

Abstract
Heparin and heparan sulfate mediated basic fibroblast growth factor (bFGF) signaling plays an important role in skeletal muscle homeostasis by maintaining a balance between proliferation and differentiation of muscle progenitor cells. In this study, we investigate the role of a synthetic mimic of heparin, poly(sodium-4-styrenesulfonate) (PSS), on myogenic differentiation of C2C12 cells. Exogenous supplementation of PSS increased the differentiation of C2C12 cells in a dose-dependent manner while the formation of multi-nucleated myotubes exhibited a nonmonotonic dependence with the concentration of PSS. Our results further suggest that one possible mechanism by which PSS promotes myogenic differentiation is by downregulating the mitogen activated extracellular regulated signaling kinase (MAPK/ERK) pathway. The binding ability of PSS to bFGF was found to be comparable to heparin through molecular docking calculations and by native PAGE. Such synthetic heparin mimics could offer a cost effective alternative to heparin and also reduce the risk associated with batch-to-batch variation and contamination of heparin.
Keywords: bFGF; heparin; synthetic mimic of heparin, poly(sodium-4-styrenesulfonate); heparan sulfate; myogenic differentiation
Introduction
Basic fibroblast growth factor (bFGF) plays an important role in skeletal muscle function and regeneration [1-3]. Continuous and elevated levels of bFGF in cultures have been shown to suppress myogenic differentiation of progenitor cells [4], while silencing of bFGF signaling promotes in vitro myogenesis of MM14 myoblasts [5]. One of the key regulators of bFGF signaling is heparin and heparan sulfate proteoglycans (HSPG) present on the cell surface and in the extracellular matrix (ECM). Heparin/HSPGs regulate bFGF signaling by directly interacting with bFGFs and FGF receptors (FGFR) to form ternary FGF-FGFR-heparin complexes [6]. While the cell surface bound HSPGs promote bFGF signaling, the ECM bound HSPGs have been shown to regulate bFGF activation by sequestering them away from the cell-surface receptors [7]. Additionally, heparin and HSPGs have been shown to protect bFGF from denaturation and proteolytic degradation [8]. The binding of FGF to cell surface HSPGs has been shown to function through the mitogen-activated protein kinase/extracellular regulated signaling kinase (MAPK/ERK) pathway [9, 10].
bFGF signaling can also be modulated by exogenous soluble heparin and its synthetic mimics, which function by sequestering bFGF away from FGFR [11,12]. Various polyanionic compounds (sulfonic and carboxylic acid polymers) show heparin mimicking ability [13-16]. A number of studies have utilized synthetic heparin mimics to support regeneration of ischemic and compromised muscles [15, 17]. One of the most studied heparin mimics is poly(sodium-4-styrenesulfonate) (PSS) [14]. Liekens et al. compared various polysulfonic compounds and found PSS to be the most potent in binding to bFGF. Additionally, PSS was also found to inhibit the proteolytic cleavage of bFGF. The heparin-mimicking ability of PSS was also harnessed to control various bFGF-mediated cellular processes such as inhibition of angiogenesis [14].
Taking cues from these studies, along with the role of bFGF signaling in myogenic differentiation of cells, we determine for the first time the effect of PSS on myogenic differentiation using C2C12 cells as a model system. As a proof of concept, we also evaluate the effect of exogenous heparin on myogenesis of C2C12 cells, and confirm the binding affinity of PSS on bFGF using molecular docking studies and native PAGE. We also propose a plausible mechanism involving MAPK/ERK pathway for the observed beneficial effects of PSS. Such synthetic mimics not only offer a cost-effective alternative to heparin but also circumvent problems associated with batch-to-batch variations and other contaminations observed in heparin [18].
