

Abstract
Endothelium of fetal or newborn arteries is atypical, displaying actin stress fibers and reduced nitric oxide (NO)-mediated dilatation. This study tested the hypothesis that Rho/Rho kinase signaling, which promotes endothelial stress fibers and inhibits endothelial dilatation, contributed to this phenotype. Carotid arteries were isolated from newborn [postnatal day 1 (P1)], P7, and P21 mice. Endothelial dilatation to acetylcholine (pressure myograph) was minimal at P1, increased at P7, and further increased at P21. Inhibition of Rho (C3 transferase) or Rho kinase (Y27632, fasudil) significantly increased dilatation to acetylcholine in P1 arteries but had no effect in P7 or P21 arteries. After inhibition of NO synthase (NG-nitro-l-arginine methyl ester), Rho kinase inhibition no longer increased acetylcholine responses in P1 arteries. Rho kinase inhibition did not affect dilatation to the NO donor DEA-NONOate. The endothelial actin cytoskeleton was labeled with phalloidin and visualized by laser-scanning microscopy. In P1 arteries, the endothelium had prominent transcytoplasmic stress fibers, whereas in P7 and P21 arteries, the actin fibers had a significantly reduced intensity and were restricted to cell borders. Phosphorylation of myosin light chains, a Rho kinase substrate, was highest in P1 endothelium and significantly reduced in P7 and P21 endothelium (laser-scanning microscopy). In P1 arteries, inhibition of Rho (C3 transferase) or Rho kinase (Y27632) significantly reduced the intensity of actin fibers, which were restricted to cell borders. Similarly, in P1 arteries, Rho inhibition significantly reduced endothelial levels of phosphorylated myosin light chains. These results indicate that the atypical function and morphology of newborn endothelium is mediated by Rho/Rho kinase signaling.
Keywords: endothelium-dependent dilatation, F-actin, cytoskeleton, developmental biology, endothelial dysfunction
the early postnatal period is associated with remarkable changes in the morphology and function of arterial endothelium. Endothelial cells lining fetal or newborn arteries are highly unusual, displaying prominent stress fibers (15, 16, 23) and reduced endothelial nitric oxide (NO)-mediated dilatation (1, 3, 8, 27). Although common in cultured cells, arterial endothelial cells in vivo generally lack stress fibers, and F-actin is restricted to a faint cortical network at the cell periphery (7, 10, 15, 16). Indeed, as newborn arteries mature in the immediate postnatal period, the endothelial actin cytoskeleton remodels into a peripheral cortical network and there is a marked increase in endothelium-dependent dilatation (1, 3, 8, 15, 16, 27).
Endothelial stress fibers and endothelial dysfunction, which is accompanied by diminished NO-mediated dilatation, are prominent features of vascular disease including atherosclerosis (10, 12). Numerous pathological and inflammatory mediators stimulate endothelial stress fiber formation by activating RhoA and Rho kinase (21, 25, 28). Rho kinase phosphorylates myosin light chains (MLCs), which are further amplified via Rho kinase-mediated inhibition of MLC phosphatase, resulting in increased actomyosin interaction and formation of stress fibers (21, 25, 28). Rho kinase also inhibits endothelium-dependent NO-mediated dilatation through multiple mechanisms (19, 28). Indeed, endothelial Rho/Rho kinase signaling contributes to the endothelial dysfunction of vascular disease including atherosclerosis but is considered to have minimal activity under normal physiological conditions (19, 28).
The present study tested the hypothesis that the atypical morphology and function of newborn arterial endothelium is mediated by endothelial Rho/Rho kinase signaling.
MATERIALS AND METHODS
Use of animals was approved by the Johns Hopkins Institutional Animal Care and Use Committee and complied with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals. Newborn [postnatal day 1 (P1)] and 1-wk (P7)-, and 3-wk (P21)- old male and female mice (C57BL6, Jackson Laboratories) were euthanized by CO2 asphyxiation. Carotid arteries were rapidly removed and placed in cold Krebs-Ringer bicarbonate (control) solution consisting of (in mM) 118.3 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 2.5 CaCl2, 25.0 NaHCO3, and 11.1 glucose.
Analysis of vascular responses.
