

Abstract
Mitochondrial depolarization following ATP-sensitive potassium (mitoKATP) channel activation has been shown to induce cerebral vasodilation by generation of mitochondrial reactive oxygen species (ROS), which sequentially promotes frequency of calcium sparks and activation of large conductance calcium-activated potassium channels (BKCa) in vascular smooth muscle (VSM). We previously demonstrated that cerebrovascular insulin resistance accompanies aging and obesity. It is unclear whether mitochondrial depolarization without the ROS generation enhances calcium sparks and vasodilation in phenotypically normal [Sprague Dawley (SD); Zucker lean (ZL)] and insulin-resistant [Zucker obese (ZO)] rats. We compared the mechanisms underlying the vasodilation to ROS-dependent (diazoxide) and ROS-independent [BMS-191095 (BMS)] mitoKATP channel activators in normal and ZO rats. Arterial diameter studies from SD, ZL, and ZO rats showed that BMS as well as diazoxide induced vasodilation in endothelium-denuded cerebral arteries. In normal rats, BMS-induced vasodilation was mediated by mitochondrial depolarization and calcium sparks generation in VSM and was reduced by inhibition of BKCa channels. However, unlike diazoxide-induced vasodilation, scavenging of ROS had no effect on BMS-induced vasodilation. Electron spin resonance spectroscopy confirmed that diazoxide but not BMS promoted vascular ROS generation. BMS- as well as diazoxide-induced vasodilation, mitochondrial depolarization, and calcium spark generation were diminished in cerebral arteries from ZO rats. Thus pharmacological depolarization of VSM mitochondria by BMS promotes ROS-independent vasodilation via generation of calcium sparks and activation of BKCa channels. Diminished generation of calcium sparks and reduced vasodilation in ZO arteries in response to BMS and diazoxide provide new insights into mechanisms of cerebrovascular dysfunction in insulin resistance.
Keywords: calcium sparks, mitochondrial membrane potential, electron spin resonance, BMS-191095, superoxide
aging, obesity, and type 2 diabetes (T2DM) are accompanied by metabolic insulin resistance. Clinical and animal studies have established a causative relationship between insulin resistance and the decline of higher cortical function accompanying aging, T2DM, and Alzheimer disease (8, 10, 58). Impairment of neurovascular coupling, which matches the demands of neuronal activity for oxygen and glucose with increased blood flow, has been implicated in age and T2DM related decline in cognitive function (20, 31, 56). Studies from our laboratory have demonstrated for the first time impaired cerebrovascular actions of insulin in aged Sprague Dawley (SD) and Zucker obese (ZO) rats with insulin resistance compared with young SD and Zucker lean (ZL) rats, respectively (35). Mitochondria act as the sensor of metabolic demand in the cells participating in the neurovascular coupling, and mitochondrial dysfunction has been implicated in the etiology of aging, Alzheimer disease, T2DM, and insulin resistance (11, 21, 47, 56, 57). Importantly, we reported diminished mitochondrial mediated vasodilation in cerebral arteries from ZO rats with insulin resistance (34).
Mitochondria responding to physical, physiological, pharmacological, and pathological factors appear to play an important role in the regulation of vascular tone in several regional circulations. Studies from our laboratory and others have clearly shown that mitochondrial mechanisms promote relaxation of endothelium-intact and endothelium-denuded cerebral arteries as well as isolated vascular smooth muscle (VSM) cells (7, 34, 36, 59, 60). Analysis of these studies has led to the concept that the integrated arterial response to mitochondrial activators such as diazoxide and BMS-191095 (BMS) involves direct VSM relaxation, which is modified by contributions of vasoactive factors such as nitric oxide and prostaglandins from endothelium (34, 36). In previous studies, Jaggar and colleagues (59) showed that diazoxide promotes reactive oxygen species (ROS) in cerebral VSM, which causes the sequential activation of ryanodine-sensitive calcium channels in sarcoplasmic reticulum, generation of calcium transients (otherwise known as ′calcium sparks′), and the opening of adjacent large-conductance calcium activated potassium channels (BKCa) on the plasma membrane. The efflux of potassium leads to hyperpolarization, decreased global intracellular calcium of VSM, and vasodilation (7, 59).
Although both diazoxide and BMS are activators of mitochondrial ATP-sensitive potassium (mitoKATP) channels, an important difference between diazoxide and BMS is that the former is associated with production of ROS, whereas the latter is not (6, 36). The ROS production by diazoxide likely arises from inhibition of succinate dehydrogenase (9); however, effects are still limited to mitochondria. In contrast, we are unaware of any off-target effects of BMS. Although, the mechanisms of action of these two agents have recently been shown to be different in cerebral vascular endothelium (36), a direct comparison has not been made in VSM. Moreover, nothing is known concerning the effects of BMS on calcium spark activity in cerebral VSM and how calcium spark activity is affected by insulin resistance. We have shown previously that dilation to diazoxide is impaired in cerebral arteries from insulin-resistant rats but the mechanisms have not been fully explored (34). Thus we hypothesized that mitochondrial depolarization enhances calcium spark activity in VSM cells independent of ROS generation. We further hypothesized that cerebral arteries with insulin resistance and mitochondrial dysfunction exhibit impaired calcium spark activity in response to mitochondrial activation.
In this study, we have examined the effects of mitochondrial depolarization induced by BMS in endothelium-denuded cerebral arteries on calcium sparks-mediated vasodilation in normal and insulin-resistant cerebral arteries. Our results have allowed us to confirm our earlier findings in cerebral vascular endothelium (36) that ROS-dependent and ROS-independent mechanisms share key elements of calcium signaling, which link mitochondrial depolarization with cellular events. Thus, our findings validate the concept that calcium spark activity subsequent to mitochondrial depolarization can occur without ROS production and thereby have illustrated the diversity of pathways linking mitochondrial activation, calcium sparks, and vasodilation in health and disease.
MATERIALS AND METHODS
The animal protocol was approved by the Institutional Animal Care and Use Committees of Tulane University School of Medicine. The investigation complies with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH; Publication No. 85-23, revised 1996). Young, male SD (n = 28) and ZL and ZO rats (n = 14 each) were obtained at 10–12 wk of age. Rats were housed in the animal care facility and received standard rat chow and tap water ad libitum. Studies were performed on arteries isolated after euthanizing the animals.
ZO rat model.
The ZO rat with a leptin receptor mutation, which is homozygous for the mutation (fa/fa), has been widely used as a model of insulin resistance and T2DM. ZL rats are heterozygous for the mutation (Fa/fa) and are genetically appropriate controls (5). Previous data from our laboratory (18, 19, 35) and others (48, 50, 54) have shown that ZO rats develop insulin resistance with a metabolic profile very similar to the human condition. As reported previously (18, 19), at 10–12 wk of age, ZO rats had significantly greater body weight and exhibited features typical of the metabolic syndrome including impaired glucose tolerance, hyperinsulinemia, hypertriglyceridemia, and hypercholesterolemia. Importantly, young ZO rats used in the present study were insulin resistant but without elevated levels of glucose and arterial blood pressure. In addition, our previous studies also established vascular insulin resistance in cerebral arteries in the ZO rats with comprehensive characterization of the mechanisms of impaired vasodilation to insulin (35). Furthermore, studies in our laboratory and others have reported impairments of vascular function in cerebral arteries (19, 32, 49, 50, 55). Notably, for the first time, we demonstrated impaired diazoxide-induced and mitochondria-mediated vasodilation in cerebral arteries in ZO rats (34).
Vascular reactivity.
Rats were euthanized under deep isoflurane inhalation anesthesia and decapitated, and rat brains were isolated. Subsequently, the posterior cerebral arteries were isolated and vasoreactivity was determined by measuring intraluminal diameter (Living Systems Instrumentation, Burlington, VT) as described previously (6, 34). Briefly, arteries were transferred to a vessel bath of oxygenated, warm, physiological salt solution (PSS), cannulated with glass pipettes and secured with nylon thread. The composition of PSS was (in mmol/l) 112 NaCl, 4.8 KCl, 26 NaHCO3, 1.2 KH2PO4·H2O, 1.8 CaCl2, 1.2 MgSO4·7H2O, and 10 glucose (pH 7.4, NaOH). Arteries were slowly pressurized to 70 mmHg with PSS until they developed stable myogenic tone, and cumulative concentration responses to drugs were determined. Endothelium was removed by injecting a bolus of 1 ml of air through the arteries. Endothelial denudation was verified by lack of dilator response to bradykinin (10 μmol/l), and viability was tested by intact vasodilator response to nitroprusside (10 μmol/l). We have validated this method by demonstrating endothelial denudation using electron microscopy (43) and have successfully used it in many of our previous studies (33–36, 44).