Materials and Methods
Materials
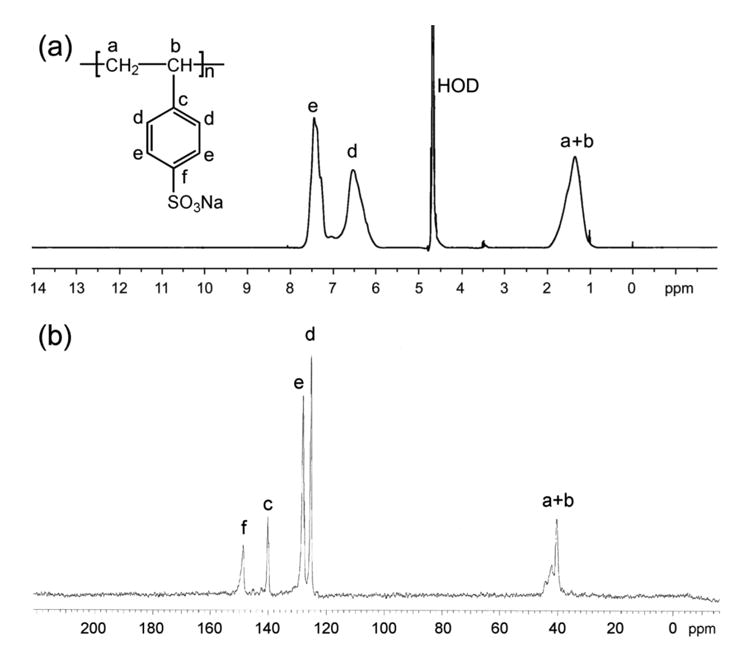
PSS (Mw: 70kDa; CAS No. 25704-18-1) was obtained from Sigma-Aldrich and heparin (Lot number PH-71609) from Celsus Laboratories Inc. According to the manufacturer, the polydispersity index of PSS is in the range 3 to 5, and the degree of sulfonation is about 75%. Sodium heparin is isolated from porcine intestinal mucosa and contains around 0.54% galactosamine. The protein content and bacterial endotoxins are less than 0.21% and 0.0015 EU/Unit, respectively. PSS and heparin were purified by dialyzing against deionized water using dialysis tubing with a molecular weight cut-off of 500 (Spectrum Labs Inc. USA) and lyophilized before use. Figure 1 shows 1H and 13C spectra of purified PSS acquired on Varian Mercury 400 MHz spectrometer using D2O as a solvent. Human recombinant bFGF (Stemfactor bFGF Cat. No. 03-002) was obtained from Stemgent Inc., USA.
Figure 1. 1H (a) and 13C (b) NMR spectra of PSS in D2O.
C2C12 Cell culture
C2C12 cells were procured from ATCC (CRL-1772), and seeded at a density of 104 cells/cm2. The cells were cultured in growth medium (GM: high glucose DMEM (Invitrogen) containing 10% fetal bovine serum (FBS, Atlanta Biologicals), 2mM L-glutamine (Invitrogen), 100 u/mL penicillin and 100μg/mL streptomycin (Gibco). Heparin or PSS solution was added at various concentrations (0, 7pg/mL, 0.7ng/mL, 7ng/mL, 70ng/mL, 700ng/mL, 7μg/mL) to 90% confluent C2C12 monolayers. The effect of PSS was also evaluated in myogenic differentiation medium (DM: similar to GM except 10% FBS is replaced with 2% horse serum). At higher PSS concentrations (e.g. 7μg/mL) a small percentage of cell death (5-15%) was observed in both GM and DM with the higher number corresponding to DM.
Immunofluorescent staining
After 72 hours of PSS or heparin incubation, the cells were stained for myosin heavy chain (MyHC), a late myogenic marker. The cells were fixed with 4% paraformaldehyde followed by permeabilization with 0.5% Triton X-100 (Fisher Scientific) in 3% bovine serum albumin (BSA, Sigma Chemicals). Cells were then stained with anti-MyHC antibody (Developmental Studies Hybridoma Bank) at 1:250 dilution with 0.1% Triton X-100 in 3% BSA for 1hr followed by the secondary antibody (goat anti-mouse - Alexa Fluor 488 - Invitrogen) for 1hr. The nuclei were stained with diamidino-2-phenylindole (DAPI, Thermo Scientific, USA) and images were taken using a fluorescent microscope (Carl Zeiss, Axio Observer A1).
Quantification of differentiation and myotube formation
Extent of myogenic differentiation and multi-nucleated myotube formation (fusion index) were calculated by analyzing images of cells stained for myosin heavy chain (MyHC), using Macbiophotonics ImageJ software (McMaster University, Ontario). The number of myotubes, i.e., MyHC positive cells, was counted manually. For counting the nuclei, the images were filtered and adjusted for threshold followed by calculating the total number of nuclei (i.e., DAPI positive cells). Four random fields of view (each with at least 800 nuclei) were analyzed per sample for each of the three independent experiments. The differentiation index was calculated as the ratio of MyHC positive cells to the total number of nuclei and the fusion index was calculated as the ratio of myotubes containing three or more nuclei to the total number of MyHC positive cells.
