

Abstract
The differential formation of excitatory (glutamatergic) and inhibitory (GABAergic) synapses is a critical step for the proper functioning of the brain. Their imbalance may lead to various neurological disorders such as autism, schizophrenia, Tourette syndrome and epilepsy1–4. Synapses are formed through the communication between the appropriate synaptic partners5–8. However, the molecular mechanisms that mediate the formation of specific synaptic types are not known. Here we show that two members of the fibroblast growth factor (FGF) family, FGF22 and FGF7, promote the organization of excitatory and inhibitory presynaptic terminals, respectively, as target-derived presynaptic organizers. FGF22 and FGF7 are expressed by CA3 pyramidal neurons in the hippocampus. The differentiation of excitatory or inhibitory nerve terminals on dendrites of CA3 pyramidal neurons is specifically impaired in mutants lacking FGF22 or FGF7. These presynaptic defects are rescued by postsynaptic expression of the appropriate FGF. FGF22-deficient mice are resistant and FGF7-deficient mice are prone to epileptic seizures, as expected from the alterations in excitatory/inhibitory balance. Differential effects by FGF22 and FGF7 involve both their distinct synaptic localizations and use of different signaling pathways. These results demonstrate that specific FGFs act as target-derived presynaptic organizers and help organize specific presynaptic terminals in the mammalian brain.
Target (postsynaptic cell)-derived “presynaptic organizers” promote the local differentiation of presynaptic axons into functional nerve terminals at sites of synaptic contact5–8. This presynaptic differentiation includes clustering of synaptic vesicles, formation of active zones, cytoskeletal restructuring and assembly of vesicle recycling machinery5–8. We have identified FGF22 and its close relatives, FGF7 and FGF10, as molecules that can promote the differentiation of presynaptic nerve terminals9. Using blocking reagents and mice deficient for their main receptor FGFR2, we showed that these FGFs are involved in presynaptic differentiation in the cerebellum9 and at the neuromuscular junction10. Here we investigate the specific synaptogenic function of FGF7 and FGF22, using FGF7-11 and FGF22-knockout (KO) mice (Supplementary Fig. 1).
In situ hybridization reveals that fgf22 and fgf7 mRNAs are strongly expressed in the mouse hippocampus at P8 (Fig. 1a), which is around the time synapses start to form12,13. They are highly expressed by CA3 pyramidal neurons, but very little expression was found in CA1 pyramidal neurons. mRNAs for FGFR2 (Fig. 1b) and FGFR114, which are possible receptors for FGF22 and FGF715, are expressed by various neurons throughout the hippocampus. Both FGF22 and FGF7 proteins are localized in the synapse-rich areas, stratum radiatum and lucidum in CA3 (Fig. 1c), supporting the idea that these FGFs are involved in synapse formation in CA3.
Figure 1. Expression of FGF22 and FGF7 in the hippocampal CA3 region during synapse formation (P8).
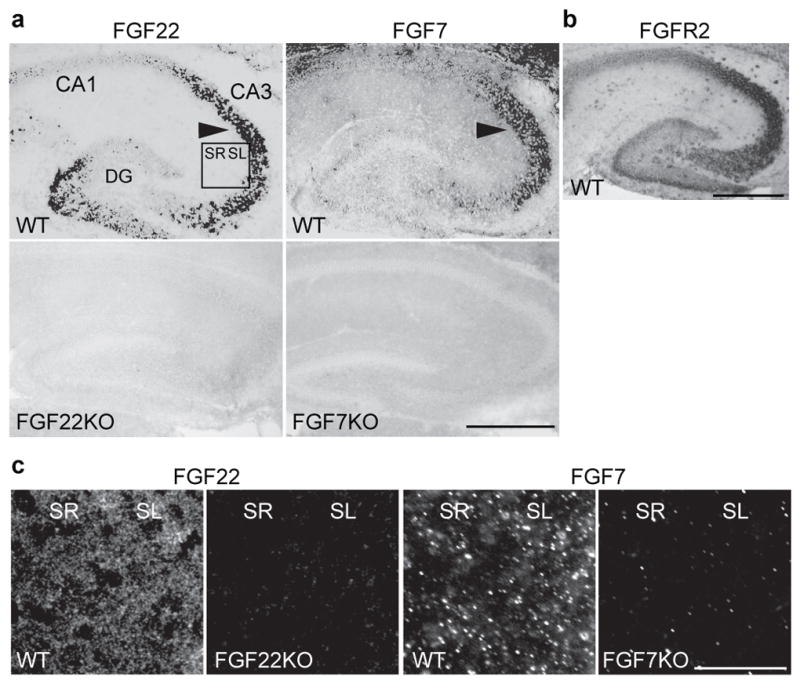
a, fgf22 and fgf7 mRNAs are highly expressed in CA3 pyramidal neurons (arrowheads), but not in CA1 pyramidal neurons. Bottom panels are negative controls using FGFKO sections. b, fgfr2 mRNA is widely expressed throughout the hippocampus. c, FGF22 and FGF7 proteins are localized in CA3 synapse-rich areas. Pictured areas correspond to the boxed area in a. Scale bars, 500 μm (a, b) and 50 μm (c). SR: stratum radiatum, SL: stratum lucidum.
