

Abstract
We have recently shown that in vivo inhibition of histone deacetylase (HDAC) stimulates endogenous myocardial regeneration in infarcted hearts (Zhang L et al. J Pharmacol Exp Ther 341: 285–293, 2012). Furthermore, our observation demonstrates that HDAC inhibition promotes cardiogenesis, which is associated with HDAC4 reduction. However, it remains unknown as to whether specific inhibition of HDAC4 modulates cardiac stem cells (CSCs) to facilitate myocardial repair and to preserve cardiac performance. c-kit+ CSCs were isolated from adult mouse hearts and were transfected with HDAC4 siRNA to knockdown HDAC4 of c-kit+ CSCs. The transfection of HDAC4 siRNA caused a marked reduction of HDAC4 mRNA and proteins in c-kit+ CSCs. Mouse myocardial infarction (MI) was created to assess the effect of HDAC4 inhibition in c-kit+ CSCs on myocardial regeneration in vivo when cells were introduced into MI hearts. Transplantation of HDAC4 siRNA-treated c-kit+ CSCs into MI hearts improved ventricular function, attenuated ventricular remodeling, and promoted CSC-derived regeneration and neovascularization. Furthermore, Ki67 and BrdU positively proliferative myocytes increased in MI hearts receiving HDAC4 siRNA-treated c-kit+ CSCs compared with MI hearts engrafted with control siRNA-treated c-kit+ CSCs. In addition, compared with MI hearts engrafted with control adenoviral GFP-infected c-kit+ CSCs, MI hearts receiving adenoviral HDAC4-infected c-kit+ CSCs exhibited attenuated cardiac functional recovery, CSC-derived regeneration, and neovascularization, which was accompanied with adverse ventricular remodeling and decrease in Ki67 and BrdU positively proliferative myocytes. HDAC4 inhibition facilitated c-kit+ CSCs into the differentiation into cardiac lineage commitments in vitro, while HDAC4 overexpression attenuated c-kit+ CSC-derived cardiogenesis. Our results indicate that HDAC4 inhibition promotes CSC-derived cardiac regeneration and improves the restoration of cardiac function.
Keywords: heart, HDAC4, regeneration, myocardial infarction, stem cells
it has now been recognized that adult hearts harbor distinct populations of cardiac progenitors (2, 25, 26, 34), which have the potential to differentiate into cardiomyocytes, endothelia, and vascular smooth muscle cells. Furthermore, several studies have shown that these cardiac progenitor cells are capable of differentiating into cardiac tissue and improving cardiac function after a myocardial injury. Exponential advances in stem cell and regenerative biology are beginning to foster a transition toward therapeutic goals for cardiac regenerative medicine (8, 14, 15, 33). However, poor cell viability, proliferation, and inefficient differentiation following transplantation have limited the reparative capacity of these cells in vivo (26, 29, 39). Thus molecular intervention strategies to enhance cardiac stem cell (CSC) proliferation and survival hold dramatic consequences for enhancing myogenesis and will empower therapeutically relevant implementation of myocardial regeneration. It was reported that genetically modified stem cells could repair infarcted myocardium, prevent remodeling, and nearly normalize cardiac performance (10, 29). Two recent clinical trials on human studies suggest that intracoronary infusion of autologous CSCs or autologous cardiosphere-derived cells is effective in improving left ventricular (LV) systolic function and reducing infarct size in patients with heart failure (3, 28).
Histone acetyltransferases (HAT) and histone deacetylases (HDAC) emerged as important mechanisms in the regulation of a variety of cellular responses (13). HDAC4 is expressed in the heart, stem cells, the endothelium, and vascular smooth muscle cells (1, 4, 12, 19, 21). Cardioprotective effects of HDAC inhibition against injury have been well identified (45, 47, 48). Our recent observation demonstrates that HDAC inhibition enhanced myocardial repair in vivo through stimulation of endogenous regeneration (46), which is in line with our observation that HDAC inhibition facilitated the embryonic stem cells' differentiation into cardiac lineages and enhanced resistance to oxidant stress (4). However, the recent evidence has indicated that HDAC4-null mice display premature ossification of developing bones due to ectopic and early onset chondrocyte hypertrophy, but overexpression of HDAC4 in proliferating chondrocytes in vivo inhibits chondrocyte hypertrophy and differentiation (40). In addition, selective loss of HDAC4 in the brain results in impairments in hippocampal-dependent learning and memory and long-term synaptic plasticity (18). These observations may suggest that HDAC4 plays a different function in different disease models.
We noted that the decrease in HDAC4 was related to cardiac differentiation of embryonic stem cells (4), but the role of HDAC4 of CSCs in modulation of myocardial regeneration remains unknown. Results presented herein demonstrate the beneficial capacity of HDAC4 inhibition to enhance cardiac regeneration, advancing the concept of ex vivo gene therapy with cultured CSCs to enhance cardiogenesis when reintroduced into infarcted myocardium.
MATERIALS AND METHODS
All animal experiments were conducted under a protocol approved by the Institutional Animal Care and Use Committee of the Roger Williams Medical Center, which conforms to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85–23, revised 1996).
c-kit+ CSC isolation, establishment of stable GFP-c-kit+ CSCs, and siRNA transfection.
The isolation of CSCs is the same as described before (31). Myocardial tissues from 3-mo-old CD-1 male mice were minced into small pieces and subjected to enzymatic dissociation, and the remaining tissue fragments were cultured as explants in explant medium [Iscove's Modified Dulbecco's IMDM with 10% fetal calf serum (FBS), 100 U/ml penicillin G, 100 μg/ml streptomycin, 2 mmol/l l-glutamine, and 0.1 mmol/l 2-mercaptoethanol] at 37°C and 5% CO2. After 2–3 wk, small-phase bright cells migrating above the fibroblast layer were formed from adherent explants. The selected CDC cells were cultured and maintained in complete media containing DMEM/F12, 10% fetal calf serum, 200 mM l-glutamine, 55 nM β-mercaptoethanol, and 1% MEM nonessential amino acids (Invitrogen). To enrich the c-kit+ CSCs, c-kit+ cells were isolated by magnetic cell sorting with CD117 magnetic beads (Miltenyi Biotec, Auburn, CA) as instructed by the manufacturer's protocols. To track the fate of transplanted CSCs in the infarcted hearts, a stable c-kit+ CSC line expressing GFP was established. Freshly isolated c-kit+ CSCs were transfected with the linearized pEGFP using Lipofectamine 2000 (Invitrogen) and maintained in the presence of G418 (500 μg/l) for 2 wk. The transfected GFP-c-kit+ CSC-positive colonies were formed, picked up under fluorescent microscopy, and expanded for subcultures. To prevent the effects of multipassages on CSCs, CSCs at passage 4 were used in this study. The method of siRNA transfection was conducted according to the manufacturer's instructions. HDAC4 siRNA (mouse) used in these studies consists of a pool of three different siRNA duplexes (mRNA accession no. NM_207225, Santa Cruz Biotech). The sequences of HDAC4 siRNA 1 duplexes are the following: HDAC4 siRNA (mRNA location: 131) 5′- CCAUCCAGAUGGACUUUCUtt-3′ (sense), 5′-AGAAAGUCCAUCUGGAUGGtt-3′ (antisense); HDAC4 siRNA (mRNA location: 2609): 5′-GGAUGUACAUCAUGGGAAUtt-3′ (sense), 5′-AUUCCCAUGAUGUACAUCCtt-3′ (antisense); HDAC4 siRNA (mRNA location: 3792) 5′-CCACUCAACUCAUCUUGUAtt-3′ (sense), 5′-UACAAGAUGAGUUGAGUGGtt-3′ (antisense). We also included additional HDAC4 siRNA (mRNA accession no. NM_207225, Sigma, St. Louis, MO), which are designated as HDAC4 siRNA 2 (mRNA location 2789) 5′-GAG UAC UUG GCA GCC UUC A-3′ (sense), 5′-UGA AGG CUG CCA AGU ACU C-3′ (antisense); HDAC4 siRNA 3 (mRNA location 856) 5′-GAA AUU ACG CUC AAG GCU U-3′ (sense), 5′-AAG CCU UGA GCG UAA UUU C-3′ (antisense).