Effect of PSS on ERK signaling
C2C12/ERK-Luciferase reporter cell line was created by permanently transfecting C2C12 cells with Cignal-ERK-Luciferase (SA Biosciences, CA) according to manufacturer's instructions. Briefly, Cignal-ERK-Luciferase virus was added to C2C12 cells growing on a 6-well plate (3×105 cells/well) at 20 MOI - multiplicity of infection. After transfection for 24hrs, the medium was replaced with fresh GM. C2C12/ERK-Luciferase reporter cell line was obtained by continually expanding these cells under puromycin selection for over three weeks. C2C12/null-Luciferase reporter cell line was created as a negative control using the same procedure.
The cells were seeded at a density of 1.5 × 104 cells/cm2 in 96-well plates. PSS solutions were added to the C2C12/ERK-luciferase reporter cell line, using C2C12 luciferase cells without reporter as a control. After adding PSS solutions, cells were allowed to incubate in GM or DM for various times (4, 24, 48 hrs). The medium was then replaced with 100 μL of ONE-Glo luciferase reagent (Promega, WI), and the luciferase expression was quantified by using a luminescence plate reader (DTX880 multimode detector, Beckman Coulter).
Statistical methods
Myogenic differentiation and myotube formation (differentiation index, and fusion index) were reported as an average of quadruplicates with standard deviation. The statistical significance was determined by using one-way ANOVA at p<0.05 with Bonferroni post-test to compare data pairs. GraphPad Prism software was used for performing all the statistical analysis.
Non-denaturing (native) PAGE
Interaction of bFGF with PSS and heparin was characterized by running native-PAGE. bFGF was reconstituted to 250ng/μL using sample buffer (50mM TRIS, 100mM NaCl, 0.05% Triton X-100, pH- 6.5 + 10% glycerol). 6% acrylamide gel prepared in running buffer (50mM TRIS, 100mM NaCl, 0.05% Triton X-100, pH- 6.5) was used for running native PAGE. 3μg of bFGF was incubated with various amounts (60ng, 120ng and 240ng) of PSS or heparin in sample buffer with a final volume of 20μL for 20 minutes at room temperature. These solutions were loaded on the gel and the gel was run at 100V for 1.45hrs with reversed polarity. Gels were stained with Coomassie blue.
Molecular docking calculations
Molecular docking of PSS on bFGF was performed using the AutoDock Vina 1.0 package [19]. This docking program is about two orders of magnitude faster in predicting binding sites than its earlier versions and it also significantly improves the accuracy of binding by allowing up to 32 rotatable bonds. The crystal structure of bFGF (1BFC) [20] minus the bound HDTH and water molecules was used for docking. A molecular model for a PSS decamer (10 monomers) was constructed using the Vega ZZ 2.3.1.2 package [21]. The 3D coordinates of PSS and bFGF were converted into the appropriate format (adding polar hydrogens, removing nonpolar hydrogens, and defining rotatable bonds) by using the AutoDockTools package [22]. The bFGF receptor was held rigid while all rotatable bonds in PSS were allowed to rotate.
We carried out 256 independent docking simulations by using default parameters in Autodock Vina 1.0 with each simulation providing one final lowest-energy binding mode. The 256 low-energy configurations were then further subjected to energy minimization and cluster analysis [23]. The energy minimization was carried out by using the Amber10 package [24], involving 500 steps of steepest-descent with the generalized Born model for the solvent. The hierarchical cluster analysis was carried out by using a root mean square deviation (RMSD) cutoff of 3Å for sulfur atoms of the docked PSS. The structure with the lowest RMSD to the average structure of the most populated cluster was determined as the most favorable docking configuration. The docked structures of the bFGF/PSS complexes were visualized using VMD package.
Electrostatic potential calculations
The electrostatic potential calculations of PSS, and bFGF were determined by using the APBS package. Hydrogen atoms were added to the crystal structures using PDB2PQR [25] and charges and radii were assigned according to PARSE force field parameters [26]. The electrostatic surface potential of the bFGF was calculated by solving the linearized Poisson–Boltzmann equation using the APBS package [27]. These calculations were performed at a temperature of 300 K, solute and solvent dielectric constants of 4 and 80, respectively, and ion concentration and exclusion radius of 0.2 M and 2.0 Å, respectively. The same procedure was carried out for the PSS ligands. APBS output including structures with 3D surface potentials were visualized using PyMol (www.pymol.org).
