

Abstract
DNA methylation is a well-studied epigenetic modification essential for efficient cellular differentiation. Aberrant DNA methylation patterns are a characteristic feature of cancer including myeloid malignancies such as acute myeloid leukemia (AML). Recurrent mutations in DNA modifying enzymes were identified in AML and linked to distinct DNA methylation signatures. In addition, discovery of Tet enzymes provided new mechanisms for the reversal of DNA methylation. Advances in base-resolution profiling of DNA methylation have enabled a more comprehensive understanding of the methylome landscape in the genome. This review will summarize and discuss the key questions in the function of DNA methylation in the hematopoietic system, including recent studies that have elucidated where and how DNA methylation regulates diverse biological processes in the genome.
The presence of 5-methylcytosine (5mC) in nucleic acid was first discovered among the hydrolysis products of tuberculinic acid in 1950 [1]. It has long been studied as a part of the genetic code with limited understanding of its importance in mammalian cells until DNA methylation reached a milestone with identified roles in transcriptional regulation of development and X chromosome inactivation in 1975 [2, 3]. The discovery of CpG islands suggested candidate regions in the genome for methylation study [4] and since then, intensive studies have expanded our understanding of the diverse effects of DNA methylation in various organisms and different tissue types, particularly in the context of CpG islands. These studies have led to the elucidation of molecular pathways required for establishing and maintaining DNA methylation, cell type specific variation in methylation patterns, and the involvement of methylation in multiple cellular processes such as transcription regulation, cellular differentiation, tumorigenesis, X chromosome-inactivation and imprinting [5–10]. Understanding the function of DNA methylation requires consideration of the distribution of methylation across the genome. Genome-wide studies of DNA methylation have begun with low resolution [11] or a reduced approaches which only capture a small fraction of the genome [12–14]. However, followed by the advent of high-throughput sequencing technology, single-base resolution genome-wide DNA methylation data is now available. In this review, we will discuss recent discoveries about genome-wide distribution of 5-methylcytosine and the role of cytosine modifying enzymes and their somatic mutations in hematopoietic malignancies to achieve a better understanding of the functional roles of DNA methylation and therapeutic applications.
DNA methylation and demethylation
DNA methylation commonly involves modification of cytosines. The mammalian DNMT family is made up of five members, DNMT1, DNMT2, DNMT3A, DNMT3B and DNMT3L. The maintenance methyltransferase, DNMT1 is responsible for maintaining the methylation pattern during replication and adds methylation to DNA when one strand is already methylated. De novo methyltransferases DNMT3A and DNMT3B create hemimethylated CpG dinucleotides to establish new patterns of methylation (Figure 1a). Their activity can be modulated by the catalytically inactive family member DNMT3L, however DNMT3L is primarily restricted to early embryogenesis, so it does not play a major role [8, 15, 16]. In mammalian genomes, 5-methylcytosine (5mC) exists mostly in the CpG dinucleotide context and about 70–80% of CpGs are methylated. Although the DNA methylation pattern in cells is generally stably maintained, DNA methylation can be removed passively by blocking methylation of newly synthesized DNA during DNA replication. Global DNA demethylation is important for resetting pluripotent states in early embryos and for erasing parental-origin-specific imprints in developing germ cells [17]. Recent compelling genetic and biochemical data indicate that genomic methylation patterns can be changed by active demethylation (Figure 1b). The discovery of the Tet family of enzymes that can modify 5mC through oxidation was another milestone in advancing our understanding of DNA demethylation mechanisms, introducing 5-hydroxymethylcytosine (5hmC) as a key intermediate and the further oxidized intermediates5-formylcytosine (5fC) and 5-carboxycytosine (5caC) in active demethylation pathways [18–20].
Figure 1. The DNA methylation and demethylation pathway.

Overview of the DNA methylation and demethylation process. Dnmt1 is responsible for maintenance methylation. A)Dnmt1 adds methylation to hemi-methylated DNA when one strand is already methylated. Dnmt3a and Dnmt3b are responsible for de novo DNA methylation. They create hemi-methylated CpG dinucleotides to establish new patterns of methylation. B) Passive demethylation occurs through loss of Dnmt1/3 (via loss of gene expression, gene mutation, or possibly via other mechanisms that inhibit protein function). Active demethylation is mediated by Tet family proteins. 5-methylcytosine (5mC) can be hydroxylated to 5-hydroxymethylcytosine (5hmC) by Tet proteins. 5hmC is not recognized by the maintenance methyltransferase (Dnmt1), so the methylated is lost during DNA replication. In addition, 5hmC can be further oxidized to 5-formylcytosines (5fC) and 5-carboxylcytosines (5caC). These latter forms may be removed by base-excision repair, in an alternate mode of active demethylation (in addition to loss of the 5hmC by cell division).
Who is the main player in hematopoiesis?