To address whether synapses in CA3 require FGF22 and FGF7 for presynaptic differentiation, we measured synaptic vesicle clustering in FGF22KO and FGF7KO mice. Their hippocampus looks anatomically normal and the fate of cells appears to be unchanged (Supplementary Fig. 2). However, clustering of the synaptic vesicle protein SV2 is significantly decreased in CA3 in both FGF22KO and FGF7KO mice compared to wild type (WT) mice (Fig. 2a and Supplementary Fig. 3b) at P14, when synaptogenesis is at its peak12,13. Synaptic vesicle clustering in CA1 is normal. Thus, FGF22 and FGF7 are involved in presynaptic differentiation in CA3, but not in CA1, which is consistent with the mRNA expression of these FGFs (Fig. 1a). The clustering of bassoon, an active zone marker, is not significantly different between the WT and FGFKO hippocampus (Fig. 2b and Supplementary Fig. 3c), suggesting that active zone formation and synaptic vesicle clustering are relatively independent.
Figure 2. Specific defects in excitatory or inhibitory presynaptic differentiation in CA3 of FGF22KO and FGF7KO mice.

a, SV2 staining in CA1 and CA3 from P14 WT, FGF22KO and FGF7KO mice demonstrates decreased synaptic vesicle (SV) clustering in CA3 of FGFKO mice. b, Normal active zone formation (bassoon clustering) in CA3 of FGFKO mice. c, d, Staining in CA3 for VGLUT1 (c) and VGAT (d), showing impaired glutamatergic and GABAergic SV clustering in CA3 of FGF22KO and FGF7KO mice, respectively. e, Normal PSD95 and gephyrin clustering in CA3 of FGFKO mice. f–k, Electron microscopic (EM) analysis of asymmetric (excitatory, f–h) and symmetric (inhibitory, i–k) synapses in CA3. Synaptic density (x1,000/mm2, f, i), representative synapses (g, j), and analysis of SVs within 400 nm from the active zone (AZ; h, k) show specific presynaptic defects in FGFKO mice. l, Western blotting of CA3 lysates, indicating no overall change in synaptic protein expression in FGFKO mice. Error bars are s.e.m. Staining data are from 15–126 fields from 5–14 mice. EM data are from 5–20 synapses. Significant difference from control at *P < 0.05; **P < 0.01; ***P < 0.001, ANOVA followed by Tukey test. Scale bars, 20 μm (c, d) and 200 nm (g, j).
The defect of synaptic vesicle clustering in either FGFKO CA3 is incomplete (Fig. 2a), suggesting that each FGF only acts on a subset of synapses. We asked whether FGF22 and FGF7 are differentially involved in glutamatergic and GABAergic presynaptic differentiation. At P14, vesicle clustering at glutamatergic synapses, monitored by vesicular glutamate transporter 1 (VGLUT1) staining, is significantly decreased in CA3 of FGF22KO mice, but not in FGF7KO mice, relative to WT mice (Fig. 2c). In contrast, GABAergic synaptic vesicle clustering, monitored by vesicular GABA transporter (VGAT) staining, is significantly decreased in FGF7KO mice, but not in FGF22KO mice (Fig. 2d). The clustering of PSD95 and gephyrin, postsynaptic scaffolding proteins at glutamatergic and GABAergic synapses, respectively, is not significantly affected in either KO mouse (Fig. 2e and Supplementary Fig. 3d, e). The morphology of CA3 pyramidal and interneurons and the projections of dentate gyrus axons in FGFKO mice are similar to those in WT mice (Supplementary Fig. 4). Thus, FGF22 and FGF7 appear to be specifically involved in glutamatergic and GABAergic presynaptic differentiation, respectively. The presynaptic defects in FGFKO mice can be detected from early stages of synapse formation12,13 (P8) to adulthood (Supplementary Fig. 5, 6), suggesting that FGF inactivation prevents presynaptic differentiation, rather than just delays it.
