlamydomonas. Plant J 31: 445–55. [DOI] [PubMed] [Google Scholar]
- 37. Meslet-Cladière L, Vallon O (2011) Novel shuttle markers for nuclear transformation of the green alga Chlamydomonas reinhardtii . Eukaryot Cell 10: 1670–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. L. incisa strain used in this work is deposited at the Culture Collection of Algae at Göttingen University under accession no. SAG 2468.