Arteries were cannulated with glass micropipettes within a microvascular chamber (Living Systems) and maintained in the absence of flow at a transmural pressure of 20 mmHg (8). The chamber was superfused with control solution (37°C, pH 7.4, 16% O2-5% CO2-balance N2) and placed on the stage of an inverted microscope (Nikon TMS-F), connected to a video camera (CCTV camera; Panasonic). The internal diameter, determined using a video dimension analyzer (Living Systems), was continuously monitored using a data acquisition system (BIOPAC) (8).
Arterial segments were constricted with the thromboxane mimic U46619. Once constriction was stable, vasodilatation to the endothelial agonist acetylcholine (10−9 to 10−6 M) or the NO donor DEA-NONOate (10−9 to 10−6 M) was determined. Concentration-response curves were generated by increasing agonist concentration in full-log increments once the response to the previous concentration stabilized. Only one cycle of constriction-vasodilatation was performed on each artery. In most experiments, responses to acetylcholine were determined in paired carotid arteries, either in the absence or presence of drug treatment. Unless stated otherwise, drugs were present in the superfusate for 60 min before and during exposure of the arteries to vasodilators. When using C3 transferase to inhibit Rho signaling, the inhibitor was present in the intraluminal perfusate for 180 min before and during vasodilator responses.
Endothelial imaging.
Carotid arteries were processed as previously described (8). Briefly, arteries were opened longitudinally during fixation with paraformaldehyde (3%, 4°C, 30 min). They were permeabilized (Triton-X, 0.5%) and incubated in donkey serum (1.5%) to reduce nonspecific binding. Arteries were incubated overnight with rhodamine phalloidin (to label F-actin) or a rabbit polyclonal antibody to PO4-MLC (Ser19, 1:200 dilution, Millipore). After removal of the primary antibody, arteries were incubated with Alexa Fluor 488-labeled donkey anti-rabbit antibody (1:200 dilution, Jackson ImmunoResearch) and with Draq5 (5 μmol/l; Biostatus) to label nuclei. Samples were viewed on a Leica AOBS-equipped SP5 laser-scanning microscope using a ×63 objective (numerical aperture, 1.4). Images (1024 × 1024 pixels) were obtained using a pinhole of 1 Airy unit, scan speed of 400 Hz, 6 line averaging, optical zoom of 3.0, and appropriate settings for Alexa Fluor 488 (excitation, 488 nm; and emission, 492–541 nm), rhodamine phalloidin (excitation, 543 nm; and emission 555–620 nm), and DRAQ (excitation, 633 nm; and emission, 650–750 nm). For quantitative comparison of fluorescence intensity, arteries were processed at the same time using the same instrument settings. When comparing fluorescent signals between different groups, the mean of the average signal intensities in P1 control arteries was set as 100%, and the intensity of all control and test images were expressed relative to that value (11).
Drugs.
Acetylcholine was from Sigma-Aldrich, cell permeable C3 transferase from Cytoskeleton, DEA-NONOate from Enzo Life Sciences, fasudil from Tocris Biosciences, U46619 from Cayman, and Y27632 from EMD Millipore.
Statistical analysis.
Vasomotor responses were expressed as a percent changes in the quiescent baseline diameters. Data are expressed as means ± SE, and n equals the number of animals from which blood vessels were studied. Statistical evaluation of the data was performed by Student's t-test for paired or unpaired observations. When more than two means were compared, analysis of variance was used. If a significant F-value was found, the Tukey-Kramer multiple comparisons test identified differences among groups. Values were considered to be statistically different when P < 0.05.
RESULTS
Endothelial dilator function in neonatal arteries.