The negative control (scrambled) siRNA, HDAC4 siRNA were mixed with Lipofectamine 2000 at a final concentration of 500 nmol/l of siRNA in medium, respectively. To examine effects of HDAC4 overexpression on c-kit+ CSC behaviors and c-kit+ CSC-derived myocardial regeneration, adenoviral vectors with the HDAC4 gene (Vector Biolabs) were used to induce HDAC4 overexpression in c-kit+ CSCs. c-kit+ CSCs were transduced overnight using dilutions of concentrated virus equivalent to 1 × 107 infectious units in non-FBS medium. c-kit+ CSC transduced with the adenoviral GFP vector served as controls. HDAC4 overexpression in c-kit+ CSCs was confirmed by Western blot. Forty-eight hours after siRNA transfection and/or HDAC4 infection, the in vivo allogeneic c-kit+ CSC transplantation was performed in MI mice.
Myocardial infarction.
Three-month-old male CD-1 mouse myocardial infarction was created following thoracotomy by applying permanent ligation to the left anterior descending artery (LAD) as previously described (38). Sham animals underwent placement of the suture without ligation. In order to evaluate the effects of HDAC4 inhibition on myocardial proliferation, animals received intraperitoneal injection of 5-bromo-2-deoxyuridine (BrdU, 50 mg/kg, Sigma, St. Louis, MO) every other day for 2 wk to pulse-chase label cardiac proliferative index into MI hearts.
In vivo allogeneic c-kit+ CSC transplantation.
Cell transplantation was conducted in MI hearts. A total of 5 × 105 GFP labeled c-kit+ CSCs were suspended in 10 μl of PBS and directly injected into five sites in the border zone of the infarcted left ventricle immediately following induction of myocardial infarction during LAD surgical operation. We have previously demonstrated that infusion of the same amount of lin(−)c-kit(+) stem cells improved myocardial functional recovery following myocardial infarction (38). Prior to surgery, animals were randomized into three groups: sham-operated animals, control siRNA-treated animals that received control siRNA-c-kit+ CSCs, and cell-treated animals that received HDAC4 siRNA-c-kit+ CSCs (n = 5–7 per group), respectively. In another set of experiments, animals were randomized into three groups: sham-operated animals, cell injected animals that received an adenoviral control vector, and HDAC4-infected c-kit+ CSCs (n = 5 per group). The investigators responsible for surgery were blinded to the treatments of injected c-kit+ CSCs. Two weeks after cell engraftments, ventricular functions were measured, and immunohistochemistry was carried out.
Measurement of left ventricular function.
The measurement of left ventricular function is described previously in detail (29, 38). Two weeks after cell engraftment, hearts were rapidly excised and arrested in ice-cold Krebs-Henseleit buffer. They were then cannulated via the ascending aorta for retrograde perfusion by the Langendorff method using Krebs-Henseleit buffer containing (in mM) 110 NaCl, 4.7 KCl, 1.2 MgSO4 7H2O, 2.5 CaCl2·2H2O, 11 glucose, 1.2 KH2PO4, 25 NaHCO3, and 0.5 EDTA. The buffer, aerated with 95% O2-5% CO2 to give a pH of 7.4 at 37°C, was perfused at a constant pressure of 55 mmHg. Left ventricular functional analysis was performed using computer software and a computer-based recording system (BIOPAC, Goleta, CA). Measured parameters include left ventricular systolic pressure, heart rate, left ventricular developed pressure (LVDP), where LVDP is systolic pressure minus left ventricular end-diastolic pressure (LVEDP), and rate-pressure product (RPP). RPP is expressed as the product of LVDP and heart rate. Left ventricular dP/dtmax and dP/dtmin were continuously recorded. The investigators responsible for ventricular functional analysis were blinded to the cell treatment groups.
Echocardiographic assessment of cardiac performance.
Mice were anesthetized with 1.5% isoflurane, and temperature was maintained at 37°C. Nair lotion (Church and Dwight Canada, Mississauga, ON, Canada) was applied on the precordial region for 3 min to cleanly remove the hair, and the region was covered with prewarmed ultrasound transmission gel (Aquasonic, Parker Laboratory, Fairfield, NJ). Transthoracic echocardiography was performed using an Acuson Sequoia C512 system with a 15L8 linear array probe. All images were acquired at a depth setting of 25 mm. Two-dimensional B-mode and M-mode echocardiographic images were obtained at the level of the papillary muscles from the parasternal short-axis view. Wall thickness and chamber dimension were determined from M-mode tracings using cardiac calcs software. All left ventricular (LV) dimensions are presented as the average of measurements using three to five consecutive selected beats.
Tissue and cellular immunocytochemistries.
Animals were killed 2 wk following cell engraftment, and cardiac tissues were collected for immunocytochemistry and histological analyses. Cardiac sections (10-μm thick) were prepared from the paraffin-embedded hearts. Tissue sections were deparaffinized for 30 min at 70°C and subsequently immersed in xylene and ethanol at decreasing concentration. Slides were then washed in distilled water. Myocytes were identified by mouse monoclonal α-sarcomeric actinin, mouse monoclonal troponin T (Sigma, St. Louis, MO), and goat-polyclonal MEF2C (Santa Cruz Biotechnology, Santa Cruz, CA). Microvessel densities were examined by anti-α-smooth muscle actin (α-SMA) monoclonal antibody (Sigma, St. Louis, MO). DNA synthesis was determined with incorporation of BrdU into the DNA of dividing cells. BrdU monoclonal antibody was used to detect the proliferation (Roche Diagnostics), polyclonal Ki67 for the cycling myocytes (Novocastra, UK). The c-kit+ CSC-derived newly formed myocytes were identified with polyclonal GFP antibody (Life Technologies) and costaining with α-sarcomeric actinin and MEF2C. The c-kit+ CSC-derived newly formed microvessels were identified with GFP and costaining with α-SMA. Sections were incubated with individual primary antibodies for 2 h at room temperature. Signals were visualized with secondary antibodies including goat anti-rabbit-IgG-Cy3, goat anti-mouse-IgG-Cy3 (Life Technologies), goat anti-rabbit FITC (Vector Laboratories, Burlingame, CA), anti-mouse-IgM-FITC (Sigma), rabbit-anti-goat FITC (Life Technologies), anti-mouse Alexa Fluor 647 (Life Technologies), respectively. 4,6-Diamidino-2-phenylindole (DAPI) was used to identify nuclei. Fluorescent imagining was performed using a high-resolution Zeiss Axioplan 2 epifluorescence microscope controlled by Zeiss Axiovision software. Confocal images were obtained with the Carl Zeiss LSM 700 laser scanning microscope equipped with the intuitive ZEN software. Stained numbers of sections were counted in approximately 20 randomized fields of the tissue sections, which were taken in the middle plane of each heart and contained infarct and border regions and were normalized to the tissue area.
Histological analysis.