We then examined the ultrastructure of synapses formed in CA3 of FGFKO mice. We identified excitatory and inhibitory synapses based on their morphology: asymmetric as excitatory and symmetric as inhibitory. In FGF22KO, FGF7KO and WT CA3, we found a similar number of asymmetric and symmetric synapses (Fig. 2f, i). At asymmetric synapses (Fig. 2g), synaptic vesicles in FGF22KO mice, but not those in FGF7KO mice, are more diffusely distributed and smaller in size compared to those in WT mice (Fig. 2h). In addition, the number of docked-vesicles is significantly decreased in FGF22KO mice. In contrast, at symmetric synapses (Fig. 2j), the number of total synaptic vesicles in each presynaptic terminal and their size are significantly decreased in FGF7KO, but not in FGF22KO mice (Fig. 2k). Thus, FGF22 and FGF7 are specifically involved in the presynaptic differentiation of excitatory and inhibitory synapses, respectively, in CA3. As Western blots show, the total level of VGLUT1 and VGAT proteins in CA3 is not changed in FGFKO mice (Fig. 2l; ± 10%), suggesting that the presynaptic defects in FGFKO mice are due to an impairment of synaptic vesicle recruitment and not its formation.
Next, we investigated whether the differentiation of glutamatergic and GABAergic terminals on a neuron is controlled by FGF expression in that neuron. Overexpression of FGF22 and FGF7 in cultured hippocampal neurons leads to an increase in the clustering of VGLUT1 and VGAT puncta, respectively, on the FGF-expressing neuron (Fig. 3a). This suggests that FGF22 and FGF7 specifically promote differentiation of glutamatergic and GABAergic presynaptic terminals, respectively, as target-derived molecules.
Figure 3. Target-derived FGF22 and FGF7 selectively promote differentiation of glutamatergic or GABAergic presynaptic terminals in CA3 through distinct localization and signaling pathways.

a, VGLUT1 and VGAT clustering on FGF-transfected WT hippocampal neurons (labeled with GFP; 7 DIV). b, Defects in the clustering of VGLUT1 and VGAT on the dendrites of FGF22KO or FGF7KO CA3 pyramidal neurons (Py-positive; 14 DIV). The number (puncta/mm neurite) and size of puncta were quantified. c, The number (x1,000 puncta/mm2) and size of VGLUT1 and VGAT puncta on non-CA3 pyramidal neurons (Py-negative). d, The presynaptic defects in FGFKO cultures are rescued by expression of the corresponding FGF in postsynaptic CA3 pyramidal neurons. Data are normalized to WT. e, f, FGF22-EGFP localizes to glutamatergic, and FGF7-DsRed to GABAergic synapses (arrowheads). g, Endogenous FGF22 and FGF7 are colocalized with PSD95 or VGAT. h, FGF22-EGFP and FGF7-DsRed exhibit differential dendritic localization. i, j, The number (x1,000 puncta/mm2) and size of VGLUT1 (i) or VGAT (j) puncta after FGF bath application to WT cultures. k, VGLUT1 and VGAT staining intensity of control and FGFR2KO CA3 sections (P8). l, Normalized staining intensity of FGFR2KO sections. Error bars are s.e.m. Data are from 50–290 neurites or 8–66 fields from at least three experiments. Significant difference from control at *P < 0.05; **P < 0.01; ***P < 0.001, t-test (d, k, l) or ANOVA followed by Tukey test; #P=0.073 (i). Scale bars, 10 μm.
If FGFs are target-derived presynaptic organizers, the differentiation of presynaptic terminals on the dendrites of CA3 pyramidal neurons, which express fgf22 and fgf7 mRNAs (Fig. 1a), should be severely and specifically impaired in FGFKO mice. To test this idea, we identified CA3 pyramidal neurons in hippocampal cultures with the antibody Py, which is specific for the dendrites and cell body of CA3 pyramidal neurons16. At 14 days in vitro (14 DIV), relative to WT cultures, the clustering of glutamatergic (VGLUT1) and GABAergic (VGAT) vesicles associated with dendrites of CA3 pyramidal neurons are dramatically reduced in FGF22KO and FGF7KO cultures, respectively (Fig. 3b and Supplementary Fig. 7). In contrast, we did not detect significant defects in presynaptic terminals associated with other neuronal populations (Fig. 3c). The presynaptic defects on CA3 pyramidal cell dendrites appear early in culture (10 DIV), and even after 21 DIV, very little apparent presynaptic differentiation is observed (Supplementary Fig. 8). Importantly, these presynaptic defects in FGF22KO and FGF7KO cultures are rescued by FGF22 or FGF7 expression in postsynaptic CA3 pyramidal cells (Fig. 3d). Together, these results support the idea that FGF22 and FGF7 are critical as target-derived molecules for the clustering of specific types of synaptic vesicles to presynaptic terminals formed onto CA3 pyramidal neurons.