Hematopoietic stem cells are the best characterized somatic stem cell, and the differentiation hierarchy that emanates from them is well characterized [21]. As epigenetic changes facilitate lineage-specific differentiation, hematopoiesis provides a well-defined model to study dynamic DNA methylation changes during cell-fate decisions. Moreover, abnormal DNA methylation patterns are characteristic feature of hematologic malignancies, further compelling us to understand the role of DNA methylation changes during normal and aberrant hematopoietic development.
The de novo methyltransferase Dnmt3a has recently been shown to be essential for hematopoietic stem cell differentiation [22] and other groups identified somatic mutation of DNMT3A in ~30% of normal karyotype acute myeloid leukemia (AML) [23, 24], pointing to the fundamental role of 5mC in hematopoietic differentiation and disease. The most common mutation of DNMT3A in AML is R882H, which is within the catalytic domain. This mutation functions as a dominant-negative inhibitor of de novo DNA methylation in an embryonic stem (ES) cell model system as well as in human AML cells [25, 26]. Active Dnmt3b and Dnmt3l are expressed in murine ES cells and contribute to methylation activities. However, in AML cells, DNMT3L is not expressed, and an inactive splice isoform is the dominant form of DNMT3B, suggesting that the de novo DNA methylation potential in hematopoiesis is largely provided by DNMT3A [26].
The methylcytosine oxidase is essential for hematopoietic stem cell homeostasis. Tet2 inactivation in the mouse resulted in multiple hematopoietic abnormalities, and ultimately in myeloproliferation and a CMML like disease [27–29]. Moreover, TET2 mutations are prevalent inhematologic disorders that result in disrupted myeloid differentiation, including AML, MDS, MPN, CMML [30–34]. In addition, the isocitrate dehydrogenase (IDH) family of enzymes catalyzes the oxidative decarboxylation of isocitrate to alpha-ketoglutarate (αKG). Mutations in IDH1/2 have been recently identified, which lead to the abnormal accumulation of 2-hydroyglutarate (2HG), which inhibits αKG-dependent enzymes, including TET-mediated DNA demethylation. Thus, mutant IDH mimics TET2 mutation, and results in increased levels of 5mC and decreased levels of 5hmC. Mutation of IDH1/2 has been found in gliomas, AMLs and MPNs [29, 35, 36] and direct measurement of 2HG in IDH1/2-mutant AML can detect 100-fold increased 2HG levels in some patients, consistent with a gain-of-function of the mutant enzyme. The incidence of these newly found mutations related to DNA methylation are considerable in hematologic malignancies, resulting in alterations in DNA methylation and aberrant gene expressions patterns (Table 1).
Table 1.
Mutations in DNA modifiers in hematologic malignancies
| Gene | Malignancy | Mutation % | References |
|---|---|---|---|
| DNMT3A | AML | 12%–22% | [22,23,92,93] |
| MDS | 8% | ||
| MPN | 7%–15% | ||
| MDS/MPN | 4% | ||
|
| |||
| TET2 | AML | 12%–34 % | [31,94–98] |
| MDS | 20%–25% | ||
| MPN | 4%–14% | ||
| CMML | 50% | ||
|
| |||
| IDH1/2 | AML | 15%–33% | [35,99–104] |
| MDS | 3.50% | ||
| T-ALL | 2.5%–5% | ||
| T-cell lymphoma | 5%–10% | ||
T-ALL=T-cell acute lymphoblastic leukemia.
Genomic distribution of DNA methylation
The human and mouse genomes have approximately 28 million and 22 million CpGs respectively. Around 7% of CpGs reside within CGIs [37] and the majority of CpG sites exist outside of CpG Island (CGI). In most cell types, CpGs have stable methylation patterns and only ~20% CpGs are dynamic [38]. Depending on the genomic location, DNA methylation may have different biological functions, it is therefore important to map the DNA methylation changes in different physiologic states, and examine the influence one xpression of nearby genes. Promoter CGIs have a low methylation ratio, which is often increased in cancer cells contributing to gene silencing. Non-promoter CGIs show variable methylation ratios and the methylation changes in these regions are often tissue-specific. Gene bodies are highly methylated and the methylation is associated with active expression and may have an impact on splicing. Repeat elements are frequently methylated and loss of methylation in these regions has been postulated to be associated with chromosome instability; thus, suppression of the expression of transposable elements by methylation may be important for genome stability. Genome-wide high resolution methylation studies have enabled us to observe more detailed methylome architectures such as CGI shores, methylation Canyons and large hypomethylated regions. Here we summarize more details about each of these regions (Figure 2), and show for murine HSCs how the DNA methylation ratios vary for given genomic features (Table 2).
Figure 2. Graphical representation of the dynamic methylome landscapes in various genomic regions.
