

Abstract
Increased activity of the renin-angiotensin system within the brain elevates fluid intake, blood pressure, and resting metabolic rate. Renin and angiotensinogen are coexpressed within the same cells of the subfornical organ, and the production and action of ANG II through the ANG II type 1 receptor in the subfornical organ (SFO) are necessary for fluid intake due to increased activity of the brain renin-angiotensin system. We generated an inducible model of ANG II production by breeding transgenic mice expressing human renin in neurons controlled by the synapsin promoter with transgenic mice containing a Cre-recombinase-inducible human angiotensinogen construct. Adenoviral delivery of Cre-recombinase causes SFO-selective induction of human angiotensinogen expression. Selective production of ANG II in the SFO results in increased water intake but did not change blood pressure or resting metabolic rate. The increase in water intake was ANG II type 1 receptor-dependent. When given a choice between water and 0.15 M NaCl, these mice increased total fluid and sodium, but not water, because of an increased preference for NaCl. When provided a choice between water and 0.3 M NaCl, the mice exhibited increased fluid, water, and sodium intake, but no change in preference for NaCl. The increase in fluid intake was blocked by an inhibitor of PKC, but not ERK, and was correlated with increased phosphorylated cyclic AMP response element binding protein in the subfornical organ. Thus, increased production and action of ANG II specifically in the subfornical organ are sufficient on their own to mediate an increase in drinking through PKC.
Keywords: adenovirus, brain, Cre-recombinase, renin-angiotensin system, subfornical organ
tight regulation of fluid and electrolyte balance is crucial for cellular function and survival. The renin angiotensin system (RAS), one of the most important physiological regulators of fluid and electrolyte homeostasis, is expressed and regulated in a tissue-specific manner (reviewed in Ref. 31). In the brain, renin is expressed primarily in neurons, while angiotensinogen, the substrate for ANG II, is widely expressed in glial cells (2, 32, 40, 64, 68). Angiotensinogen is also expressed in neurons, but its expression in neurons is restricted to those nuclei important for cardiovascular regulation, such as the subfornical organ (SFO), paraventricular nucleus of the hypothalamus (PVN), and rostral ventrolateral medulla (20, 32, 39, 68). This expression profile suggests the potential for the de novo production of ANG II in the brain. Indeed, studies in transgenic mice using sensitive reporter genes controlled by the renin and angiotensinogen promoters have revealed areas of the brain where renin and angiotensinogen are expressed in adjacent cells (i.e., where there is a potential for local extracellular generation of angiotensin peptides) and regions where both renin and angiotensinogen are coexpressed in the same neuron (i.e., where there is a potential for intracellular generation of angiotensin peptides) (39, 40, 68).
The SFO has been implicated as an important nucleus for the control of fluid intake (reviewed in Ref. 62). There is strong experimental support for the local action of ANG II in the SFO. ANG-II AT1 receptors (AT1R) are expressed on SFO neurons and on postsynaptic neurons in the PVN and supraoptic nuclei, which receive projections from the SFO (9, 24, 28, 42, 47). Direct injection of ANG II into the brain causes increased fluid intake in rats through an AT1R-dependent pathway, which is abolished by lesions of the SFO (56–58). In mice, peripheral injection of ANG II stimulates c-Fos immunoreactivity in the SFO, as it does in rats, but in mice, the magnitude of the drinking response is diminished (54) or absent (36). Central injection of ANG II in mice induces drinking (19, 36), doing so via an AT1R-dependent pathway (17), and overexpression of either ANG II (55) or AT1R in neurons of mice induces water intake (41). Genetic ablation of endogenous AT1AR selectively in the SFO of mice carrying a conditional allele of the AT1AR gene (AT1ARflox) attenuates the polydipsia and hypertension induced by DOCA-salt (33). This suggests that DOCA-salt-induced polydipsia and hypertension has an SFO AT1R-dependent component. It should be noted, however, that studies also implicate other nuclei located in the anteroventral third ventricle (AV3V), the organum vasculosum of the lamina terminalis (OVLT), for example, in mediating responses to ANG II, because lesions of the SFO do not effectively ablate ANG II-induced drinking when ANG II is injected directly into the AV3V region (7, 8).