Vascular responses to 10, 50, and 100 μmol/l BMS were determined in endothelium-denuded arteries. In addition, responses to 50 μmol/l of BMS or diazoxide were evaluated in endothelium-denuded arteries pretreated with 100 μmol/l manganese(III) tetrakis(4-benzoic acid) porphyrin chloride (MnTBAP; a cell-permeant SOD mimetic) (1, 34, 37), and 100 nmol/l iberiotoxin (a selective inhibitor of BKCa channels) (1, 59).
Mitochondrial membrane potential measurements.
Mitochondrial membrane potential was determined by using 100 nmol/l tetramethylrhodamine ethyl ester (TMRE; λex: 532 nm, λem: >550 nm long pass filter). TMRE (1 mg/ml) stock solution was prepared in methanol and a final solution (100 nmol/l) prepared in phenol-free DMEM with 10 mmol/l HEPES was used to load the arteries for 15 min at 37°C. Fluorescence images were acquired both before and after application of vehicle (DMSO) or BMS (50 μmol/l) or diazoxide (100 μmol/l) for TMRE fluorescence measurements. Imaging conditions such as gain levels and laser power were held constant. Imaging studies of Zucker rats were performed with paired ZO and ZL arteries in an alternating sequence. Offline analysis of images to determine the average pixel intensity of smooth muscle cells in each field (n = 20–30) was performed using ImageJ software (NIH, Bethesda, MD), and the results were expressed in relative fluorescence units (RFUs). Fluorescence measurements in RFUs were expressed as percent change from the baseline images before administration of vehicle or BMS or diazoxide. Representation of TMRE data as percent change from baseline is consistent with previous reports and the concentration of TMRE used. The n value represents the average fluorescence intensity of all the cells from the number of arteries from each treatment group. We used two different approaches for the color presentation of the representative images to aid the reader in the assessment of the data.
Mitochondrial ROS measurements.
Mitochondrial ROS, specifically superoxide, was determined by using 5 μmol/l MitoSOX (λex: 405 nm, λem: >550 nm), based on the method reported by Robinson et al. (52). MitoSOX (5 mmol/l) stock solution was prepared in DMSO, and final solution prepared in phenol-free DMEM with 10 mmol/l HEPES was used to load the arteries for 15 min at 37°C. MitoSOX fluorescence was captured by Leica SP2 AOB laser confocal microscope with a C-Apochromat 63×/NA 1.2 oil immersion objective. We also used an alternate method for measuring MitoSOX fluorescence with λexcitation of 405 nm and λemission >550 nm. Fluorescence images were acquired both before and after application of vehicle (DMSO) or 50 μmol/l BMS or 100 μmol/l diazoxide, maintaining imaging conditions such as gain levels and laser power constant. Offline analysis of images was performed using ImageJ software (NIH). Fluorescence images were captured from each artery at an average 6–9 nonoverlapping fields of view with each containing 40–50 VSM cells. Average MitoSOX fluorescence measurements were determined by choosing region of interest (ROI) of all the VSM cells in each image. The average fluorescence measurements from all arterial segments were expressed as means ± SE RFU for each treatment.
Calcium spark imaging.
Fluo-4 AM (λex: 488 nm, λem: 505 nm long pass filter) was used to study [Ca2+]i. The arteries were loaded in the dark with a 1:1 mixture of 5 μmol/l Fluo-4 AM and 20% (wt/vol) pluronic F-127 diluted in HEPES-buffered PSS (in mmol/l) of 134 NaCl, 6 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 0.026 EDTA, and 10 glucose; pH 7.4, NaOH for 60 min at room temperature before cannulation as previously described (59). Confocal microscopy was performed using a laser scanning confocal system (7 Live; Zeiss, Jena, Germany) attached to an inverted microscope with a Zeiss C-Apochromat 63×/NA 1.2 water immersion objective. A 512×512 pixel area was imaged at 50–60 frames/s using the identical laser power attenuation and settings for all experiments. Collected images were analyzed using LC_Pro plugin to ImageJ (NIH), developed by Michael Francis and Mark S. Taylor (University of South Alabama, Mobile, AL). Each spark site was analyzed as a ROI (25 pixels2). A minimum F/F0 of 1.2 was required for an event to be considered a spark. Each image contained 15–25 cells, and spark frequency and amplitude were averaged for all ROIs in all visible cells to provide a single value for each of these parameters for each artery (22, 29). These values were then averaged for all arteries in the study.
Electron spin resonance studies.
Our previous studies using dihydroethidine and MitoSOX fluorescence in endothelial cells (36) and neurons (23, 24) have clearly demonstrated that diazoxide induces ROS generation, whereas BMS fails to promote ROS generation. In the present study, electron spin resonance (ESR) spectroscopy was used to provide additional evidence of diazoxide-induced ROS generation and lack of ROS induction in response to BMS. Endothelium denuded aortic segments were used instead of cerebral arteries because of the greater quantity of tissue required for the ESR experiments. We believe that VSM cells in the endothelium denuded aorta are comparable with the VSM cells from cerebral arteries in terms of mitochondrial responses (36). ESR spectroscopy measurements of superoxide in aortic homogenates in place of cerebral arteries have been reported in animals with cerebrovascular oxidative stress (40). Arterial ROS production was measured using ESR as described previously (14) using the spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethyl-pyrrolidine (CMH). Diethyldithiocarbamate (DETC; 2.5 μmol/l) and 25 μmol/l desferoxamine were dissolved under nitrogen gas bubbling in ice-cold modified Krebs-Hepes (KH) buffer containing (in mmol/l) 99.01 NaCl, 4.69 KCl, 1.87 CaCl2, 1.20 MgSO4, 25 NaHCO3, 1.03 K2HPO4, 20 sodium-HEPES, and 11.1 d-glucose, at pH 7.35. Thoracic aortas from SD rats were rapidly isolated and removed from animals euthanized under deep anesthesia. The aortas were cleaned of excessive adventitial tissues and fat in cold PSS as previously described. Aortic endothelium was denuded by gently rubbing with a cotton tip. Subsequently, 3–5 mm segments of aorta were cut and 2 segments were placed in a single well of a 96-well plate containing 150 μl/well freshly made 200 μmol/l CMH solution of KH buffer containing DETC and desferoxamine. Later, the aortic segments were treated with vehicle (DMSO) or 50 μmol/l BMS-191095 or 100 μmol/l diazoxide in the presence and absence of polyethylene glycol-SOD (200 units/ml; a ROS scavenger) or rotenone + antimycin A (1 μmol/l each; respiratory complex I and III inhibitors, respectively) and incubated at 37°C for 60 min. Aliquots of incubated CMH containing KH buffer solutions were then taken in 50 μl disposable glass capillary tubes (Noxygen Science Transfer and Diagnostics) for determination of ROS production. ESR measurements were performed using an EMX ESR eScan BenchTop spectrometer (Bruker, Germany). Time-dependent formation of ROS was determined using ESR settings: center field, 1.99 g; microwave power, 20 mW; modulation amplitude, 2 G; sweep time, 10 s; number of scans, 10; and field sweep, 60 G. The amplitude measurements of the ESR spectra (arbitrary units) were normalized to the dry weight of the corresponding aortic segments of each treatment. N represents the number of experiments (2 aortic segments each) that include two experiments per treatment per each rat.
Western blot analysis.
Proteins were harvested by placing the isolated cerebral arteries (anterior, middle, posterior cerebral, and basilar arteries) from Zucker rats in ice-cold NP40 lysis buffer (Invitrogen) supplemented with proteinase inhibitor cocktail (Cat No. P8340; Sigma) and phosphatase inhibitor cocktail (Cat No. P2850; Sigma) (each 5 μl/ml). Cell lysates were incubated with SDS/β-mercaptoethanol sample buffer at 100°C for 5 min. Protein samples were separated by electrophoresis on a 4–20% SDS-PAGE gradient gel, and proteins were transferred onto a polyvinylidene difluoride (0.22 μm) or nitrocellulose membrane. Membranes were then incubated in a blocking buffer (Tris-buffered saline, 0.1% Tween 20, and 1% skimmed milk powder) for 1 h at room temperature followed by incubation with primary antibodies against markers of endoplasmic reticulum (ER) stress, overnight at 4°C in the blocking solution. Antibodies against total and phosphorylated eukaryotic initiation factor 2α (38 kDa, 1:400; Cell Signaling), 78 kDa glucose regulated protein also known as Binding Protein (1.1,000; Cell Signaling), C/EBP-homologous protein that inhibits C/EBP (27 kDa, 1:1,000; Cell Signaling), and X box binding protein 1 (42 kDa, 1:1,000; abcam) were used. The membranes were washed three times in Tris-buffered saline with 0.1% Tween 20 and incubated for 1 h in the blocking buffer with goat anti-rabbit IgG (1:5,000; Santa Cruz) or goat anti-mouse IgG (1:5,000; Santa Cruz) conjugated to horseradish peroxidase. The final reaction products were visualized using enhanced chemiluminescence (SuperSignal West Pico; Pierce, Rockford, IL) and developed on an X-ray film. For quantitative analysis, the bands were scanned and band densities were quantified using ImageJ 1.3.1 software. The band intensities were normalized to that of β-actin.