Endothelium-dependent dilatation to acetylcholine was minimal in P1 arteries, increased dramatically by P7, and increased further by P21 (Fig. 1) (8). Maximal dilatation to acetylcholine was 21.5 ± 7.3% (n = 7) of the constriction to U46619 in P1 arteries, 78.4 ± 4.8% in P7 arteries (n = 7, P < 0.001 compared with P1), and 101.1 ± 3.2% in P21 arteries (n = 7, P < 0.001 compared with P1, P < 0.05 compared with P7). Inhibition of Rho (cell permeable C3 transferase, 1 μg/ml) significantly increased dilatation to acetylcholine in P1 arteries, with the maximal dilatation increasing from 24.1 ± 9.7 to 69.5 ± 8.5% of the constriction to U46619 (n = 6, P < 0.01) (Fig. 1). Likewise, inhibition of Rho kinase (fasudil, 3 μM; or Y27632, 0.3 μM) significantly increased dilatation to acetylcholine in P1 arteries, with the maximal dilatation increasing from 18.5 ± 6.8% of the constriction to U46619 (n = 13, combined control) to 70.2 ± 9.0% after fasudil (n = 7, P < 0.001) and 70.8 + 5.8% after Y27632 (n = 6, P < 0.01) (Fig. 2A). Inhibition of NO synthase (NOS) with NG-nitro-l-arginine methyl ester (100 μM) abolished the amplified dilatations to acetylcholine following inhibition of Rho (C3 transferase) or Rho kinase (fasudil, Y27632) in P1 arteries (data not shown). Rho kinase inhibition (fasudil, Y27632) did not significantly affect dilatation to the NO donor DEA-NONOate in P1 arteries (Fig. 2). In contrast to P1 arteries, inhibition of Rho kinase (fasudil, Y27632) did not significantly affect dilatations to acetylcholine in P7 or P21 arteries (Fig. 3).
Fig. 1.

Postnatal maturation of endothelium-dependent dilatation. Carotid arteries were isolated from [postnatal day 1 (P1)], P7, and P21 mice and analyzed in a microperfusion system at a transmural pressure (PTM) of 20 mmHg. Vasomotor responses are expressed as a percentage of the baseline diameter of the arteries (B) and presented as means ± SE. To observe dilatation, arteries were constricted to ∼80% of baseline diameter with the thromboxane mimic U46619 (U4). Top: acetylcholine caused minimal dilatation in P1 arteries, which was increased in P7 and P21 arteries. ***P < 0.001, significant difference with P1; #P < 0.05 and ###P < 0.001, significant difference between P7 and P21 (n = 7). Bottom: Rho inhibition with C3 transferase (1 μg/ml) increased responses to acetylcholine in P1 carotid arteries. **P < 0.001 indicates significant difference with control arteries (n = 6).
Fig. 2.

Effects of Rho kinase inhibition with fasudil (3 μM) or Y27632 (0.3 μM) on dilator responses to acetylcholine (top; n = 7 for fasudil; and n = 6 for Y27632) or the nitric oxide donor DEA-NONOate (bottom; n = 5 for fasudil; and n = 4 for Y27632) in P1 carotid arteries. Isolated arteries were analyzed in a microperfusion system at a PTM of 20 mmHg and treated with or without a Rho kinase inhibitor. To observe dilation, arteries were constricted to ∼80% of baseline diameter with the thromboxane mimic U46619 (U4). Functional responses are expressed as a percentage of the baseline diameter (B) and presented as means ± SE. ***P < 0.001, significant difference with arteries treated with fasudil; #P < 0.05, significant difference with arteries treated with Y27632. The inhibitors significantly increased dilatation to acetylcholine but did not affect responses to the nitric oxide donor.
Fig. 3.

Effects of Rho kinase inhibition with fasudil (3 μM) or Y27632 (0.3 μM) on dilator responses to acetylcholine in P7 (top; n = 6 for fasudil; and n = 7 for Y27632) and P21 carotid arteries (bottom; n = 5 for fasudil; and n = 6 for Y27632). Paired isolated arteries were analyzed in a microperfusion system at a PTM of 20 mmHg and treated with or without a Rho kinase inhibitor. To observe dilation, arteries were constricted to ∼80% of baseline diameter with the thromboxane mimic U46619 (U4). Responses are expressed as a percentage of the baseline diameter (B) and presented as means ± SE. Rho kinase inhibition did not significantly affect dilatation to acetylcholine in P7 or P21 arteries.
Endothelial actin cytoskeleton in neonatal arteries.