We also performed a series of control experiments, which suggest that FGF-deficiency specifically affects synaptic vesicle clustering. (1) The number and morphology of Py-positive neurons, interneurons and dentate gyrus neurons are similar in FGFKO and WT cultures (Supplementary Fig. 9). (2) The clustering of bassoon, postsynaptic receptors and scaffolding proteins is not impaired in FGFKO cultures (Supplementary Fig. 7 and 10). (3) VGLUT1 and VGAT proteins appear to be properly localized and functional at synaptic vesicles, because i) they are not stuck in cell bodies (Supplementary Fig. 11) and ii) quantal size, as measured by electrophysiological recordings, is normal in FGFKO neurons (Fig. 4a–d).
Figure 4. Altered synaptic transmission and seizure susceptibility in FGFKO mice, and a model for the role of FGF22 and FGF7 in specific presynaptic differentiation.

a–d, Representative whole-cell recordings of mEPSCs (a) and mIPSCs (c) from WT and FGFKO hippocampal neurons; frequency and amplitude of mEPSCs (b) and mIPSCs (d) in the indicated groups (18–24 neurons/group). e, Representative time-course of seizure development during kindling experiments. f, Percentage of mice kindled when about half of the control mice are kindled (~21 PTZ injections) from four independent experiments (4–6 mice/experiment). Error bars are s.e.m. Significant difference from control at *P < 0.05 and ***P < 0.001, ANOVA followed by Tukey test. g, Summary model. FGF22 and FGF7 from CA3 pyramidal neurons promote the differentiation of excitatory and inhibitory presynaptic terminals, respectively. FGF22 and FGF7 are localized at corresponding synapses and activate differential signaling pathways for specific presynaptic differentiation.
To understand the mechanisms by which FGF22 and FGF7 mediate differential presynaptic effects, we examined their synaptic localization. When FGF22-EGFP and FGF7-DsRed were transfected into hippocampal cultures, both FGFs localized to MAP2-positive dendrites and not to neurofilament-positive axons (Supplementary Fig. 12). In dendrites, >85% of FGF22-EGFP is co-localized with PSD95 (Fig. 3e), but not with gephyrin (<6%). In contrast, >85% of FGF7-DsRed is co-localized with gephyrin (Fig. 3f) and not with PSD95 (<8%). Endogenous FGF22 and FGF7 puncta are also associated with PSD95 and VGAT puncta, respectively (>85% and >75%; Fig. 3g). FGF22-EGFP and FGF7-DsRed show non-overlapping expression (Fig. 3h), which confirms that FGF22 and FGF7 are targeted to different dendritic sub-domains. Thus, in hippocampal neurons, FGF22 and FGF7 are specifically localized at glutamatergic and GABAergic synapses, respectively, which likely accounts for their differential effects on excitatory and inhibitory synapse development.
We then tested the additional possibility that FGF22 and FGF7 activate different signaling pathways for their differential effects. When we applied the maximum effective concentration of recombinant FGFs9,15 to WT hippocampal cultures, only FGF22 significantly increased the number of VGLUT1-positive puncta (Fig. 3i). In contrast, both FGF22 and FGF7 are able to increase the number and size of VGAT-positive puncta (Fig. 3j); however, the effect of FGF7 is more dramatic than that of FGF22. Thus, even when exogenously applied, FGF22 and FGF7 show limited specificity on presynaptic differentiation, suggesting that they activate different sets of signaling pathways. Different signaling pathways might be mediated by distinct FGF receptors. An in vitro assay showed that FGF7 preferentially signals through FGFR2b, whereas FGF22 signals through both FGFR2b and FGFR1b15. The analysis of conditional FGFR2KO mice, in which fgfr2 is inactivated postnatally using the Cre-ER system9,10, is consistent with this receptor specificity. We found that the clustering of both glutamatergic and GABAergic vesicles is decreased in FGFR2KO CA3 (Fig. 3k), but the decrease is more pronounced at GABAergic relative to glutamatergic synapses (Fig. 3l). These results suggest that the differential effects of FGF22 and FGF7 are achieved by their distinct synaptic localization and use of different receptors.
To investigate the functional consequences of the impaired presynaptic differentiation in FGFKO neurons, we recorded synaptic currents. We found that the frequency, but not amplitude, of miniature excitatory and inhibitory postsynaptic currents (mEPSCs and mIPSCs) is specifically decreased in FGF22KO and FGF7KO hippocampal cultures, respectively (Fig. 4a–d and Supplementary Fig. 13a, b). Paired-pulse facilitation and synaptic depression in response to a stimulus train are both increased in FGF22KO relative to WT neurons (Supplementary Fig. 13c–f), suggesting that release probability and the readily-releasable vesicle pool are both diminished in the absence of FGF22. Therefore, FGF22KO and FGF7KO neurons appear to have specific defects in excitatory and inhibitory presynaptic function, respectively. These results also suggest that postsynaptic differentiation is relatively independent of presynaptic differentiation, which is similar to findings from FGF14 (FHF4) KO mice17.