Results and Discussions
bFGF signaling has been shown to play an important role in the formation of skeletal muscle and their homeostasis. bFGF promotes proliferation of muscle progenitor cells and maintain them in an undifferentiated state [28,29]. Therefore, one of the commonly used approaches to promote myogenesis is by repressing bFGF activity [30]. Heparin and HSPGs have been shown to play an important role in bFGF signaling mediated myogenesis [31]. In this study, we have evaluated the effect of soluble heparin and a heparin-mimicking polymer, PSS, on myogenic differentiation and myotube formation of C2C12 cells in two different culture conditions, GM and DM. GM containing higher serum components promotes proliferation of C2C12 cells, while DM containing lower serum components promotes myogenic differentiation. The myogenic differentiation of C2C12 cells was quantified in terms of differentiation and fusion indices. The differentiation index is the fraction of total nuclei that are MyHC positive while fusion index is the fraction of MyHC positive cells containing three or more nuclei within them [32].
Before examining the effects of PSS, we have first confirmed that soluble heparin promotes myogenic differentiation. Exogenous supplementation of heparin both in GM and DM showed myogenic differentiation of C2C12 cells, as characterized by MyHC positive cells (Figure 2 and Figure 4a-b). As expected, the number of differentiated cells was higher in DM as compared to GM at all concentrations of heparin. We next examined the effect of PSS on promoting myogenic differentiation of C2C12 cells in GM and DM. The PSS-mediated increase in myogenic differentiation was evident after 24 hrs and was found to increase further with culture time. Figure 3 shows MyHC staining images of C2C12 cells at different culture conditions after 72 hrs of culture. Compared to control cultures devoid of any PSS, cultures containing PSS promoted both myogenic differentiation and myotube formation. Similar to heparin, the PSS-mediated myogenesis was more prominent in DM (Figure 4d, 5d) as compared to GM (Figure 4c and 5c).
Figure 2.

Dose dependent effect of soluble heparin (hep) on myogenic differentiation of C2C12 cells: MyHC immunofluorescent staining of C2C12 cultured with different concentrations of heparin in growth medium (top row) and differentiating medium (bottom row). Higher number of MyHC positive cells indicating differentiated cells, was observed in cultures containing higher heparin concentrations, while culture medium containing lower heparin concentration promoted multi-nucleated myotube formation. (Scale bar: 100μm)
Figure 4.
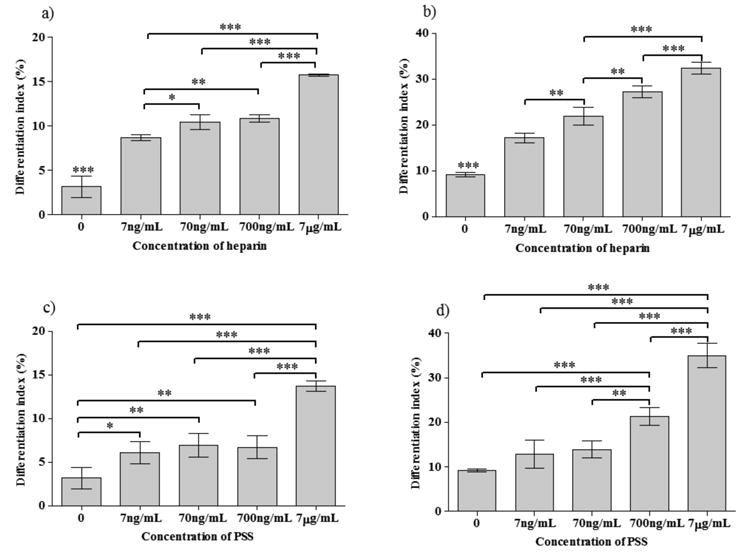
Differentiation indices (a-d) of C2C12 cells at different concentrations of heparin (a,b) and PSS (c,d) (a,c-in GM; b,d-in DM); Statistical significance is shown as * for p<0.05, ** for p<0.01 and *** for p<0.001
Figure 3.

Dose dependent effect of PSS on myogenic differentiation of C2C12 cells: MyHC immunofluorescent staining of C2C12 cultured with different concentrations of PSS in growth medium (top row) and differentiating medium (bottom row). Similar to heparin containing culture, higher PSS concentration resulted in higher number of MyHC positive cells while culture medium containing lower PSS concentration promoted multi-nucleated myotube formation. (Scale bar: 100μm)
Figure 5.