Endothelial cells lining newborn carotid arteries (P1) had prominent transcytoplasmic F-actin stress fibers running through the cell interior parallel to the long axis of the cells (Fig. 4). In P7 and P21 arteries, the intensity of the endothelial F-actin fibers was significantly reduced compared with P1 arteries (P7, 55.5 ± 6.3% of P1, n = 11, P < 0.001; and P21, 45.4 ± 4.7% of P1, n = 7, P < 0.001) and the fibers were restricted to the cell periphery (Fig. 4). In P1 arteries, inhibition of Rho (C3 transferase, 1 μg/ml) or Rho kinase signaling (Y27632, 0.3 μM) significantly reduced the intensity of endothelial F-actin fibers (C3 transferase, 63.4 ± 6.8% of controls, n = 8, P < 0.001; and Y27632, 65.0 ± 4.0% of control arteries, n = 4, P < 0.001) and reorganized the fibers to the cell periphery (Fig. 4).
Fig. 4.
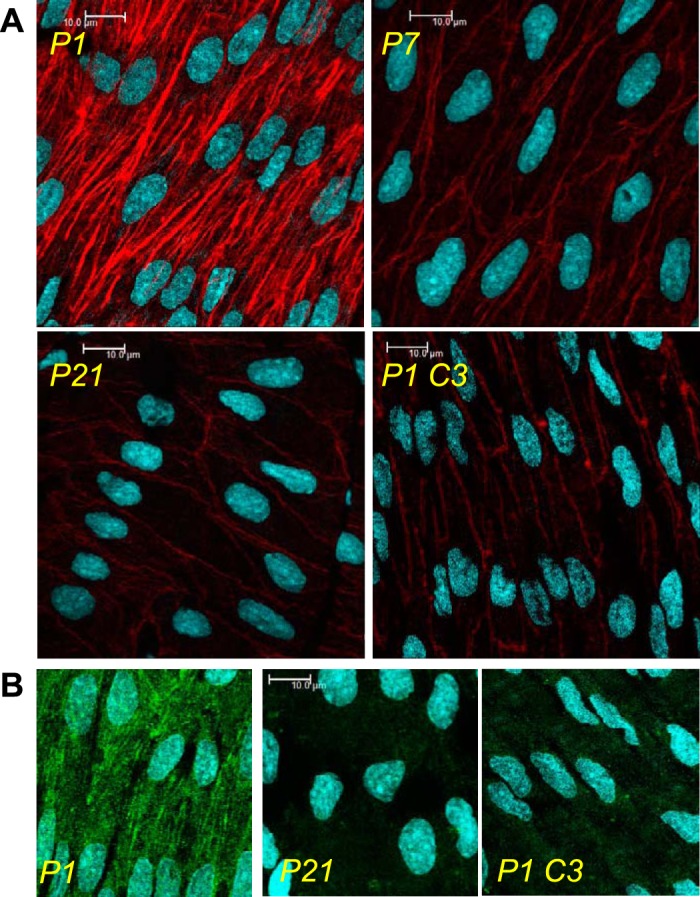
Representative laser-scanning microscopy fluorescent images, demonstrating postnatal maturation of arterial endothelium. Carotid arteries were isolated from P1, P7, and P21 mice and processed to visualize the endothelial actin cytoskeleton (rhodamine phalloidin, red; A) or PO4-myosin light chain (MLC; Alexa-488, green; B) using laser-scanning microscopy analysis. The endothelial actin cytoskeleton displayed prominent stress fibers in P1 arteries but had significantly reduced intensity and was restricted to a cortical actin structure delineating cell borders in P7 and P21 arteries (A). This was accompanied by a significant reduction in endothelial PO4-MLC levels in maturing arteries (B). In P1 arteries, Rho inhibition (C3 transferase, 1 μg/ml, P1 C3) reduced endothelial PO4-MLC (B) and the intensity of endothelial actin fibers, which were restricted to the cell periphery (A).
In parallel with the postnatal reduction in endothelial stress fibers, staining intensity for PO4-MLC, a Rho kinase substrate, was reduced in endothelial cells lining P7 (54.8 ± 3.2%, n = 4, P < 0.001) and P21 arteries (44.3 ± 1.7%, n = 7, P < 0.001), compared with P1 arteries (Fig. 4). Inhibition of Rho signaling (C3 transferase, 1 μg/ml) significantly reduced PO4-MLC levels in P1 arteries (to 36.3 ± 3.1% of controls, n = 6, P < 0.001) (Fig. 4).