A synaptic imbalance in the hippocampus may contribute to various neurological disorders, including epilepsy4. Hence, we subjected FGFKO and control animals to the kindling protocol, an animal model of epilepsy18,19. We injected adult animals with pentylenetetrazol, a GABA receptor antagonist, every 48 hours to induce epileptic seizures18,19. Most FGF7KO mice developed major seizures (kindled) sooner than control mice (Fig. 4e). In contrast, none of the FGF22KO mice were kindled within the time period examined (30 injections). When around half of control mice were kindled (~21 injections; 52.5 ± 3.8%), no FGF22KO mice were kindled, while most FGF7KO mice were kindled (83.3 ± 4.4%; Fig. 4f). Thus, FGF22KO mice are resistant and FGF7KO mice are prone to epileptic seizures, indicating that synaptogenic effects by FGFs have life-long consequences.
Using in vivo and in vitro approaches, we have identified FGF22 and FGF7 as target-derived presynaptic organizers for excitatory and inhibitory synapses in the hippocampal CA3 region (Fig. 4g). How FGFs work together with other synaptogenic molecules such as neuroligins, Ephs/ephrins, BDNF, WNTs, SynCAM, NGL, thrombospondins6–8, SIRP20 and LRRTM21, activity-dependent regulators such as Npas422, and glial cells6,7 to coordinate appropriate synapses will be an important next question to be answered.
Selective synaptic targeting of FGFs might also contribute to the spatial restriction of FGF effects. Indeed, we did not find significant defects in stratum lacunosum moleculare in CA3 of FGFKO mice (not shown). Such spatial specificity was also observed in neuroligin-2 KO mice23. It will be interesting to address the mechanisms by which FGF22 and FGF7 differentially localize in hippocampal neurons.
The impairment of presynaptic differentiation in FGFKO mice has a significant consequence on brain function - altered seizure susceptibility. Future inquiries should address whether these FGFs are abnormally expressed in epileptic patients, and whether the application or blockade of FGFs/FGFRs alleviate seizures. These studies will help design appropriate strategies for the treatment and prevention of epilepsy and other neurological disorders with abnormal synapse formation.
METHODS SUMMARY
Knockout mice
All studies reported in this manuscript were performed on littermate offsprings derived from heterozygote matings. FGF7KO mice were from the Jackson Laboratory11. FGF22KO mice were generated by replacing the exon 1 and a part of the exon 2 of the fgf22 gene with the neomycin resistant gene (Supplementary Fig. 1) by a homologous recombination (performed by Deltagen, San Mateo, CA). Actin-Cre-ER transgenic and conditional FGFR2KO mice were described previously9,10. Tamoxifen (100 μg) was injected at P0. All animal care and use was in accordance with the institutional guidelines and approved by the University Committee on Use and Care of Animals.
Histology
Hippocampal sections and neurons were immunostained and imaged with an epifluorescent (Olympus) or a confocal microscope (FV1000, Olympus). Synaptic protein clustering was measured using MetaMorph software as previously described9. For quantification in brain sections, areas were randomly chosen and the average staining intensity (on a 0–4,095 scale) was quantified for each area. The staining intensity of the fimbria hippocampus in the same section was subtracted. For quantification in cultures, neurites or fields were randomly selected, thresholded (see Supplementary Fig. 7), and the number and average size of puncta were quantified. Colocalization indices were determined using MetaMorph software. Images of single fluorescence channels were thresholded and binarized. An object was considered to colocalize if >25% of its area were covered by the signal in the second channel.
Electrophysiology
Whole-cell patch clamp recording was conducted in cultured hippocampal neurons at 14–19 DIV (mEPSCs) or 18–23 DIV (mIPSCs) with an Axopatch 200B amplifier and Clampex 8.0 software (Axon Instruments). Neurons with a pyramidal-like shape were selected for recording. Miniature postsynaptic currents were obtained at a holding potential of −70 mV and analyzed using Minianalysis (Synaptosoft).
METHODS
In situ hybridization
In situ hybridizations were performed as described24 using digoxigenin-labeled riboprobes (Roche). The probes were previously described9.
Primary neuronal cultures
Dissociated hippocampal cultures were prepared from E16.5-P0 WT, FGF22KO and FGF7KO mice as previously described25. 0.2–3 × 105 neurons were plated on a poly-D-lysine coated glass coverslip (12–14 mm diameter), and maintained in neurobasal medium with B27 supplement (Invitrogen). Transfections were carried out using the calcium phosphate method (CalPhos MammalianTransfection Kit, Clontech). 0.5–1 × 105 hippocampal neurons from E16.5-P0 mice were cultured for 3 days on coverslips and transfected with 1 μg of plasmids for 1 hr. The transfected cells were further cultured for 4–25 days in neurobasal medium with B27 supplement before imaging.