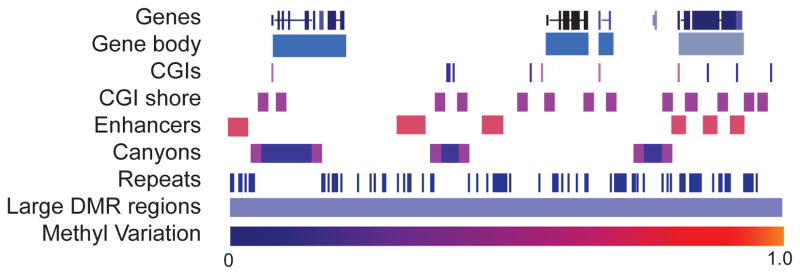
DNA methylation level variation across each feature represented using colors from level 0 (blue) to 1.0 (red). Enhancer regions, CGI shores, Canyon edges represent the most differentially methylated features [38, 60].
Table 2.
Mouse hematopoietic stem cell methylation ratio in various genomic regions
| Genomic Regions | Methylation Ratio |
|---|---|
| All | 83.52% |
| Canyons | 4.33% |
| CGI | 7.71% |
| CGI shore | 48.02% |
| Gene | 82.68% |
| Intron | 84.45% |
| Exon | 72.40% |
| Promoter | 23.78% |
| 5′UTR | 68.61% |
| 3′UTR | 83.07% |
| LINE | 88.53% |
| LTR | 89.79% |
| SINE | 90.03% |
| CTCF binding regions | 41.04% |
| Bivalent domains | 4.70% |
| H3K4me3 binding regions | 9.35% |
| H3K27me3 binding regions | 43.04% |
| H3K36me3 binding regions | 94.03% |
| Gata2 binding regions | 47.26% |
| PU.1 binding regions | 17.48% |
Gata2=xx; H3K27me3=xx; H3K36me3=xx; H3K4me3=xx; LINE=xx; LTR=xx; SINE =xx; UTR=xx.
All data from Sun et al. [54].
Promoter CGIs
Approximately 70% of the genes in the genome contain short CpG-rich regions known as CGIs, where as the remainder of the genome is depleted for CpGs [39]. Most studies have focused on 5mCs in a CpG context, and much of the work on DNA methylation focused on CGIs at promoter regions at the single-gene level. Most promoter CGIs are largely unmethylated in normal tissues, regardless of their differentiation state [40]. When genes with CGIs at the promoter are unmethylated, their promoters are usually characterized by nucleosome-free regions at the transcriptional start site (TSS). These nucleosome-free regions are often marked with H3K4me3 [41], and the levels of transcription are controlled by associated transcription factors. Transcription at some promoters is repressed by various mechanisms such the polycomb complex and H3K27me3 [42]. Methylation in promoter CGI regions in normal cells is usually restricted to genes at which there is long-term silencing such as the inactive X chromosome and imprinted genes and genes that are exclusively expressed in germ cells but not in somatic cells [43]. However, in hematological malignancies, many promoter CGIs become aberrantly hypermethylated. In particular, hypermethylation of cell cycle regulators, apoptosis and DNA repair genes are thought to contribute to reduced expression and the promotion of transformation [44, 45].
CGI shores
In recent years, genome-wide approaches have facilitated the anlaysis of regions outside of promoters and CGIs, and are thus expanding our understanding of DNA methylation in different cell types (including stem and differentiated cells). Comprehensive high-throughput arrays for relative methylation (CHARM) assays revealed that tissue- and cancer-specific differentially methylated regions occur more frequently within CGI shores and regions of relatively low CpG density that flank traditional CGIs (upto 2 kb in distance), than within CGIs themselves, suggesting the involvement of CGI shore methylation in tissue differentiation, epigenetic reprogramming and cancer [46, 47]. Methylome studies in hematopoietic lineages showed differential methylation regions (DMRs) in numerous genes known to play a role in lymphoid or myeloid fate specification and differential methylation occurs more frequently in CGI shores than CGIs during the differentiation process [48].
Enhancers
Little is known about DNA methylation in intergenic regions. These regions contain functionally important elements such as enhancers. Recently, low methylation regions (LMRs) and unmethylated regions (UMRs) have been suggested to function as enhancers [49]. Transcriptional enhancers support tissue-specific expression profiles through physical interactions with gene promoters. Unmethylated promoters are permissive but not necessary for transcription initiation. Enhancer methylation associates with cell-specific transcription levels, even when the promoter is constantly unmethylated. These sites bind chromatin-modulating factors, interact with distal promoters through DNA loops, and demonstrate a unique pattern of DNA methylation in different cell types. Global mapping of DNA methylation at different stages of hematopoiesis shows that differential methylated regions are enriched for transcription factor binding sites (TFBSs), and specific hypomethylation at myeloid TFBSs in lymphoid progenitors is observed [50]. Bock et al also observed binding sites of key myeloid-specific factors (Gata2, Tal1 and Lmo2) became robustly methylated during lymphoid differentiation [51]. Similarly, Sun et al found that TFBS of key HSC-associated transcription factors (e.g. Scl/Tal1) became hypomethylated in aging HSCs, while those of differentiation-associated transcription factors (e.g. Pu.1) were more likely to become hypermethylated, likely contributing to enhancing self-renewal and inhibiting differentiation with age [52, 53] [54].