In addition to the local action of ANG II, there is experimental support for the concept that ANG II is locally generated within the SFO. Renin and angiotensinogen are transcriptionally coexpressed within the same cells in the SFO, and angiotensin peptide immunoreactivity is found within the soma and processes of SFO neurons (39, 44). Ferguson et al. (24) have shown that ANG II released from synaptic terminals of SFO neurons can activate neurons in the PVN, suggesting that ANG II produced in the SFO can induce neuronal signaling in other nuclei that project from the SFO (reviewed in Ref. 23).
Double transgenic mice (termed sRA), expressing human renin in all neurons via the synapsin promoter (sR mice), and human angiotensinogen (A mice), driven by its endogenous promoter, exhibit robust angiotensinogen and angiotensin peptide expression in the SFO and exhibit markedly increased fluid intake (30, 55). AT1R blockade blunts the increase in drinking in this model, as does selectively eliminating the expression of human angiotensinogen in the SFO in double-transgenic mice carrying a conditional allele of human angiotensinogen (sRAflox) (55, 59). Thus, production and action of ANG II in the SFO are necessary to increase fluid intake. What remains unknown is whether selective ANG II production only in the SFO is sufficient on its own to increase fluid intake and BP. We tested this directly by generating and analyzing a unique mouse model in which production of ANG II can be genetically induced in any region of the brain. Herein, we used an adenovirus encoding Cre-recombinase to specifically target ANG II production within the SFO.
METHODS
Generation of the ARed construct.
The ARed construct contains: 1) the ubiquitous CAG promoter consisting of the cytomegalovirus (CMV) early enhancer element and the chicken β-actin gene promoter, first exon and intron, and the rabbit β-globin gene splice acceptor; 2) a transgene encoding the dsRed fluorescent protein and a transcriptional and translational STOP signal surrounded by loxP sites; and 3) a human angiotensinogen transgene (Fig. 1A). In response to Cre-recombinase, the dsRed gene and STOP signal are eliminated, and the expression of human angiotensinogen is induced wherever Cre-recombinase is expressed. In practice, dsRed was easily detectable in cultured cells, whereas it was not detectable in the mouse brain.
Fig. 1.

Inducible expression of human angiotensinogen. A: map of the ARed construct. Cre-mediated deletion of the dsREN and STOP signal will allow transcription of human angiotensinogen. B: dsRED fluorescence in ARed-transfected COS cells infected with either AdGFP or AdCRE. C: expression of human angiotensinogen protein in response to AdCRE in ARed-transfected COS cells. D: map of the Cre-reporter construct. Cre-mediated deletion of the STOP signal will allow transcription of tdTomato. E: 1 mo after intracerebroventricular injection of AdCRE in tdTomato reporter mice (ROSA) mice, fluorescence was found exclusively in the subfornical organ (SFO) (SFO is indicated by arrow; 3V denotes third ventricle; * indicates corpus callosum). F: ARed construct induces the expression of human angiotensinogen cDNA after Cre-mediated recombination. Expression of human angiotensinogen (copies normalized to actin) in the median preoptic nucleus (MnPO), SFO, and paraventricular nucleus (PVN) of ARed mice [Adenovirus expressing eGFP (AdGFP), n = 10]; [Adenovirus expressing Cre-recombinase (AdCRE), n = 20] compared with control mice (AdGFP, n = 8; AdCRE, n = 8) after intracerebroventricular injection of AdCRE. White bar is 100 μm. *P < 0.01, Bonferroni post hoc comparison.
The ARed construct was created by first subcloning the CAG promoter from pDRIVE-CAG into PCR-Blunt-II-TOPO to yield pTOPO-CAG. It was then subcloned from pTOPO-CAG into pSTEC1 to yield pSTEC1-CAG (63). The human angiotensinogen (hAGT) gene was removed from pTOPO-hAGT and cloned into pSTEC1-CAG to yield pSTEC1-CAG-hAGT. The loxP-DsRed-STOP-loxP was removed from the Cre-Stoplight and cloned into pSTEC1-CAG-hAGT, which yielded ARed (69). The ARed construct was transfected into Cos-7 cells. DsRed fluorescence and hAGT expression were assayed. The final construct was isolated after cutting with BamH1 and injected into the pronuclei of C57BL/6J × SJL/J 1-cell fertilized mouse embryos. The mice were first backcross-bred to C57BL/6 and then to sR mice to generate the sRARed double-transgenic strain. The sR strain of transgenic mice carries the neuron-specific synapsin promoter driving a secreted form of active human renin due to replacement of the prorenin-converting enzyme cleavage site with a furin cleavage site (50).
