

Abstract
Variable survival outcomes are seen following treatment for aggressive non-Hodgkin lymphoma (NHL). This study examined whether outcomes for aggressive B-cell NHL are associated with single nucleotide polymorphisms (SNPs) in oxidative stress-related genes, which can alter drug metabolism and immune responses. Genotypes for 53 SNPs in 29 genes were determined for 337 patients given anthracycline-based therapies. Their associations with progression-free survival (PFS) and overall survival (OS) were estimated by Cox proportional hazard regression; associations with hematologic toxicity were estimated by logistic regression. To validate the findings, the top 3 SNPs were tested in an independent cohort of 572 DLBCL patients. The top SNPs associated with PFS in the discovery cohort were the rare homozygotes for MPO rs2243828 (hazard ratio [HR]=1.87, 95% confidence interval [CI]=1.14–3.06, P = 0.013), AKR1C3 rs10508293 (HR=2.09, 95% CI=1.28–3.41, P=0.0032) and NCF4 rs1883112 (HR=0.66, 95% CI=0.43–1.02, P=0.06). The association of the NCF4 SNP with PFS was replicated in the validation dataset (HR=0.66, 95% CI=0.44–1.01, P=0.05) and the meta-analysis was significant (HR=0.66, 95% CI=0.49–0.89, P<0.01). The association of the MPO SNP was attenuated in the validation dataset, while the meta-analysis remained significant (HR=1.64, 95% CI=1.12–2.41). These two SNPs showed similar trends with OS in the meta-analysis (for NCF4, HR=0.72, 95% CI 0.51–1.02, P=0.07 and for MPO, HR=2.06, 95% CI 1.36–3.12, P<0.01). In addition, patients with the rare homozygote of the NCF4 SNP had an increased risk of hematologic toxicity. We concluded that genetic variations in NCF4 may contribute to treatment outcomes for patients with aggressive NHL.
Keywords: lymphoma, pharmacogenomic, oxidation, outcomes research
Introduction
Despite treatment advances, many patients with aggressive non-Hodgkin B-cell lymphoma (NHL) will not be cured [1]. The international prognostic index (IPI) that is based on age (greater than 60), lower performance status, higher tumor stage, more than one extranodal site of involvement and elevated serum lactate dehydrogenase has been used to predict the patients who are most likely to fail treatment [2]. A limitation of the IPI risk score is that it provides little insight into the molecular mechanisms underlying the variable survival outcomes seen following treatment for aggressive NHL. Gene expression profiling studies have led to models for predicting treatment response based on molecular signatures and biological pathways, which work independently of the IPI [3–6]. In diffuse large B-cell lymphoma (DLBCL), the most common of the aggressive B-cell NHL subtypes accounting for 30–40% of newly-diagnosed lymphomas in the U.S. [7], gene expression profiling studies have identified both tumor and host cell properties contributing to treatment resistance [4–6].
An important host property in dictating treatment response is oxidative stress. Our previous gene expression profiling study showed that DLBCLs from patients with the worst prognosis following treatment had significantly decreased expression of oxidative stress-related genes in the tumor at the time of diagnosis [8]. Furthermore, the gene expression pattern seen in the patients with the worst prognosis indicates an increase in thioredoxin system function and in the expression of genes upregulated in response to oxidative stress. This spectrum of changes supports the hypothesis that an altered redox environment contributes to treatment resistance in DLBCL. Corroborating with our above findings is the fact that the generation of reactive oxygen species (ROS) and the resulting cellular damage is a major cytotoxic mechanism used by many chemotherapeutic drugs, including the standard chemotherapeutic regimen for DLBCL (cyclophosphamide, doxorubicin, vincristine and prednisone, or CHOP) [9].
Inherited genetic variations, mainly in the form of single nucleotide polymorphisms (SNPs), are a potential source of host variability in oxidative stress. SNPs in redox-related genes, such as those encoding myeloperoxidase (MPO) and the antioxidant enzymes manganese superoxide dismutase (SOD2) and catalase (CAT), have been studied in relation to treatment outcomes in other cancers including breast cancer [10, 11]. There have been few such studies, however, involving patients with aggressive lymphomas. One prior pharmacogenetic study of DLBCL patients found significant associations between SNPs in genes encoding for the p22phox subunit of NADPH oxidase (CYBA) and alpha1 class glutathione S-transferase (GSTA1) and PFS in patients treated with rituximab-CHOP [12]. That study involved only 106 patients, raising the question of whether the findings would hold up with a larger patient cohort.
In the current study, we investigated 53 SNPs in 29 oxidative stress-related genes for associations with treatment outcomes for aggressive B-cell NHL. The discovery phase of our study involved 337 patients enrolled in seven Phase II or Phase III SWOG clinical trials who received anthracycline-based combination chemotherapy. We then attempted to validate the top 3 SNPs in an independent, prospective study of 572 DLBCL patients from the Molecular Epidemiology Resource of the Iowa/Mayo Lymphoma Specialized Program of Research Excellence (SPORE), who were treated with anthracyline-based immunochemotherapy.
Methods
SWOG Patient population
Clinical data and diagnostic aggressive B-cell NHL tissue used in the discovery phase of the study had been collected for the 7 SWOG clinical trials listed in Table I. We chose these trials based on recruitment of patients with aggressive B-cell NHL, curative-intent, anthracycline-based therapies and sufficient time having elapsed for follow-up analyses. S8736 and S0014 enrolled patients with Ann Arbor stage I or II disease, while the remaining trials enrolled patients with stage IIB to IV disease. Further details on eligibility criteria, treatment protocols and outcomes are provided in the previous reports of these trials [13–17].
Table I.