Large Hypomethylated regions and methylation Canyons
Genome-wide approaches have identified additional large regions with important alterations in methylation in cancer and cell fate decisions. Epigenetic deregulation can occur not only at single genes, but can also encompass large chromosomal domains during differentiation and tumorigenesis. Hansen et al found large hypomethylated blocks to be enriched for genes with hyper variable expression in colon cancer, which could drive tumor cell heterogeneity [55].
Large hypo-methylated regions have been identified by comparing differentiated fibroblasts to human ESCs. These have been termed partially methylated domains (PMDs) [56]. The loss of methylation in these regions is accompanied by acquisition of repressive histone marks and genes in these domain are down-regulated [55, 57]. Long-range epigenetic activation domains (LREA) are large regions that typically span 1Mb and harbor key oncogenes and cancer biomarker genes, while long-range epigenetic silencing domains (LRES)harbor key tumor suppressors and miRNAs and were discovered in cancer cells [58, 59].
Jeong et al identified exceptionally large regions with very low levels of methylation (DNA methylation Canyons) inhematopoietic stem cells, which showed novel epigenomic features [60]. They are conserved across species and cell types, and dynamic DNA methylation changes occur at the edge of Canyons in the absence of Dnmt3a. Similar features have been reported in ES cells and termed DNA methylation valleys (DMVs) [61]. Altered large domain methylome architectures are associated with changes in transcriptional output and altered genomic stability that may be responsible for key gene-set regulation in cancer progression.
Gene body methylation
Most gene bodies are CpG-poor and extensively methylated. While most CGIs are located in promoter regions, CGIs also exist within the gene bodies. Gene body methylation is not associated with gene repression; instead, positive correlations between active transcription and gene body methylation have been reported [62]. DNA methylation in human cells has identified hypermethylation in the gene bodies of actively transcribed genes [14, 56]. Study of chronic lymphocytic leukemia (CLL) patients revealed that sites of DNA hypomethylation in the gene body are mostly enhancer sites and recognized a DNA methylation signature that distinguishes new clinico-biological subtypes of CLL [63]. Gene body methylation may also regulate tissue-specific expression from alternative promoters [64]. Distinctive epigenetic patterns in the gene body, including DNA methylation and nucleosome positioning have been identified around exons and exon–intron borders, suggesting that chromatin structure is also important to exon selection [65]. DNA methylation in gene bodies may facilitate exon exclusion via recruitment of the multifunctional CpG binding proteins [66]. DNA methylation inhibits CTCF binding to exons and this prevents CTCF-mediated Pol II pausing and spliceosome assembly [67]. Thus, DNA methylation in gene bodies has distinct functions from that of promoter methylation. How gene-body methylation levels are regulated and the underlying mechanisms through which it exerts an influence on gene expression are just beginning to be elucidated.
Repeats and Ribosomal DNA
Repetitive elements are DNA sequences that are present in multiple copies in the genomes in which they reside. Methylation in repeat regions such as centromeres is important for chromosomal stability during mitosis [68] and is also likely to suppress the expression of transposable elements and thus to have a role in genome stability. Recent data have also identified a role for tissue-specific retro element hypomethylation in association with enhancer activity [61]. Whole genome bisulfate sequencing (WGBS) data from DNMT3B-mutant immunodeficiency, centromeric instability, facial anomalies (ICF) patients showed profound changes in inactive heterochromatic regions, satellite repeats and transposons, which causes aberrant expression of immune genes and hypomethylation of pericentromeric regions accompanied by chromosomal instability. But interestingly, transcriptionally active loci and ribosomal RNA (rRNA) repeats escaped global hypomethylation [69]. The genes encoding rRNA are the most abundant genes in the genome. They reside in tandem repetitive clusters, in some cases totaling hundreds of copies. Due to their repetitive structure and highly active transcription, the rRNA gene repeats are some of the most fragile sites in the chromosome. CD34+ hematopoietic progenitor cells (HPCs) from MDS patients showed reduced rRNA expression and increased rDNA promoter methylation compare to controls. Treatment of myeloid cell lines with 5-aza-2′-deoxycytidine resulted in a significant decrease in the methylation of the rDNA promoter and an increase in rRNA levels [70].
The role of 5-Hydroxymethylcytosine
After the discovery of different levels of 5hmC in various mouse and human cells, several studies suggested there is a fine balance between 5mC and 5hmC that is critical for maintaining the normal state of cells. [18, 20, 71, 72].5hmC is not recognized by Dnmt1, so as the DNA is replicated, the methylation at that site is lost- thus offering a passive mechanism for DNA de-methylation [73]. However, active DNA de-methylation in the presence of 5hmC has also been proposed to occur via the base excision repair pathway [74]. Finally, 5hmC may have other specific functional roles in gene expression, aside from facilitating removal of DNA methylation. In order to understand these possible roles, the location of 5hmC must first be mapped at the base-resolution level.