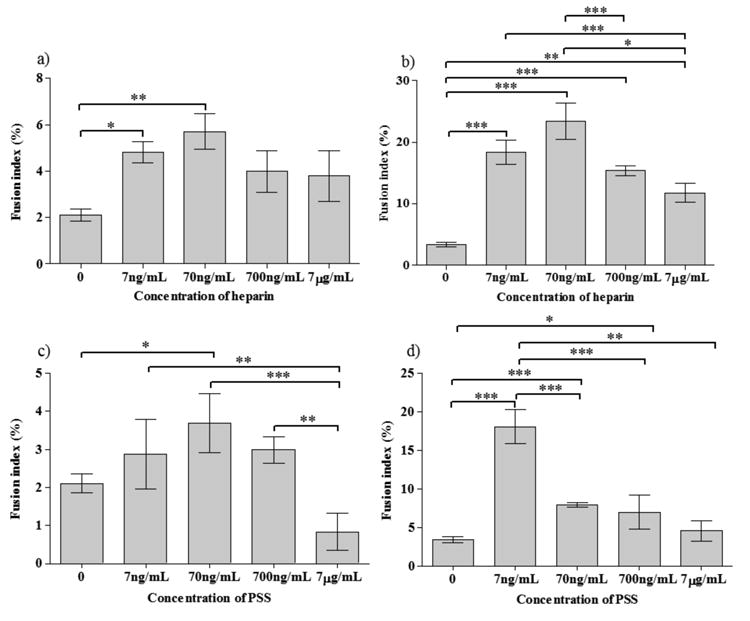
Fusion indices (a-d) of C2C12 cells at different concentrations of heparin (a,b) and PSS (c,d) (a,c-in GM; b,d-in DM); Statistical significance: * for p<0.05, ** for p<0.01 and *** for p<0.001
Both in heparin as well as PSS containing cultures, an increase in differentiation index with increasing concentration of heparin and PSS was observed in both GM and DM, though the trends were different (Figure 4). For all cases, except PSS in GM, the myogenic differentiation showed a continuous increase with the PSS and heparin concentrations, while PSS in GM showed an initial increase, then a plateau in the range 7ng/mL – 700 ng/mL, followed by another increase with increasing PSS concentration. At the highest concentration (7μg/mL) of PSS in DM, the differentiation index was about 3.8 and 2.5 times higher than that of control and GM containing the same PSS concentration, respectively. We observed a similar trend for heparin in DM with a 3.5 and 2.0 fold increases in the differentiation index compared to control and GM, respectively. Lower concentrations of PSS (0.7ng/mL and 7pg/mL) did not exhibit any noticeable effect on C2C12 myogenesis (data not shown). The slight differences observed between heparin and PSS could be attributed to differences in their chemical structure, molecular weight and the extent of sulfation.
In contrast to differentiation, the fusion index of C2C12 cells showed a non-monotonic dependence with the concentration of PSS and heparin, exhibiting a peak at 70 ng/mL in GM (Figure 5). At higher PSS concentrations (7μg/mL), most of the differentiated cells were mono-nucleated and the fusion index was lower than that of PSS-deficient control in GM. On the other hand, in DM, at all PSS concentrations, the fusion index was higher than that of the control (Figure 5d), again exhibiting a peak at lower concentrations, 7ng/mL. A similar trend was observed for heparin in DM with a slightly higher fusion index at higher concentrations of heparin (70ng/ml). The reason for the observed decrease in fusion index at high PSS and heparin concentrations despite the differentiation of C2C12 is not clear and could be due to various factors such as the interference of exogenous PSS and heparin on other signaling pathways involved in fusion of differentiated myoblasts. The higher differentiation and fusion indices observed in DM compared to GM could be attributed to the low serum (and hence low bFGF) present in DM.
We next determined the effect of PSS on ERK pathway, given the importance of MAPK/ERK pathway in myogenesis [10, 33]. ERK activity of C2C12 cells exposed to various concentrations of PSS was examined using the C2C12/ERK-luciferase reporter cell line, with the C2C12 luciferase cell line without reporter and the untreated ERK reporter as controls. Exogenous supplementation of PSS decreased ERK activity in a dose dependent manner (Figure 6). We did not observe any time-dependent effect on ERK down-regulation (data not shown). Down regulation of MAPK/ERK pathway is known to promote myogenesis of C2C12 cells (33).
Figure 6.