Cardiac samples were obtained 2 wk after cell engraftments. Masson's trichrome staining was performed according to the manufacturer's protocol (Sigma). Images of the three sections from the base to the apex of the left ventricle in each heart were taken using an Olympus BX51 microscope with Spot Advanced software. Infarct scar area and total area of left ventricle were traced manually and measured using image software (NIH ImageJ). Wall thickness of left ventricle, viable myocardium, and scar size were measured as described before (5). To quantitate the degree of left ventricle dilation, the LV expansion index was calculated using a modification of the method: expansion index = (LV cavity area/total area) × (noninfarcted region wall thickness/risk region wall thickness). To measure the size of myocytes in the remote area of infarcted hearts, sections (10 μm) were prepared from paraffin-embedded tissues. Myocyte cross-sectional area was measured from images captured from the sections obtained middistance from the base to the apex. Wheat germ agglutinin (WGA) staining was carried out using immunofluorescent staining to measure cell size. Suitable cross-sections were defined as having nearly circular-to-oval myocyte sections. The outline of myocytes was traced in the LV of each animal, using NIH ImageJ software to determine myocyte cross-sectional area. A value from each heart was calculated by the measurements of approximately 400–600 cells in remote area from infarction of an individual heart.
Terminal deoxynucleotidyl transferase mediated dUTP nick end labeling assay (TUNEL).
Survival of transplanted cells was detected with TUNEL labeling using an in situ cell death detection kit from Roche following the manufacturer's instructions (38). Transplanted cells were labeled with GFP and nuclei were stained with DAPI. TUNEL positive cells were observed using confocal laser scanning microscopy LSM 700 (Carl Zeiss). The numbers of GFP positive/TUNEL cells were determined and were normalized to the tissue area.
In vitro analysis of cardiac differentiation.
The c-kit+ CSCs were cultured in the Millicell EZ slides (Millipore) for cellular immunostaining analysis. The c-kit+ CSCs at 60% confluency were transfected with the negative control (scrambled) siRNA and HDAC4 siRNA using Lipofectamine 2000 at a final concentration of 500 nmol/l of siRNA in medium, respectively. In another set of experiments, c-kit+ CSCs were transduced overnight using dilutions of concentrated virus equivalent to 1 × 107 infectious units in non-FBS medium to determine the effects of overexpression of HDAC4 on cardiac proliferation and cardiac linage specification. To induce the differentiation of c-kit+ CSC, leukemia inhibitory factor was withdrawn from the culture medium and 10−8 M dexamethasone was added for 5 days in in vitro culture. The proliferation was evaluated with anti-rabbit polyclonal Ki67 antibody, and polyclonal phosphorylated histone 3 antibody was used to assess mitosis. Cardiac lineage specifications were determined by anti-rabbit polyclonal MEF2C antibody (Santa Cruz Biotech, CA). Cells were fixed in 3.7% (vol/vol) paraformaldehyde for 15 min and then permeabilized in 0.5% Triton X-100 in PBS for 10 min. The cells were then incubated with primary antibodies for 2 h. Secondary anti-rabbit IgG (H+L)-Cy3 (Vector Laboratories, Burlingame, CA) and/or anti-mouse IgM-FITC antibodies were applied at room temperature. To examine the effect of knockdown of HDAC4 on the protein levels in c-kit+ CSCs, an identical experimental culture protocol as above was carried out in 6-well culture plates to detect the protein expression of HDAC4.
Real-time polymerase chain reaction (PCR).
Total RNA was extracted from CSCs from different groups with Trizol reagent (Life Technologies, Grand Island, NY). cDNA was synthesized from 5 μg of total RNA. The reverse transcribed cDNA (5 μl) was amplified to a final volume of 50 μl by PCR under standard conditions. Real-time PCR experiments were performed on a Mastercycler Realplex4 (Eppendorf North America) system using qPCR Kit master mix. (Kapa Biosystems, Boston, MA). Primer sequences of HDAC4 used in these studies are as follows: forward 5-CTG CAA GTG GCC CCT ACA G-3, reverse 5-CTG CTC ATG TTG ACG CTG GA-3. GAPDH was used as the internal control.
Western blot analysis.
Proteins (50 μg/lane) were separated by SDS-PAGE and then transferred onto a nitrocellulose membrane. The membrane was blocked with 5% non-fat dry milk in 1X Tris-buffered saline containing 0.5% Tween 20 for 1 h. The blots were incubated with their respective polyclonal antibodies HDAC4, Ki67, phosphorylated histone 3, MEF2C, and β-actin (1:1,000) for 2 h and visualized by incubation with anti-rabbit horseradish peroxidase-conjugated secondary antibody (1:5,000) for 1 h and developed with ECL Chemiluminescence detection reagent (Amersham Pharmacia Biotech).
HDAC activity assay.
Measurement of HDAC activity of c-kit+ CSCs was conducted using the colorimetric HDAC activity assay kit (BioVision Research, Mountain View, CA).
Statistics.
All data are expressed as means ± SE. Differences among multigroups were analyzed by one-way analysis of variance (ANOVA), followed by Bonferroni correction. Student's unpaired t-test was used when there were two groups. A probability of P < 0.05 was considered to be a significant difference.
RESULTS
Genetic knockdown of HDAC4 in c-kit+ CSCs reduces protein expression.
In order to see whether siRNA HDAC4 led to the knockdown of HDAC4 in c-kit+ CSCs, HDAC4 protein contents and mRNA were examined at 5 days posttransduction in the transfected c-kit+ CSCs. As shown in Fig. 1A, HDAC4 mRNA contents were significantly reduced following the transfection of HDAC4 siRNA duplex c-kit+ CSCs, suggesting that HDAC4 siRNA duplexes effectively knockdown HDAC4 in c-kit+ CSCs. In order to rule out the potential off-target effect and demonstrate the efficiencies of HDAC4 siRNA duplexes in this study, we included two additional oligonucleotides to further confirm the effect of HDAC4 siRNA. All of these HDAC4 siRNA dramatically attenuated the HDAC4 mRNA in c-kit+ CSCs. We also included HDAC4 shRNA as a control for confirming the effect of knockdown of HDAC4 (data not shown). Likewise, as shown in Fig. 1B, HDAC4 siRNA duplex-transfected c-kit+ CSCs showed a reduction in HDAC4 protein. Therefore, HDAC4 siRNA duplexes were employed for both subsequent in vivo and in vitro experiments. Furthermore, as shown in Fig. 1C, an overexpression of HDAC4 increased HDAC4 protein at 5 days posttransduction of adenoviral HDAC4 in c-kit+ CSCs. As shown in Fig. 2, A and B, HDAC4 siRNA significantly attenuated HDAC activity, but an overexpression of HDAC4 resulted in the marked increase in HDAC activity.
Fig. 1.

Effect of HDAC4 inhibition on HDAC4 proteins in cultured c-kit+ cardiac stem cells (CSCs). A: c-kit+ CSCs were transiently transfected with HDAC4 siRNA 1 (duplexes), HDAC4 siRNA2 and HDAC4 siRNA3 (500 nmol/l), or control siRNA (500 nmol/l) for 5 days to prepare RNA for real-time quantitative PCR to evaluate mRNA of HDAC4. B: protein lysates were prepared and HDAC4 proteins were measured. Densitometric analysis showing HDAC4 proteins in HDAC4 siRNA-transfected c-kit+ CSCs (n = 3 per group). Values represent means ± SE. *P < 0.05 vs. control siRNA. C: effect of HDAC4 overexpression on HDAC4 proteins in cultured c-kit+ CSCs. c-kit+ CSCs were infected with adenoviral GFP or adenoviral HDAC4 for 5 days to prepare protein lysates, and following immunoblotting assay, HDAC4 proteins were measured. D: densitometric analysis showing HDAC4 protein expression in adenoviral GFP or adenoviral HDAC4-infected c-kit+ CSCs (n = 3 per group).Values represent means ± SE. *P < 0.05 vs. adenoviral GFP.
Fig. 2.