DISCUSSION
The present study confirms a remarkable transformation in endothelial structure and function during the immediate postnatal period. Endothelial cells lining newborn carotid arteries displayed prominent transcytoplasmic stress fibers and minimal endothelium-dependent dilatation. However, during the first week of postnatal life, the endothelial actin cytoskeleton remodeled from stress fibers to a peripheral cortical network, and there was a dramatic emergence of endothelial dilator function. This endothelial maturation was paralleled by a significant reduction in endothelial levels of PO4-MLC, which would be consistent with decreasing activity of Rho/Rho kinase signaling. Indeed, inhibition of Rho and Rho kinase in newborn arteries mimicked the maturation process, reducing actin stress fibers, reducing PO4-MLC levels, and increasing endothelium-dependent dilatation. The results suggest that the atypical morphology and function of newborn endothelial cells is mediated by heightened endothelial Rho/Rho kinase signaling.
Endothelial levels of PO4-MLC, a Rho kinase substrate, were highest in newborn arteries and reduced to similar low levels by acute inhibition of Rho signaling or by developmental maturation in P7 and P21 arteries. This suggests that endothelial Rho/Rho kinase signaling is highest in newborn arteries, is dramatically reduced by P7, and maintained at a low level in P21 arteries. Similarly, the functional activity of Rho/Rho kinase signaling, as defined by the influence of inhibitors on endothelial dilatation, was highest in newborn arteries and not evident in P7 and P21 arteries. Acute inhibition of Rho or Rho kinase caused a marked increase in dilation to acetylcholine in P1 arteries, which was abolished by inhibition of NOS with L-NAME and was not accompanied by a significant change in smooth muscle responsiveness to exogenous NO. This suggests that the amplifying effect of Rho/Rho kinase inhibition on newborn endothelial dilatation is mediated by increased activity of endothelium-derived NO. Inhibition of Rho kinase did not affect dilator responses to acetylcholine in P7 and P21 arteries. Furthermore, after Rho kinase inhibition, there was no longer any difference in the magnitude of responses to acetylcholine between P1 and P7 arteries. These results demonstrate that inhibition of Rho/Rho kinase mimics the maturation process and suggest that heightened activity of endothelial Rho/Rho kinase signaling is entirely responsible for impaired endothelial dilation in newborn arteries.
Endothelial Rho/Rho kinase signaling promotes the incorporation of F-actin into stress fibers (21, 25, 28). Although a common feature of cultured cells, these structures are not normally present in vivo, consistent with a low activity of endothelial Rho/Rho kinase signaling. Indeed, the endothelium of P7 and P21 arteries displayed the normal pattern of actin cytoskeleton, a cortical network delineating the cell borders. The atypical intense stress fibers in newborn endothelium was dependent on the heightened Rho/Rho kinase signaling in these cells, with acute inhibition of Rho or Rho kinase, causing a marked reduction in their activity and revealing a more mature cortical actin system. Endothelial stress fibers are a prominent feature in vascular disease and in areas susceptible to atherosclerosis (10, 12). This is thought to result from alterations in the subendothelial matrix (10, 16), most importantly an increased deposition of fibronectin (6, 13, 20). Indeed, endothelial adhesion to fibronectin stimulates stress fiber formation, which is prevented by inhibiting Rho kinase (9). The subendothelial matrix of endothelial cells lining fetal or newborn arteries is enriched in fibronectin, with endothelial stress fibers connecting via focal adhesion complexes to fibronectin fibrils (4, 5, 14, 23). As endothelial cells mature during the immediate postnatal period, they reorganize their underlying matrix, decreasing the levels of fibronectin and generating a normal subendothelial matrix (4, 5, 15, 16). Therefore, the presence of fibronectin in the matrix of newborn endothelium likely contributes to heightened activity of Rho/Rho kinase signaling in these cells.
The emergence of effective dilatation to acetylcholine in the immediate postnatal period appears to reflect an increase in NOS activation by phosphoinositide 3-kinase (PI3K)/Akt signaling (8). Although Rho kinase can inhibit endothelial NO activity through multiple mechanisms, a key inhibitory step appears to be activation of phosphatase and tensin homolog (PTEN), which counteracts PI3K activity and reduces Akt activity (22, 24, 26). Therefore, the increased activity of PI3K/Akt/NOS signaling in the immediate postnatal period may reflect the diminution in Rho/Rho kinase signaling. The postnatal maturation in endothelial dilator activity may be triggered by increased clustering of vascular endothelial-cadherin at adherens junctions (8). Indeed, clustering of vascular endothelial cadherin initiates assembly of a signaling complex that inhibits RhoA/Rho kinase and amplifies PI3K/Akt signaling (2, 17, 18). Therefore, this key process may contribute to the developmental changes in signaling, function, and morphology of maturing endothelial cells.