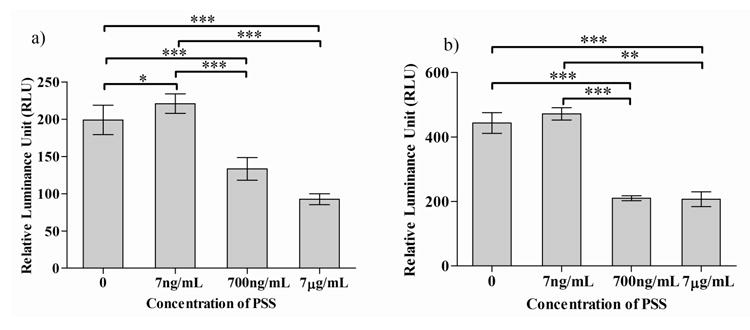
Effect of PSS on ERK signaling of C2C12 cells cultured in GM (a) and DM (b); Statistical significance is shown as * for p<0.05 and *** for p<0.001
We next confirmed the ability of PSS and heparin to interact with bFGF by subjecting bFGF to non-denaturing polyacrylamide gel electrophoresis (native PAGE) in the presence and absence of PSS and heparin (Figure.7). bFGF alone and bFGF treated with PSS or heparin exhibited significant differences in their mobility under identical gel running conditions (pH = 6.5 and reverse polarity), reflecting changes in their relative molecular mass and net charge. As anticipated, bFGF treated with PSS and heparin exhibited a retarded migration compared to bFGF alone. Similarly, in contrast to the intense band observed for bFGF alone, bFGF-PSS and bFGF-heparin samples showed a reduction in band intensity (for the unbound bFGF) in a dose-dependent manner. Taken together, these findings suggest that both PSS and heparin complex with bFGF.
Figure 7.

Interaction of bFGF with heparin and PSS as characterized by native PAGE. 3μg of bFGF was incubated with varying amount of PSS or heparin for 20minutes at room temperature before loading on the gel.
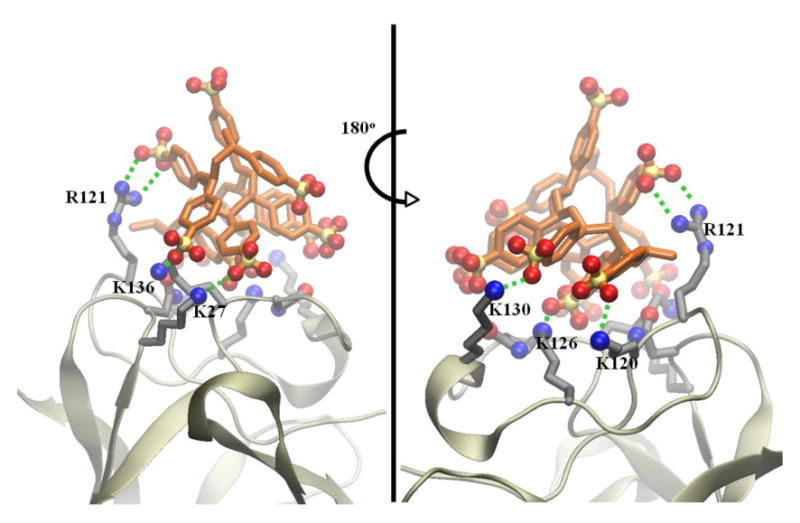
Finally, we have performed docking calculations of a PSS decamer onto bFGF to further examine their interactions and compared it with results from heparin binding [34]. Our calculations reveal that PSS can bind to bFGF with a binding free energy of −5 kcal/mol, similar to that of heparin [34]. Figure 8 shows the most favorable docked configuration of PSS on bFGF and highlights the key residues involved in the binding. The binding results primarily from cooperative salt bridges, hydrogen bonds, and electrostatic interactions between the acidic groups of PSS (SO3-) and basic residues of bFGF (K27, K120, R121, K126, K130, and K136). By comparing the docked configuration of PSS with that of heparin obtained from the heparin-bFGF complex crystal structure [20], we observe that the same bFGF surface involved in heparin binding is also involved in the binding of PSS. However, we note that PSS binding has a stronger van der Waals component compared to heparin, though both bindings are dominated by electrostatic interactions.
Figure 8. Front and side views of the most favorable docked configuration of PSS on bFGF, illustrating salt bridges and hydrogen bonding interactions (green dashed lines) between the PSS and bFGF residues.