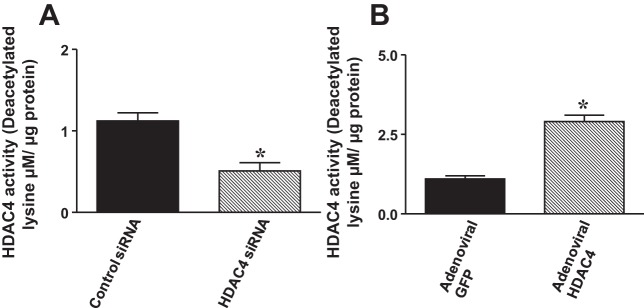
HDAC activities were measured in c-kit+ CSCs. A: HDAC activities in c-kit+ CSCs transfected with control siRNA and HDAC4 siRNA 1 (duplexes), respectively. B: HDAC activities in c-kit+ CSCs were infected with adenoviral GFP or adenoviral HDAC4, respectively. Values are expressed as means ± SE (n = 5 per group). *P < 0.05 vs. control siRNA or adenoviral GFP.
Inhibition of HDAC4 in c-kit+ CSCs improves cardiac functional restoration.
Left ventricular functional parameters were measured in isovolumetric perfused hearts, which avoids the confusing effects of preload, afterload, and in vivo sympathetic activity. Left ventricular developed pressure in MI hearts decreased compared with that in sham-operated mice (Fig. 3A). However, there were greater improvements in LV dP/dtmax and LV dP/dtmin restoration in MI hearts that received HDAC4 siRNA treated c-kit+ CSCs compared with MI hearts that received control siRNA-treated c-kit+ CSCs. After 2 wk of myocardial infarction, echocardiographic assessment showed that MI mice receiving HDAC4 siRNA-treated CSCs demonstrated improvement in ejection fraction and fractional shortening compared with MI hearts receiving control siRNA-treated CSCs (Fig. 3B). Likewise, MI mice receiving HDAC4 siRNA-treated CSCs prevented the increase in LVID in the infarcted mice (Fig. 3B).
Fig. 3.

HDAC4 inhibition improved cardiac functional improvement in c-kit+ CSCs engrafted infarcted hearts. Left ventricular (LV) function was assessed in isovolumetric hearts after 2 wk of MI (A). The measured parameters include LV developed pressure (LVDP), heart rate (HR), and rate-pressure product (RPP), where LVDP is systolic pressure minus LV end-diastolic pressure (LVEDP). Left ventricular dP/dtmax and dP/dtmin were continuously recorded. Values represent means ± SE. *P < 0.05 vs. control siRNA groups (n = 5–7 per group). Echocardiographic analyses of cardiac function after 2 wk of MI (B). EF, ejection fraction; FS, fractional shortening; LVID;d, left ventricular internal dimension in diastole; LVID;s, left ventricular internal dimension in systole; MI, myocardial infarction. Values represent means ± SE. **P < 0.01 vs. control siRNA groups (n = 5–7 per group).
Furthermore, overexpression of HDAC4 in c-kit+ CSCs diminished the restoration of functional recovery in c-kit+ CSC-engrafted MI hearts compared with MI hearts receiving adenoviral GFP-treated c-kit+ CSCs (Fig. 4A). As shown in Fig. 4B, echocardiographic measurement demonstrated that MI mice receiving adenoviral HDAC4-infected CSCs had diminished ejection fraction and fractional shortening compared with MI hearts receiving adenoviral-GFP-treated CSCs. Likewise, MI mice receiving adenoviral HDAC4-treated CSCs resulted in the increase in LVID in the infarcted mice (Fig. 4B). The results indicate that HDAC4 inhibition in c-kit+ CSCs improves myocardial functional recovery in c-kit+ CSC-engrafted hearts.
Fig. 4.

Overexpression of HDAC4 in c-kit+ CSCs attenuated cardiac functional improvement in MI hearts engrafted with adenoviral HDAC4-infected c-kit+ CSCs (A). LV functional parameters include LVDP, LV dP/dtmax, LV dP/dtmin, RPP, and HR. The values represent means ± SE. *P < 0.05 vs. sham and control adenoviral GFP-infected c-kit+ CSCs (n = 5 per group). Echocardiographic analyses of cardiac function after 2 wk of MI (B). Values represent means ± SE. **P < 0.01 vs. sham and control adenoviral GFP-infected c-kit+ CSCs (n = 5 per group).
Inhibition of HDAC4 attenuates cardiac remodeling in c-kit+ CSC engrafted MI hearts.
Morphometric analysis of infarcted hearts shows severe LV chamber dilatation and infarct wall thinning in all MI hearts. MI hearts that engrafted HDAC4 siRNA-treated c-kit+ CSCs demonstrated attenuated LV remodeling, which was shown by a more viable myocardium and thicker infarcted wall but smaller scar size and less LV expansion (Fig. 5, A and B). However, MI heart engrafted with adenoviral HDAC4-infected c-kit+ CSCs exacerbated myocardial remodeling compared with MI hearts that were engrafted with adenoviral GFP-infected c-kit+ CSCs (Fig. 5, C and D). Furthermore, the increase in heart/tibia length ratio and heart/body weight ratio was attenuated in MI hearts engrafted with HDAC4 siRNA-treated c-kit+ CSCs (Fig. 6, A and C), whereas overexpression of HDAC4 exacerbated the heart/tibia length ratio and heart/body ratio in MI heart engrafted with c-kit+ CSCs (Fig. 6, B and D). Likewise, increase in cross-sectional area of myocytes was also mitigated in MI heart receiving HDAC4 siRNA-treated c-kit+ CSCs (Fig. 6, E and G). However, MI hearts engrafted with adenoviral HDAC4-infected CSCs resulted in the enlargement in cross-sectional area of myocytes (Fig. 6, F and H). These results indicate that HDAC4 inhibition of CSCs improves myocardial remodeling in CSC-engrafted MI hearts.
Fig. 5.

Effects of HDAC inhibition improved myocardial remodeling in c-kit+ CSC-engrafted MI hearts. Viable myocardium, wall thickness of infarcted myocardium, scar sizes and the expansion index were determined in MI hearts engrafted with c-kit+ CSCs. A: quantitative analyses of remodeling index in MI hearts that received c-kit+ CSCs transfected with control siRNA, HDAC4 siRNA, respectively. B: representative Masson-trichrome-stained myocardial left ventricular sections in MI hearts that received c-kit+ CSCs transfected with control siRNA, HDAC4 siRNA, respectively. C: quantitative analyses of remodeling index in MI hearts that received c-kit+ CSCs infected with adenoviral HDAC4 and adenoviral GFP. D: representative Masson-trichrome-stained myocardial left ventricular sections in MI hearts that received c-kit+ CSCs infected with adenoviral HDAC4 and adenoviral GFP. Values represent means ± SE (n = 3 per group). *P < 0.05 vs. control siRNA in A; *P < 0.05 vs. adenoviral GFP in C. Scale bars represent 500 μm.
Fig. 6.

Effects of HDAC inhibition attenuate hypertrophic responses in c-kit+ CSC-engrafted MI hearts. A: quantitative analyses of heart/tibia length ratio in MI hearts that received c-kit+ CSCs transfected with control siRNA, HDAC4 siRNA, respectively. B: quantitative analyses of heart/tibia length ratio in MI hearts that received c-kit+ CSCs infected with adenoviral HDAC4 and adenoviral GFP. C: quantitative analyses of heart/body weight ratio in MI hearts that received c-kit+ CSCs transfected with control siRNA, HDAC4 siRNA, respectively. D: quantitative analyses of heart/body weight ratio in MI hearts that received c-kit+ CSCs infected with adenoviral HDAC4 and adenoviral GFP. E: relative myocyte areas were determined with WGA staining in MI hearts that received c-kit+ CSCs transfected with control siRNA, HDAC4 siRNA, respectively. F: relative myocyte areas were determined with WGA staining in MI hearts that received c-kit+ CSCs infected with adenoviral HDAC4 and adenoviral GFP. G: representative image of WGA in MI hearts that received c-kit+ CSCs transfected with control siRNA, HDAC4 siRNA, respectively. H: representative image of WGA in MI hearts that received c-kit+ CSCs infected with adenoviral HDAC4 and adenoviral GFP. Values represent means ± SE (n = 3 per group). *P < 0.05 vs. control siRNA; #P < 0.05 vs. sham in A, C, E; *P < 0.05 vs. adenoviral GFP; #P < 0.05 vs. sham in B, D, F. Scale bar represents 50 μm.
