

Abstract
Background
Glioblastoma (GBM) is a highly aggressive type of glioma with poor prognosis. However, a small number of patients live much longer than the median survival. A better understanding of these long-term survivors (LTSs) may provide important insight into the biology of GBM.
Methods
We identified 7 patients with GBM, treated at Memorial Sloan-Kettering Cancer Center (MSKCC), with survival >48 months. We characterized the transcriptome of each patient and determined rates of MGMT promoter methylation and IDH1 and IDH2 mutational status. We identified LTSs in 2 independent cohorts (The Cancer Genome Atlas [TCGA] and NCI Repository for Molecular Brain Neoplasia Data [REMBRANDT]) and analyzed the transcriptomal characteristics of these LTSs.
Results
The median overall survival of our cohort was 62.5 months. LTSs were distributed between the proneural (n = 2), neural (n = 2), classical (n = 2), and mesenchymal (n = 1) subtypes. Similarly, LTS in the TCGA and REMBRANDT cohorts demonstrated diverse transcriptomal subclassification identities. The majority of the MSKCC LTSs (71%) were found to have methylation of the MGMT promoter. None of the patients had an IDH1 or IDH2 mutation, and IDH mutation occurred in a minority of the TCGA LTSs as well. A set of 60 genes was found to be differentially expressed in the MSKCC and TCGA LTSs.
Conclusions
While IDH mutant proneural tumors impart a better prognosis in the short-term, survival beyond 4 years does not require IDH mutation and is not dictated by a single transcriptional subclass. In contrast, MGMT methylation continues to have strong prognostic value for survival beyond 4 years. These findings have substantial impact for understanding GBM biology and progression.
Keywords: gene expression, glioblastoma, prognostic genes, TCGA
See the editorial by Bi and Beroukhim, on pages 1159–1160.
Glioblastomas (GBMs) are the most common primary brain tumor in adults.1 Despite advances in therapy over the past few decades, prognosis remains poor with a median survival of 12–15 months.2 Long-term survivors (LTSs) of this disease exist but are rare, and the factors that predict prolonged survival are still incompletely defined.
Historically, prognostic factors consisted of clinical variables including age, performance status, and extent of resection.3 More recently, methylation of the MGMT promoter has been identified as a favorable prognostic factor, particularly in patients treated with concomitant and adjuvant temozolomide (TMZ).4–6 Furthermore, recent studies have demonstrated the prognostic significance of mutations in IDH1 and IDH2.7–10 Mutations in IDH1 and 2 occur frequently in intermediate-grade gliomas and secondary glioblastomas and are associated with better prognosis than IDH wild-type gliomas. The presence of IDH mutation imparts the glioma hypermethylator phenotype (G-CIMP).9
With the advent of large-scale gene expression studies of GBM, the disease has been classified into molecular subtypes.11,12 The Cancer Genome Atlas (TCGA) project identified 4 distinct subgroups: proneural, neural, classical, and mesenchymal.11,13 Though other gene expression profiling studies have found an association between subtype and prognosis,12,14 the TCGA failed to show a correlation between subtype and survival with the exception of the finding that IDH-mutant/G-CIMP tumors were associated with better survival.7,11,13
In this study, we sought to identify the molecular characteristics of GBM patients who survived at least 48 months. Forty-eight months represents extreme long-term survival in this disease with a median survival of 12–15 months. Unlike other studies, we focused on very LTSs with a median survival in our cohort of more than 5 years. Importantly, it is not yet known what the transcriptomal subclass characteristics of such GBM LTSs are and how known prognostic factors such as MGMT relate to transcriptomal identity in these LTS patients. We identified 7 patients with GBM treated at Memorial Sloan-Kettering Cancer Center (MSKCC) with survival >48 months. We examined MGMT promoter methylation and IDH1 and IDH2 mutation status in our LTS cohort. We characterized the transcriptomes of each patient and correlated gene expression data from our patients with the data from the TCGA and NCI Repository for Molecular Brain Neoplasia Data (REMBRANDT). We used the data from the TCGA and LTSs at our institution to identify genes prognostic of survival and validated these genes using an independent dataset.
Materials and Methods
Clinical Characteristics
Clinical and survival data for the MSKCC LTSs were obtained by retrospective review of patients' charts. Patients' age, sex, treatment, extent of resection, Karnofsky performance status (KPS), and survival were determined. Overall survival (OS) was calculated using the Kaplan-Meier method. The 7 patients represented consecutive patients at MSKCC who met the survival criteria. All patients were consented and approved by the institutional review board for inclusion in this study.