Detecting genome-wide 5hmC distribution is challenging because of its low abundance. Through the use of available technologies, which include cytosine-5-methylenesulfonate (CMS-seq) [75], hydroxymethyl DNA immunoprecipitation sequencing (hMeDIP-seq) [76], oxidative bisulfate sequencing (oxBS-seq) [77] and Tet-assisted bisulfite sequencing (TAB-seq) [78], some conserved features of the 5hmC landscape have emerged. 5hmC is present at 1% of the total level of 5mC in immune cell populations [34], 5–10% of the level of 5mC in ES cells and 40% of 5mC in neuronal cells [18, 20, 72]. Genome-wide mapping of Tet1 and 5hmC in ESC genomic DNA indicates that Tet1 and 5hmC are enriched at transcription start sites with specific histone modifications known to be associated with inactive genes suggesting that 5hmC may contribute to the poised chromatin signatures at developmentally regulated genes. [79, 80]. Other studies showed the opposite results such as correlation between 5hmC and histone modifications in enhancer regions of human ES cells, and 5hmC in promoters and exon regions with increased levels of transcription [81–83]. It is possible that an independent mechanism can directly cause hydroxymethylation of the cytosines in a site-specific manner [84]. Tissue-specific differentially hydroxymethylated regions are located in the intragenic regions of the genome with intermediate GC content [83]. Studies of 5hmC in human CD34+ and in several erythroid developmental stages showed dynamic changes of 5hmC during differentiation to the erythroid lineages [85]. In addition, gain of 5hmC at the genomic loci of erythroid-specific transcription factor binding sites, and loss of 5hmC at transcriptionally repressed genes such as CD34, was shown [85]. Finally, rapid DNA demethylation occurs during erythropoiesis [86], and this is likely to be via a Tet-mediated mechanism. These findings suggests that 5hmC influences cell-specific transcriptional programs during differentiation, thereby facilitating gene expression. Overall, the discovery of the importance of the Tet family of proteins has transformed our views of DNA demethylation and underscored the importance of dynamic DNA methylation in cell fate decisions and gene regulation. We are at the very start of understanding the detailed mechanisms through which these proteins act in hematopoiesis.
The mechanism of DNA methyltransferase inhibitors in malignancies
The two approved DNA demethylating drugs, decitabine (DAC) and its analog azacitidine (AZA)are irreversible inhibitors of the DNA methyltransferase enzymes DNMT1 and DNMT3 [87, 88]. These drugs become incorporated into DNA, trap DNMTs and target these enzymes for degradation. They are potent drugs for MDS, leukemias and multiple types of solid tumors [89–91] and the clinical data suggest responses in about half of patients [92, 93]. Despite the clinical efficacy of DNA methyltransferase inhibitors (DNMTIs), there is stilla lack of understanding of the mechanism through which they function. Earlier studies reported that the activity of DNMTIs in cancers is via their ability to induce a DNA-damage response and apoptosis [94–96]. More recent studies indicated that treatment of cancer cells with clinically relevant low doses of DAC and AZA can selectively hypomethylate aberrantly methylated CpGs and reactivate repressed genes without inducing immediate cytotoxic effects such as DNA damage, apoptosis, and cell cycle arrest [97]. A number of studies have shown that DAC maintains normal HSC self-renewal but induces terminal differentiation in leukemia cells [87, 98–100]. However the mechanisms of sensitivity and resistance to DNMTIs are still open with questions. One of the important goals of genome-wide profiling of DNA methylation is to identify differences between malignant cells and normal cells that can be exploited for therapy.
Conclusions and perspectives
Recent advances in DNA methylation mapping have altered our view of the most dynamic sites of de novo methylation and demethylation, revealing that these changes occur more frequently in distal regions such as CGI shores, Canyon edges and enhancers with low CpG densities, rather than CGIs with high CpG density. In addition,5hmC and Tet proteins are detected at these regions [60, 82, 101]. How CpGs in these genomic regions are selectively targeted by Dnmts and Tets, and the mechanisms through which they impact gene expression and cancer, are critical remaining questions.
Recent discoveries of the importance to normal and malignant hematopoiesis of the proteins involved in DNA methylation and demethylation have transformed our outlook on gene regulation during hematopoiesis. The discoveries of the importance of proteins such as TET2 and DNMT3A have fortuitously come at a time when our capacity to identify DNA methylation modifications at base-pair resolution have been enormously facilitated by the dramatic drop in the cost of high-throughput sequencing. Thus, we are presented with a new opportunity to study DNA methylation changes in normal and malignant hematopoiesis by careful mapping. While it is possible that the proteins involved in DNA methylation also have alternative functions perturbed by mutation, it is essential to map the DNA methylation changes, and correlate these to changes in gene expression and cellular function. Through this focused approach, we will eventually shed light on how these mutations exert their powerful influence on cellular physiology.