To further demonstrate that the PSS-bFGF binding is dominated by electrostatic interactions, we have computed the electrostatic potential map of heparin, PSS, and bFGF under physiological salt concentrations (Figure 9). The atomic coordinates for PSS for these calculations were taken from the most favorable docked configuration and those for heparin and bFGF were taken from their X-ray crystallographic structures [27]. bFGF exhibits a strong positively charged pocket that is involved in PSS and heparin binding, and both heparin and PSS exhibit a strong negative charge. We have also utilized these electrostatic calculations to roughly determine contribution to binding from solvation and Coulombic forces. In fact, PSS binding exhibited a more favorable solvation plus Coulombic contribution (−20 kcal/mol) compared to heparin binding (−16.7 kcal/mol).
Figure 9.
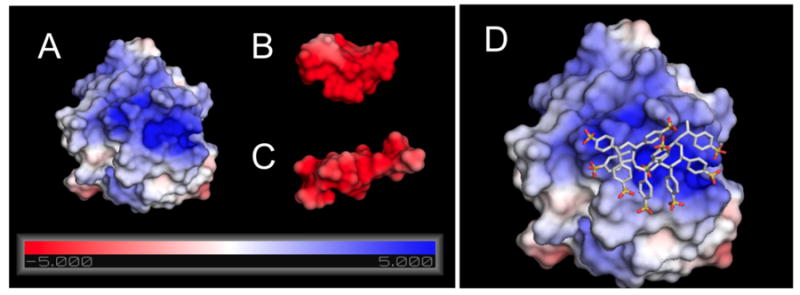
Electrostatic potential maps (in units of kT/e) for bFGF (A), PSS (B), and heparin (C) at their respective solvent accessible surfaces, computed using APBS; (D) The docked configuration of PSS decamer, shown in atomic detail, on the bFGF surface.
Taken together, our results suggest that cost-effective synthetic heparin mimics like PSS, which bind in an analogous manner to bFGF as heparin, can promote myogenic differentiation of muscle progenitor cells through down-regulating the ERK pathway. Therefore, PSS would be an ideal alternative to heparin for modulating activities of heparin binding growth factors such as bFGF, especially given the recent observations of contaminants and batch-to-batch variations in heparin [18].
Acknowledgments
This research was supported in part by the California Institute of Regenerative Medicine (RN2-00945) to S.V. C-W.C was supported by the UCSD California Institute of Regenerative Medicine Postdoctoral fellowship program. Authors thank Prof. Uli Muller for technical advice and comments. Authors thank Chao Zhang for help with NMR. The MyHC antibody developed by Helen Blau was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology.
References
- 1.Eswarakumar VP, Lax I, Schlessinger J. Cytokines & growth factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Scata KA, Bernard DW, Fox J, Swain JL. Exp Cell Res. 1999;250:10–21. doi: 10.1006/excr.1999.4506. [DOI] [PubMed] [Google Scholar]
- 3.Brunetti A, Goldfine ID. J Bio Chem. 1990;265:5960–5963. [PubMed] [Google Scholar]
- 4.Moore JW, Dionne C, Jaye M, Swain JL. Development. 1999;111:741–748. doi: 10.1242/dev.111.3.741. [DOI] [PubMed] [Google Scholar]
- 5.Olwin BB, Rapraeger A. J Cell Bio. 1992;118:631–639. doi: 10.1083/jcb.118.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pellergrini L. Curr Op Struct Bio. 2001;11:629–634. doi: 10.1016/s0959-440x(00)00258-x. [DOI] [PubMed] [Google Scholar]
- 7.Gospodarowicz D, Cheng J. J Cell Physi. 1986;128:475–484. doi: 10.1002/jcp.1041280317. [DOI] [PubMed] [Google Scholar]
- 8.Vlodavsky I, Miao HQ, Medalion B, Danagher P, Ron D. Cancer & Metastasis Rev. 1996;15:177–186. doi: 10.1007/BF00437470. [DOI] [PubMed] [Google Scholar]
- 9.Chua CC, Rahimi N, Forsten-Williams K, Nugent MA. Circulation Res. 2004;91:316–323. doi: 10.1161/01.RES.0000112965.70691.AC. [DOI] [PubMed] [Google Scholar]
- 10.Delehedde M, Seve M, Sergeant N, Wartelle I, Lyon M, Rudland PS, Fernig DG. J Bio Chem. 2000;275:33905–33910. doi: 10.1074/jbc.M005949200. [DOI] [PubMed] [Google Scholar]