Drugs, chemicals and solutions.
All chemicals were purchased from Sigma (St. Louis, MO) except MnTBAP (Calbiochem, San Diego, CA), DETC (Noxygen, Elzach, Germany), TMRE, MitoSOX, Fluo-4 AM, Pluronic F-127 (Molecular Probes, Eugene, OR), and DMEM (Gibco, Grand Island, NY).
Data analysis and statistics.
Results were expressed as means ± SE; n indicates the number of independent experiments. Means were compared by one-way ANOVA. Post hoc analysis was done by Tukey's test. P <0.05 was considered as statistically significant.
RESULTS
Mitochondrial depolarization.
Treatment with BMS depolarized the mitochondria of VSM cells from SD rats indicated by reduction of TMRE fluorescence compared with vehicle-treated arteries (Fig. 1, A and B). The decrease in TMRE fluorescence (percent change from baseline; 1.00) in VSM cells was 23.4 ± 5.3% in response to 50 μmol/l BMS (n = 8; P < 0.05) vs. 0.99 ± 0.9% in response to vehicle (n = 7).
Fig. 1.

BMS-191095 (BMS) induced mitochondrial-depolarization and vasodilation. Mitochondrial depolarization and vasodilation in response to BMS in endothelium-denuded cerebral arteries from SD rats are shown. A: typical images of tetramethylrhodamine ethyl ester (TMRE) fluorescence in the presence of vehicle (DMSO; n = 7 arteries) and BMS (50 μmol/l; n = 8 arteries) showing decreased TMRE fluorescence in response to BMS vs. vehicle indicating mitochondrial depolarization. B: cumulative data of percent change from TMRE fluorescence at baseline before the application of BMS/vehicle are represented in the bar graph. C: vascular responses to BMS (10, 50, and 100 μmol/l) showing dose-dependent increase in vasodilation to BMS (n = 6–14 arteries). *Significant difference in vasodilation to BMS response (P < 0.05) compared with vehicle treatment, which had no effect arterial diameter. D: vascular responses to BMS (50 μmol/l) in the presence of iberiotoxin (n = 6 arteries) and MnTBAP (n = 5 arteries). Data are presented as means ± SE. *Significant difference in corresponding baseline BMS response (P < 0.05).
Intraluminal diameter measurements.
The resting diameters of the cerebral arteries from SD rats were similar for each group of experiments (154 ± 6, n = 17, in endothelium-denuded; 142 ± 19, n = 6 in iberiotoxin treated; and 142 ± 15, n = 5 in MnTBAP treated), and they were constricted to a similar degree (51 ± 4%, n = 17, in endothelium-denuded; 56 ± 2% n = 6 in iberiotoxin treated; and 52 ± 4%, n = 5 in MnTBAP treated) upon pressurization.
The BMS elicited a dose-dependent vasodilation in endothelium denuded cerebral arteries with 8.1 ± 2.3%, 31.6 ± 2.1%, and 39.5 ± 3.2% relaxation in response to 10, 50, and 100 μmol/l, respectively (n = 6–14; P < 0.05) (Fig. 1C). Scavenging of the ROS with MnTBAP did not affect vasodilation to 50 μmol/l BMS (34.9 ± 6.7%, n = 5) confirming that BMS-induced vasodilation was independent of ROS generation (Fig. 1D). Inhibition of BKCa channels with iberiotoxin decreased vasodilation to 50 μmol/l BMS in endothelium-denuded arteries (17.7 ± 1.9%, n = 6; P < 0.05) (Fig. 1D), suggesting that BKCa channels mediate approximately one-half of endothelium-independent vasodilation induced by BMS. The mechanisms underlying the mitochondria mediated vasodilation that is independent of BKCa channels have to be determined in future studies.
ROS measurements by ESR.
A characteristic ESR signal with three peaks (12, 13) was detected in rat aortas from SD rats incubated with CMH and various drugs. The magnitude of this signal (arbitrary units normalized to dry weight of tissue) was greatly increased by stimulation with diazoxide (1.69 × 106 ± 0.6 × 106, n = 17; P < 0.05) and rotenone+antimycin A combination (1.96 × 106 ± 0.27 × 106, n = 8; P < 0.05) compared with vehicle-treated aortas (0.56 × 106 ± 0.6 × 106, n = 15), indicating increased generation of ROS. However, treatment with BMS failed to enhance the ROS compared with vehicle-treated aortic segments (0.49 × 106 ± 0.57 × 106, n = 15), confirming that BMS-induced mitochondrial depolarization did not result in generation of ROS in vascular wall (Fig. 2, A and B). Cotreatment with cell-permeable ROS scavenger SOD diminished baseline (vehicle treated) (0.35 × 106 ± 0.61 × 106, n = 6; P < 0.05) and diazoxide-induced (0.73 × 106 ± 0.55 × 106, n = 8; P < 0.05) increase in ESR signal amplitude confirming the generation of superoxide. However, ESR signal amplitude was unchanged by SOD and BMS cotreatment (0.33 × 106 ± 0.43 × 106, n = 7; P = not significant), suggesting that BMS did not promote superoxide generation in the vascular wall (Fig, 2B).
Fig. 2.

Mitochondrial depolarization with and without reactive oxygen species (ROS) generation. A: representative electron spin resonance (ESR) spectra of SD rat aortas incubated for 60 min at 37°C with 200 μmol/l 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethyl-pyrrolidine (CMH), a spin trap for superoxide radical. The vertical axis represents signal intensity in arbitrary units, and the horizontal axis represents the magnetic field (G). A characteristic CMH signal with 3 peaks was detected in rat aortas incubated with CMH and various drugs. Representative ESR spectra of the basal (vehicle treated, n = 7), 50 μmol/l BMS (n = 7), and 100 μmol/l diazoxide (n = 9) are shown in A. B: cumulative data normalized to the dry weight of aortic segments and expressed as arbitrary units as a bar graph are shown. Cotreatment with ROS scavenger SOD reduced the ESR signal amplitude in response to vehicle and diazoxide. Mitochondrial superoxide generation following treatment with the combination of antimycin A and rotenone (inhibitors of respiratory chain complexes) was used as positive control to confirm the superoxide generation. An n represents the number of experiments (2 aortic segments each) that include 2 experiments per treatment per each rat. C: fluorescence images of endothelium-denuded cerebral arteries loaded with MitoSOX, a fluoroprobe for mitochondrial superoxide, are shown. Typical spindle-shaped MitoSOX fluorescence with a central hollow space is seen in the smooth muscle cell around the fluorescence free nucleus. D: bar graph of cumulative MitoSOX fluorescence data. Diazoxide (100 μmol/l) treatment enhanced MitoSOX fluorescence in the vascular smooth muscle (VSM) cells of endothelium-denuded cerebral arteries vs. vehicle (DMSO) treatment indicating increased generation of mitochondrial superoxide. However, treatment with BMS-19195 did not increase MitoSOX fluorescence in VSM cells, indicating that mitochondrial depolarization by BMS-191095 was not accompanied by generation of superoxide from mitochondria. The n value represents average fluorescence intensity of all the cells from n number of arteries that were treated with each treatment. *Significant difference in corresponding response following vehicle treatment (P < 0.05).
Mitochondrial ROS generation.
We observed that MitoSOX fluorescence was uniform along the length of the arterial segment. Treatment with diazoxide, our positive control, increased MitoSOX fluorescence from the mitochondria of VSM cells, indicating increased mitochondrial ROS generation compared with vehicle-treated arteries (Fig. 2, C and D). In contrast, MitoSOX fluorescence was not changed by BMS when compared with vehicle-treated arteries. MitoSOX fluorescence in cerebral artery segments expressed as RFU was 71 ± 3 in response to 100 μmol/l diazoxide (n = 5; P < 0.05) and 40 ± 2 in response to 50 μmol/l BMS (n = 5) vs. 42 ± 1 in response to vehicle-treated arterial segments (n = 4). N represents the number of arterial segments.
Calcium spark generation.
To determine whether mitochondrial depolarization activates calcium spark generation independent of ROS, arteries from SD rats were treated with ROS-independent BMS or ROS-dependent diazoxide and calcium spark frequency was assessed. Representative traces of calcium spark activity under basal (vehicle) and BMS-stimulated conditions are shown in Fig. 3A. Both diazoxide and BMS robustly increased the frequency of calcium sparks in VSM cells. Calcium spark frequency (in sparks/cell/s) were 0.08 ± 0.01 at basal levels (vehicle, n = 6 arteries) compared with 0.22 ± 0.03 in diazoxide (n = 8 arteries; P < 0.05) and 0.26 ± 0.02 in BMS (n = 8 arteries; P < 0.05)-treated arteries (Fig. 3B). N represents the number of arterial segments, and 1 to 2 arterial segments from each rat was used for each experiment.
Fig. 3.