Clinical studies of DNMTIs have demonstrated that targeting DNA methylation is selective and an efficient strategy for malignant cells but not for normal HSCs. Despite these findings, there are still many unanswered questions including the distribution of oxidized 5mC bases (5hmC/5fC/5caC) in the genome and their role during cellular processes, determination of the exact genes or loci that are important in their pathophysiology, and identification of the signature of DNA methylation that is predictive of therapeutic response. Ongoing genome-wide studies with advanced bioinfomatic analysis and rapid and cost-effective sequencing techniques will allow us to address many remaining questions.
Acknowledgments
This work is supported by NIH grants DK092883, AG036562, and CA183252, and the Waxman Cancer Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copy editing, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Mira Jeong, Email: mjeong@bcm.edu.
Margaret A. Goodell, Email: goodell@bcm.edu.
REFERNCES
- 1.Wyatt GR. Occurrence of 5-methylcytosine in nucleic acids. Nature. 1950;166:237–8. doi: 10.1038/166237b0. [DOI] [PubMed] [Google Scholar]
- 2.Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–32. [PubMed] [Google Scholar]
- 3.Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet. 1975;14:9–25. doi: 10.1159/000130315. [DOI] [PubMed] [Google Scholar]
- 4.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–82. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 5.Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:2395–402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 6.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–26. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 7.Lippman Z, Gendrel AV, Black M, et al. Role of transposable elements in heterochromatin and epigenetic control. Nature. 2004;430:471–6. doi: 10.1038/nature02651. [DOI] [PubMed] [Google Scholar]
- 8.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 9.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–32. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 10.Straussman R, Nejman D, Roberts D, et al. Developmental programming of CpG island methylation profiles in the human genome. Nat Struct Mol Biol. 2009;16:564–71. doi: 10.1038/nsmb.1594. [DOI] [PubMed] [Google Scholar]
- 11.Rauch TA, Wu X, Zhong X, Riggs AD, Pfeifer GP. A human B cell methylome at 100-base pair resolution. Proc Natl Acad Sci U S A. 2009;106:671–8. doi: 10.1073/pnas.0812399106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meissner A, Mikkelsen TS, Gu H, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–70. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng J, Shoemaker R, Xie B, et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol. 2009;27:353–60. doi: 10.1038/nbt.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ball MP, Li JB, Gao Y, et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27:361–8. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 16.Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19:219–20. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 17.Feng S, Jacobsen SE, Reik W. Epigenetic reprogramming in plant and animal development. Science. 2010;330:622–7. doi: 10.1126/science.1190614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–33. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–30. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ema H, Morita Y, Suda T. Heterogeneity and hierarchy of hematopoietic stem cells. Exp Hematol. 2014;42:74–82. e2. doi: 10.1016/j.exphem.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 22.Challen GA, Sun D, Jeong M, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2012;44:23–31. doi: 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–33. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan XJ, Xu J, Gu ZH, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011;43:309–15. doi: 10.1038/ng.788. [DOI] [PubMed] [Google Scholar]
- 25.Kim SJ, Zhao H, Hardikar S, Singh AK, Goodell MA, Chen T. A DNMT3A mutation common in AML exhibits dominant-negative effects in murine ES cells. Blood. 2013;122:4086–9. doi: 10.1182/blood-2013-02-483487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Russler-Germain DA, Spencer DH, Young MA, et al. The R882H DNMT3A Mutation Associated with AML Dominantly Inhibits Wild-Type DNMT3A by Blocking Its Ability to Form Active Tetramers. Cancer Cell. 2014 doi: 10.1016/j.ccr.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moran-Crusio K, Reavie L, Shih A, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quivoron C, Couronne L, Della Valle V, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 29.Li Z, Cai X, Cai CL, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118:4509–18. doi: 10.1182/blood-2010-12-325241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delhommeau F, Dupont S, Della Valle V, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 31.Kosmider O, Gelsi-Boyer V, Ciudad M, et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica. 2009;94:1676–81. doi: 10.3324/haematol.2009.011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langemeijer SM, Kuiper RP, Berends M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41:838–42. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- 33.Tefferi A, Lim KH, Abdel-Wahab O, et al. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia. 2009;23:1343–5. doi: 10.1038/leu.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ko M, Huang Y, Jankowska AM, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–43. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sonoda Y, Kumabe T, Nakamura T, et al. Analysis of IDH1 and IDH2 mutations in Japanese glioma patients. Cancer Sci. 2009;100:1996–8. doi: 10.1111/j.1349-7006.2009.