HDAC4 inhibition of c-kit+ CSC enhances myocardial regeneration.
We estimated the survival of cells in the cell-engrafted infarcted hearts. As shown in Fig. 7, A and C, HDAC4 siRNA treatment led to an increase in CSC retention and survival in cell-engrafted MI heart. Likewise, the retention and survival of CSCs in cell-engrafted MI hearts were mitigated by overexpression of HDAC4 (Fig. 7, B and D). In the CSC-treated MI hearts, GFP was utilized to recognize the CSC progeny and α-sarcomeric actinin and α-smooth muscle actin were employed to identify regenerated myocytes and microvessels, respectively. Following acute MI, cells were injected into the border and infarcted areas of hearts. As shown in Fig. 8, A and B, CSC-derived myocytes and microvessels were demonstrated in MI hearts that received cell transplantation. Knockdown of HDAC4 of c-kit+ CSCs resulted in an increase in CSC-derived myocytes and microvessels in cell-engrafted MI heart compared with MI hearts that received control siRNA-treated CSCs (Fig. 8C). c-kit+ CSC-derived myocytes and microvessels were located in the border and infarcted areas. In the cell-injected MI heart, CSC-derived cardiomyocytes and microvessels both are present in some areas of CSC-engrafted infarcted heart. However, As shown in Fig. 8D, MI hearts receiving adenoviral HDAC4-infected c-kit+ CSCs displayed a decrease in CSC-derived myocytes and microvessels compared with MI hearts that received control adenoviral-GFP-infected CSCs. Taken together, the data indicate that inhibition of HDAC4 facilitates c-kit+ CSC-derived cardiac regeneration and angiogenesis, but overexpression of HDAC4 attenuated CSC-derived cardiac regeneration and angiogenesis.
Fig. 7.

HDAC4 inhibition increases the retention and survival of transplanted cells in the post-MI heart. The transplanted cell retention and deaths in MI hearts that received HDAC4 siRNA treated c-kit+ CSCs and control siRNA-treated c-kit+ CSCs are shown in A and C. The transplanted cell retention and deaths in MI hearts that received adenoviral HDAC4 infected-c-kit+ CSCs and MI hearts that received control adenoviral-GFP treated-c-kit+ CSCs are shown in B and D. The cell retention of c-kit+ CSCs in MI hearts was determined by examining GFP positive stained populations, and cell deaths were determined by TUNEL analysis, which is described in materials and methods. The cell retention and deaths of transplanted cells were normalized to the tissue area. The values represent means ± SE (n =3 hearts per group). *P < 0.05 vs. MI hearts that received control siRNA-treated c-kit+ CSCs or control adenoviral GFP-treated c-kit+ CSCs.
Fig. 8.

HDAC4 inhibition affects c-kit+ CSC-derived myocardial regeneration in infarcted hearts. A: the representative images of c-kit+ CSC-derived cardiomyocytes in MI hearts that received HDAC4 siRNA-treated and control siRNA-treated c-kit+ CSCs, respectively. c-kit+ CSCs were labeled with GFP, cardiomyocytes were stained with α-sarcomeric actinin (green), GFP was stained with anti-rabbit-IgG-Cy-3 (red), and nuclei were stained with DAPI (blue). B: the representative images of c-kit+ CSC-derived microvessels in MI hearts received HDAC4 siRNA-treated and control siRNA-treated c-kit+ CSCs, respectively. Microvessels were stained with α-smooth muscle actin (α-SMA) (red), GFP was stained with anti-rabbit-IgG-FITC (green), and nuclei were stained with DAPI (blue). C: quantitative analyses of c-kit+ CSC-derived myocytes and microvessels in MI hearts that received HDAC4 siRNA-treated and control siRNA-treated c-kit+ CSCs, respectively. D: quantitative analyses of c-kit+ CSC-derived myocytes and microvessels in MI hearts that received adenoviral HDAC4 and adenoviral GFP infected c-kit+ CSCs, respectively. The details of the immunostaining procedure are described in materials and methods. The number of c-kit+ CSC-derived myocytes and microvessels were counted in 5–6 randomized fields of 3 tissue sections for each heart. These sections, which contained infarct and border regions, were taken in the middle plane of each heart and were normalized to the tissue area. The values represent means ± SE (n = 3–5 per group). *P < 0.05 vs. MI heart that received control siRNA-treated c-kit+ CSCs in C. *P < 0.05 vs. MI hearts that received adenoviral GFP-infected c-kit+ CSCs in D. Scale bars represent 50 μm.
HDAC4 inhibition increases myocyte proliferation in c-kit+ CSC-engrafted MI hearts.
The dividing amplifying cells from CSC-engrafted MI hearts were evaluated by Ki67; the regenerated myocytes were detected with BrdU. Myocytes positive for Ki67 and BrdU were demonstrated in the infarct areas (Fig. 9, A and B). There are rare detectable BrdU and Ki67 positive myocytes in sham control hearts (Fig. 9C). As shown in Fig. 9D, compared with MI hearts receiving control siRNA-treated c-kit+ CSCs, there were significantly more BrdU positive myocytes in the MI hearts transplanted with HDAC4 siRNA-treated c-kit+ CSCs. Likewise, the percentage of Ki67 positive myocytes was increased in the MI hearts engrafted with HDAC4 siRNA-treated c-kit+ CSCs. We further tested whether overexpression of HDAC4 of CSCs could eliminate the proliferative capacity of c-kit+ CSCs-engrafted MI hearts. As shown in Fig. 9E, compared with MI hearts engrafted with control adenoviral GFP-infected c-kit+ CSCs, c-kit+ CSCs overexpressing HDAC4 resulted in a remarkable reduction in BrdU and Ki67 positive myocytes.
Fig. 9.

HDAC4 inhibition affects myocyte proliferation in c-kit+ CSC-engrafted MI hearts. A: the representative images of Ki67 and BrdU positive myocytes in the infarcted and border areas of MI hearts that received HDAC4 siRNA and control siRNA-treated c-kit+ CSCs, respectively. B: the representative images of Ki67 and BrdU positive myocytes in MI hearts that received adenoviral HDAC4 and control adenoviral GFP-infected c-kit+ CSCs. Cardiomyocytes were stained with α-sarcomeric actinin (green), Ki67 and BrdU were labeled with anti-rabbit and anti-mouse-Cy3, respectively (red), and nuclei were stained with DAPI (blue). C: the representative images of Ki67 and BrdU positive myocytes in sham control heart. Cardiomyocytes were stained with α-sarcomeric actinin (green); Ki67 and BrdU were labeled with anti-rabbit and anti-mouse-Cy3, respectively (red); and nuclei were stained with DAPI (blue). D: quantitative analyses of BrdU and Ki67 positive myocytes in MI hearts that received HDAC4 siRNA and control siRNA-treated c-kit+ CSCs, respectively. E: quantitative analyses of BrdU and Ki67 in MI hearts that received adenoviral HDAC4 and control adenoviral GFP-infected c-kit+ CSCs. Details of immunostaining procedures are described in materials and methods. The number of Ki67 and BrdU positive myocytes was counted in 5–6 randomized fields of the 3 tissue sections of each heart. These sections, which contained infarct and border regions, were taken in the middle plane of each heart and were normalized to the tissue area. The values represent means ± SE (n = 3–5 hearts per group). *P < 0.05 vs. MI heart that received control siRNA-treated c-kit+ CSCs in D; *P < 0.05 vs. MI hearts that received control adenovirus GFP-treated c-kit+ CSCs in E. Scale bars represent 50 μm.