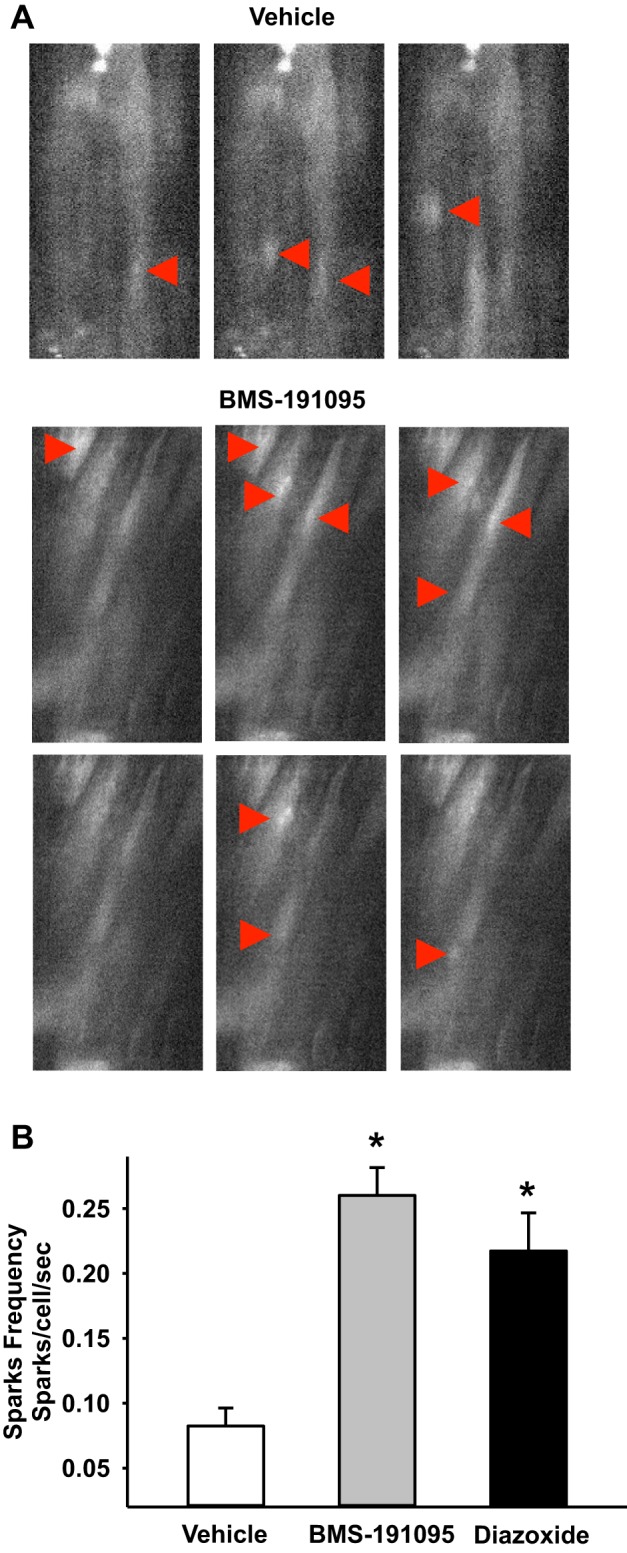
Calcium sparks generation in response to BMS-191095 and diazoxide. A: selected fluorescence images of VSM cells loaded with 5 μmol/l Fluo-4AM from time series image stacks are shown. Region of interest sites of calcium sparks are shown (arrow heads) from vehicle (DMSO) and BMS (50 μmol/l)-treated endothelium-denuded cerebral arteries from SD rats. B: bar graph showing the cumulative data of calcium sparks frequency in response to vehicle, diazoxide, and BMS. *Significant difference in response vs. vehicle (P < 0.05).
ROS-independent effects of mitochondrial depolarization in ZO arteries.
Our previous studies demonstrated impaired diazoxide-induced mitochondrial depolarization dependent on ROS generation (35). To determine whether mitochondrial depolarization independent of ROS generation is altered by insulin resistance, we determined the responses to BMS in the cerebral arteries of Zucker rats. BMS-induced mitochondrial depolarization was diminished in VSM cells from ZO arteries compared with ZL arteries similar to diazoxide (Fig. 4A). Mitochondrial depolarization indicated by the decrease in TMRE fluorescence (in percentage) was 10.7 ± 2, 12.1 ± 0.5, and 18.5 ± 0.5 in ZL arteries in response to 25 μmol/l BMS, 50 μmol/l BMS, and 100 μmol/l diazoxide, respectively (n = 4, 1 artery per animal). In contrast, the decrease in TMRE fluorescence (in percentage) was 0.92 ± 0.7, 3.7 ± 1.5, and 9.2 ± 1.3 in ZO arteries in response to 25 μmol/l BMS, 50 μmol/l BMS, and 100 μmol/l diazoxide, respectively (Fig. 4B). However, consistent with our previous observations (35) baseline TMRE fluorescence was similar in both ZO and ZL arteries (data not shown).
Fig. 4.
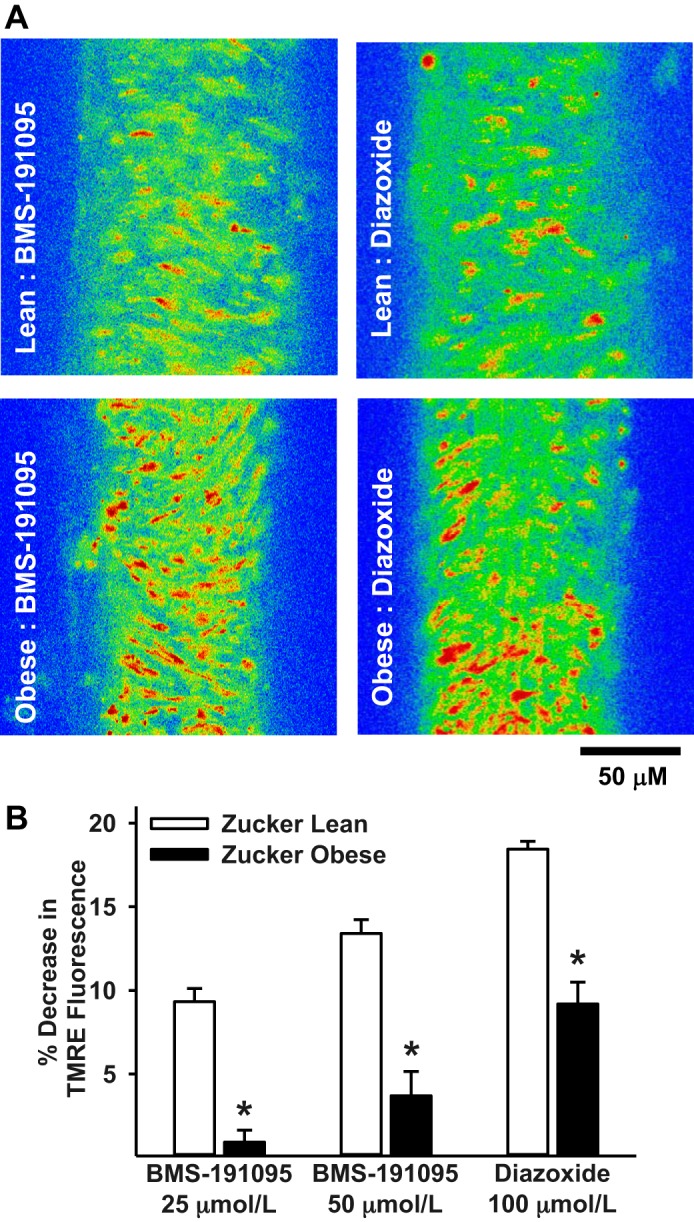
Mitochondrial depolarization in Zucker rat arteries. A: representative images of VSM cells loaded with TMRE are shown from BMS (50 μmol/l) and diazoxide (100 μmol/l) treated cerebral arteries of Zuker lean (ZL) and Zucker obese (ZO) rats. The color range from red to yellow indicates the range of TMRE fluorescence intensity from fully polarized (red) to depolarized (yellow) mitochondria. BMS and diazoxide elicited robust mitochondrial depolarization in ZL arteries indicated by greater yellow relative to red. In contrast, BMS and diazoxide elicited relatively diminished mitochondrial depolarization in ZO arteries compared with ZL arteries, indicated by less yellow relative to red suggestive of impaired mitochondrial depolarization. B: bar graph showing cumulative data of percent decrease in TMRE fluorescence in response to BMS (25 and 50 μmol/l) and diazoxide (100 μmol/l) from baseline before the application of drugs. Data are means ± SE of 6–14 experiments. *Significant difference in response to corresponding treatment in ZL arteries (P < 0.05). An n represents the number of arterial segments and single arterial segment per experiment from each rat was used.