Tumors
All tumors (n = 7) were obtained following surgical resection at MSKCC as part of routine clinical care and snap-frozen. Tumors were obtained in accordance with institutional review board policies at MSKCC. Each sample was examined histologically by 3 independent neuropathologists and confirmed to be World Health Organization (WHO) grade IV glioma (GBM). Before analysis, tumors were sectioned and microdissected. Genomic DNA or RNA was extracted using the DNeasy kit (Qiagen) or RNeasy Lipid Tissue Mini Kit (Qiagen) according to the manufacturers' instructions. Nucleic acid quality was determined using the Agilent 2100 Bioanalyzer.
IDH1 and IDH2 Mutation Analysis
Sanger sequencing was performed to identify mutations in R132 of IDH1 and R172 of IDH2, as previously described.9 The exons of these residues were sequenced. Standard M13 tails were added to the tails to facilitate Sanger sequencing. PCR reactions were carried out in 384-well plates in a Duncan DT-24 water bath thermal cycler with 10 ng of whole genome-amplified DNA (REPLI-g Midi, Qiagen) as a template, using a touchdown PCR protocol with KAPA Fast HotStart (Kapa Biosystems). The touchdown PCR method consisted of: 1 cycle of 95°C for 5 minutes; 3 cycles of 95°C for 30 seconds, 64°C for 15 seconds, 72°C for 30 seconds; 3 cycles of 95°C for 30 seconds, 62°C for 15 seconds, 72°C for 30 seconds; 3 cycles of 95°C for 30 seconds, 60°C for 15 seconds, 72°C for 30 seconds; 37 cycles of 95°C for 30 seconds, 58°C for 15 seconds, 72°C for 30 seconds; and 1 cycle of 70°C for 5 minutes. Templates were purified using AMPure (Agencourt Biosciences). The purified PCR reactions were split in two and sequenced bidirectionally with M13 forward and reverse primer and the Big Dye Terminator Kit v.3.1 (Applied Biosystems) at Agencourt Biosciences. Dye terminators were removed using the CleanSEQ kit (Agencourt Biosciences), and sequence reactions were run on ABI PRISM 3730xl sequencing apparatus (Applied Biosystems). Passing reads were assembled against reference sequences containing all coding exons, including 5 kb upstream and downstream of the gene, using command line Consed 16.0. Geneious R7 (version 7.0.4) was used to check for the presence or absence of point mutations at R132 in IDH1 and R172 in IDH 2.7,9,10,13
Analysis of MGMT Promoter Methylation
Extracted DNA was subjected to bisulphite modification using the EZ DNA Methylation Kit (Zymo Research) according to the manufacturer's instructions. Primer sequences of MGMT were for the unmethylated reaction 5′-TTTGTGTTTTGATGTTTGTAGGTTTTTGT-3′ (forward primer) and 5′-AACTCCACACTCTTCCAAAAACAAAACA-3′ (reverse primer) and for the methylated reaction 5′-TTTCGACGTTCGTAGGTTTTCGC-3′ (forward primer) and 5′-GCACTCTTCCGAAAACGAAACG-3′ (reverse primer), as have been previously described.6 The annealing temperature was 59°C. In vitro methylated DNA (IVD) was used as a positive control, and whole genome amplified DNA (WGA) was used as a negative control. Each PCR reaction (20 μl) was directly loaded onto 2% agarose gels, stained with ethidium bromide, and visualized under UV illumination.
Genomic and Data Analysis
Expression analysis of tumors was performed using the Affymetrics U133 2.0 microarray (Affymetrix). Affymetrix CEL files were imported into the Partek Genomics Suite (Partek). The TCGA gene expression data (HT-HG-U133A) were accessed from the TCGA data repositories (http://cancergenome.nih.gov; downloaded December 12, 2013). In total, 560 tumors were analyzed for gene expression. We performed analysis of variance (ANOVA) to identify genes that were differentially expressed between MSKCC and TCGA LTSs and TCGA patients with survival <1 year. Differential expression was defined as a P value <.05 with concordant fold change of at least 1.25 fold which corresponds to at least a 20% decrease and a 25% increase. Functional analysis of this gene list was performed using DAVID bioinformatics resource and categories with adjusted P values (Benjamini) <.05 considered significant. ANOVA was also performed to identify genes that were differentially expressed between the TCGA LTSs and the TCGA patients with survival <1 year.
We performed unsupervised hierarchical clustering of the MSKCC LTS and TCGA core tumors with survival information (n = 177) using Kendall dissimilarity to classify the MSKCC LTSs into the 4 subtypes identified using the TCGA data.
For the validation set, expression datasets of 219 cases were identified from the NCI Repository for Molecular Brain Neoplasia Data (REMBRANDT; http://rembrandt.nci.nih.gov). We classified the REMBRANDT dataset into LTSs >48 months (n = 16) and patients with survival <12 months (n = 73). We performed ANOVA to identify genes that were differentially expressed using the same criteria as in the TCGA dataset. Hypergeometric analysis was performed to assess the significance of overlap of the 2 gene lists.