Detection of tdTomato.
tdTomato reporter mice (ROSA) mice were injected intracerebroventricularly with AdCRE alongside each cohort of sRARed experimental mice that was injected. Three to six weeks after injection, they were anesthetized with ketamine/xylazine and were intracardially perfused with 4% paraformaldehyde in PBS. The fixed brains were then removed and sectioned at 40 μm on a vibratome for visualization of tdTomato fluorescence.
Quantitative RT-quantitative PCR.
The median preoptic nucleus (MnPO), SFO, and bilateral PVN were collected from individual sRARed and littermate control mice (33). Three to six weeks after intracerebroventricular injection of AdCRE, the mice were killed via CO2, their brains were removed, sunk and frozen in optimal cutting tool (OCT; Sakura). The brain was sectioned on a cryostat, and the MnPO, SFO, and bilateral PVN were identified, according to The Mouse Brain Atlas in Stereotaxic Coordinates (53). These brain areas were punched with a 0.5-mm punch (Stoeling). RNA was isolated from the punches and converted into cDNA for RT-quantitative PCR. hAGT-FAM and mouse actin-VIC-labeled TaqMan probes (Applied Biosystems) were used, so that hAGT and actin could be measured within the same reaction. Each sample was run with a no-reverse transcriptase control. If the CT value of the reverse transcriptase sample were greater than or equal to the no-reverse transcriptase control CT, it was considered not to be expressed. Expression of hAGT was quantified via a standard curve from 30 to 3 × 105 copies of recombinant hAGT cDNA (OriGene Technologies) and was normalized to actin.
Animals, surgery, and pharmacology.
Male and female mice 12–20 wk of age were used in this study. All procedures were approved by the University of Iowa Animal Care and Use Committee in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Double-transgenic sRARed mice consist of transgenic mice expressing human renin selectively in all neurons via the synapsin promoter (sR mice) bred with ARed transgenic mice (50, 55). There were no differences in survival comparing sRARed mice with single transgenic or nontransgenic littermates before or after AdCRE. Experimental mice were sRARed, whereas control mice consisted of either single transgenic sR, single transgenic ARed mice, or nontransgenic littermates. There was no increase in drinking when these control mice were given AdCRE intracerebroventricularly. Cre-recombinase reporter mice [B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J] mice were obtained from the Jackson Laboratory (stock no. 007914).
Intracerebroventricular injection or placement of a chronic intracerebroventricular cannula was performed, as described previously (18). For acute intracerebroventricular injection or placement of a chronic intracerebroventricular cannula, anesthesia was induced with 5% isoflurane in O2 and maintained with 2 ± 1% isoflurane in O2 (33). Mice were placed in a stereotaxic apparatus, and the lateral ventricle was located at the following coordinates: AP: 0.3 mm; ML: 1.0 mm; DV: 3.0 mm. One microliter of adenovirus was injected via a Hamilton syringe over 5 min. To selectively activate the ARed transgene in the SFO, 1 μl of Ad5CMV-Cre or Ad5-CMV-eGFP (1 × 107 pfu/μl; University of Iowa Gene Transfer and Vector Core) was injected intracerebroventricularly via a Hamilton syringe over 5 min (60, 61). Mice recovered from anesthesia in their home cage on a hot pad for 24 h after surgery. After intracerebroventricular injection of adenovirus, mice were placed in a special room in the vivarium for 1 wk as they were shedding virus, after which time, they were transferred to their normal room or were used for experiments. For intracerebroventricular cannula placement, 26-gauge guide cannulas were stereotaxically placed into the lateral ventricle, held in place with cranial screws and dental acrylic, and capped until injection with a 33-gauge internal cannula (PlasticsOne). Intracerebroventricular injections through the cannula were performed with a cannula injector attached to a gas-tight Hamilton syringe via tubing. Mineral oil was loaded into the tubing, and an air bubble was present between the drug and mineral oil to visualize movement.