HDAC4 inhibition increases cell proliferation and cardiac commitments in c-kit+ CSCs in vitro.
We next assessed whether HDAC4 induces CSCs to adopt cardiac specification in vitro. Knockdown of HDAC4 of c-kit+ CSCs resulted in an increase in MEF2C positive cardiac progenitors, which was associated with an increase in Ki67 and phosphorylated-histone 3 cycling index (Fig. 10, A and C). However, c-kit+ CSCs infected with adenoviral HDAC4 decreased the magnitude of MEF2C positive cardiac progenitors compared with control adenoviral HDAC4 infected CSCs, which was associated with the reduction in Ki67 and phosphorylated histone 3 positive cardiac progenitors (Fig. 10B). In addition, specific knockdown of HDAC4 led to the increase in Ki67 and phosphorylated histone 3 (Fig. 10D). In contrast, the overexpression of HDAC4 inhibited Ki67 and phosphorylated histone 3 of c-kit+ CSCs (Fig. 10E).
Fig. 10.

Effects of HDAC4 inhibition on c-kit+ CSC proliferation and cardiac commitments in vitro. A: quantitative analyses of MEF2C, Ki67, and phosphorylated histone 3 (pH3) positive CSC lineages in cultured c-kit+ CSCs transfected with control siRNA, HDAC4 siRNA, respectively. MEF2C was used to label cardiac transcriptional factor, while Ki67 and pH3 were utilized to label proliferative cells. Cardiac lineages were labeled with anti-α-sarcomeric actinin (green). Detailed procedures for in vitro c-kit+ CSC culture are described in materials and methods. B: quantitative analyses of MEF2, Ki67, and pH3 positive CSC lineages in adenoviral HDAC4 or control adenoviral GFP-infected c-kit+ CSC in vitro cultures. C: representative images for Ki67, phosphorylated-histone 3, and MEF2C positive c-kit+ CSCs-derived cardiac progenitors that received HDAC4 siRNA and control siRNA treatments, respectively. Nuclei were stained with DAPI (blue), and cardiomyocytes with α-sarcomeric actinin (green); MEF2C, Ki67 and phosphorylated-histone 3 were labeled in red; pH3, phosphorylated-histone 3. Each group represents 13 individual experiments. Values represent means ± SE. *P <0.05 vs. control siRNA-treated c-kit+ CSCs in A (n = 13 per group). *P < 0.05 vs. control adenoviral GFP-treated c-kit+ CSCs in B (n = 13 per group). D: representative images of Ki67 and phosphorylated histone 3 protein expressions in cultured c-kit+ CSC transfected with control siRNA, HDAC4 siRNA, respectively (top panel); densitometric analysis of Ki67 and phosphorylated histone 3 proteins (bottom panel). E: representative images of Ki67 and phosphorylated histone 3 protein expressions in adenoviral HDAC4 or control adenoviral GFP-infected c-kit+ CSCs in vitro (top panel); densitometric analysis of Ki67 and phosphorylated histone 3 proteins (bottom panel). Each group represents 3 individual experiments. *P < 0.05 vs. control siRNA in D (n = 3 per group); *P < 0.05 vs. adenoviral GFP in E (n = 3 per group). Scale bars represent 50 μm.
DISCUSSION
The availability of these well-characterized heart progenitor cells allows for a direct examination of their biological function and specific pathway that drives cardiogenesis in the developmental stage and regeneration infarcted myocardium (6, 30, 37). Epigenetic intervention and/or HDAC inhibition have been recently identified as the critical determinant for cell programming in in vitro studies (16, 42, 44). Although we have recently demonstrated that HDAC inhibition serves as a central mechanism to trigger endogenous regeneration and repair infarcted tissue (46), it is not clear which specific HDAC isoform determines CSC-derived myocardial regeneration following MI. In this study, we demonstrated that HDAC4 inhibition plays a major role in controlling cardiac commitment of CSCs, inducing myocardial regeneration and restoration of cardiac functional recovery.
Mouse genetics have demonstrated an essential role of HDAC in embryogenesis (9, 11, 24, 32). In this study, using the established siRNA approach, which is widely performed to knockdown specific genes in progenitor cells (17), we demonstrated that the treatment of HDAC4 siRNA significantly induced the knockdown of HDAC4 in c-kit+ CSCs, which is also accompanied by the reduction of HDAC activity. Knockdown of HDAC4 of c-kit+ CSCs exhibited an increase in newly formed myocytes and microvessels in CSC engrafted MI hearts. In contrast, overexpression of HDAC4 antagonized this event, indicating that the suppression of HDAC4 of CSCs is required for cardiac lineage commitment in vivo. This is supported by a previous observation that the overexpression of HDAC4 inhibited cardiomyogenesis, shown by the downregulation of cardiac muscle genes in P19 cells (21). Activation of HDAC4 suppresses MEF2-dependent gene expression and contributes to progressive muscle dysfunction observed in neuromuscular diseases (7). Molecular analysis reveals that through the NH2-terminal domain of HDAC4, HDAC4 interacts with the MADS-box transcription factor MEF2C to negatively regulate gene expression (41). In our observation, we demonstrated that inhibition of HDAC4 increased the MEF2 positive progenitors. It has been demonstrated that class II HDAC proteins suppress the formation of slow twitch, oxidative myofibers through the repression of MEF2 activity. However, expression of a hyperactive form of MEF2 in skeletal muscle of transgenic mice promotes the formation of slow fibers and enhances running endurance, providing a mechanism for skeletal muscle function by augmenting the transcriptional activity of MEF2 (35). It is not clear whether regulation of MEF2 following HDAC4 inhibition may also be attributed for the formation of cardiac muscle in CSC-engrafted MI heart, which will need further investigation.
The enhanced myocardial regeneration derived from c-kit+ CSCs by the suppression of HDAC4 was associated with an improvement in functional restoration. In contrast, the overexpression of HDAC4 abolished the c-kit+ CSC-derived regenerative capacity and functional restoration, suggesting that the effect of HDAC4 inhibition on CSC-derived myocardial regeneration determines the restoration of cardiac functional recovery. This is consistent with our previous observation that ESC-derived cardiac lineage commitment was associated with HDAC4 reduction (4).
Neovascularizations were significantly increased in MI hearts receiving c-kit+ CSCs in which HDAC4 was specifically inhibited, indicating that HDAC inhibition mediated in microvessels in CSC-engrafted hearts might also be responsible for the improvement of myocardial function and the reduction of myocardial remodeling. This is supported by observations that the augmentation of neovascularizations in MI hearts was closely associated with the prevention of cardiac remodeling (20, 43). It is very interesting to evaluate the volume of infarct size over time following the transplantation of CSCs in the MI heart, which is the limitation in our observation. In this study, our finding indicates that cardiogenesis was enhanced following specific inhibition of HDAC4. However, it is not clear whether HDAC4 inhibition can facilitate c-kit+ CSCs to capture features of embryonic stem cells such as immortality or teratogenicity in vivo. The induction of pluripotent stem cells by defined factors has been shown to cause the development of teratomas. Cardiac sections from infarcted hearts receiving siRNA HDAC4-treated c-kit+ CSCs did not demonstrate the formation of tumorigenesis in hearts 6 mo after cell engraftment (data not shown), suggesting that HDAC4 inhibition in c-kit+ CSCs does not increase the risk of developing teratomas. In addition, c-kit+ CSCs-engrafted MI hearts antagonized cardiac hypertrophy, which is in line with previous observations that inhibition of HDACs showed cardioprotection and blocks cardiac hypertrophy (22, 23).