Results
Clinical Characteristics of Glioma Long-term Survivors
The clinical characteristics of the MSKCC LTSs are detailed in Table 1. Patients had a mean age of 57 years (range, 32–70 years). All patients received radiation and chemotherapy, and all but one patient (86%) had a gross total resection of the tumor. The vast majority of patients (86%) had a KPS of 90%–100% at presentation. Mean and median survival for the patients was 75.5 months and 62.5 months, respectively (range, 50.6–138.3 months). All patients were confirmed to have GBM with representative hematoxylin-and-eosin stained slides pictured (Fig. 1). Figure 2A shows the Kaplan-Meier plot of overall survival of the LTS cohort.
Table 1.
Clinical information for Memorial Sloan Kettering Cancer Center long-term survivors
| Variable | Patients (%) |
|---|---|
| Age at diagnosis, mean (range) | 57 (32–70) |
| Sex | |
| Male | 5 (71) |
| Female | 2 (29) |
| Radiation therapy, any | 7 (100) |
| 6000–6290 cGy | 4 (57) |
| Gamma knife, 1100 cGy | 1 (14) |
| Unknown dose | 2 (29) |
| Chemotherapy | 7 (100) |
| Temodar | 6 (86) |
| Carmustine (BCNU) | 1 (14) |
| Extent of resection | |
| Gross | 6 (86) |
| Subtotal | 1 (14) |
| KPS at diagnosis | |
| 90%–100% | 6 (86) |
| 80% | 1 (14) |
| Survival (months) | |
| Mean | 75.5 |
| Median | 62.5 |
| Range | 50.6–138.3 |
Abbreviation: KPS, Karnofsky performance status.
Fig. 1.
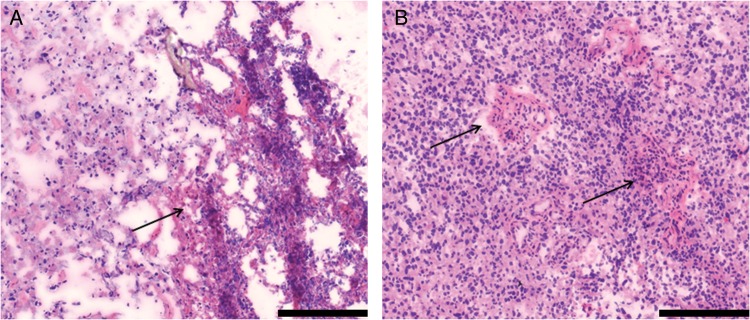
Representative slides from the tumors of MSKCC long-term survivors. Staining was performed with hematoxylin and eosin. Scale bars represent 200μM. (A) Sample LTS 6 with arrow indicating area of necrosis. (B) Sample LTS 3 with arrows indicating areas of endothelial proliferation.
Fig. 2.

Kaplan–Meier plot of overall survival of (A) the MSKCC long-term survivors and (B) TCGA dataset used for analysis. Black line indicates 48-month cutoff for long-term survivors used in this series.
Transcriptional Diversity of Tumors From Glioma Long-term Survivors
In order to characterize the transcriptome of GBM tumors from MSKCC LTSs, we obtained gene expression data using the Affymetrics U133 2.0 microarray. These data were imported along with TCGA expression data (U133A). Both datasets contained identical probes (n = 22 277). The data were normalized using quantile normalization, and ANOVA was performed to correct for batch effects. Mean values were obtained for duplicate tumors and/or duplicate probes. Survival data for the MSKCC and TCGA cohorts were obtained and used to classify patients as LTSs (MSKCC, n = 7; TCGA, n = 26) and those with survival <1 year (TCGA, n = 209). Figure 2B shows the Kaplan-Meier plot for the TCGA cohort used in the analysis (n = 533 with survival data).
Unsupervised hierarchical clustering was performed to classify our samples into the subtypes identified in the TCGA data. In order to do so, we used the well-validated 840 gene signature described by Verhaak et al.11 and the core samples from the TCGA dataset (n = 177 with survival data). Interestingly, instead of uniformly classifying into a single subgroup, our patients classified into all 4 subgroups; 2 patients belonged to the proneural subgroup, 2 to the neural subtype, 2 to the classical subtype, and 1 to the mesenchymal subtype. Figure 3 shows unsupervised hierarchical clustering of the MSKCC and TCGA samples. The transcriptomal diversity of the MSKCC LTSs is further illustrated and verified by comparing expression levels of genes representative of each subtype (Supplementary Fig. S1).11,15
Fig. 3.