To determine whether impaired mitochondrial depolarization independent of ROS generation elicits diminished vasodilation in cerebral arteries from ZO rats, we compared the vasodilator responses to BMS with those to diazoxide in arteries from Zucker rats. BMS-induced vasodilation was diminished in endothelium denuded arteries from ZO arteries compared with ZL arteries similar to diazoxide (Fig. 5, A and B). BMS induced an increase in diameter (in μm) from 105 ± 5.6 at baseline (preconstricted) to 156 ± 10 in ZL arteries (n = 9; P < 0.05). However, the BMS-induced change in diameter was diminished from 105 ± 2 at a preconstricted level to 121 ± 2.8 in ZO arteries (n = 9; P < 0.05). Similarly, diazoxide-induced an increase in diameter (in μm) from 110 ± 6.4 at a preconstricted level to 149 ± 9 in ZL arteries (n = 9; P < 0.05). In contrast, the diazoxide-induced change in diameter was diminished from 104 ± 8 at preconstricted level to 119 ± 2.8 in ZO arteries (n = 11). However, both BMS and diazoxide-induced diameters increases in ZO arteries were significantly decreased when compared with responses in ZL arteries (P < 0.05; Fig. 5, A and B). N represents the number of arterial segments and single arterial segment from each rat was used for the experiment.
Fig. 5.
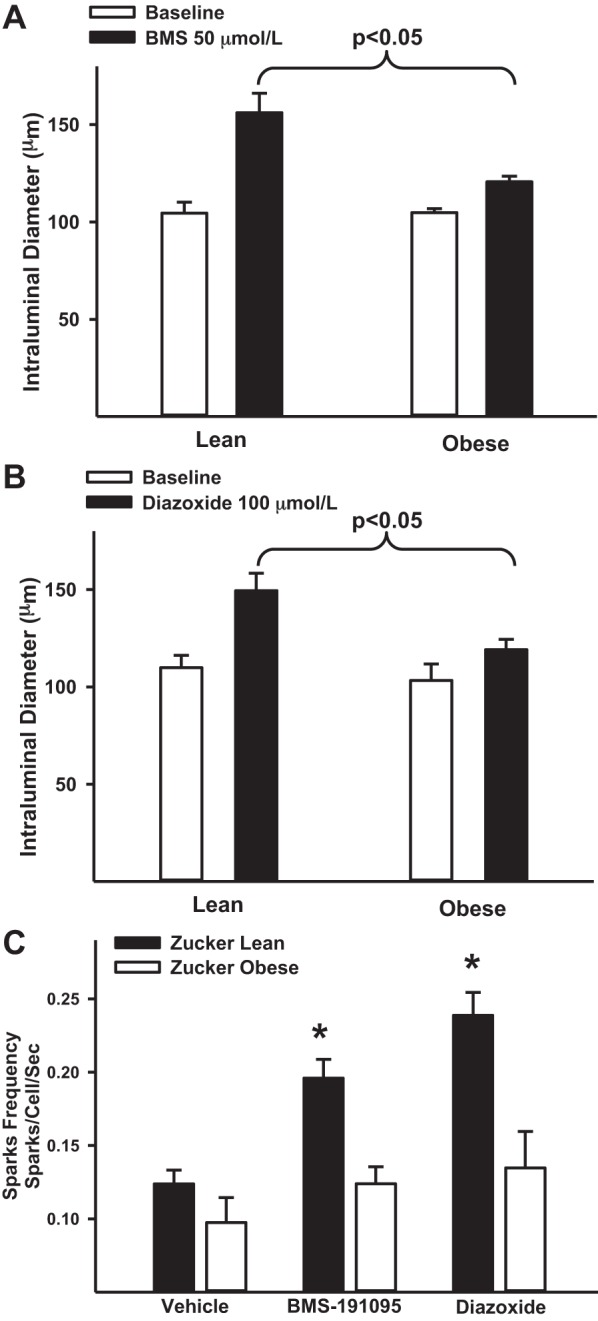
Mitochondria-mediated vasodilation in Zucker rats. A: diameter measurements in response to BMS (50 μmol/l) in endothelium-denuded cerebral arteries of ZL and ZO rats are shown. B: diameter measurements in response to diazoxide (100 μmol/l) in endothelium-denuded cerebral arteries of ZO and ZL are shown. C: bar graph showing cumulative data of calcium sparks frequency in response to vehicle, BMS, and diazoxide in ZL and ZO arteries. *Significant difference in response vs. corresponding response in ZL arteries (P < 0.05). An n represents the number of arterial segments and single arterial segment per experiment from each rat was used.
To determine whether mitochondrial depolarization independent of ROS generation elicits diminished calcium spark generation in VSM cells of cerebral arteries from ZO rats, we compared the calcium spark generation in response to BMS with that of diazoxide in arteries from Zucker rats. Both BMS and diazoxide promoted increased frequency of calcium sparks in VSM cells from ZL arteries. Calcium spark frequency increased from 0.12 ± 0.01 at basal levels (vehicle, n = 9) to 0.2 ± 0.01 in diazoxide (n = 9; P < 0.05) and 0.24 ± 0.02 in BMS (n = 4; P < 0.05)-treated ZL arteries (Fig. 5C). In contrast, calcium spark frequency did not significantly increase in VSM cells from ZO arteries. Calcium spark frequency was 0.12 ± 0.01 at basal levels (n = 9), 0.134 ± 0.03 following diazoxide (n = 8) and 0.124 ± 0.01 in response to BMS (n = 5) in ZO arteries (Fig. 5C). N represents the number of arterial segments and single arterial segment from each rat was used for the experiment. Thus, similar to diazoxide, BMS-induced mitochondrial depolarization failed to elicit an increase in calcium spark generation in ZO arteries with impaired mitoKATP channel function.
ZO arteries exhibit ER stress.
Immunoblot analysis of various markers of ER stress showed increased phosphorylated and total eukaryotic initiation factor 2α ratio and protein levels of binding protein/78 kDa glucose regulated protein, C/EBP-homologous protein that inhibits C/EBP, and X box binding protein 1 in ZO arteries compared with ZL arteries (Fig. 6).
Fig. 6.
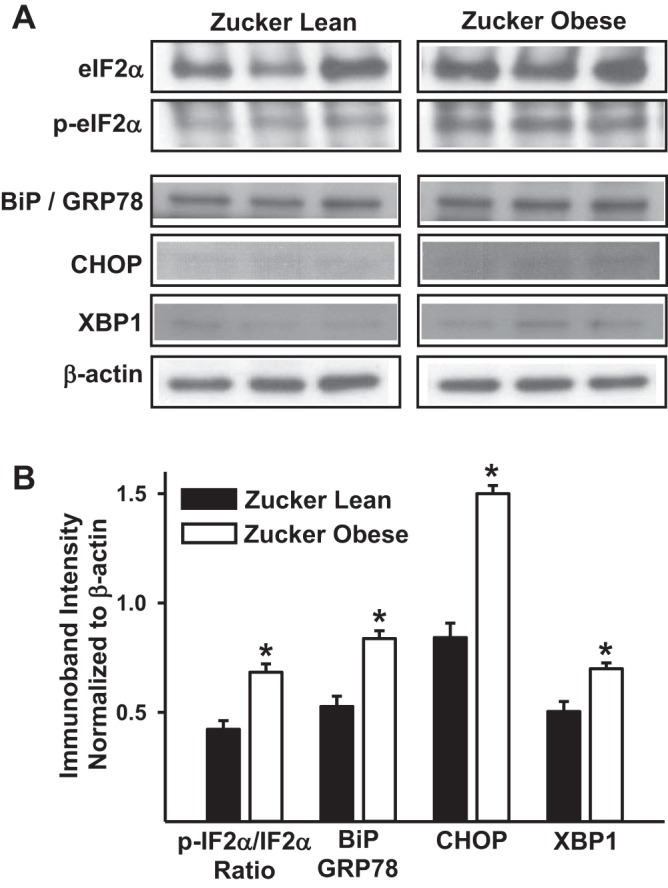
Increased levels of endoplasmic reticulum (ER) stress markers in cerebral arteries from Zucker rats. A: representative images of immunoblots determining the total and phosphorylated binding protein/78 kDa glucose regulated protein (BiP/GRP78), C/EBP-homologous protein that inhibits C/EBP (CHOP), and X box binding protein 1 (XBP1) in the cellular lysates from the cerebral arteries of ZL and ZO rats are shown. B: bar graphs showing cumulative data of immunoband intensity normalized to β-actin in arbitrary units are shown. *Significant differences in immunoband intensity of given protein compared with the immunoband intensity the same protein in ZL arteries (P < 0.05; n = 6 animals per group). IF2α, initiation factor 2α.