In addition to the effect of HDAC4 inhibition on CSC-derived cardiogenesis following transplantation, it is likely that an increase in CSC survival may also have contributed to cardiac performance preservation and attenuation of remodeling. It has been reported that the engraftment of exogenous stem cells activated cardiac resident stem cells to increase endogenous myocardial repair (27). It is not clear whether inhibition of HDAC4 in engrafted CSCs could affect resident CSCs to repair damaged hearts. It was reported that there were conflicting observations in cell fusion in cell engrafted host myocardium (3, 34); the role of cell fusion on functional improvement remains unknown. We did not find cell fusion of engrafted stem cells with resident cardiomyocytes in infarcted heart. The present study did not show whether cell fusion constitutes a major mechanism(s) by which HDAC4 mediates myocardial repair, an interesting topic to investigate in the future. Our previous observation indicates that HDAC inhibition stimulates endogenous angiomyogenesis, and preconditioning of CSCs with HDAC inhibitor dramatically increases newly formed myocytes and microvessels when reintroduced into infarcted myocardium, which support our observation that HDAC4 inhibition promotes cardiac stem cells to enhance myocardial repairs in this investigation (46, 49). However, it is not clear whether HDAC4 could mediate endogenous angiomyogenesis through a similar pathway or paracrine mechanism, a line which could be an interesting to investigate in the future.
Conclusions.
Taken together, our results indicate that specific inhibition of HDAC4 promoted c-kit+ CSC-derived myocardial repair, functional restoration, and improved myocardial remodeling in MI hearts engrafted with HDAC4 siRNA-treated c-kit+ CSCs. Conversely, the overexpression of HDAC4 of c-kit+ CSCs mitigates myocardial regeneration and functional recovery in CSC-engrafted MI hearts. Furthermore, in vitro evidence facilitated c-kit+ CSCs into cardiac commitment and proliferation, while the overexpression of HDAC4 antagonizes this event. Our results demonstrate that HDAC4 inhibition plays a crucial role in mediating c-kit+ CSCs to induce cardiac regeneration. Our study not only provides new insight into our understanding of myocardial repair, but also holds promise in developing a novel therapeutic approach for cardiac regeneration.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S. Z., Y. T., T. C. Z. conception and design of research; L. X. Z., M. D., X. Q., J. D., J. M. performed experiments; M. D., J. D. analyzed data; L. W., G. Q., interpreted results of experiments; M. D., prepared figures; T. C. Z. drafted manuscript; T. Y. Z., S. Z., P. Y. L., L. W., edited and revised manuscript; T. C. Z. approved final version of manuscript.
ACKNOWLEDGMENTS
This work was supported by National Heart, Lung, and Blood Institute Grants R01 HL-089405 and R01 HL-115265 and the American Heart Association National Center Grant 0735458N (to T. C. Zhao).
REFERENCES
- 1.Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest 116: 1853–64, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114: 763–776, 2003 [DOI] [PubMed] [Google Scholar]
- 3.Bolli R, Chugh AR, D'Amario Loughran D, JH, Stoddard MF, Ikram S, Beache GM, Wagner SG, Leri A, Hosoda T, Sanada F, Elmore JB, Goichberg P, Cappetta D, Solankhi NK, Fahsah I, Rokosh DG, Slaughter MS, Kajstura J, Anversa P. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet 378: 1847–1857, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4.Chen H, DeNicola M, Qin X, Zhao Y, Zhang L, Long X, Zhuang S, Liu P, Zhao T. HDAC inhibition promotes cardiogenesis and the survival of embryonic stem cells through proteasome-dependent pathway. J Cell Biochem 112: 3246–3255, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng K, Li TS, Malliaras K, Davis DR, Zhang Y, Marbán E. Magnetic targeting enhances engraftment and functional benefit of iron-labeled cardiosphere-derived cells in myocardial infarction. Circ Res 106: 1570–1581, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chimenti I, Smith RR, Li TS, Gerstenblith G, Messina E, Giacomello A, Marbán E. Relative roles of direct regeneration versus paracrine effects of human cardiosphere-derived cells transplanted into infarcted mice. Circ Res 106: 971–980, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen TJ, Barrientos T, Hartman ZC, Garvey SM, Cox GA, Yao TP. The deacetylase HDAC4 controls myocyte enhancing factor-2-dependent structural gene expression in response to neural activity. FASEB J 23: 99–106, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dimmeler A, Zeiher AM, Schneider MD. Unchain my heart: the scientific foundations of cardiac repair. J Clin Invest 115: 572–583, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dovey OM, Foster CT, Cowley SM. Histone deacetylase 1 (HDAC1), but not HDAC2, controls embryonic stem cell differentiation. Proc Natl Acad Sci USA 107: 8242–8247, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer KM, Cottage CT, Wu W, Din S, Gude NA, Avitabile D, Quijada P, Collins BL, Fransioli J, Sussman MA. Enhancement of myocardial regeneration through genetic engineering of cardiac progenitor cells expressing Pim-1 kinase. Circulation 120: 2077–2087, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foster CT, Dovey OM, Lezina L, Luo JL, Gant TW, Barlev N, Bradley A, Cowley SM. Lysine-specific demethylase 1 regulates the embryonic transcriptome and CoREST stability. Mol Cell Biol 30: 4851–4863, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gordon JW, Pagiatakis C, Salma J, Du M, Andreucci JJ, Zhao J, Hou G, Perry RL, Dan Q, Courtman D, Bendeck MP, McDermott JC. Protein kinase A-regulated assembly of a MEF2. HDAC4 repressor complex controls c-Jun expression in vascular smooth muscle cells. J Biol Chem 284: 19027–19042, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hansen JC, Tse C, Wolffe AP. Structure and function of the core histone N-termini: more than meets the eye. Biochemistry 37: 17637–17644, 1998 [DOI] [PubMed] [Google Scholar]
- 14.Hattori F, Fukuda K. Strategies for ensuring that regenerative cardiomyocytes function properly and in cooperation with the host myocardium. Exp Mol Med 42: 155–165, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu S, Huang M, Nguyen PK, Gong Y, Li Z, Jia F, Lan F, Liu J, Nag D, Robbins RC, Wu JC. Novel microRNA prosurvival cocktail for improving engraftment and function of cardiac progenitor cell transplantation. Circulation 124: S27–S34, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huangfu D, Osafune K, Maehr R, Guo W, Eijkelenboom A, Chen S, Muhlestein W, Melton DA. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol 26: 1269–1275, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Huang J, Zhang Z, Guo J, Ni A, Deb A, Zhang L, Mirotsou M, Pratt RE, Dzau VJ. Genetic modification of mesenchymal stem cells overexpressing CCR1 increases cell viability, migration, engraftment, and capillary density in the injured myocardium. Circ Res 106: 1753–1762, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim MS, Akhtar MW, Adachi M, Mahgoub M, Bassel-Duby R, Kavalali ET, Olson EN, Monteggia LM. An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J Neurosci 32: 10879–10886, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Illi B, Dello Russo C, Colussi C, Rosati J, Pallaoro M, Spallotta F, Rotili D, Valente S, Ragone G, Martelli F, Biglioli P, Steinkuhler C, Gallinari P, Mai A, Capogrossi MC, Gaetano C. Nitric oxide modulates chromatin folding in human endothelial cells via protein phosphatase 2A activation and class II histone deacetylases nuclear shuttling. Circ Res 102: 51–58, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Jujo K, Hamada H, Iwakura A, Thorne T, Sekiguchi H, Clarke T, Ito A, Misener S, Tanaka T, Klyachko E, Kobayashi K, Tongers J, Roncalli J, Tsurumi Y, Hagiwara N, Losordo DW. CXCR4 blockade augments bone marrow progenitor cell recruitment to the neovasculature and reduces mortality after myocardial infarction. Proc Natl Acad Sci USA 107: 11008–11013, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karamboulas C, Swedani A, Ward C, Al-Madhoun AS, Wilton S, Boisvenue S, Ridgeway AG, Skerjanc IS. HDAC activity regulates entry of mesoderm cells into the cardiac muscle lineage. J Cell Sci 119: 4305–4314, 2006 [DOI] [PubMed] [Google Scholar]