Classification of GBM long-term survivors (LTSs) into TCGA subgroups. Unsupervised hierarchical clustering was used to classify the 7 MSKCC LTS patients and 15 TCGA LTS patients into the TCGA subgroups. One hundred sixty-one TCGA core samples and 7 MSKCC samples are shown in the heat map above. Color scale indicates normalized relative expression. TCGA subgroup identity for each cluster is noted and color coded along the Y-axis.
We then sought to determine which genes were differentially expressed between the LTSs and patients with survival <1 year. In order to do so, we performed ANOVA between the LTSs (MSKCC and TCGA) and the TCGA patients with survival <1 year. We did not restrict the analysis to the 840 gene signature but rather included all of the genes. Sixty genes were differentially expressed between the 2 groups. None of these genes remained significant after multiple test correction, which was likely due to the relatively small numbers. A list of these genes can be found in Table 2. We also performed this analysis using only the TCGA data, examining differential gene expression between TCGA LTSs and TCGA patients with survival <1 year. When the MSKCC samples were excluded from the analysis, all 60 genes from the initial analysis remained significant for differential expression. A list of genes that were differentially expressed when the MSKCC samples were excluded from the analysis can be found in Supplementary Table S1. Among the 60 genes were CHI3L1, EMP3, IGFBP2, IGFBP3, LGALS3, MAOB, PDPN, SERPING1, and TIMP1, which were found (in an independent analysis) to be associated with survival with decreased expression correlated with longer survival.16 Also among this list were 7 genes (CBR1, CHI3L1, F3, FMOD, LGALS3, PDPN, and RBP1) that are among the top 50 most differentially hypermethylated and downregulated genes in proneural G-CIMP-positive tumors.8,9 Gene ontology pathway analysis was performed using the DAVID bioinformatics resource. The LTSs were found to have downregulation of genes involved in response to inflammation including response to wounding, acute inflammatory response, and humoral inflammatory response (Supplementary Table S2).
Table 2.
Genes differentially expressed between MSKCC and TCGA long-term survivors versus TCGA patients with survival less than 1 year
| Gene Symbol | P value | Percent Change | Increase or Decrease in LTS vs. patients with survival <1 year | Description |
|---|---|---|---|---|
| EFEMP2* | 0.000091 | 31.31 | Decrease | EGF containing fibulin-like extracellular matrix protein 2 |
| CHI3L1* | 0.004373 | 44.95 | Decrease | chitinase 3-like 1 (cartilage glycoprotein-39) |
| MSN | 0.004389 | 25.28 | Decrease | Moesin |
| PDPN* | 0.004496 | 26.12 | Decrease | Podoplanin |
| LOC390940* | 0.004600 | 20.55 | Decrease | Protein LOC390940 |
| IGFBP2* | 0.004785 | 39.00 | Decrease | Insulin-like growth factor binding protein 2 |
| SPP1* | 0.004798 | 35.29 | Decrease | Secreted phosphoprotein 1 |
| TIMP1 | 0.004898 | 37.38 | Decrease | Tissue inhibitor of metalloproteinase 1 |
| FKBP9* | 0.007392 | 23.47 | Decrease | FK506 binding protein 9, 63 kDa |
| C1R* | 0.007482 | 31.76 | Decrease | Complement C1r subcomponent |
| WWTR1 | 0.007926 | 20.50 | Decrease | WW domain containing transcription regulator 1 |
| FMOD* | 0.007932 | 26.67 | Decrease | fibromodulin |
| DUSP6 | 0.009469 | 23.63 | Decrease | Dual specificity protein phosphatase 6 |
| CLEC5A* | 0.009478 | 20.59 | Decrease | C-type lectin domain family 5 member A |
| CHST2 | 0.009603 | 23.15 | Decrease | Carbohydrate sulfotransferase 2 |
| CHL1 | 0.009900 | 34.00 | Decrease | cell adhesion molecule L1-like protein |
| EMP3* | 0.010202 | 32.50 | Decrease | epithelial membrane protein 3 |
| AEBP1* | 0.012936 | 31.90 | Decrease | Adipocyte enhancer-binding protein 1 |
| HIST3H2A | 0.014230 | 32.37 | Increase | Histone H2A type 3 |
| DIRAS3* | 0.014387 | 24.36 | Decrease | GTP-binding protein Di-Ras3 |