Treatment protocols, total enrollment and number of participants from the SWOG trials analyzed in this study
| SWOG Trial | Study Title | Total Enrolled | Number Analyzed |
|---|---|---|---|
| S8516 | A Phase III Comparison of CHOP Versus m-BACOD Versus Pro-MACE-CytaBOM Versus MACOP-B in Patients with Intermediate or High-Grade Non-Hodgkin’s Lymphoma | 952 | 31 |
| S8736 | Treatment of Localized Non-Hodgkin’s Lymphoma: Comparison of Chemotherapy (CHOP) to Chemo-therapy Plus Radiation Therapy | 401 | 71 |
| S9125 | A Phase II Trial of CVAD/Verapamil/Quinine for Treatment of Non-Hodgkin’s Lymphoma | 89 | 14 |
| S9240 | A Phase II Trial of CVAD for Treatment of Non-Hodgkin’s Lymphoma | 89 | 36 |
| S9349 | A Randomized Phase II Trial of CHOP with G-CSF Support or ProMACE-CytaBOM with G-CSF Support for Treatment of Non Hodgkin’s Lymphoma | 186 | 95 |
| S9704 | A Randomized Phase III Trial Comparing Early High Dose Chemoradiotherapy and an Autologous Stem Cell Transplant to Conventional Dose CHOP Chemotherapy Plus Rituximab for CD20+ B Cell Lymphomas for Patients with Diffuse Aggressive Non-Hodgkin’s Lymphoma in the High-Intermediate and High Risk International Classification Prognostic Groups | 388 | 60 |
| S0014 | Evaluation of CHOP Plus Rituximab Plus Involved Field Radiotherapy for Stages I, IE, and Non-Bulky Stages II and IIE, CD20 Positive, High Risk Localized Aggressive Histologies of Non-Hodgkin’s Lymphoma | 61 | 30 |
|
| |||
| Total | 2166 | 337 | |
Abbreviations: m-BACOD, low-dose methotrexate with leucovorin rescue, bleomycin, doxorubicin, cyclophosphamide, vincristine, dexamethasone; Pro-MACE-CytaBOM, prednisone, doxorubicin, cyclophosphamide, etoposide followed by cytarabine, bleomycin, vincristine, methotrexate with leucovorin rescue; CVAD, cyclophosphamide, and infusional vincristine and doxorubicin, dexamethasone
Peripheral blood was not available for genotyping from these trials and formalin-fixed, paraffin-embedded (FFPE) tumor specimens were used as the source of DNA. Previous studies comparing genotypes derived from tumor tissues and matched normal tissues showed very high concordance rates [18–20]. All specimens were archived in the SWOG Lymphoma Bank located at the University of Arizona and maintained under the same storage conditions. Classification into the aggressive B-cell NHL subtypes of DLBCL, grade 3 follicular lymphoma (FL), Burkitt or Burkitt-like lymphoma (BL/BLL) and mantle cell lymphoma (MCL) was based on the World Health Organization (WHO) guidelines for hematopoietic tumors [21]. Specimens that had been collected prior to implementation of this classification system, and for which the WHO criteria diagnosis was not available, were prepared for immunohistochemistry, stained using a CD20 staining kit and the Benchmark XT automated immunostainer from Ventana Medical Systems, and examined by an expert hematopathologist (CMS) to establish the diagnosis according to the WHO guidelines. The number of patients with each type of aggressive B-cell NHL is given in Table II.
Table II.
Demographic and clinical characteristics of participants in the discovery and validation cohorts
| Discovery – N (%) by SWOG trial
|
Validation N (%) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Variable | S8516 | S8736 | S9125 | S9240 | S9349 | S9704 | S0014 | Total | |
| Sex | |||||||||
| Female | 9 (29) | 27 (38) | 4 (29) | 14 (39) | 39 (41) | 20 (33) | 17 (57) | 130 (39) | 273 (48) |
| Male | 22 (71) | 44 (62) | 10 (71) | 22 (61) | 56 (59) | 40 (67) | 13 (43) | 201 (61) | 299 (52) |
| Age, years | |||||||||
| Median | 55 | 61 | 63 | 53 | 54 | 49 | 72 | 57 | 62 |
| Range | 24–78 | 20–87 | 36–74 | 28–71 | 21–74 | 20–64 | 37–84 | 20–87 | 18–92 |
| >60 | 12 (39) | 36 (51) | 8 (57) | 12 (33) | 28 (30) | 8 (6) | 25 (83) | 129 (38) | 315 (55) |
| Race | |||||||||
| Black | 2 (6) | 3 (4) | 1 (7) | 2 (5) | 11 (12) | 2 (3) | 2 (7) | 23 (7) | 5 (1) |
| White | 26 (84) | 69 (96) | 13 (93) | 33 (92) | 83 (87) | 55 (92) | 28 (93) | 306 (91) | 528 (92) |
| Other | 0 | 0 | 0 | 1 (3) | 1 (1) | 2 (3) | 0 | 4 (1) | 5 (1) |
| Unknown | 3 (10) | 0 | 0 | 0 | 0 | 1 (2) | 0 | 4 (1) | 34 (6) |
| Histology | |||||||||
| DLBCL | 29 (94) | 60 (85) | 11 (79) | 29 (80) | 70 (74) | 60 (100) | 30 (100) | 289 (86) | 572 (100) |
| FL | 0 | 6 (8) | 2 (14) | 4 (11) | 6 (6) | 0 | 0 | 18 (5) | 0 |
| B/BLL | 0 | 5 (7) | 0 | 1 (3) | 7 (7) | 0 | 0 | 13 (4) | 0 |
| MCL | 0 | 0 | 0 | 0 | 8 (9) | 0 | 0 | 8 (2) | 0 |
| NOS | 2 (6) | 0 | 1 (7) | 2 (6) | 4 (4) | 0 | 0 | 9 (3) | 0 |
| IPI Score | 0 | ||||||||
| 0–1 | 7 (23) | 61 (86) | 3 (21) | 10 (28) | 23 (24) | 10 (17) | 26 (87) | 140 (41) | 209 (37) |
| 2–3 | 22 (71) | 10 (14) | 10 (72) | 22 (61) | 63 (66) | 50 (83) | 4 (13) | 181 (54) | 294 (51) |
| 4–5 | 2 (6) | 0 | 1 (7) | 4 (11) | 9 (9) | 0 | 0 | 16 (5) | 69 (12) |
| Hematologic Toxicity >3 | 24 (77) | 38 (54) | 14 (100) | 24 (67) | 85 (90) | 23 (38) | 20 (67) | 228 (68) | |
Abbreviation: NOS, aggressive B-cell NHL, not otherwise specified
All participants gave written informed consent. The study was approved by the Institutional Review Boards at the University of Arizona and Roswell Park Cancer Institute in accordance with an assurance filed with and approved by the U.S. Department of Health and Human Services.
SNP selection, DNA extraction, and genotyping
Candidate genes important in oxidative stress-related pathways and implicated in previous cancer pharmacogenetics studies were selected by Medline search and cross-checking of references. For each gene, SNPs associated with cancer susceptibility, prognosis and treatment outcomes were identified by interrogating the HuGE Navigator, a public database curating published population-based genetic associations from PubMed [22]. We reasoned that SNPs implicated in previous studies were potentially functional in drug cytotoxicity pathways commonly shared in different types of cancer. Coding SNPs in candidate genes that were not previously studied were queried in dbSNP and those with a minor allele frequency ≥0.05 in populations of European ancestry were selected for genotyping. For statistical power consideration, only SNPs with minor allele frequency greater than 0.05 in the population of European descent were included. As a result, 71 SNPs in 33 oxidative stress-related genes were assembled for genotyping (Supplemental Table I). Genotyping was performed in the Genomics Shared Resource Core at Roswell Park Cancer Institute utilizing the MassARRAY® technology and iPLEX Gold assay (Sequenom), which has been shown to be suitable for DNA derived from FFPE samples [23]. For SNPs that failed the assay design or validation, neighboring SNPs in perfect linkage disequilibrium (r2=1.0) in the HapMap CEU population were identified and added to the multiplex pools of Sequenom assays. If multiple SNPs were in perfect linkage disequilibrium with the SNP of interest, the one that had been used in previous studies was chosen (rs2243828 as a proxy for rs2333227 (−463G/A) in MPO), or one was randomly chosen if none had been used in previous studies (rs12232410 as a proxy for rs1800566 (C609T) in NQO1). For three other failed SNPs without perfect proxies, including rs4880 in SOD2, rs1800566 in NQO1 and rs1799983 in NOS3, pre validated Taqman® SNP genotyping assay were available and used instead.