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rollins RA, Haghighi F, Edwards JR, et al. Large-scale structure of genomic methylation patterns. Genome Res. 2006;16:157–63. doi: 10.1101/gr.4362006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ziller MJ, Gu H, Muller F, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500:477–81. doi: 10.1038/nature12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–22. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 41.Kelly TK, Miranda TB, Liang G, et al. H2A.Z maintenance during mitosis reveals nucleosome shifting on mitotically silenced genes. Mol Cell. 2010;39:901–11. doi: 10.1016/j.molcel.2010.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taberlay PC, Kelly TK, Liu CC, et al. Polycomb-repressed genes have permissive enhancers that initiate reprogramming. Cell. 2011;147:1283–94. doi: 10.1016/j.cell.2011.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 44.Khan H, Vale C, Bhagat T, Verma A. Role of DNA methylation in the pathogenesis and treatment of myelodysplastic syndromes. Semin Hematol. 2013;50:16–37. doi: 10.1053/j.seminhematol.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 45.Toyota M, Issa JP. Epigenetic changes in solid and hematopoietic tumors. Semin Oncol. 2005;32:521–30. doi: 10.1053/j.seminoncol.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 46.Doi A, I, Park H, Wen B, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41:1350–3. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Irizarry RA, Ladd-Acosta C, Wen B, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ji H, Ehrlich LI, Seita J, et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010;467:338–42. doi: 10.1038/nature09367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stadler MB, Murr R, Burger L, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–5. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- 50.Hodges E, Molaro A, Dos Santos CO, et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol Cell. 2011;44:17–28. doi: 10.1016/j.molcel.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bock C, Beerman I, Lien WH, et al. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol Cell. 2012;47:633–47. doi: 10.1016/j.molcel.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ziliotto R, Gruca MR, Podder S, et al. PU.1 promotes cell cycle exit in the murine myeloid lineage associated with downregulation of E2F1. Exp Hematol. 2014;42:204–217. e1. doi: 10.1016/j.exphem.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 53.Staber PB, Zhang P, Ye M, et al. Sustained PU.1 levels balance cell-cycle regulators to prevent exhaustion of adult hematopoietic stem cells. Mol Cell. 2013;49:934–46. doi: 10.1016/j.molcel.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun D, Luo M, Jeong M, et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell. 2014 doi: 10.1016/j.stem.2014.03.002. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hansen KD, Timp W, Bravo HC, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43:768–75. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show wide spread epigenomic differences. Nature. 2009;462:315–22. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hon GC, Hawkins RD, Caballero OL, et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012;22:246–58. doi: 10.1101/gr.125872.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Coolen MW, Stirzaker C, Song JZ, et al. Consolidation of the cancer genome into domains of repressive chromatin by long-range epigenetic silencing (LRES) reduces transcriptional plasticity. Nat Cell Biol. 2010;12:235–46. doi: 10.1038/ncb2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bert SA, Robinson MD, Strbenac D, et al. Regional activation of the cancer genome by long-range epigenetic remodeling. Cancer Cell. 2013;23:9–22. doi: 10.1016/j.ccr.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 60.Jeong M, Sun D, Luo M, et al. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat Genet. 2014;46:17–23. doi: 10.1038/ng.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xie W, Schultz MD, Lister R, et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell. 2013;153:1134–48. doi: 10.1016/j.cell.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–3. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- 63.Kulis M, Heath S, Bibikova M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44:1236–42. doi: 10.1038/ng.2443. [DOI] [PubMed] [Google Scholar]
- 64.Maunakea AK, Nagarajan RP, Bilenky M, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–7. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anastasiadou C, Malousi A, Maglaveras N, Kouidou S. Human epigenome data reveal increased CpG methylation in alternatively spliced sites and putative exonic splicing enhancers. DNA Cell Biol. 2011;30:267–75. doi: 10.1089/dna.2010.1094. [DOI] [PubMed] [Google Scholar]
- 66.Maunakea AK, Chepelev I, Cui K, Zhao K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013;23:1256–69. doi: 10.1038/cr.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shukla S, Kavak E, Gregory M, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74–9. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moarefi AH, Chedin F. ICF syndrome mutations cause a broad spectrum of biochemical defects in DNMT3B-mediated de novo DNA methylation. J Mol Biol. 2011;409:758–72. doi: 10.1016/j.jmb.2011.04.050. [DOI] [PubMed] [Google Scholar]