| SERPING1* | 0.015011 | 28.54 | Decrease | Serin peptidase inhibitor, clade G, member 1 |
| CBR1* | 0.015149 | 20.98 | Decrease | Carbonyl reductase [NADPH] 1 |
| FABP7 | 0.015658 | 32.75 | Decrease | Fatty acid-binding protein, brain |
| FZD7* | 0.016326 | 22.39 | Decrease | frizzled family receptor 7 |
| RBP1 | 0.016362 | 35.50 | Decrease | Retinol-binding protein 1 |
| SCG5* | 0.017010 | 31.36 | Decrease | Secretogranin V |
| F3 | 0.017753 | 25.51 | Decrease | coagulation factor III (thromboplastin, tissue factor) |
| C1S* | 0.017854 | 31.36 | Decrease | Complement C1s subcomponent |
| FAM46A | 0.018064 | 21.56 | Decrease | family with sequence similarity 46, member A |
| PLSCR1 | 0.018198 | 21.25 | Decrease | Phospholipid scramblase 1 |
| TMEM158* | 0.019466 | 30.96 | Decrease | Transmembrane protein 158 |
| GBP1 | 0.023537 | 27.14 | Decrease | Interferon-induced guanylate-binding protein 1 |
| CD24 | 0.024976 | 48.86 | Increase | CD24 molecule |
| DYNLT3 | 0.026435 | 20.29 | Decrease | Dynein, light chain, Tctex-type 3 |
| NAMPT* | 0.028738 | 29.91 | Decrease | Nicotinamide phosphoribosyltransferase |
| LGALS3* | 0.028989 | 31.57 | Decrease | lectin, galactoside-binding, soluble, 3 |
| CD14 | 0.030171 | 28.67 | Decrease | CD14 molecule |
| PDGFA* | 0.032116 | 20.34 | Decrease | Platelet-derived growth factor subunit A |
| ADM | 0.033162 | 32.99 | Decrease | adrenomedullin |
| SEC61G* | 0.033389 | 33.20 | Decrease | Protein transport protein Sec61 subunit gamma |
| SERPINA3 | 0.034414 | 37.00 | Decrease | Serpin peptidase inhibitor, clade A, member 3 |
| A2M | 0.034520 | 24.66 | Decrease | Alpha-2-macroglobulin |
| PMP22 | 0.035318 | 23.11 | Decrease | Peripheral myelin protein 22 |
| DPYD | 0.037046 | 24.90 | Decrease | Dihydropyrimidine dehydrogenase [NADP(+)] |
| CSRP1 | 0.037825 | 22.15 | Decrease | Cysteine and glycine-rich protein 1 |
| IGFBP3* | 0.038283 | 30.79 | Decrease | Insulin-like growth factor binding protein 3 |
| MAOB | 0.038425 | 32.38 | Decrease | Monoamine oxidase B |
| MT1M | 0.039683 | 30.55 | Decrease | Metallothionein 1M |
| TMEM176B | 0.039863 | 25.14 | Decrease | Transmembrane protein 176B |
| AKAP12 | 0.041885 | 25.59 | Decrease | A-kinase anchor protein 12 |
| TRIM22 | 0.042183 | 22.67 | Decrease | E3 ubiquitin-protein ligase TRIM22 |
| BST2* | 0.042421 | 22.07 | Decrease | Bone marrow stromal antigen 2 |
| BASP1 | 0.042542 | 39.41 | Increase | Brain acid soluble protein 1 |
| FCGR2B | 0.043519 | 20.62 | Decrease | Low affinity immunoglobulin gamma Fc region receptor II-b |
| CD99 | 0.044209 | 21.97 | Decrease | CD99 molecule |
| TNFAIP6 | 0.044390 | 23.09 | Decrease | Tumor necrosis factor-inducible gene 6 protein |
| TMEM176A | 0.047687 | 23.46 | Decrease | Transmembrane protein 176A |
| POSTN | 0.047970 | 27.83 | Decrease | Periostin |
| SLC1A3 | 0.048246 | 29.60 | Decrease | Excitatory amino acid transporter 1 |
| CCL2 | 0.048957 | 32.36 | Decrease | C-C motif chemokine 2 |
*also significant in REMBRANDT data set.
In the updated TCGA analysis,13 the LTSs (>3 years) were analyzed and found to have somewhat fewer amplifications of CDK4 and EGFR and fewer deletions of CDKN2A. Though we were unable to assess copy number, we did look at expression level for these genes and found no statistical difference between the MSKCC LTSs and the TCGA patients with survival <1 year (See Supplementary Fig. S2A and B).
Molecular Genetic Analysis
After determining that there was transcriptomal diversity among the LTSs from our institution, we sought to determine whether all of our LTSs were IDH1 or IDH2 mutant and/or positive for hypermethylation of the MGMT promoter, 2 known indicators of good prognosis. On targeted sequencing of R132 for IDH1 and R172 for IDH2, none of the MSKCC LTSs were found to possess mutant IDH1 or 2.
To determine MGMT methylation status, methylation-specific PCR was used and revealed that 5 of the 7 MSKCC LTSs (71%) showed methylation positivity, while 2 were unmethylated (as shown in Figure 4).
Fig. 4.