- 11.Rusnati M, Presta M. Int J Clin Lab Res. 1996;26:15–23. doi: 10.1007/BF02644769. [DOI] [PubMed] [Google Scholar]
- 12.Ludin L, Ronnstrand L, Cross M, Hellberg C, Lindahl U, Claesson-Welsh L. Expt Cell Res. 2003;287:190–198. doi: 10.1016/s0014-4827(03)00125-3. [DOI] [PubMed] [Google Scholar]
- 13.Tardieu M, Gamby C, Avramoglou T, Jozefonvicz J, Barritault D. J Cell Physi. 1992;150:194–203. doi: 10.1002/jcp.1041500126. [DOI] [PubMed] [Google Scholar]
- 14.Liekens S, Leali D, Neyts J, Esnouf R, Rusnati M, Dell'era P, Maudgal PC, De Clercq E, Presta M. Mol Pharma. 1999;56:204–213. doi: 10.1124/mol.56.1.204. [DOI] [PubMed] [Google Scholar]
- 15.Desgranges P, Barbaud C, Caruelle JP, Barritault D, Gautron J. FASEB J. 1999;13:761–766. doi: 10.1096/fasebj.13.6.761. [DOI] [PubMed] [Google Scholar]
- 16.Benezra M, Ben-Sasson SA, Regan J, Chang M, Bar-Shavit R, Vlodavsky I. Arterioscler Thromb Vasc Bio. 1994;14:1992–1999. doi: 10.1161/01.atv.14.12.1992. [DOI] [PubMed] [Google Scholar]
- 17.Nozaki M, Li Y, Zhu J, Ambrosio F, Uehara K, Fu FH, Huard J. Am J Sports Med. 2008;36:2354–2362. doi: 10.1177/0363546508322886. [DOI] [PubMed] [Google Scholar]
- 18.Guerrini M, Beccati D, Shriver Z, Naggi A, Viswanathan K, Bisio A, Capila I, Lansing JC, Guglieri S, Fraser B, Al-Hakima A, Gunay NS, Zhang Z, Robinson L, Buhse L, Nasr M, Woodcok J, Langer R, Venkataraman G, Linhardt RJ, Casu B, Torri G, Sasisekharan R. Nat Biotech. 2008;26:669–675. doi: 10.1038/nbt1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trott O, Olson AJ. J Comput Chem. 2009;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faham S, Hileman RE, Fromm JR, Linhardt RJ, Rees DC. Science. 1996;271:1116–1120. doi: 10.1126/science.271.5252.1116. [DOI] [PubMed] [Google Scholar]
- 21.Pedretti A, Villa L, Vistoli G. J Comp Aided Mol Design. 2004;18:167–173. doi: 10.1023/b:jcam.0000035186.90683.f2. [DOI] [PubMed] [Google Scholar]
- 22.Sanner MF. J Mol Graphics Mod. 1999 Feb;17:57–61. [PubMed] [Google Scholar]
- 23.Takaoka T, Mori K, Okimoto N, Neya S, Hoshino T. J Chem Theor Comput. 2007;3:2347–2356. doi: 10.1021/ct700029q. [DOI] [PubMed] [Google Scholar]
- 24.Case DA, Cheathan TE, III, Darden T, Gohike H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. Nucleic Acids Res. 2007;35:W522–5. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sitkoff D, Sharp KA, Honig B. J Phy Chem. 1994;98:1978–1988. [Google Scholar]
- 27.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olwin BB, Hannon K, Hein P, Mcfall A, Riley B, Szebenyi G, Zhou Z, Zuber ME, Rapraeger AC, Fallon JF, Kudla AJ. Mol Reprod Develop. 1994;39:90–101. doi: 10.1002/mrd.1080390114. [DOI] [PubMed] [Google Scholar]
- 29.Hannon K, Kudla AJ, McAvoy MJ, Clase KL, Olwin BB. J Cell Bio. 1996;132:1151–9. doi: 10.1083/jcb.132.6.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clegg CH, Linkhart TA, Olwin BB, Hauschka SD. J Cell Bio. 1987;105:949–956. doi: 10.1083/jcb.105.2.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brandan E, Larrain J. Basic Appl Myol. 1998;8:107–113. [Google Scholar]
- 32.Crescenzi C, Crouch DH, Tato F. J Cell Bio. 1994;125:1137–1145. doi: 10.1083/jcb.125.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tortorella LT, Milasincic DJ, Pilch PF. J Bio Chem. 2001;276:13709–13717. doi: 10.1074/jbc.M100091200. [DOI] [PubMed] [Google Scholar]
- 34.Takaoka T, Mori K, Okimoto N, Neya S, Hoshino TJ. Chem Theo Comput. 2007;3:2347–2356. doi: 10.1021/ct700029q. [DOI] [PubMed] [Google Scholar]