Genomic DNA was extracted from three 5-μm sections of each FFPE tissue specimen using the QIAamp DNA FFPE Tissue Kit (Qiagen) according to the manufacturer’s instructions. DNA (500 ng/sample) was aliquoted into 96-well microtiter plates. For quality control purpose, 5% duplicates were included in each plate, as well as DNA from an in-house CEPH trio used to control for Mendelian inheritance error. The genotyping facility was blinded to the locations of duplicates and treatment received by patients. DNA samples with call rates less than 80% were excluded. There were 19 samples with a call rate of 85–89% and 10 samples with a call rate of 80–84% that were included in the analyses. The average successful genotyping rate was 91.9% for each DNA and 91.7% for each SNP. The concordance rate among duplicates was 98.4%. One SNP had a call rate less than 80% (rs511895 in CAT), another violated Mendelian inheritance (rs1801282 in PPARG), and genotype frequencies of 6 SNPs departed from Hardy Weinberg equilibrium (rs699473 in SOD3, rs552105 in PRDX4, rs4485648 in TXNRD2, rs3957356 in GSTA1, rs8483 in AKR1C3, and rs1060826 in NOS2) (P<1 × 10−4). Cluster plots of these 8 failed SNPs were manually inspected; no substantial improvement could be made by manually re clustering and these SNPs were therefore excluded. Also excluded were 10 SNPs with less than 6 patients with the rare homozygous genotype (rs1050828 and rs1050829 in G6PD, rs11548 in GPX3, rs7208693 in MPO, rs2297518 in NOS2, rs2234694 in SOD1, rs2842958 in SOD2, rs1799895 in SOD3, rs7221 in TXNIP and rs6518591 in TXNRD2).
Genotyping in the validation patient population
We attempted to validate the top 3 SNPs from the PFS analysis (rs2243828 in MPO, rs10508293 in AKR1C3 and rs1883112 in NCF4) using data and biospecimens from the Molecular Epidemiology Resource of the Iowa/Mayo Lymphoma SPORE [24, 25]. The validation study was reviewed and approved by the Human Subjects Review Boards at the University of Iowa and Mayo Clinic in accordance with an assurance filed with and approved by the U.S. Department of Health and Human Services. Briefly, all newly diagnosed DLBCL patients (within 9 months of first diagnosis) have been prospectively offered enrollment into an observational cohort study initiated in 2002 at the University of Iowa and Mayo Clinic. All pathology was centrally reviewed, and baseline clinical, laboratory and treatment data were abstracted using a standard protocol. Participants provided a peripheral blood sample, and DNA was extracted using a standard procedure (Gentra, Minneapolis, MN). All patients were systematically followed every 6 months for the first 3 years, then annually thereafter; disease progression, retreatment, and deaths were verified through medical record review. For this analysis, we used Taqman assays to genotype the 3 SNPs in 572 DLBCL patients enrolled from 2002–2009 who were treated with anthracycline-based immunochemotherapy and who had available DNA. There were 4 internal control samples per 96 well plate (3 CEPH and a no template control). All 3 SNPs were in Hardy Weinberg equilibrium (P>0.05) and all call rates were >80%: 88.1% for rs1883112, 92.8% for rs10508293 and 98.8% for rs2243828. The concordance rate for internal control samples was 100% for all 3 SNPs.
Statistical analyses
For genotype analysis, the codominant model, i.e., three genotypes with independent effects, was initially assumed and tested using the common homozygote as a reference group. Depending on the direction of the hazard ratios, the number of patients in each genotype and the model used in previous studies, the most suitable dominant, recessive or codominant model was then selected for each SNP. Patient survival outcomes assessed in this study included progression free survival (PFS) and overall survival (OS), with PFS defined from the date of registration to the date of progression or death due to any cause and OS defined from the date of registration until the date of death due to any cause. For the discovery phase of the study, patient follow-up was truncated at 10 years in order to reduce potential bias due to differential follow-up lengths between studies. PFS and OS estimates were calculated using the method of Kaplan and Meier [26] and associations of genotypes with survival outcomes were assessed with Cox proportional hazard regression models. The associations between genotypes and hematologic toxicity were also examined using unconditional logistic regression. Patients were classified according to whether they experienced any Grade 4–5 hematologic adverse event during protocol treatment. All the toxicities were graded according to the NCI CTCAE version used at that time. The definitions of grade 4–5 hematologic toxicities have remained consistent throughout the trials. All regression models were adjusted by treatment trials. Specifically, the SWOG trials were grouped into 3 strata according to disease stage (S8736/S0014 versus S9349/S9704 versus S8516/S9125/S9240). The regression models were further adjusted for IPI risk score, to test whether the effects of genotypes were independent from IPI score. A permutation-based resampling procedure drawn from 1,000 permutated samples was used to control the family-wise error rate due to multiple comparisons [27, 28]. A meta-analysis approach was used to combine results of the discovery and validation datasets [29], and study heterogeneity was tested by Cochran’s Q-test and Higgins’ I2 index. All analyses were performed using R 2.12. Results from this study are reported following the REMARK criteria [30].
Results
Identification and inclusion of eligible SWOG patients
The 7 SWOG trials used for the discovery phase of this study involved curative-intent, anthracycline-based therapies and recruited 2,166 patients (Table I). Rituximab was part of the treatment regimen in the two most recent trials, S9704 and S0014. Archived diagnostic tissues, maintained under the same storage conditions, were available in the SWOG Lymphoma Bank from 546 of the patients for potential use in this study. However, we restricted the analyses to tissues from patients who had: (i) a diagnosis of aggressive B-cell NHL; (ii) received anthracycline-based treatment; (iii) sufficient archived diagnostic tissue for DNA extraction, and (iv) a genotyping rate for the tissue of at least 80%. A total of 337 patients fit these criteria and were included in the final analyses. The flow of SWOG patient identification and inclusion through the discovery phase of the study is diagrammed in Figure 1 and the final numbers of patients included in the analyses are shown in Table I.
Figure 1.