- 22.Kee HJ, Sohn IS, Nam KI, Park JE, Yin Z, Ahn Y, Jeong MH, Bang YJ, Kim N, Kim JK, Kim KK, Epstein JA, Kook H. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation 113: 51–59, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Kong Y, Tannous P, Lu G, Berenji K, Rothermel BA, Olson EN, Hill JA. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 113: 2579–2588, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lagger M, O'Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, Seiser C. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J 21: 2672–2681, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laugwitz KL, Moretti A, Lam J, Gruber P, Chen Y, Woodard S, Lin LZ, Cai CL, Lu MM, Reth M, Platoshyn O, Yuan JX, Evans S, Chien KR. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature 433: 647–653, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, Lee A, Huang M, Chun H, Chung J, Chu P, Hoyt G, Yang P, Rosenberg J, Robbins RC, Wu JC. Imaging survival and function of transplanted cardiac resident stem cells. J Am Coll Cardiol l53: 1229–1240, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loffredo FS, Steinhauser ML, Gannon J, Lee RT. Bone marrow-derived cell therapy stimulates endogenous cardiomyocyte progenitors and promotes cardiac repair. Cell Stem Cell 8: 389–398, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LE, Berman D, Czer LS, Marbán L, Mendizabal A, Johnston PV, Russell SD, Schuleri KH, Lardo AC, Gerstenblith G, Marbán E. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet 379: 895–904, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mangi AA, Noiseux N, Kong D, He H, Rezvani M, Ingwall JS, Dzau VJ. Mesenchymal stem cells modified with Akt prevent remodeling and restore performance of infarcted hearts. Nat Med 9: 1195–1201, 2003 [DOI] [PubMed] [Google Scholar]
- 30.Matsuura K, Honda A, Nagai T, Fukushima N, Iwanaga K, Tokunaga M, Shimizu T, Okano T, Kasanuki H, Hagiwara N, Komuro I. Transplantation of cardiac progenitor cells ameliorates cardiac dysfunction after myocardial infarction in mice. J Clin Invest 119: 2204–2217, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyamoto S, Kawaguchi N, Ellison GM, Matsuoka R, Shin'oka T, Kurosawa H. Characterization of long-term cultured c-kit+ cardiac stem cells derived from adult rat hearts. Stem Cells Dev 19: 105–116, 2010 [DOI] [PubMed] [Google Scholar]
- 32.Montgomery L, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev 21: 1790–1802, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Musunuru K, Domian IJ, Chien KR. Stem cell models of cardiac development and disease. Annu Rev Cell Dev Biol 26: 667–687, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y, Pocius J, Michael LH, Behringer RR, Garry DJ, Entman ML, Schneider MD. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci USA 100: 12313–12318, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Potthoff MJ, Wu H, Arnold MA, Shelton JM, Backs J, McAnally J, Richardson JA, Bassel-Duby R, Olson EN. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J Clin Invest 117: 2459–2467, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rota M, Kajstura J, Hosoda T, Bearzi C, Vitale S, Esposito G, Iaffaldano G, Padin-Iruegas ME, Gonzalez A, Rizzi R, Small N, Muraski J, Alvarez R, Chen X, Urbanek K, Bolli R, Houser SR, Leri A, Sussman MA, Anversa P. Bone marrow cells adopt the cardiomyogenic fate in vivo. Proc Natl Acad Sci USA 104: 17783–17788, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang XL, Rokosh G, Sanganalmath SK, Yuan F, Sato H, Mu J, Dai S, Li C, Chen N, Peng Y, Dawn B, Hunt G, Leri A, Kajstura J, Tiwari S, Shirk G, Anversa P, Bolli R. Intracoronary administration of cardiac progenitor cells alleviates left ventricular dysfunction in rats with a 30-day-old infarction. Circulation 121: 293–305, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tseng A, Stabila J, McGonnigal B, Yano N, Yang MJ, Tseng YT, Davol PA, Lum LG, Padbury JF, Zhao TC. Effect of disruption of Akt-1 of lin(−)c-kit(+) stem cells on myocardial performance in infarcted heart. Cardiovasc Res 87: 704–712, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toma C, Pittenger MF, Cahill KS, Byrne BJ, Kessler PD. Human mesenchymal stem cells differentiate to a cardiomyocyte phenotype in the adult murine heart. Circulation 105: 93–98, 2002 [DOI] [PubMed] [Google Scholar]
- 40.Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, McAnally J, Pomajzl C, Shelton JM, Richardson JA, Karsenty G, Olson EN. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 119: 555–566, 2004 [DOI] [PubMed] [Google Scholar]
- 41.Wang AH, Bertos NR, Vezmar M, Pelletier N, Crosato M, Heng HH, Th'ng J, Han J, Yang XJ. HDAC4, a human histone deacetylase related to yeast HDA1, is a transcriptional corepressor. Mol Cell Biol 19: 7816–7827, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ware CB, Wang L, Mecham BH, Shen L, Nelson AM, Bar M, Lamba DA, Dauphin DS, Buckingham B, Askari B. Histone deacetylase inhibition elicits an evolutionarily conserved self-renewal program in embryonic stem cells. Cell Stem Cell 4: 359–369, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu Y, Ip JE, Huang J, Zhang L, Matsushita K, Liew CC, Pratt RE, Dzau VJ. Essential role of ICAM-1/CD18 in mediating EPC recruitment, angiogenesis, and repair to the infarcted myocardium. Circ Res 99: 315–322, 2006 [DOI] [PubMed] [Google Scholar]
- 44.Yuan X, Wan H, Zhao X, Zhu S, Zhou Q, Ding S. Combined chemical treatment enables Oct4-induced reprogramming from mouse embryonic fibroblasts. Stem Cells 29: 549–553, 2011 [DOI] [PubMed] [Google Scholar]
- 45.Zhang LX, Zhao Y, Cheng G, Guo TL, Chin YE, Liu PY, Zhao TC. Targeted deletion of NF-kappaB p50 diminishes the cardioprotection of histone deacetylase inhibition. Am J Physiol Heart Circ Physiol 298: H2154–H2163, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang L, Qin X, Zhao Y, Fast L, Zhuang S, Liu P, Cheng G, Zhao TC. Inhibition of histone deacetylases preserves myocardial performance and prevents cardiac remodeling through stimulation of endogenous angiomyogenesis. J Pharmacol Exp Ther 341: 285–293, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao TC, Cheng G, Zhang LX, Tseng YT, Padbury JF. Inhibition of histone deacetylases triggers pharmacologic preconditioning effects against myocardial ischemic injury. Cardiovasc Res 76: 473–481, 2007 [DOI] [PubMed] [Google Scholar]
- 48.Zhao TC, Zhang LX, Cheng G, Liu JT. gp-91 mediates histone deacetylase inhibition-induced cardioprotection. Biochim Biophys Acta 1803: 872–880, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang L, Chen B, Zhao Y, Dubielecka PM, Wei L, Qin GJ, Chin YE, Wang Y, Zhao TC. Inhibition of histone deacetylase-induced myocardial repair is mediated by c-kit in infarcted hearts. J Biol Chem 287: 39338–39348, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]