DISCUSSION
The novel findings from our studies establish an important new concept that both ROS-independent and ROS-dependent mechanisms can mediate the mitochondrial depolarization-induced generation of calcium sparks in cerebral VSM and subsequent vasodilation. First, VSM mitochondrial depolarization induced by BMS, which is not associated with ROS generation, elicits enhanced calcium spark activity and vasodilation of endothelium-denuded cerebral arteries. Second, both ROS-independent and -dependent vasodilation induced by the mitochondrial depolarization is mediated by activation of BKCa channels in the VSM. Third, vascular mitochondrial dysfunction accompanying insulin resistance displays impaired mitochondrial depolarization, reduced calcium spark activity, and associated attenuated vasodilation to both diazoxide and BMS, thereby demonstrating shared signaling pathways. Thus our findings demonstrate that 1) increased calcium spark frequency subsequent to mitochondrial depolarization can occur without ROS production and 2) pathways linking and/or impairing mitochondrial activation, calcium sparks, and vasodilation in health and disease are diverse.
Mitochondrial depolarization not accompanied by ROS generation.
Studies in our laboratory and others have demonstrated the role of activation of mitoKATP channels in mediating the effects of diazoxide (39, 51, 59) and BMS (2, 6, 24, 26, 27, 34, 36, 38, 42, 45) in isolated mitochondria, neurons, endothelium, vasculature, and cardiomyocytes. Scavenging of ROS failed to reduce BMS-induced vasodilation in endothelium-denuded arteries, and measurements of mitochondrial or cellular ROS failed to demonstrate an increase in ROS with BMS. Thus the ability of mitochondrial depolarization by BMS to promote vasodilation independent of ROS generation in VSM is consistent with our previous observations concerning the production of vasoactive factors in cerebral endothelium (36). Furthermore, our laboratory has characterized the distinct ROS-independent mechanisms of action of BMS compared with diazoxide in isolated mitochondria and cultured cortical neurons, cultured endothelial cells, and endothelium intact arteries (6, 24, 36). The present study has confirmed our previous findings of ROS-independent actions of BMS in promoting mitochondria-mediated vasodilation via increased frequency of calcium sparks in VSM. Direct measurements of mitochondrial ROS by MitoSOX fluorescence from cerebral VSM cells has confirmed that mitochondrial depolarization, unlike diazoxide, does not result in ROS generation. Furthermore, measurements of ROS by ESR have provided additional supportive evidence of the inability of BMS, unlike diazoxide, to promote ROS generation in vascular tissues. Because regulation of ROS generation and the sources of ROS vary depending on the species, tissues, and cell types studied, we confirmed the inability of BMS to promote ROS production in VSM cells by using complementary methods of ESR spectroscopy and fluorescence microscopy. Consistent with a previous report by Jaggar et al. (59), we observed increased mitochondrial and cellular ROS generation in response to diazoxide in our MitoSOX fluorescence and ESR experiments.
BKCa channels and calcium sparks.
BKCa channels have been shown by us (34) and others (59) to mediate part of the mitochondria-induced vasodilation. The mechanisms contributing to the remaining mitochondria-mediated vasodilation are still not completely known. Inhibition of BKCa channels diminished vasodilation to BMS in endothelium-denuded arteries, indicating that part of the mitochondria-mediated vasodilation was BKCa channel dependent. This finding is consistent with previous observations from us and others, which showed that diazoxide-induced vasodilation partly involved BKCa channels (34, 36, 59). Interestingly, previous studies in our laboratory failed to detect the activation of whole cell K+ currents in isolated VSM cells in response to BMS-191095 (42). It is possible that BKCa channels were more readily activated in the isolated pressurized arteries used in our study than those in patch clamp experiments in which VSM cells were isolated by enzymatic digestion. Myogenic response in cerebral arteries has been shown to be regulated by BKCa channels through hyperpolarization of VSM membrane potential and inactivation of voltage-gated calcium channels (25, 30, 46). Generation of calcium sparks is the primary mechanism of activation of cerebral artery BKCa channels; however, the exact mechanisms underlying the increased frequency of calcium sparks in VSM cells are not fully understood. Recent evidence has implicated mitochondrial depolarization and subsequent ROS formation in increased generation of calcium sparks via redox regulation of RyR channels (59). Consistent with previous reports, we observed that mitochondrial depolarization by diazoxide enhanced generation of calcium sparks in VSM cells of endothelium-denuded arteries. In addition, we observed that mitochondrial depolarization by BMS was capable of enhancing the generation of calcium sparks in VSM cells independent of ROS. Thus BMS and diazoxide share many vascular actions including depolarization of VSM mitochondria, increased frequency of calcium sparks, activation of BKCa channels, and promotion of vasodilation in cerebral arteries (Fig. 6). However, the exact mechanism by which mitochondrial depolarization independent of ROS activates calcium sparks is not clear and needs further investigation. Many studies have identified close physical and functional communication between ER/sarcoplasmic reticulum (SR) and mitochondria, which facilitates the transfer of ions, nucleotides, radicals, and yet unknown factors that aid in the bidirectional regulation of organelle (15, 41). We speculated that close SR-mitochondrial communication may facilitate mitochondrial depolarization to activate RyR through a mechanism that involves electrical coupling of the organelle (Fig. 7). Further studies are needed to identify the nature of interorganelle communication leading to the increased frequency of calcium sparks.
Fig. 7.
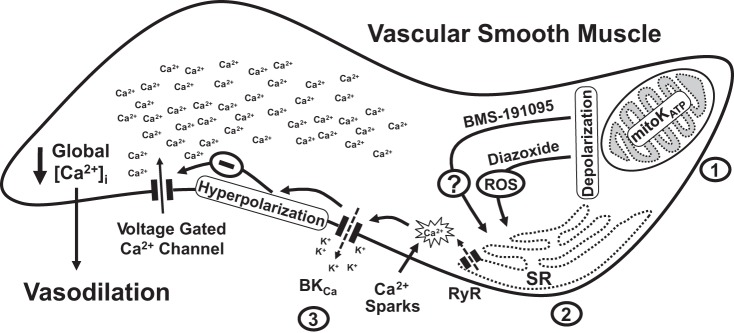
Mechanisms of mitochondrial-depolarization-induced vasodilation. A schematic of the proposed mechanisms underlying the vasodilation induced by the mitochondrial depolarization in the VSM is shown. BMS and diazoxide cause mitochondrial depolarization by activating mitochondrial KATP-sensitive (mitoKATP) channels. Depolarization of mitochondria leads to generation of calcium sparks by activation of ryanodine-sensitive calcium channels (RyR) in the sarcoplasmic reticulum (SR) in microdomains adjacent to mitochondria. Calcium sparks generation in response to diazoxide is ROS dependent. However, an unknown ROS-independent mechanism mediates calcium spark generation in response to BMS-191095. Generation of calcium sparks results in localized elevation of calcium as calcium transients lasting for a few milliseconds leading to activation of adjacent large-conductance calcium-activated potassium channels (BKCa). Potassium efflux through BKCa channels hyperpolarizes the VSM membrane resulting in inactivation of voltage-gated calcium channels leading to decreased global intracellular calcium ([Ca2+]i) and vasodilation. The likely sites affected by the insulin resistance in cerebral arteries of ZO rats are indicated by the numbers 1–3. 1, Previous findings from our studies demonstrated impaired activation of mitoKATP channels in ZO arteries resulting in impaired vasodilation to diazoxide (34). 2, Our current Western blot analysis revealed that ZO arteries display ER stress that may likely contribute to impaired calcium spark generation from intracellular calcium stores. 3, Our previous studies also demonstrated impaired activation of BKCa channels and plasma membrane ATP-sensitive potassium channels in cerebral arteries of ZO rats (16, 17).
ROS-independent mitochondria mediated vasodilation in insulin resistance.
We further evaluated the ROS-independent effects of mitochondrial depolarization by comparing the BMS and diazoxide induced mitochondrial depolarization, calcium spark generation, and vasodilation in cerebral arteries of ZO rats. Our previous studies have established impaired cerebrovascular responses to insulin (vascular insulin resistance) and characterized the underlying mechanisms in cerebral arteries of aged SD and ZO rats with metabolic insulin resistance (35). Moreover, we demonstrated impaired mitochondrial mediated vasodilation in response to diazoxide in the cerebral arteries from ZO rats, describing vascular mitochondrial dysfunction accompanying insulin resistance for the first time (34). Specifically, we observed impaired diazoxide-induced mitochondrial depolarization and subsequent ROS generation in ZO arteries (34). Our previous studies also demonstrated impaired activation of KATP and BKCa channels in cerebral arteries of ZO rats (17, 18, 34) and high fructose-fed rats (16) with insulin resistance. In the present study, similar to diazoxide, BMS elicited diminished mitochondrial depolarization, calcium spark generation, and vasodilation in ZO arteries compared with ZL arteries. The current studies confirm and extend our previous results showing that endothelium and VSM-dependent vasodilation to diazoxide was diminished in ZO arteries. Reduced mitochondrial depolarization mediated by mitochondrial KATP channels leading to impaired generation of mitochondrial ROS was found to be the cause of diminished diazoxide-mediated vasodilation in ZO arteries (34). The present studies also confirm and extended our previous observations that BKCa channels as well as other types of potassium channels are inhibited by insulin resistance and demonstrate that a reduction in diazoxide and BMS-induced enhanced generation of calcium sparks also contribute to impaired mitochondria-dependent vasodilation in ZO arteries. It is apparent that impaired activation of mitochondrial KATP channels by two mechanistically different activators resulted in identical deficits in vasodilation in ZO arteries, suggesting that shared signaling pathways mediate ROS-dependent and ROS-independent effects of mitochondrial depolarization. Furthermore, findings from ZO and ZL arteries validate the hypothesis that ROS-independent actions of BMS similar to ROS-dependent generation of calcium sparks rely on mitochondria-SR functional coupling.