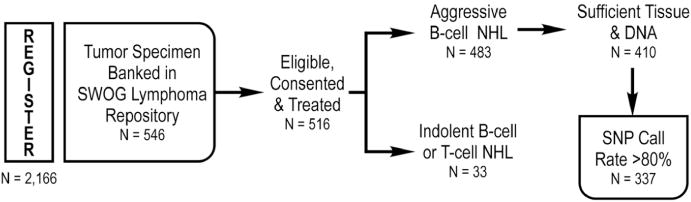
Identification and inclusion of patients from SWOG trials.
Patient characteristics
The demographic and clinical characteristics of the 337 SWOG patients included in the analyses are summarized in Table II. The demographic and clinical characteristics of the subset of patients that we were able to use from each trial did not deviate significantly from the total enrolled population for the respective treatment trial. White males made up the majority (61%) of the patient population and the median age was 57 (range, 20 – 87). More than 95% of the population had an IPI risk score between 0 and 3. Approximately 86% of the patients were diagnosed with DLBCL, consistent with the representation of this subtype in aggressive B-cell NHLs.
Associations between SNPs and PFS and OS
Tests for associations of genotypes with PFS and OS were performed on the 53 SNPs in 29 oxidative stress-related genes listed in Supplemental Table I. We found the strongest associations with survival outcomes for SNPs in genes encoding aldo-ketose reductase 1C3 (AKR1C3 rs10508293), MPO (rs2243828) and neutrophil cytosolic factor 4 (NCF4, rs1883112) (see Supplemental Table II for complete results). The minor allele frequencies (MAF) of these SNPs were 0.23, 0.18 and 0.40, respectively. The MAF for all SNPs in the analysis are given in Supplemental Table II. Table III gives the hazard ratios and 95% confidence intervals from Cox regression analyses for PFS and OS by genotypes of these three SNPs, after adjustment for IPI and stratification by treatment trial (see Methods section for details). Previous pharmacogenetic analyses among breast cancer patients examined the rs2333227 MPO (−463G>A) SNP in the promoter region. We used rs2243828 SNP as a surrogate for rs2333227, due to technical difficulties with detecting the latter. This approach has been used previously [11] and is justified due to complete linkage between the two SNPs (r2=1.0; the A allele of rs2243828 is linked with the G allele of rs2333227). The rare homozygous genotypes for both the AKR1C3 (CC genotype) and MPO (GG genotype) were associated with a significantly increased risk of disease progression and mortality. Kaplan-Meier curves of OS by genotypes for these SNPs are shown in Figure 2. The rare homozygous genotype for NCF4 was associated with a marginally significantly decreased risk of disease progression and mortality (Table III).
Table III.
Associations between AKR1C3, MPO and NCF4 genotypes, PFS and OS
| SWOG
|
Molecular Epidemiology Resource
|
Meta-Analysis
|
Study Heterogeneity
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene/SNP | Genotype | Events/Patients | HR* (95% CI) | P | Events/Patients | HR** (95% CI) | P | HR (95% CI) | P | P for Q-test | I2 |
| PFS | |||||||||||
| AKR1C3 | TT/CT | 164/305 | 1.00 (reference) | 199/520 | 1.00 (reference) | 1.00 (reference) | |||||
| rs10508293 | CC | 19/24 | 2.09 (1.28–3.41) | <0.01 | 4/11 | 0.98 (0.36–2.65) | 0.97 | 1.80 (1.16–2.80) | <0.01 | 0.16 | 48% |
| MPO | AA/AG | 168/311 | 1.00 (reference) | 206/543 | 1.00 (reference) | 1.00 (reference) | |||||
| rs2243828 | GG | 18/22 | 1.87 (1.14–3.06) | 0.01 | 10/22 | 1.35 (0.72–2.56) | 0.35 | 1.64 (1.12–2.41) | 0.01 | 0.44 | 0% |
| NCF4 | GG/AG | 157/274 | 1.00 (reference) | 164/423 | 1.00 (reference) | 1.00 (reference) | |||||
| rs1883112 | AA | 24/53 | 0.66 (0.43–1.02) | 0.06 | 26/81 | 0.66 (0.44–1.01) | 0.05 | 0.66 (0.49–0.89) | <0.01 | 1 | 0% |
| OS | |||||||||||
| AKR1C3 | TT/CT | 140/305 | 1.00 (reference) | 140/520 | 1.00 (reference) | 1.00 (reference) | |||||
| rs10508293 | CC | 18/24 | 2.14 (1.29–3.55) | <0.01 | 4/11 | 1.12 (0.41–3.03) | 0.83 | 1.88 (1.19–2.95) | <0.01 | 0.25 | 25% |
| MPO | AA/AG | 143/311 | 1.00 (reference) | 145/543 | 1.00 (reference) | 1.00 (reference) | |||||
| rs2243828 | GG | 17/22 | 2.34 (1.40–3.90) | <0.01 | 8/22 | 1.61 (0.79–3. 82) | 0.19 | 2.06 (1.36–3.12) | <0.01 | 0.47 | 0% |
| NCF4 | GG/AG | 135/274 | 1.00 (reference) | 110/424 | 1.00 (reference) | 1.00 (reference) | |||||
| rs1883112 | AA | 20/53 | 0.70 (0.44–1.12) | 0.14 | 19/81 | 0.75 (0.46–1.23) | 0.26 | 0.72 (0.51–1.02) | 0.07 | 0.85 | 0% |
Adjusted for IPI and stratified by treatment trial
Adjusted for IPI
Figure 2.
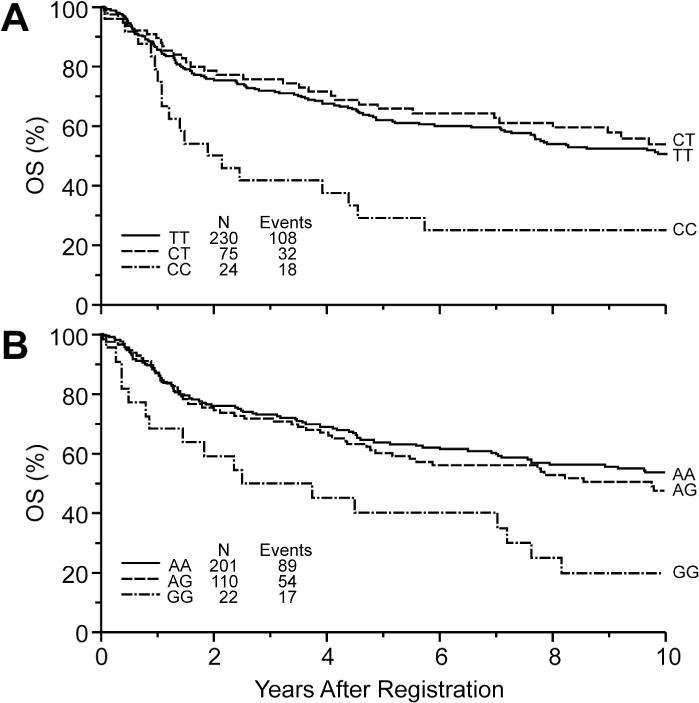
Kaplan Meier curves of overall survival for NHL patients by (A) AKR1C3 and (B) MPO genotypes.