Immunohistochemistry
Generally, brains were fixed in 4% paraformaldehyde (PFA) for overnight, infiltrated with 30% sucrose, frozen and sagittally sectioned at 20 μm in a cryostat. Sections were blocked in 2% BSA, 2% normal goat serum and 0.1% TritonX-100 for 1 hr, followed by the incubation with primary antibodies for overnight at 4°C. For goat polyclonal primary antibodies, sections were blocked in 2% BSA and 0.1% TritonX-100 for 1 hr, and then incubated with primary antibodies. Secondary antibodies were applied for 1 hr at room temperature, and slides were mounted with p-phenylenediamine. For immunostaining for SV2, bassoon, PSD95, FGF22 and FGF7, brains were fresh frozen in Neg-50 freezing compound (Richard-Allan Scientific) and sectioned at 20 μm in a cryostat. Sections were fixed with methanol for 2 min at −20°C for SV2, bassoon and PSD95 staining, with 1% PFA for 10 min at room temperature for FGF22 staining, or with acetone for 10 min at 4°C followed by methanol for 2 min at −20°C for FGF7 staining.
Cultured neurons were generally fixed with methanol for 5 min at −20°C and stained as described above. For immunostaining for FGF22, FGF7, MAP2, neurofilament, GFP and DsRed, neurons were fixed with 1% PFA for 10 min at 37°C.
Dilutions and sources of antibodies used are as follows: anti-VGLUT1 (1:4,500; Millipore), anti-VGAT (1:1,000; Synaptic Systems), anti-PSD95 (1:700; Affinity Bioreagants), anti-gephyrin (1:500; Synaptic Systems), anti-SV2 (1:50; Developmental Studies Hybridoma Bank), anti-bassoon (1:500; Assay Designs), anti-MAP2 (1:3,000; Sigma), anti-neurofilament (1:1,000; Covance), anti-FGF22 (1:100; Santa Cruz, V-17), anti-FGF7 (1:100; Santa Cruz, H-73 and C-19), anti-GFP (1:1,000; Millipore or 1:100; BD), anti-DsRed (1:500; Clontech) and the Py antibody (1:50; a kind gift from M. Webb and P.L. Woodhams).
Imaging
12-bit images were acquired on an epi-fluorescence microscope (Olympus, BX61) using 4x, 20x and 40x objective lenses and F-View II CCD Camera (Soft Imaging System) at 1,376 × 1,032 pixel resolution, or on a confocal microscope (Olympus, FV1000) using 20x and 40x objective lenses at 1,024 × 1,024 pixel resolution. For each experiment, all images were acquired with identical settings for the laser power, detector gain and amplifier offset. Images were acquired as a z-stack (15–75 optical sections, 0.3–1.2 μm step size). For images of hippocampal sections, background was calculated from the staining intensity in the lateral ventricle for VGLUT1 and VGAT, and from the intensity in the fimbria hippocampus for SV2, bassoon, PSD95 and gephyrin. This background was subtracted from each image. The background intensity was not significantly different between FGFKO and WT mice. For images of cultured neurons, which are stained for synaptic proteins, the staining intensity of the dendritic shaft in WT cultures was calculated as the background fluorescence. This background was subtracted from images of WT and FGFKO cultures for each experiment (see Supplementary Fig. 7).
Electron microscopy
For electron microscopic analysis, P14 WT, FGF22KO and FGF7KO mice were transcardially perfused with fixative (4% PFA and 2.5% glutaraldehyde in 0.1 M Sorensen’s buffer, pH 7.4). Hippocampi were removed and 250 μm thick cross sections were prepared with a tissue slicer (Stoelting). Small pieces (~0.6 × 0.6 × 0.25 mm) of the stratum lucidum layer of the CA3 region were dissected, postfixed in the fixative for 3 days, washed and osmicated for 1 hr (1% osmium tetroxide in 0.1 M Sorensen’s buffer, pH 7.4). The samples were then rinsed in water to remove phosphate salt, en bloc stained with 3% uranyl acetate for 1 hr, dehydrated through a graded series of ethanol, rinsed in propylene oxide, and embedded in epoxy resin. Ultrathin sections (70 nm) were cut, contrasted with uranyl acetate and lead citrate, and observed with Philips CM100 electron microscope at 60 kV. Digital images were captured with a Hamamatsu ORCA-HR digital camera system operated with the AMT software (Advanced Microscopy Techniques Corp.).
Plasmid
Expression plasmids for FGF22 and FGF7 were generated by subcloning the full-length mouse fgf22 or fgf7 cDNA into the NheI-XhoI sites of APtag-5 (GenHunter).