Methylation status of the MGMT promoter in the MSKCC long-term survivors. A methylation-specific PCR assay was used, with in vitro methylated DNA (IVD) used as a positive control, whole genome amplified DNA (WGA) used as a negative control for methylation, and water as a negative PCR control. “U” denotes the presence of unmethylated alleles and “M” the presence of methylated alleles. Five glioblastoma samples (201, 202, 205, 206, and 204) show methylation, and 2 samples (207, 203) are unmethylated.
We then reviewed the association of IDH1 and LTS in the TCGA dataset. The mutation status of IDH1 was available in 22 of the LTSs in the TCGA dataset. Of these 22 patients, 6 (27%) were mutant for IDH1. Of the remaining 295 patients with IDH1 mutation status who were not LTSs (living patients excluded since survival status not yet determined), 12 were mutant for IDH1 (4%). Table 3 summarizes the molecular features of the MSKCC and TCGA LTS patients.
Table 3.
Summary of molecular features of Memorial Sloan Kettering Cancer Center long-term survivors
| Molecular feature | MSKCC LTSs % | TCGA LTSs (%) |
|---|---|---|
| Transcriptional subgroup | n = 7 | n = 27 |
| Proneural | 2 (29) | 13 (48) |
| Classical | 2 (29) | 4 (15) |
| Mesenchymal | 1 (14) | 7 (26) |
| Neural | 2 (29) | 3 (11) |
| MGMT methylated | 5 (71) | 10 (67) (n = 15 with available data) |
| IDH 1/2 mutated | 0 (0) | 6 (27) (n = 22 with available data) |
Abbreviations: LTSs, long-term survivors; MSKCC, Memorial Sloan Kettering Cancer Center; TCGA, The Cancer Genome Atlas.
Validation of Long-term Survivor Gene Expression Patterns Using the REMBRANDT Cohort
In order to further validate the findings found in the TCGA and MSKCC cohorts, we sought to establish the reproducibility of our observations by using the independent REMBRANDT dataset. After accounting for duplicate samples and duplicate probes, we were left with gene expression data from 219 patients, 186 of whom had survival data. Among these 186 patients, there were 16 LTSs and 73 with survival <1 year. We performed ANOVA to compare genes that were differentially expressed between the LTSs and those patients with survival <1 year. Twenty-five of the 60 genes found to be significant in the TCGA and MSKCC datasets were also significant in the REMBRANDT data, many of which were downregulated in the LTS tumors. These genes are denoted by an asterisk in Table 2. These genes include CHI3L1, EFEMP1, EMP3, NAMPT, C1S, and PDGFA. We performed a hypergeometric analysis of the genes that overlapped between the TCGA and MSKCC data and the REMBRANDT data. The P value obtained was 1.83 × 10–19, demonstrating statistically significant overlap. There was no difference in expression levels of EGFR and CDKN2A between the MSKCC and REMBRANDT LTSs and the REMBRANDT patients with survival <1 year (Supplementary Fig. S2C and D).
Finally, we sought to classify the REMBRANDT dataset into the TCGA transcriptional subtypes in order to determine if the LTSs in the validation cohort were also diverse with regard to subtype. In order to do so, we performed unsupervised hierarchical clustering using the 840 gene signature identified by Verhaak et al.11 of the TCGA core samples and the REMBRANDT LTSs (Fig. 5). As with the LTSs in the MSKCC cohort, the LTSs in the REMBRANDT dataset belonged to all 4 subtypes, with 44% classified as proneural and the remainder divided among classical, mesenchymal, and neural subtypes. The distribution of the LTSs from all 3 datasets is pictured in Figure 6. We also performed unsupervised hierarchical clustering on the REMBRANDT samples alone (Supplementary Fig. S3) and found that the LTSs in the REMBRANDT dataset were similarly distributed among all 4 subtypes.
Fig. 5.

Classification of REMBRANDT long-term survivors (LTSs) and TCGA samples into TCGA subgroups. Unsupervised hierarchical clustering of TCGA samples and REMBRANDT LTSs. Color scale indicates normalized relative expression.
Fig. 6.
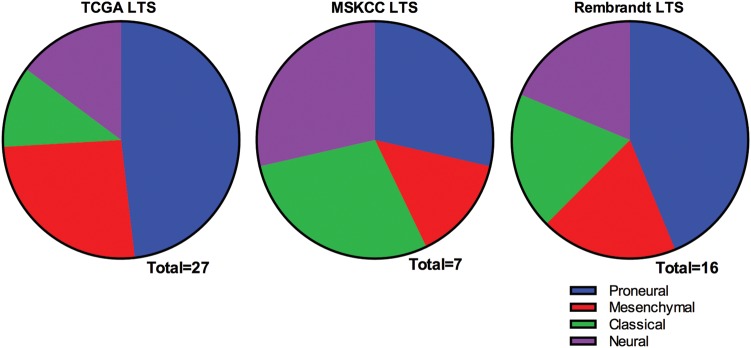
Distribution of long-term survivors from 3 datasets among the The Cancer Genome Atlas transcriptional subtypes.