The analyses described above included all 337 patients in the SWOG cohort. Thus, aggressive histologies of DLBCL, grade 3 FL, B/BLL and MCL were combined. Given that DLBCL patients constituted 86% of this cohort, we conducted a sensitivity analysis excluding all of the non-DBCL patients. The results were consistent with the overall analysis, showing a significant association between the GG genotype for the MPO rs2243828 SNP and inferior PFS (HR=2.00 and 95% CI=1.2–3.33) and OS (HR=2.52, 95% CI=1.48–4.29). Likewise, DLBCL patients with the CC genotype for the AKR1C3 rs10508293 SNP had inferior PFS (HR=1.94 and 95% CI=1.14–3.29) and OS (HR=2.07 and 95% CI=1.21–3.54). The results for NCF4 had similar point estimates to the overall analysis OS, NCF4 (AA vs GG/AG): HR=.82 (.48–1.40), PFS, NCF4 (AA vs GG/AG): HR=.73 (.45–1.20) but the confidence intervals show that they did not achieve significance.
Associations between SNPs and hematologic toxicity
Analysis of the Grade 4 and 5 hematologic toxicity data showed no significant findings, except for the rare homozygote for the NCF4 SNP rs1883112 (OR=1.81, 95% CI=0.86–3.77, P = 0.12) and manganese superoxide dismutase (SOD2, rs4880; OR=1.70, 95% CI=0.92–3.14, P = 0.09), which were marginally significant. The complete results of tests for associations between SNPs and hematologic toxicity are given in Supplemental Table III. No associations between toxicity and SNP genotypes remained significant after applying a permutation-based resampling procedure drawn from 1,000 permutated samples to control the family-wise error rate due to multiple comparisons [27, 28].
Validation of top SNPs for PFS
The replication dataset consisted of 572 immunochemotherapy treated DLBCL patients with a median age at diagnosis of 62 years (range, 18–92 years); 52% were male and 88% had an IPI of 0–3. Full details of the patient population are provided in Table I. The MAF of AKR1C3 rs10508293 (0.17), MPO rs2243828 (0.22) and NCF4 rs1883112 (0.43) were consistent with the discovery dataset. During a median follow-up of 59 months, there were 219 events (progression, retreatment or death) and 154 deaths. As shown in Table III, AKR1C3 rs10508293 was not replicated and MPO rs2243828 showed an increased risk but more modest than the discovery dataset, which did not reach statistical significance. NCF4 rs1883112 was replicated (HR=0.66, 95% CI=0.44–1.01, P=0.05), with essentially the same hazard ratio for PFS as in the discovery dataset. Adjustment for IPI did not change these results. Kaplan-Meier curves of PFS by NCF4 rs1883112 genotypes in the discovery and validation dataset are shown in Figure 3A and 3B, respectively. In meta-analysis combining the results from the two datasets, the AKR1C3 and MPO SNPs were significantly associated with PFS and OS (Table III). The meta-analysis of the NCF4 SNP was highly significant with PFS (meta HR=0.66, 95% CI=0.49–0.89, P<0.01), and marginally significant with OS (HR=0.72, 95% CI=0.51–1.02, P=0.07). There was no evidence of study heterogeneity found by either Q-test or I2 index (Table III).
Figure 3.
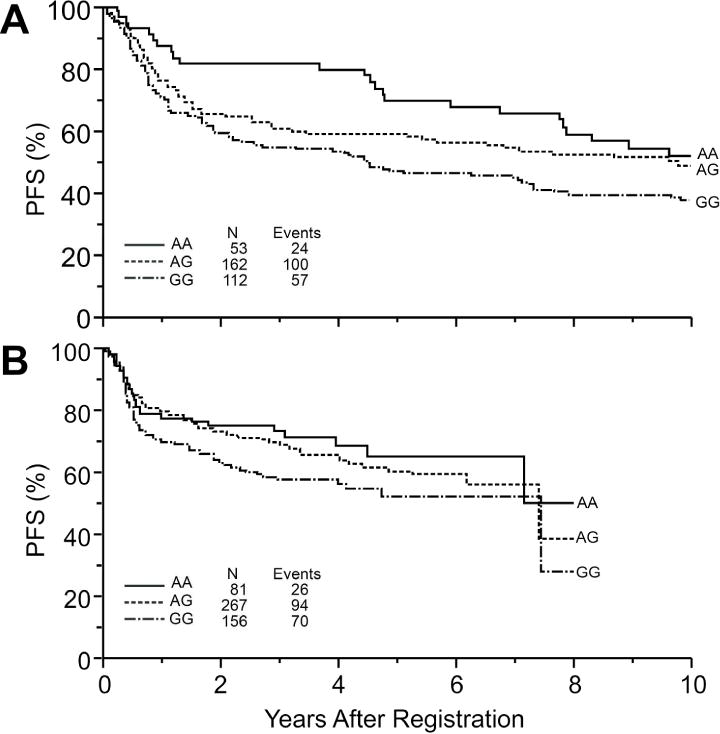
Kaplan Meier curves of PFS for NHL patients by NCF4 genotype in the (A) discovery and (B) validation cohorts.
Discussion
The clinical trial process has gradually improved the treatment of DLBCL, with ~60% of patients now being cured with R-CHOP. Ongoing research efforts are aimed at understanding the biological basis behind de novo or acquired resistance in the remaining 40% of patients. Previous pharmacogenomics analyses have linked polymorphisms in genes involved in drug metabolism [12], DNA repair [31] and immune function [32] to variable survival outcomes seen in DLBCL. This is the first study examining the impact of a broad range of inherited variations within oxidative stress-related pathways on treatment outcomes for aggressive B-cell NHL. In the discovery phase of the study, we found the rare genotypes of MPO rs2243838 and AKR1C3 rs10508293 SNPs associated with worse PFS and OS, while the rare genotype of NCF4 rs188312 was marginally associated with improved PFS. In addition, the same NCF4 SNP associated with better survival was also associated with a higher risk of severe hematologic toxicity. The association with NCF4 rs188312 was successfully replicated in an independent patient population, while the associations for MPO only marginally replicated.
The association of treatment outcomes in DLBCL with gene polymorphisms in redox-related genes supports the concept that the cellular redox environment influences the efficacy of treatments for this aggressive lymphoma. NCF4 encodes for p40phox, a regulatory subunit of NADPH oxidase. This multi-subunit complex generates ROS by catalyzing the one-electron reduction of oxygen to superoxide anion radical [reviewed in 33]. NADPH oxidases were originally identified in neutrophils; they are now known to be present in a wide range of cell types. The p40phox gene that we have found associated with NHL treatment outcome is expressed in hematopoietic cells [34]. ROS generated by NADPH oxidases may induce apoptosis by acting on redox-sensitive survival signaling pathways or may generate death-signaling molecules by oxidizing lipids, proteins or DNA [33].