Expression plasmids for FGF22-EGFP and FGF7-DsRed were generated by subcloning the full-length mouse fgf22 or fgf7 cDNA (minus the stop codon) into the NheI-XhoI or EcoRI-ApaI sites of pEGFP-N1 or pDsRed2-N1 (Clontech) in frame.
Western blotting
CA3 regions were dissected from P14 WT, FGF22KO and FGF7KO mice and were lysed in lysis buffer (1% NP-40, 50 mM Tris buffer, pH 7.5) with a freshly added protein inhibitor cocktail tablet (Roche). Western blotting assays were performed as described previously20. An equal amount of protein for each group was applied to the gel. Equal loading was confirmed by testing the level of α-tubulin by Western blotting. Films were scanned and the total intensity of each band was measured with MetaMorph software. Dilutions of antibodies used for blotting are: anti-VGLUT1, VGAT, PSD95 and gephyrin, 1:500; anti-α-tubulin (Sigma), 1:5,000.
Electrophysiological Recording
During recording, neurons were bathed in HEPES buffered saline (HBS), containing 119 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 30 mM glucose, and 10 mM HEPES (pH 7.4). HBS was supplemented with 1 μM tetrodotoxin and 20 μM bicuculline (for mEPSCs) or 10 μM NBQX and 20 μM APV (for mIPSCs). Whole-cell internal solution for mEPSC and eEPSC recording includes 100 mM gluconic acid, 0.2 mM EGTA, 5 mM MgCl2, 2 mM ATP, 0.3 mM GTP, 40 mM HEPES (pH 7.2), and 0.15% Lucifer yellow. Whole-cell internal solution for mIPSC recording includes 135 mM CsCl2, 1 mM EGTA, 5 mM MgCl2, 4 mM ATP, 1 mM GTP, 10 mM HEPES (pH 7.2), and 0.15% Lucifer yellow. Recording pipettes had a resistance of 4–6 MΩ.
For paired-pulse and synaptic depletion experiments, recordings were conducted in cultured hippocampal neurons at DIV14–17. HBS plus 40 μM bicuculline was used externally, and 1mM QX-314 was included in the internal solution. Pulses were stimulated extracellularly; once an evoked EPSC (eEPSC) had been elicited, stimulus strength was adjusted to the lowest amplitude that still evoked a response. Stimuli were spaced by 25 ms, 50 ms, 100 ms, 200 ms, and 1 s. To study synaptic depletion, 20 EPSCs were evoked at a frequency of 5 Hz. eEPSCs were analyzed using Clampfit (Axon Instruments).
Kindling
FGF22KO and FGF7KO mice were backcrossed with C57/BL6 mice for at least 5 generations to minimize background effects. 4–6 mice (2 to 3 months old) of each genotype were used per experiment. Control (WT, FGF22+/− and FGF7+/− mice; these mice did not show significant differences in seizure development), FGF22KO, and FGF7KO mice were injected i.p. with 35 mg/kg of pentylenetetrazol (PTZ, Sigma) every 48 hours to induce kindled seizures18. Seizure activity, which is usually induced within 15 min of PTZ injections, was observed for 1 hr after each PTZ injection. Seizures were classified according to the criterion by Racine19: Class 0, no seizure; Class 1, head nodding; Class 2, sporadic full-body shaking, spasms; Class 3, chronic full-body spasms; Class 4, jumping, shrieking, falling over; Class 5, violent convulsions, falling over, death. Major seizures defined as a single Class 5 or two consecutive Class 4 seizures are the indicative of kindled seizures (epileptic state). Experiments were repeated four times.
Supplementary Material
Acknowledgments
We thank J. Sanes and M. Hortsch for critical comments on the manuscript; M. Webb and P. Woodhams for the antibody Py; D. Sorenson for help with electron microscopy; A. Murayama for plasmid construction; M. De Freitas for help with histology; and M. Zhang for technical assistance. This work was supported by the Ester A. & Joseph Klingenstein Fund, the Edward Mallinckrodt Jr. Foundation, the March of Dimes Foundation and the Whitehall Foundation (H.U.).
Footnotes
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions
A.T. and H.U. conceived and designed the experiments, performed or participated in each of the experiments and wrote the manuscript. E.M.J.V. and M.A.S. performed the electrophysiological recordings. A.B.T. participated in the culture and histological experiments. D.J. performed the seizure-related experiments.
Author Information
Reprints and permissions information is available at www.nature.com/nature.