Discussion
In this study, we describe the transcriptomal and molecular features of patients with GBM with survival >4 years. One strength of this study is the long mean and median survival of our cohort (75.5 months and 62.5 months, respectively; >3 standard deviations above the mean survival for all-comers), highlighting the fact that our cohort consists of extreme outliers with long-term survival among patients with GBM. Our data add to an important body of literature examining genomic predictors of outcome in GBM.
In identifying patients with survival >4 years, we selected the extreme outliers with this deadly disease. Surprisingly, these LTSs fell within all 4 TCGA transcriptomal subgroups, and none harbored an IDH mutation. Since IDH-mutant/CIMP-positive proneural tumors have been linked to improved survival (at least in the short term), it was reasonable to expect that the vast majority of the extreme LTSs would be of the proneural subtype or harbor IDH mutation.7–9,17–19 Using 3 independent cohorts, our study shows that long-term survival can be achieved in patients with nonproneural tumors and no IDH mutations. Nevertheless, in 2 of the 3 cohorts (TCGA and REMBRANDT), ∼50% of the LTSs were proneural, and in the TCGA cohort, 27% of the LTSs harbored IDH mutations. Previous studies have also shown higher incidence of IDH1 mutations in LTSs as compared with short-term survivors (STS)20 or non-LTSs.21 Therefore, it appears that these features are associated with survival out to 4 years and beyond. The discrepancy between our data and prior series may reflect a selection bias or a difference in the biological distribution of the MSKCC patients. Nevertheless, this study demonstrates that GBM survivorship out to 4 years or longer can occur in patients of all subgroups and in patients without IDH mutations.
The high rate of MGMT promoter hypermethylation in our group of LTS is consistent with prior data showing frequent hypermethylation of MGMT in LTSs.20,22 It is also consistent with data that LTSs with wild-type IDH1/2 have a higher rate of MGMT promoter methylation compared with STS with wild-type IDH ½.21 Our data indicate that MGMT methylation remains a powerful predictor of long-term survivorship to beyond 4 years.
In the most recent update of the TCGA data,13 Brennan et al., examined the LTSs (>3 year survival). They reported no specific genomic alteration that was significantly over- or underrepresented in this cohort, although they noted fewer amplifications of CDK4 and EGFR and fewer deletions of CDKN2A in the LTSs. Using expression as a proxy for copy number, we did not observe differences in expression of these specific genes in our data. However, we did find a list of genes that were differentially expressed between the LTSs and those with survival <1 year, which was found to have significant overlap with a gene list generated using the independent REMBRANDT dataset. Furthermore, there was overlap between our gene signature and previous signatures identified by Colman et al.16 and Kim et al.,23 as being prognostic of survival, although we note that some of this overlap might have been due to use of the same TCGA data in the latter study. Interestingly, some of the genes we identified as being downregulated in our signature are also known to be among the most differentially hypermethylated and downregulated genes in proneural G-CIMP positive tumors,8 which further implicate these genes in survivorship. Our definition of LTSs of >4 years and our comparison group as having survival <1 year was more restrictive than the definition used in other studies16,23 (survival > vs. <2 years). Thus, we hypothesize that the gene expression changes observed in our study better predict very long survival for GBM patients.
Though our LTSs did not classify into a single subgroup, many of the genes that were differentially downregulated in the LTS tumors in this study (eg, CHI3L1 (YKL40), EFEMP2, EMP3, LGALS3, PDPN, and TIMP1) are associated with mesenchymal tissues and/or the extracellular matrix (ie, the mesenchymal subgroup).11–13 The MSKCC and TCGA LTSs showed lower levels of expression of these genes as compared with the TCGA STS. This finding is consistent with previous observations attributing a poorer prognosis with higher expression of mesenchymal-angiogenic associated-genes.12,16,23 Further analysis of the differentially expressed genes revealed a downregulation of genes involved in response to inflammation such as A2M, CCL2, PDPN, CHST2, SERPING1, C1R, C1S, F3, SERPINA3, and SPP1. Eighteen of the 60 differentially expressed genes (30%) were involved in response to wounding. Other significant pathways included defense response, acute inflammatory response, and regulation of response to external stimuli. This downregulation of inflammatory and immune markers was consistent with a recent 14-gene prognostic signature that found active inflammatory response pathway in a high-risk group of GBM patients.24 The association of a reduced inflammatory response with LTSs may have been due to reduction in an active inflammatory environment leading to decreased tumor growth and progression.25
In conclusion, we have demonstrated that extreme LTSs of GBM do not represent a single molecular subtype but are transcriptomally diverse. Furthermore, MGMT methylation was common in our cohort and continues to be prognostic for very long-term survival. IDH1 and IDH2 mutations were not present in any of the MSKCC LTSs examined in this study and may not be required to achieve survivorship to beyond 4 years. These data have significant implications for understanding the determinants of survival in GBM patients.