Previous studies have associated the NCF4 rs1883112 SNP with adverse effects of DLBCL treatments [12, 35, 36]. In a cohort of 106 Italian DLBCL patients receiving R-CHOP, carriers of the NCF4 common rs1883112 G allele had a reduced risk of grade 3–4 hematologic toxicity (HR=0.45, P = 0.018) and grade 2–4 cardiac toxicity (HR=0.37, P = 0.023) [12]. The German non-Hodgkin Lymphoma Study Group reported an association of the rare AA genotype of NCF4 rs1883112 with an increased risk of chronic anthracycline induced cardiotoxicity based on a study of 1,697 patients with aggressive NHL who were treated with CHOP or CHOP with the addition of etoposide (CHOEP; OR = 2.5, 95% CI, 1.3 – 5) [35]. These results are consistent with our findings that the rare homozygous genotype was associated with increased risk of grade 4–5 hematologic toxicity. In collaboration with the German Study Group, Hoffman et al. found an association between the p22phox subunit of NADPH oxidase and PFS and OS in 878 DLBCL patients treated with CHOP or CHOEP [36]. p22phox is a membrane protein that binds to the catalytic (gp91phox) subunit of NADPH oxidase. The less favorable p22phox genotype results in decreased stability of the encoded mRNA and lower NADPH enzyme activity as measured in isolated peripheral blood mononuclear cells from healthy subjects [36]. Together with these previous findings, our results point to NADPH oxidase as mediating treatment effects on tumor and healthy normal cells in DLBCL patients. Genotyping of the NCF4 rs1883112 SNP would identify DLBCL patients who are likely to benefit from standard therapy, but who should be monitored closely for treatment-related adverse events. Alternatively, patients with the NCF4 AA genotype might receive sufficient benefit from lower dose regimens and be spared the toxic side-effects. Additional clinical trials would be needed to confirm the potential clinical use of NCF4 genotyping. A trial to assign patients into lower dose or standard dose, for example, six cycles of regular-strength R-CHOP versus R-miniCHOP based on the NCF4 genotype, could test whether patients with the rare homozygote have equivalent or superior survival and fewer adverse events than patients with the other genotypes, particularly in elderly patients.
More favorable NADPH oxidase genotypes with respect to survival outcomes in DLBCL may enhance immune responses contributing to effective therapy in patients receiving rituximab. In the analyses conducted by Hoffman et al. through the German non-Hodgkin Lymphoma Study Group, the treatment regimens did not include rituximab and there was no association between NCF4 rs1883112 and PFS (P=0.5) [36]. The majority of patients in the discovery phase of our study did not receive rituximab and we observed only a marginally significant association between NCF4 rs1883112 and PFS (P=0.06). In the validation cohort, wherein a significant association was detected, all of the patients received rituximab. Tumor regression in patients treated with rituximab may be due, at least in part, to antibody-dependent cell-mediated cytotoxicity. Rituximab bound to CD20 antigen can be recognized by Fcγ receptor on NK cells and macrophages to mediate killing or phagocytosis, respectively, of the tumor cells. Studies of neutrophils have shown that p40phox regulates Fcγ receptor-induced superoxide generation following internalization of phagosomes [37]. Inherited differences in the activity of NADPH oxidases could also influence the development of antitumor antibodies. Presentation of antigen to CD4+ T cells is impaired in human B cells expressing reduced levels of p40phox; increasing p40phox through gene transfection restores antigen presentation and also increases intracellular ROS generation [38]. Our findings with respect to the NCF rs1883112 genotypes and survival outcomes may, therefore, be generally applicable to the use of immunotherapy in cancer treatments.
A caveat in the interpretation of our study findings is the lack of direct evidence that different genotypes of the NCF4 rs1883112 SNP result in variable levels of NADPH oxidase activity. Further studies are needed to test the functional consequences of this polymorphism. Negative results from such studies could indicate that NCF4 rs1883112 is a proxy for a nearby SNP, which has functional consequences on NADPH oxidase activity and subsequently on treatment outcomes for patients with aggressive NHL.
We found the rare genotype of MPO rs2243838 associated with worse PFS and OS in the discovery phase of our study and in the meta-analyses of all patients. Inherited variations in the MPO gene are associated with treatment outcome in esophageal [39] and breast cancers [10, 11, 40]. Notably, the latter association is significant only for breast cancer patients who received adjuvant therapy, the majority of whom were treated with combination chemotherapy that included cyclophosphamide and/or doxorubicin. MPO produces the potent oxidant, hypochlorous acid, and catalyzes other oxidation reactions [41, 42]. Decreased MPO expression may lead to lower levels of oxidants needed for the full activity of chemotherapeutic drugs.
The association of the rare homozygous genotype for AKR1C3 rs10508293 with a significantly increased risk of disease progression and mortality, seen in the discovery phase of the study, was not replicated in the validation study. A failure to reproduce results, particularly when small patient cohorts have been analyzed, is a common shortcoming in SNP studies [30]. It is possible that our initial findings with AKR1C3 rs10508293 were the result of chance associations. An alternative explanation is the difference in therapy; rituximab was given to all patients in the validation study, but only a minority in the SWOG trials. AKR1C3 is one of thirteen known human aldo-keto reductases (AKRs) (reviewed in [43]). These enzymes metabolize a diverse group of compounds, including doxorubicin. In a direct comparison of eight human aldo-keto reductases, the AKR1C3 enzyme showed the highest catalytic efficiency with doxorubicin as a substrate, which it converts to an inactive metabolite [44]. The addition of rituximab may negate or mask any affect that AKR1C3 rs10508293 SNP genotype has on the efficacy of doxorubicin-based treatment regimens.
The MPO and AKR1C3 SNP genotypes that were associated with worse outcomes were found in <5% of the study population. There are several ways in which relatively rare genotypes can have clinical relevance. As discussed above for the NCF4 SNP, patients with the rare MPO or AKR1C3 SNP genotypes could be excellent candidates for studies testing whether higher dose treatments provide a survival benefit without adverse toxicity. Second, it is likely that treatment response, as a complex phenotype, is determined by multiple variants. The SNPs in oxidative stress genes examined here may be combined with other SNPs identified in the future as a polygenic score for risk prediction in clinics. Although the frequency of each SNP is relatively low, they may be important contributors to the polygenic score. Finally, genetic heterogeneity in DLBCL is well-recognized. Patient subsets are already being tested with different therapies based on molecular features. Genotypes of SNPs in oxidative stress-related genes may define disease subsets that will respond to more personalized treatment regimens.