References
- 1.Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wassef A, Baker J, Kochan LD. GABA and schizophrenia: a review of basic science and clinical studies. J Clin Psychopharmacol. 2003;23:601–640. doi: 10.1097/01.jcp.0000095349.32154.a5. [DOI] [PubMed] [Google Scholar]
- 3.Singer HS, Minzer K. Neurobiology of Tourette’s syndrome: concepts of neuroanatomic localization and neurochemical abnormalities. Brain Dev. 2003;25:S70–S84. doi: 10.1016/s0387-7604(03)90012-x. [DOI] [PubMed] [Google Scholar]
- 4.Möhler H. GABAA receptors in central nervous system disease: anxiety, epilepsy, and insomnia. J Recept Signal Transduct Res. 2006;26:731–740. doi: 10.1080/10799890600920035. [DOI] [PubMed] [Google Scholar]
- 5.Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- 6.Fox MA, Umemori H. Seeking long-term relationship: axon and target communicate to organize synaptic differentiation. J Neurochem. 2006;97:1215–1231. doi: 10.1111/j.1471-4159.2006.03834.x. [DOI] [PubMed] [Google Scholar]
- 7.Waites CL, Craig AM, Garner CC. Mechanisms of vertebrate synaptogenesis. Annu Rev Neurosci. 2005;28:251–274. doi: 10.1146/annurev.neuro.27.070203.144336. [DOI] [PubMed] [Google Scholar]
- 8.Dalva MB, McClelland AC, Kayser MS. Cell adhesion molecules: signalling functions at the synapse. Nat Rev Neurosci. 2007;8:206–220. doi: 10.1038/nrn2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Umemori H, Linhoff MW, Ornitz DM, Sanes JR. FGF22 and its close relatives are presynaptic organizing molecules in the mammalian brain. Cell. 2004;118:257–270. doi: 10.1016/j.cell.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 10.Fox MA, et al. Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell. 2007;129:179–193. doi: 10.1016/j.cell.2007.02.035. [DOI] [PubMed] [Google Scholar]
- 11.Guo L, Degenstein L, Fuchs E. Keratinocyte growth factor is required for hair development but not for wound healing. Genes Dev. 1996;10:165–175. doi: 10.1101/gad.10.2.165. [DOI] [PubMed] [Google Scholar]
- 12.Steward O, Falk PM. Selective localization of polyribosomes beneath developing synapses: a quantitative analysis of the relationships between polyribosomes and developing synapses in the hippocampus and dentate gyrus. J Comp Neurol. 1991;314:545–557. doi: 10.1002/cne.903140311. [DOI] [PubMed] [Google Scholar]
- 13.Danglot L, Triller A, Marty S. The development of hippocampal interneurons in rodents. Hippocampus. 2006;16:1032–1060. doi: 10.1002/hipo.20225. [DOI] [PubMed] [Google Scholar]
- 14.Gonzalez AM, Berry M, Maher PA, Logan A, Baird A. A comprehensive analysis of the distribution of FGF-2 and FGFR1 in the rat brain. Brain Res. 1995;701:201–226. doi: 10.1016/0006-8993(95)01002-x. [DOI] [PubMed] [Google Scholar]
- 15.Zhang X, et al. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281:15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woodhams PL, Webb M, Atkinson DJ, Seeley PJ. A monoclonal antibody, Py, distinguishes different classes of hippocampal neurons. J Neurosci. 1989;9:2170–2181. doi: 10.1523/JNEUROSCI.09-06-02170.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao M, et al. Impaired hippocampal synaptic transmission and plasticity in mice lacking fibroblast growth factor 14. Mol Cell Neurosci. 2007;34:366–77. doi: 10.1016/j.mcn.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 18.Morimoto K, Fahnestock M, Racine RJ. Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog Neurobiol. 2004;73:1–60. doi: 10.1016/j.pneurobio.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 19.Racine RJ. Modification of seizure activity by electrical stimulation. II Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 20.Umemori H, Sanes JR. Signal regulatory proteins (SIRPS) are secreted presynaptic organizing molecules. J Biol Chem. 2008;283:34053–34061. doi: 10.1074/jbc.M805729200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Linhoff MW, et al. An unbiased expression screen for synaptogenic proteins identifies the LRRTM protein family as synaptic organizers. Neuron. 2009;61:734–749. doi: 10.1016/j.neuron.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin Y, et al. Activity-dependent regulation of inhibitory synapse development by Npas4. Nature. 2008;455:1198–204. doi: 10.1038/nature07319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibson JR, Huber KM, Südhof TC. Neuroligin-2 deletion selectively decreases inhibitory synaptic transmission originating from fast-spiking but not from somatostatin-positive interneurons. J Neurosci. 2009;29:13883–13897. doi: 10.1523/JNEUROSCI.2457-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schaeren-Wiemers N, Gerfin-Moser A. A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry. 1993;100:431–440. doi: 10.1007/BF00267823. [DOI] [PubMed] [Google Scholar]
- 25.Goslin K, Asmussen H, Banker G. Rat hippocampal neurons in low-density culture. In: Banker G, Goslin K, editors. Culturing Nerve Cells. Cambridge: MIT Press; 1998. pp. 339–370. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.