Cerebral arteries from ZO rats also displayed increased levels of ER stress markers compared with that of ZL arteries. Many studies have demonstrated impaired calcium filling of ER/SR and abnormal calcium release from intracellular stores as a result of ER stress (3, 4). Although we have not measured ER/SR calcium dynamics in the arteries, ER stress in ZO arteries may likely contributed to the impaired calcium spark generation. However, further studies are needed to establish this potential mechanism of impaired mitochondrial-mediated calcium spark generation in ZO arteries.
Limitations.
Our studies used only pharmacological approaches to promote mitochondrial depolarization without ROS formation, and this approach reflects the stage of development of this field. Nonetheless, there are advantages to this approach. For example, we and others have extensively studied diazoxide and BMS in a variety of cell types and experimental conditions and we are aware of potential nonspecific effects. Diazoxide has been shown to exhibit some nontarget and nonspecific mitochondrial effects, especially at higher doses than those we used, and, despite thorough investigations, we are unaware of BMS causing any effects other than activation of mitochondrial KATP channels. Many physiological/pathological stimuli (hypoxia) (28, 53) that promote mitochondrial depolarization are similarly plagued by multiple sites of action. Unfortunately, there are no other pharmacological agents currently available to induce mitochondrial depolarization not accompanied by ROS generation other than BMS. In addition, the present studies provide evidence supporting ROS-independent communication between mitochondria and SR; however, the nature of this communication needs further investigation.
In summary, our study has revealed a novel ROS-independent signaling pathway mediating VSM mitochondrial-depolarization induced vasodilation in cerebral arteries, which involves the production of calcium sparks and subsequent activation of BKCa channels. Furthermore, we have shown that insulin resistance-induced VSM mitochondrial dysfunction elicits a similar but reduced level of mitochondrial depolarization and frequency of calcium sparks, which has underscored the essential role of mitochondria-SR interorganelle communication. Thus the present study provides the mechanistic evidence for impaired vascular mitochondrial function in insulin resistance that can potentially explain the impairments of neurovascular coupling observed in aging and T2DM.
GRANTS
This work was supported by the National Heart, Lung, and Blood Institute Grants HL-077731, HL-030260, HL-093554, and HL-065380 (to D. W. Busija).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: P.V.G.K. conception and design of research; P.V.G.K., A.O.G., and V.N.S. performed experiments; P.V.G.K., A.O.G., V.N.S., and I.R. analyzed data; P.V.G.K., I.R., and D.W.B. interpreted results of experiments; P.V.G.K. prepared figures; P.V.G.K. drafted manuscript; P.V.G.K., A.O.G., V.N.S., I.R., and D.W.B. edited and revised manuscript; P.V.G.K., A.O.G., V.N.S., I.R., and D.W.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Nancy Busija for editing the manuscript. We also thank Dr. Mark S. Taylor and Dr. Michael Francis for providing the LC_Pro plugin to ImageJ to help analyze calcium sparks data.
REFERENCES
- 1.Adebiyi A, McNally EM, Jaggar JH. Sulfonylurea receptor-dependent and -independent pathways mediate vasodilation induced by ATP-sensitive K+ channel openers. Mol Pharmacol 74: 736–743, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmad N, Wang Y, Haider KH, Wang B, Pasha Z, Uzun O, Ashraf M. Cardiac protection by mitoKATP channels is dependent on Akt translocation from cytosol to mitochondria during late preconditioning. Am J Physiol Heart Circ Physiol 290: H2402–H2408, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Alder NN, Shen Y, Brodsky JL, Hendershot LM, Johnson AE. The molecular mechanisms underlying BiP-mediated gating of the Sec61 translocon of the endoplasmic reticulum. J Cell Biol 168: 389–399, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bodalia A, Li H, Jackson MF. Loss of endoplasmic reticulum Ca2+ homeostasis: contribution to neuronal cell death during cerebral ischemia. Acta Pharmacol Sin 34: 49–59, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bray GA. The Zucker-fatty rat: a review. Fed Proc 36: 148–153, 1977 [PubMed] [Google Scholar]
- 6.Busija DW, Katakam P, Rajapakse NC, Kis B, Grover G, Domoki F, Bari F. Effects of ATP-sensitive potassium channel activators diazoxide and BMS-191095 on membrane potential and reactive oxygen species production in isolated piglet mitochondria. Brain Res Bull 66: 85–90, 2005 [DOI] [PubMed] [Google Scholar]
- 7.Cheranov SY, Jaggar JH. Mitochondrial modulation of Ca2+ sparks and transient KCa currents in smooth muscle cells of rat cerebral arteries. J Physiol 556: 755–771, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cholerton B, Baker LD, Craft S. Insulin resistance and pathological brain ageing. Diabet Med 28: 1463–1475, 2012 [DOI] [PubMed] [Google Scholar]
- 9.Coetzee WA. Multiplicity of effectors of the cardioprotective agent, diazoxide. Pharmacol Ther 140: 167–175, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Craft S. Insulin resistance syndrome and Alzheimer's disease: age- and obesity-related effects on memory, amyloid, and inflammation. Neurobiol Aging 26 Suppl 1: 65–69, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res 110: 1109–1124, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dikalov S, Fink B. ESR techniques for the detection of nitric oxide in vivo and in tissues. Methods Enzymol 396: 597–610, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Dikalov SI, Harrison DG. Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid Redox Signal 20: 372–382, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dikalov SI, Kirilyuk IA, Voinov M, Grigor′ev IA. EPR detection of cellular and mitochondrial superoxide using cyclic hydroxylamines. Free Radic Res 45: 417–430, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eisner V, Csordas G, Hajnoczky G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle—pivotal roles in Ca2+ and reactive oxygen species signaling. J Cell Sci 126: 2965–2978, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erdos B, Miller AW, Busija DW. Alterations in KATP and KCa channel function in cerebral arteries of insulin-resistant rats. Am J Physiol Heart Circ Physiol 283: H2472–H2477, 2002 [DOI] [PubMed] [Google Scholar]
- 17.Erdos B, Simandle SA, Snipes JA, Miller AW, Busija DW. Potassium channel dysfunction in cerebral arteries of insulin-resistant rats is mediated by reactive oxygen species. Stroke 35: 964–969, 2004 [DOI] [PubMed] [Google Scholar]
- 18.Erdos B, Snipes JA, Miller AW, Busija DW. Cerebrovascular dysfunction in Zucker obese rats is mediated by oxidative stress and protein kinase C. Diabetes 53: 1352–1359, 2004 [DOI] [PubMed] [Google Scholar]
- 19.Erdos B, Snipes JA, Tulbert CD, Katakam P, Miller AW, Busija DW. Rosuvastatin improves cerebrovascular function in Zucker obese rats by inhibiting NAD(P)H oxidase-dependent superoxide production. Am J Physiol Heart Circ Physiol 290: H1264–H1270, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Fabiani M, Gordon BA, Maclin EL, Pearson MA, Brumback-Peltz CR, Low KA, McAuley E, Sutton BP, Kramer AF, Gratton G. Neurovascular coupling in normal aging: a combined optical, ERP and fMRI study. Neuroimage 85: 592–607, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fisher-Wellman KH, Neufer PD. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol Metab 23: 142–153, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Francis M, Qian X, Charbel C, Ledoux J, Parker JC, Taylor MS. Automated region of interest analysis of dynamic Ca2+ signals in image sequences. Am J Physiol Cell Physiol 303: C236–C243, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaspar T, Domoki F, Lenti L, Institoris A, Snipes JA, Bari F, Busija DW. Neuroprotective effect of adenoviral catalase gene transfer in cortical neuronal cultures. Brain Res 1270: 1–9, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaspar T, Snipes JA, Busija AR, Kis B, Domoki F, Bari F, Busija DW. ROS-independent preconditioning in neurons via activation of mitoKATP channels by BMS-191095. J Cereb Blood Flow Metab 28: 1090–1103, 2008 [DOI] [PubMed] [Google Scholar]