The present study employed quantitative immunofluorescent imaging to assess endothelial F-actin cytoskeleton and endothelial PO4-MLC levels. Although this approach localized signaling changes to the endothelium and avoided contamination from other vascular cells (including smooth muscle), a limitation of the study is that these results were not confirmed by immunoblot analyses. Likewise, although the study used a comprehensive pharmacological approach using agents with proven selectivity and efficacy at the designated concentrations, the approach did not directly confirm inhibition of individual enzyme activity.
The results of the present study demonstrate that the atypical phenotype of newborn endothelial cells is mediated by heightened endothelial Rho/Rho kinase signaling, resulting in prominent endothelial stress fibers and a marked inhibition of endothelial dilatation. In many respects, the unusual phenotype of newborn endothelium resembles dysfunctional endothelial cells, which contribute to the progression of vascular disease. Retention or reemergence of the newborn endothelial phenotype may therefore contribute to the development of vascular disease by negating the normal protective role of this important cell layer.
GRANTS
This study was supported by National Institutes of Health Grants HL-102715 and HD-078639 (to N. A. Flavahan).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.F. and N.A.F. performed experiments; S.F. and N.A.F. analyzed data; S.F. and N.A.F. edited and revised manuscript; S.F. and N.A.F. approved final version of manuscript; N.A.F. conception and design of research; N.A.F. interpreted results of experiments; N.A.F. prepared Figs.; N.A.F. drafted manuscript.
REFERENCES
- 1.Abman SH, Chatfield BA, Rodman DM, Hall SL, McMurtry IF. Maturational changes in endothelium-derived relaxing factor activity of ovine pulmonary arteries in vitro. Am J Physiol Lung Cell Mol Physiol 260: L280–L285, 1991 [DOI] [PubMed] [Google Scholar]
- 2.Birukova AA, Tian Y, Dubrovskyi O, Zebda N, Sarich N, Tian X, Wang Y, Birukov KG. VE-cadherin trans-interactions modulate Rac activation and enhancement of lung endothelial barrier by iloprost. J Cell Physiol 227: 3405–3416, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boegehold MA. Endothelium-dependent control of vascular tone during early postnatal and juvenile growth. Microcirculation 17: 394–406, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis EC. Endothelial cell connecting filaments anchor endothelial cells to the subjacent elastic lamina in the developing aortic intima of the mouse. Cell Tissue Res 272: 211–219, 1993 [DOI] [PubMed] [Google Scholar]
- 5.Davis EC. Immunolocalization of microfibril and microfibril-associated proteins in the subendothelial matrix of the developing mouse aorta. J Cell Sci 107: 727–736, 1994 [DOI] [PubMed] [Google Scholar]
- 6.Feaver RE, Gelfand BD, Wang C, Schwartz MA, Blackman BR. Atheroprone hemodynamics regulate fibronectin deposition to create positive feedback that sustains endothelial inflammation. Circ Res 106: 1703–1711, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flavahan NA, Bailey SR, Flavahan WA, Mitra S, Flavahan S. Imaging remodeling of the actin cytoskeleton in vascular smooth muscle cells after mechanosensitive arteriolar constriction. Am J Physiol Heart Circ Physiol 288: H660–H669, 2005 [DOI] [PubMed] [Google Scholar]
- 8.Flavahan S, Mozayan MM, Lindgren I, Flavahan NA. Pressure-induced maturation of endothelial cells on newborn mouse carotid arteries. Am J Physiol Heart Circ Physiol 305: H321–H329, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujiwara H, Gu J, Sekiguchi K. Rac regulates integrin-mediated endothelial cell adhesion and migration on laminin-8. Exp Cell Res 292: 67–77, 2004 [DOI] [PubMed] [Google Scholar]