In conclusion, we have provided further proof-of-concept for the cellular redox environment as a determinant of treatment efficacy in aggressive NHL. Our previous gene expression profiling-based study shows that a redox signature score is predictive of outcome for DLBCL patients in the pre-rituximab era [8]. This redox score is based on the expression of antioxidant defense and thioredoxin system genes that scavenge ROS or repair proteins that have been damaged by ROS. The data reported here, linking an NADPH oxidase subunit to treatment outcome, indicate that inherited variations in ROS-generating proteins may also contribute to treatment efficacy and be particularly relevant to immunotherapy. Future prospective studies with large DLBCL study populations receiving R-CHOP are warranted to further validate the clinical usefulness of SNPs in redox-related genes for predicting treatment outcomes. These studies could be designed to establish the extent to which the SNPs in oxidative stress-related genes identified here and previously are predictive of treatment outcome within IPI-based risk groups, across all subtypes of aggressive B-cell NHL and for molecular subtypes within DLBCL.
Supplementary Material
Acknowledgments
The authors would like to thank Yvette Frutiger and Catherine Rangel, Department of Pathology, University of Arizona for technical assistance. We would also like to thank the funding agencies that supported this work: Hope foundation, Predolin Foundation and National Cancer Institute (R01-CA71768, T32-CA009213-31, U54-CA143924, P30 CA016056 27-support for the RPCI Genomics Core Facility as a CCSG Shared Resource, P50 CA97274, R01 CA129539, and P30 CA15083 support for the MCCC Genomics Shared Resource). Additional support was provided by the following PHS Cooperative Agreement grant numbers awarded by the National Cancer Institute, DHHS: CA32102, CA38926, CA13612, CA11083 and CA46282.
References
- 1.Sehn LH, Donaldson J, Chhanabhai M, Fitzgerald C, et al. Introduction of combined CHOP plus rituximab therapy dramatically improved outcome of diffuse large B-cell lymphoma in British Columbia. J Clin Oncol. 2005;23:5027–5033. doi: 10.1200/JCO.2005.09.137. [DOI] [PubMed] [Google Scholar]
- 2.The International Non Hodgkin’s Lymphoma Prognostic Factors Project. A predictive model for aggressive non-Hodgkin’s lymphoma. N Engl J Med. 1993;329:987–994. doi: 10.1056/NEJM199309303291402. [DOI] [PubMed] [Google Scholar]
- 3.Alizadeh AA, Eisen MB, Davis RE, Ma C, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 4.Rosenwald A, Wright G, Chan WC, Connors JM, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–1947. doi: 10.1056/NEJMoa012914. [DOI] [PubMed] [Google Scholar]
- 5.Monti S, Savage KJ, Kutok JL, Feuerhake F, et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood. 2005;105:1851–1861. doi: 10.1182/blood-2004-07-2947. [DOI] [PubMed] [Google Scholar]
- 6.Rimsza LM, Roberts RA, Miller TP, Unger JM, et al. Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: a follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood. 2004;103:4251–4258. doi: 10.1182/blood-2003-07-2365. [DOI] [PubMed] [Google Scholar]
- 7.Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol. 1998;16:2780–2795. doi: 10.1200/JCO.1998.16.8.2780. [DOI] [PubMed] [Google Scholar]
- 8.Tome ME, Johnson DB, Rimsza LM, Roberts RA, et al. A redox signature score identifies diffuse large B-cell lymphoma patients with a poor prognosis. Blood. 2005;106:3594–3601. doi: 10.1182/blood-2005-02-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minotti G, Menna P, Salvatorelli E, Cairo G, et al. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 10.Ambrosone CB, Ahn J, Singh KK, Rezaishiraz H, et al. Polymorphisms in genes related to oxidative stress (MPO, MnSOD, CAT) and survival after treatment for breast cancer. Cancer Res. 2005;65:1105–1111. [PubMed] [Google Scholar]
- 11.He C, Tamimi RM, Hankinson SE, Hunter DJ, et al. A prospective study of genetic polymorphism in MPO, antioxidant status, and breast cancer risk. Breast Cancer Res Treat. 2009;113:585–594. doi: 10.1007/s10549-008-9962-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rossi D, Rasi S, Franceschetti S, Capello D, et al. Analysis of the host pharmacogenetic background for prediction of outcome and toxicity in diffuse large B-cell lymphoma treated with R-CHOP21. Leukemia. 2009;23:1118–1126. doi: 10.1038/leu.2008.398. [DOI] [PubMed] [Google Scholar]
- 13.Fisher RI, Gaynor ER, Dahlberg S, Oken MM, et al. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin’s lymphoma. N Engl J Med. 1993;328:1002–1006. doi: 10.1056/NEJM199304083281404. [DOI] [PubMed] [Google Scholar]
- 14.Miller TP, Dahlberg S, Cassady JR, Adelstein DJ, et al. Chemotherapy alone compared with chemotherapy plus radiotherapy for localized intermediate and- high-grade non-Hodgkin’s lymphoma. N Engl J Med. 1998;339:21–26. doi: 10.1056/NEJM199807023390104. [DOI] [PubMed] [Google Scholar]
- 15.Gaynor ER, Unger JM, Miller TP, Grogan TM, et al. Infusional CHOP chemotherapy (CVAD) with or without chemosensitizers offers no advantage over standard CHOP therapy in the treatment of lymphoma: a Southwest Oncology Group Study. J Clin Oncol. 2001;19:750–755. doi: 10.1200/JCO.2001.19.3.750. [DOI] [PubMed] [Google Scholar]
- 16.Blayney DW, Leblanc ML, Grogan T, Gaynor ER, et al. Dose-intense chemotherapy every 2 weeks with dose intense cyclophosphamide, doxorubicin, vincristine, and prednisone may improve survival in intermediate- and high-grade lymphoma: a phase II study of the Southwest Oncology Group (SWOG 9349) J Clin Oncol. 2003;21:2466–2473. doi: 10.1200/JCO.2003.06.137. [DOI] [PubMed] [Google Scholar]
- 17.Persky DO, Unger JM, Spier CM, Stea B, et al. Phase II study of rituximab plus three cycles of CHOP and involved-field radiotherapy for patients with limited-stage aggressive B-cell lymphoma: Southwest Oncology Group study 0014. J Clin Oncol. 2008;26:2258–2263. doi: 10.1200/JCO.2007.13.6929. [DOI] [PubMed] [Google Scholar]
- 18.Rae JM, Cordero KE, Scheys JO, Lippman ME, et al. Genotyping for polymorphic drug metabolizing enzymes from paraffin-embedded and immunohistochemically stained tumor samples. Pharmacogenetics. 2003;13:501–507. doi: 10.1097/00008571-200308000-00008. [DOI] [PubMed] [Google Scholar]