- 25.Gollasch M, Lohn M, Furstenau M, Nelson MT, Luft FC, Haller H. Ca2+ channels, Ca2+ sparks, and regulation of arterial smooth muscle function. Z Kardiol 89 Suppl 2: 15–19, 2000 [DOI] [PubMed] [Google Scholar]
- 26.Grover GJ, D′Alonzo AJ, Darbenzio RB, Parham CS, Hess TA, Bathala MS. In vivo characterization of the mitochondrial selective KATP opener (3R)-trans-4-((4-chlorophenyl)-N-(1H-imidazol-2-ylmethyl)dimethyl-2H-1-ben zopyran-6-carbonitril monohydrochloride) (BMS-191095): cardioprotective, hemodynamic, and electrophysiological effects. J Pharmacol Exp Ther 303: 132–140, 2002 [DOI] [PubMed] [Google Scholar]
- 27.Grover GJ, D′Alonzo AJ, Garlid KD, Bajgar R, Lodge NJ, Sleph PG, Darbenzio RB, Hess TA, Smith MA, Paucek P, Atwal KS. Pharmacologic characterization of BMS-191095, a mitochondrial KATP opener with no peripheral vasodilator or cardiac action potential shortening activity. J Pharmacol Exp Ther 297: 1184–1192, 2001 [PubMed] [Google Scholar]
- 28.Gupte SA, Wolin MS. Oxidant and redox signaling in vascular oxygen sensing: implications for systemic and pulmonary hypertension. Antioxid Redox Signal 10: 1137–1152, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jackson-Weaver O, Osmond JM, Riddle MA, Naik JS, Gonzalez Bosc LV, Walker BR, Kanagy NL. Hydrogen sulfide dilates rat mesenteric arteries by activating endothelial large-conductance Ca2+-activated K+ channels and smooth muscle Ca2+ sparks. Am J Physiol Heart Circ Physiol 304: H1446–H1454, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. Am J Physiol Cell Physiol 278: C235–C256, 2000 [DOI] [PubMed] [Google Scholar]
- 31.Kalaria RN. Vascular basis for brain degeneration: faltering controls and risk factors for dementia. Nutr Rev 68 Suppl 2: S74–S87, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karagiannis J, Reid JJ, Darby I, Roche P, Rand MJ, Li CG. Impaired nitric oxide function in the basilar artery of the obese Zucker rat. J Cardiovasc Pharmacol 42: 497–505, 2003 [DOI] [PubMed] [Google Scholar]
- 33.Katakam PV, Domoki F, Lenti L, Gaspar T, Institoris A, Snipes JA, Busija DW. Cerebrovascular responses to insulin in rats. J Cereb Blood Flow Metab 29: 1955–1967, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katakam PV, Domoki F, Snipes JA, Busija AR, Jarajapu YP, Busija DW. Impaired mitochondria-dependent vasodilation in cerebral arteries of Zucker obese rats with insulin resistance. Am J Physiol Regul Integr Comp Physiol 296: R289–R298, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katakam PV, Snipes JA, Steed MM, Busija DW. Insulin-induced generation of reactive oxygen species and uncoupling of nitric oxide synthase underlie the cerebrovascular insulin resistance in obese rats. J Cereb Blood Flow Metab 32: 792–804, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katakam PV, Wappler EA, Katz PS, Rutkai I, Institoris A, Domoki F, Gaspar T, Grovenburg SM, Snipes JA, Busija DW. Depolarization of mitochondria in endothelial cells promotes cerebral artery vasodilation by activation of nitric oxide synthase. Arterioscler Thromb Vasc Biol 33: 752–759, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim EY, Anderson M, Dryer SE. Insulin increases surface expression of TRPC6 channels in podocytes: role of NADPH oxidases and reactive oxygen species. Am J Physiol Renal Physiol 302: F298–F307, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kis B, Nagy K, Snipes JA, Rajapakse NC, Horiguchi T, Grover GJ, Busija DW. The mitochondrial KATP channel opener BMS-191095 induces neuronal preconditioning. Neuroreport 15: 345–349, 2004 [DOI] [PubMed] [Google Scholar]
- 39.Kis B, Rajapakse NC, Snipes JA, Nagy K, Horiguchi T, Busija DW. Diazoxide induces delayed pre-conditioning in cultured rat cortical neurons. J Neurochem 87: 969–980, 2003 [DOI] [PubMed] [Google Scholar]
- 40.Lynch CM, Kinzenbaw DA, Chen X, Zhan S, Mezzetti E, Filosa J, Ergul A, Faulkner JL, Faraci FM, Didion SP. Nox2-derived superoxide contributes to cerebral vascular dysfunction in diet-induced obesity. Stroke 44: 3195–3201, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta 1837: 461–469, 2014 [DOI] [PubMed] [Google Scholar]
- 42.Mayanagi K, Gaspar T, Katakam PV, Kis B, Busija DW. The mitochondrial KATP channel opener BMS-191095 reduces neuronal damage after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab 27: 348–355, 2007 [DOI] [PubMed] [Google Scholar]
- 43.Miller AW, Dimitropoulou C, Han G, White RE, Busija DW, Carrier GO. Epoxyeicosatrienoic acid-induced relaxation is impaired in insulin resistance. Am J Physiol Heart Circ Physiol 281: H1524–H1531, 2001 [DOI] [PubMed] [Google Scholar]
- 44.Miller AW, Katakam PV, Lee HC, Tulbert CD, Busija DW, Weintraub NL. Arachidonic acid-induced vasodilation of rat small mesenteric arteries is lipoxygenase-dependent. J Pharmacol Exp Ther 304: 139–144, 2003 [DOI] [PubMed] [Google Scholar]
- 45.Neckar J, Szarszoi O, Koten L, Papousek F, Ost′adal B, Grover GJ, Kolar F. Effects of mitochondrial KATP modulators on cardioprotection induced by chronic high altitude hypoxia in rats. Cardiovasc Res 55: 567–575, 2002 [DOI] [PubMed] [Google Scholar]
- 46.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science 270: 633–637, 1995 [DOI] [PubMed] [Google Scholar]
- 47.Newsholme P, Gaudel C, Krause M. Mitochondria and diabetes. An intriguing pathogenetic role. Adv Exp Med Biol 942: 235–247, 2012 [DOI] [PubMed] [Google Scholar]
- 48.Oltman CL, Richou LL, Davidson EP, Coppey LJ, Lund DD, Yorek MA. Progression of coronary and mesenteric vascular dysfunction in Zucker obese and Zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol 291: H1780–H1787, 2006 [DOI] [PubMed] [Google Scholar]
- 49.Osmond JM, Mintz JD, Dalton B, Stepp DW. Obesity increases blood pressure, cerebral vascular remodeling, and severity of stroke in the Zucker rat. Hypertension 53: 381–386, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Phillips SA, Sylvester FA, Frisbee JC. Oxidant stress and constrictor reactivity impair cerebral artery dilation in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol 288: R522–R530, 2005 [DOI] [PubMed] [Google Scholar]
- 51.Rajapakse N, Shimizu K, Kis B, Snipes J, Lacza Z, Busija D. Activation of mitochondrial ATP-sensitive potassium channels prevents neuronal cell death after ischemia in neonatal rats. Neurosci Lett 327: 208–212, 2002 [DOI] [PubMed] [Google Scholar]
- 52.Robinson KM, Janes MS, Beckman JS. The selective detection of mitochondrial superoxide by live cell imaging. Nat Protoc 3: 941–947, 2008 [DOI] [PubMed] [Google Scholar]
- 53.Shimoda LA, Undem C. Interactions between calcium and reactive oxygen species in pulmonary arterial smooth muscle responses to hypoxia. Respir Physiol Neurobiol 174: 221–229, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stepp DW. Impact of obesity and insulin resistance on vasomotor tone: nitric oxide and beyond. Clin Exp Pharmacol Physiol 33: 407–414, 2006 [DOI] [PubMed] [Google Scholar]
- 55.Stepp DW, Pollock DM, Frisbee JC. Low-flow vascular remodeling in the metabolic syndrome X. Am J Physiol Heart Circ Physiol 286: H964–H970, 2004 [DOI] [PubMed] [Google Scholar]
- 56.Toth P, Tucsek Z, Sosnowska D, Gautam T, Mitschelen M, Tarantini S, Deak F, Koller A, Sonntag WE, Csiszar A, Ungvari Z. Age-related autoregulatory dysfunction and cerebromicrovascular injury in mice with angiotensin II-induced hypertension. J Cereb Blood Flow Metab 33: 1732–1742, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ungvari Z, Sonntag WE, Csiszar A. Mitochondria and aging in the vascular system. J Mol Med (Berl) 88: 1021–1027, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Williamson R, McNeilly A, Sutherland C. Insulin resistance in the brain: an old-age or new-age problem? Biochem Pharmacol 84: 737–745, 2012 [DOI] [PubMed] [Google Scholar]
- 59.Xi Q, Cheranov SY, Jaggar JH. Mitochondria-derived reactive oxygen species dilate cerebral arteries by activating Ca2+ sparks. Circ Res 97: 354–362, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang DX, Gutterman DD. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am J Physiol Heart Circ Physiol 292: H2023–H2031, 2007 [DOI] [PubMed] [Google Scholar]