- 10.Gabbiani G, Gabbiani F, Lombardi D, Schwartz SM. Organization of actin cytoskeleton in normal and regenerating arterial endothelial cells. Proc Natl Acad Sci USA 80: 2361–2364, 1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goel A, Su B, Flavahan S, Lowenstein CJ, Berkowitz DE, Flavahan NA. Increased endothelial exocytosis and generation of endothelin-1 contributes to constriction of aged arteries. Circ Res 107: 242–251, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guyton JR, Shaffer DR, Henry PD. Stress fibers in endothelial cells overlying atherosclerotic lesions in rabbit aorta. Am J Med Sci 298: 79–82, 1989 [DOI] [PubMed] [Google Scholar]
- 13.Hahn C, Orr AW, Sanders JM, Jhaveri KA, Schwartz MA. The subendothelial extracellular matrix modulates JNK activation by flow. Circ Res 104: 995–1003, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jinguji Y. Developmental stage dependent expression of the endothelial stress fibers and organization of fibronectin fibrils in the aorta of chick embryos. Zoolog Sci 20: 1359–1366, 2003 [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi N, Sakai T. Postnatal reorganization of actin filaments and differentiation of intercellular boundaries in the rat aortic endothelial cells. Cell Tissue Res 278: 471–482, 1994 [DOI] [PubMed] [Google Scholar]
- 16.Kocher O, Skalli O, Cerutti D, Gabbiani F, Gabbiani G. Cytoskeletal features of rat aortic cells during development. An electron microscopic, immunohistochemical, and biochemical study. Circ Res 56: 829–838, 1985 [DOI] [PubMed] [Google Scholar]
- 17.Lampugnani MG, Dejana E. Adherens junctions in endothelial cells regulate vessel maintenance and angiogenesis. Thromb Res 120, Suppl 2: S1–S6, 2007 [DOI] [PubMed] [Google Scholar]
- 18.Lampugnani MG, Zanetti A, Breviario F, Balconi G, Orsenigo F, Corada M, Spagnuolo R, Betson M, Braga V, Dejana E. VE-cadherin regulates endothelial actin activating Rac and increasing membrane association of Tiam. Mol Biol Cell 13: 1175–1189, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loirand G, Pacaud P. The role of Rho protein signaling in hypertension. Nat Rev Cardiol 7: 637–647, 2010 [DOI] [PubMed] [Google Scholar]
- 20.Orr AW, Sanders JM, Bevard M, Coleman E, Sarembock IJ, Schwartz MA. The subendothelial extracellular matrix modulates NF-kappaB activation by flow: a potential role in atherosclerosis. J Cell Biol 169: 191–202, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prasain N, Stevens T. The actin cytoskeleton in endothelial cell phenotypes. Microvasc Res 77: 53–63, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchez T, Skoura A, Wu MT, Casserly B, Harrington EO, Hla T. Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscler Thromb Vasc Biol 27: 1312–1318, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Sugimoto K, Fujii S, Takemasa T, Yamashita K. Factors inducing codistribution of marginal actin fibers and fibronectin in rat aortic endothelial cells. Am J Physiol Heart Circ Physiol 272: H2188–H2194, 1997 [DOI] [PubMed] [Google Scholar]
- 24.Tamguney T, Stokoe D. New insights into PTEN. J Cell Sci 120: 4071–4079, 2007 [DOI] [PubMed] [Google Scholar]
- 25.van Nieuw Amerongen GP, Musters RJ, Eringa EC, Sipkema P, van Hinsbergh VW. Thrombin-induced endothelial barrier disruption in intact microvessels: role of RhoA/Rho kinase-myosin phosphatase axis. Am J Physiol Cell Physiol 294: C1234–C1241, 2008 [DOI] [PubMed] [Google Scholar]
- 26.Wolfrum S, Dendorfer A, Rikitake Y, Stalker TJ, Gong Y, Scalia R, Dominiak P, Liao JK. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vasc Biol 24: 1842–1847, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zellers TM, Vanhoutte PM. Endothelium-dependent relaxations of piglet pulmonary arteries augment with maturation. Pediatr Res 30: 176–180, 1991 [DOI] [PubMed] [Google Scholar]
- 28.Zhou Q, Gensch C, Liao JK. Rho-associated coiled-coil-forming kinases (ROCKs): potential targets for the treatment of atherosclerosis and vascular disease. Trends Pharmacol Sci 32: 167–173, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]