- 19.Lips EH, Dierssen JW, van ER, Oosting J, et al. Reliable high-throughput genotyping and loss-of-heterozygosity detection in formalin-fixed, paraffin-embedded tumors using single nucleotide polymorphism arrays. Cancer Res. 2005;65:10188–10191. doi: 10.1158/0008-5472.CAN-05-2486. [DOI] [PubMed] [Google Scholar]
- 20.Weiss JR, Baer MR, Ambrosone CB, Blanco JG, et al. Concordance of pharmacogenetic polymorphisms in tumor and germ line DNA in adult patients with acute myeloid leukemia. Cancer Epidemiol Biomarkers Prev. 2007;16:1038–1041. doi: 10.1158/1055-9965.EPI-06-0964. [DOI] [PubMed] [Google Scholar]
- 21.Jaffe ES, Harris NL, Stein H, Vardiman JW, World Health Organization Classification of Tumours . Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2001. [Google Scholar]
- 22.Yu W, Gwinn M, Clyne M, Yesupriya A, et al. A navigator for human genome epidemiology. Nat Genet. 2008;40:124–125. doi: 10.1038/ng0208-124. [DOI] [PubMed] [Google Scholar]
- 23.Horn H, Pott C, Kalla J, Dreyling M, et al. A multiplex MALDI-TOF MS approach facilitates genotyping of DNA from formalin-fixed paraffin-embedded tumour specimens. Pharmacogenet Genomics. 2010;20:598–604. doi: 10.1097/FPC.0b013e32833deb16. [DOI] [PubMed] [Google Scholar]
- 24.Drake MT, Maurer MJ, Link BK, Habermann TM, et al. Vitamin D insufficiency and prognosis in non-Hodgkin’s lymphoma. J Clin Oncol. 2010;28:4191–4198. doi: 10.1200/JCO.2010.28.6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Charbonneau B, Maurer MJ, Ansell SM, Slager SL, et al. Pretreatment circulating serum cytokines associated with follicular and diffuse large B-cell lymphoma: a clinic-based case-control study. Cytokine. 2012;60:882–889. doi: 10.1016/j.cyto.2012.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 27.Hilsenbeck SG, Clark GM. Practical p-value adjustment for optimally selected cutpoints. Stat Med. 1996;15:103–112. doi: 10.1002/(SICI)1097-0258(19960115)15:1<103::AID-SIM156>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 28.LeBlanc M, Crowley J. Step-function covariate effects in the proportional-hazards model. Can J Stat. 1995;23:109–129. [Google Scholar]
- 29.Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta-analyses of the published literature for survival endpoints. Stat Med. 1998;17:2815–2834. doi: 10.1002/(sici)1097-0258(19981230)17:24<2815::aid-sim110>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 30.McShane LM, Altman DG, Sauerbrei W, Taube SE, et al. Reporting recommendations for tumor Marker prognostic studies (REMARK) Breast Cancer Res Treat. 2006;100:229–235. doi: 10.1007/s10549-006-9242-8. [DOI] [PubMed] [Google Scholar]
- 31.Rossi D, Rasi S, Di Rocco A, Fabbri A, et al. The host genetic background of DNA repair mechanisms is an independent predictor of survival in diffuse large B-cell lymphoma. Blood. 2011;117:2405–2413. doi: 10.1182/blood-2010-07-296244. [DOI] [PubMed] [Google Scholar]
- 32.Habermann TM, Wang SS, Maurer MJ, Morton LM, et al. Host immune gene polymorphisms in combination with clinical and demographic factors predict late survival in diffuse large B-cell lymphoma patients in the pre-rituximab era. Blood. 2008;112:2694–2702. doi: 10.1182/blood-2007-09-111658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 34.Zhan S, Vazquez N, Zhan S, Wientjes FB, et al. Genomic structure, chromosomal localization, start of transcription, and tissue expression of the human p40-phox, a new component of the nicotinamide adenine dinucleotide phosphate-oxidase complex. Blood. 1996;88:2714–2721. [PubMed] [Google Scholar]
- 35.Wojnowski L, Kulle B, Schirmer M, Schluter G, et al. NAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicity. Circulation. 2005;112:3754–3762. doi: 10.1161/CIRCULATIONAHA.105.576850. [DOI] [PubMed] [Google Scholar]
- 36.Hoffmann M, Schirmer MA, Tzvetkov MV, Kreuz M, et al. A functional polymorphism in the NAD(P)H oxidase subunit CYBA is related to gene expression, enzyme activity, and outcome in non-Hodgkin lymphoma. Cancer Res. 2010;70:2328–2338. doi: 10.1158/0008-5472.CAN-09-2388. [DOI] [PubMed] [Google Scholar]
- 37.Tian W, Li XJ, Stull ND, Ming W, et al. Fc gamma R-stimulated activation of the NADPH oxidase: phosphoinositide-binding protein p40phox regulates NADPH oxidase activity after enzyme assembly on the phagosome. Blood. 2008;112:3867–3877. doi: 10.1182/blood-2007-11-126029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crotzer VL, Matute JD, Arias AA, Zhao H, et al. Cutting edge: NADPH oxidase modulates MHC class II antigen presentation by B cells. J Immunol. 2012;189:3800–3804. doi: 10.4049/jimmunol.1103080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu X, Gu J, Wu TT, Swisher SG, et al. Genetic variations in radiation and chemotherapy drug action pathways predict clinical outcomes in esophageal cancer. J Clin Oncol. 2006;24:3789–3798. doi: 10.1200/JCO.2005.03.6640. [DOI] [PubMed] [Google Scholar]
- 40.Ambrosone CB, Barlow WE, Reynolds W, Livingston RB, et al. Myeloperoxidase genotypes and enhanced efficacy of chemotherapy for early-stage breast cancer in SWOG-8897. J Clin Oncol. 2009;27:4973–4979. doi: 10.1200/JCO.2009.21.8669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Winterbourn CC, Vissers MCM, Kettle AJ. Myeloperoxidase. Curr Opin Hematol. 2000;7:53–58. doi: 10.1097/00062752-200001000-00010. [DOI] [PubMed] [Google Scholar]
- 42.Arnhold J, Flemmig J. Human myeloperoxidase in innate and acquired immunity. Arch Biochem Biophys. 2010;500:92–106. doi: 10.1016/j.abb.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 43.Jin Y, Penning TM. Aldo-keto reductases and bioactivation/detoxication. Annu Rev Pharmacol Toxicol. 2007;47:263–292. doi: 10.1146/annurev.pharmtox.47.120505.105337. [DOI] [PubMed] [Google Scholar]
- 44.Bains OS, Grigliatti TA, Reid RE, Riggs KW. Naturally occurring variants of human aldo-keto reductases with reduced in vitro metabolism of daunorubicin and doxorubicin. J Pharmacol Exp Ther. 2010;335:533–545. doi: 10.1124/jpet.110.173179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.