Primary Accessions
Microarray data have been accessioned with the Gene Expression Omnibus under series GSE54077.
Supplementary Material
Funding
This work was supported in part by the Frederick Alder fund (TAC) and the Memorial Sloan Kettering Cancer Center Brain Tumor Center (TAC).
Supplementary Material
Acknowledgments
We thank Kety Huberman, Agnes Viale, the MSKCC Genomics Core, and the MSKCC Pathology Core for excellent technical support. This work was supported in part by the Frederick Alder fund (TAC) and the MSKCC Brain Tumor Center (TAC).
Conflict of interest statement. None declared.
References
- 1.Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310(17):1842–1850. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 2.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 3.Curran WJ, Jr., Scott CB, Horton J, et al. Recursive partitioning analysis of prognostic factors in three Radiation Therapy Oncology Group malignant glioma trials. J Natl Cancer Inst. 1993;85(9):704–710. doi: 10.1093/jnci/85.9.704. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 5.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 6.Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350–1354. doi: 10.1056/NEJM200011093431901. [DOI] [PubMed] [Google Scholar]
- 7.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17(5):510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9(3):157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 13.Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freije WA, Castro-Vargas FE, Fang Z, et al. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004;64(18):6503–6510. doi: 10.1158/0008-5472.CAN-04-0452. [DOI] [PubMed] [Google Scholar]
- 15.Huse JT, Holland E, DeAngelis LM. Glioblastoma: molecular analysis and clinical implications. Annu Rev Med. 2013;64:59–70. doi: 10.1146/annurev-med-100711-143028. [DOI] [PubMed] [Google Scholar]
- 16.Colman H, Zhang L, Sulman EP, et al. A multigene predictor of outcome in glioblastoma. Neuro Oncol. 2010;12(1):49–57. doi: 10.1093/neuonc/nop007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thota B, Shukla SK, Srividya MR, et al. IDH1 mutations in diffusely infiltrating astrocytomas: grade specificity, association with protein expression, and clinical relevance. Am J Clin Pathol. 2012;138(2):177–184. doi: 10.1309/AJCPZOIY3WY4KIKE. [DOI] [PubMed] [Google Scholar]
- 18.Carrillo JA, Lai A, Nghiemphu PL, et al. Relationship between tumor enhancement, edema, IDH1 mutational status, MGMT promoter methylation, and survival in glioblastoma. AJNR Am J Neuroradiol. 2012;33(7):1349–1355. doi: 10.3174/ajnr.A2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mellai M, Monzeglio O, Piazzi A, et al. MGMT promoter hypermethylation and its associations with genetic alterations in a series of 350 brain tumors. J Neurooncol. 2012;107(3):617–631. doi: 10.1007/s11060-011-0787-y. [DOI] [PubMed] [Google Scholar]
- 20.Zhang GB, Cui XL, Sui DL, et al. Differential molecular genetic analysis in glioblastoma multiforme of long- and short-term survivors: a clinical study in Chinese patients. J Neurooncol. 2013;113(2):251–258. doi: 10.1007/s11060-013-1102-x. [DOI] [PubMed] [Google Scholar]
- 21.Hartmann C, Hentschel B, Simon M, et al. Long-Term Survival in Primary Glioblastoma With Versus Without Isocitrate Dehydrogenase Mutations. Clin Cancer Res. 2013;19(18):5146–5157. doi: 10.1158/1078-0432.CCR-13-0017. [DOI] [PubMed] [Google Scholar]
- 22.Martinez R, Schackert G, Yaya-Tur R, et al. Frequent hypermethylation of the DNA repair gene MGMT in long-term survivors of glioblastoma multiforme. J Neurooncol. 2007;83(1):91–93. doi: 10.1007/s11060-006-9292-0. [DOI] [PubMed] [Google Scholar]
- 23.Kim YW, Koul D, Kim SH, et al. Identification of prognostic gene signatures of glioblastoma: a study based on TCGA data analysis. Neuro Oncol. 2013;15(7):829–839. doi: 10.1093/neuonc/not024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amimappamagan A, Somasundaram K, Thennarasu K, et al. A fourteen gene GBM prognostic signature identifies association of immune response pathway and mesenchymal subtype with high risk group. PLOS One. 2013;8(4):1–14. doi: 10.1371/journal.pone.0062042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mantovani A, Allavena P, Sica A, et al. Cancer-related inflammation. Nature. 2008;454(7203):436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.