- 69.Heyn H, Vidal E, Sayols S, et al. Whole-genome bisulfite DNA sequencing of a DNMT3B mutant patient. Epigenetics. 2012;7:542–50. doi: 10.4161/epi.20523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Raval A, Sridhar KJ, Patel S, Turnbull BB, Greenberg PL, Mitchell BS. Reduced rRNA expression and increased rDNA promoter methylation in CD34+ cells of patients with myelodysplastic syndromes. Blood. 2012;120:4812–8. doi: 10.1182/blood-2012-04-423111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koh KP, Yabuuchi A, Rao S, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–13. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Globisch D, Munzel M, Muller M, et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One. 2010;5:e15367. doi: 10.1371/journal.pone.0015367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Valinluck V, Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67:946–50. doi: 10.1158/0008-5472.CAN-06-3123. [DOI] [PubMed] [Google Scholar]
- 74.Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–9. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang Y, Pastor WA, Zepeda-Martinez JA, Rao A. The anti-CMS technique for genome-wide mapping of 5-hydroxymethylcytosine. Nat Protoc. 2012;7:1897–908. doi: 10.1038/nprot.2012.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tan L, Xiong L, Xu W, et al. Genome-wide comparison of DNA hydroxymethylation in mouse embryonic stem cells and neural progenitor cells by a new comparative hMeDIP-seq method. Nucleic Acids Res. 2013;41:e84. doi: 10.1093/nar/gkt091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Booth MJ, Branco MR, Ficz G, et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–7. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- 78.Yu M, Hon GC, Szulwach KE, et al. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat Protoc. 2012;7:2159–70. doi: 10.1038/nprot.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pastor WA, Pape UJ, Huang Y, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–7. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu H, D’Alessio AC, Ito S, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389–93. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Song CX, Szulwach KE, Fu Y, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ficz G, Branco MR, Seisenberger S, et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- 83.Szulwach KE, Li X, Li Y, et al. Integrating 5-hydroxymethylcytosine into the epigenomic landscape of human embryonic stem cells. PLoS Genet. 2011;7:e1002154. doi: 10.1371/journal.pgen.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liutkeviciute Z, Lukinavicius G, Masevicius V, Daujotyte D, Klimasauskas S. Cytosine-5-methyltransferases add aldehydes to DNA. Nat Chem Biol. 2009;5:400–2. doi: 10.1038/nchembio.172. [DOI] [PubMed] [Google Scholar]
- 85.Madzo J, Liu H, Rodriguez A, et al. Hydroxymethylation at gene regulatory regions directs stem/early progenitor cell commitment during erythropoiesis. Cell Rep. 2014;6:231–44. doi: 10.1016/j.celrep.2013.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shearstone JR, Pop R, Bock C, Boyle P, Meissner A, Socolovsky M. Global DNA demethylation during mouse erythropoiesis in vivo. Science. 2011;334:799–802. doi: 10.1126/science.1207306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 88.Yang X, Lay F, Han H, Jones PA. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol Sci. 2010;31:536–46. doi: 10.1016/j.tips.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Blum W, Klisovic RB, Hackanson B, et al. Phase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemia. J Clin Oncol. 2007;25:3884–91. doi: 10.1200/JCO.2006.09.4169. [DOI] [PubMed] [Google Scholar]
- 90.Cashen AF, Shah AK, Todt L, Fisher N, DiPersio J. Pharmacokinetics of decitabine administered as a 3-h infusion to patients with acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS) Cancer Chemother Pharmacol. 2008;61:759–66. doi: 10.1007/s00280-007-0531-7. [DOI] [PubMed] [Google Scholar]
- 91.Issa JP, Garcia-Manero G, Giles FJ, et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004;103:1635–40. doi: 10.1182/blood-2003-03-0687. [DOI] [PubMed] [Google Scholar]
- 92.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223–32. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kantarjian H, Oki Y, Garcia-Manero G, et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2007;109:52–7. doi: 10.1182/blood-2006-05-021162. [DOI] [PubMed] [Google Scholar]
- 94.Valdez BC, Li Y, Murray D, Corn P, Champlin RE, Andersson BS. 5-Aza-2′-deoxycytidine sensitizes busulfan-resistant myeloid leukemia cells by regulating expression of genes involved in cell cycle checkpoint and apoptosis. Leuk Res. 2010;34:364–72. doi: 10.1016/j.leukres.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lavelle D, DeSimone J, Hankewych M, Kousnetzova T, Chen YH. Decitabine induces cell cycle arrest at the G1 phase via p21(WAF1) and the G2/M phase via the p38 MAP kinase pathway. Leuk Res. 2003;27:999–1007. doi: 10.1016/s0145-2126(03)00068-7. [DOI] [PubMed] [Google Scholar]
- 96.Tsujioka T, Yokoi A, Uesugi M, et al. Effects of DNA methyltransferase inhibitors (DNMTIs) on MDS-derived cell lines. Exp Hematol. 2013;41:189–97. doi: 10.1016/j.exphem.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 97.Tsai HC, Li H, Van Neste L, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21:430–46. doi: 10.1016/j.ccr.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Milhem M, Mahmud N, Lavelle D, et al. Modification of hematopoietic stem cell fate by 5aza 2′ deoxycytidine and trichostatin A. Blood. 2004;103:4102–10. doi: 10.1182/blood-2003-07-2431. [DOI] [PubMed] [Google Scholar]
- 99.Pinto A, Attadia V, Fusco A, Ferrara F, Spada OA, Di Fiore PP. 5-Aza-2′-deoxycytidine induces terminal differentiation of leukemic blasts from patients with acute myeloid leukemias. Blood. 1984;64:922–9. [PubMed] [Google Scholar]
- 100.Broske AM, Vockentanz L, Kharazi S, et al. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat Genet. 2009;41:1207–15. doi: 10.1038/ng.463. [DOI] [PubMed] [Google Scholar]
- 101.Stroud H, Feng S, Morey Kinney S, Pradhan S, Jacobsen SE. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 2011;12:R54. doi: 10.1186/gb-2011-12-6-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]