Fluid intake was measured via a burette in either a home or metabolic cage (Nalgene). In some experiments, mice housed in metabolic cages were given a two-bottle comparator choice between deionized water and different molarities of NaCl made in deionized water (30). For these experiments, mice were intracerebroventricularly injected with AdCRE. Eighteen days after injection, they were placed into metabolic cages (Nalgene) and were given a two-bottle choice between water and 0.15 M NaCl. After 2 days of acclimation, 24-h fluid and food intake and body weight were measured and averaged for 2 days. The comparator drink was then changed to water and 0.3 M NaCl, and they were given a day of acclimation, after which time, similar measurements were taken for 2 days; this was repeated for 0.5 M NaCl.
Losartan (dose per figure legend; Sigma), a blocker of ANG II AT1R, bisindolylmaleimide I (BIM; 4 μg in 2 μl of aCSF; Cell Signaling), a blocker of PKC, and FR180204 (4 μg in 2 μl of DMSO; Santa Cruz Biotechnology), a blocker of ERK1/2, were injected into sRARed and littermate control mice, as described previously (18). The maximum change in water intake of losartan from aCSF was calculated by three-parameter, sigmoidal regression.
Blood pressure and metabolism.
Tail-cuff (Visitech Systems BP-2000) and radiotelemeter (TA11PA-C10; Data Sciences International) measurements of blood pressure were performed, as described previously (29). For tail cuff, mice were acclimated to the apparatus for 1 wk. After acclimation, 2 wk of baseline measurements were taken from the average of 30 measurements daily. Once baseline measurements were obtained, mice were injected with AdCRE intracerebroventricularly, as described above, and then daily measurements resumed 1 wk later and continued for 62 days. At day 24 or 25 after intracerebroventricular injection of AdCRE, water intake was measured from their home cage, as described above. For radiotelemetry, mice were implanted with radiotelemeters (TA11PA-C10; Data Sciences International) into the common carotid artery while under ketamine/xylazine anesthesia (33). The mice underwent intracerebroventricular placement of a cannula, and they recovered from the surgery for at least 5 days. After recovery, spontaneous physical activity, blood pressure, and heart rate were recorded every 10 s for 5 min using Dataquest software (Data Science International). Baseline measurements were taken for 1 wk. The mice then underwent intracerebroventricular injection of AdCRE, recovered for 1 wk, and daily measurements were taken for 5 wk. Any mouse with a poor radiotelemeter signal, pulse pressure below 15 mmHg, or a systolic BP above 200 mmHg (indicative of an occluded catheter) was excluded from further analysis. To ensure transgene activation in the cohort of mice that underwent blood pressure measurement, water intake was measured 24 days after AdCRE.
Mice were placed into temperature-controlled, insulated chambers for estimation of heat production by respirometry (10). First, the CO2 (model CD-3A, AEI) and O2 analyzers (model S-3A/II, AEI) were calibrated to standardized air containing 5,000 ppm CO2 and 20.50% O2 (Praxair), respectively. Mice were then placed into water-jacketed, temperature-controlled, and air-sealed chambers, and the change in effluent O2 and CO2 concentrations were recorded using a PowerLab and Chart software (AD Instruments). Flow was determined by mass flow meters (EM1; Sensiron) to STP-correct flow values. Heat production was estimated using the equation based on Lusk (1928): Heat = V̇o2 [1.232 respirtory exchange ratio (RER) + 3.815], where V̇o2 = [ΔO2%]·[STP-corrected flow] and RER = ΔCO2%/ΔO2%.
Serum chemistry.
Mice were euthanized with CO2 and truncal blood was collected into nonheparinized tubes stored on ice. Whole blood from the nonheparinized tubes was taken into a syringe, and 95 μl was injected into a CHEM-8+ I-Stat cartridge (Abbott Point of Care). The cartridges were run on a VetScan i-Stat1 (Abbott Point of Care).
Immunohistochemistry.
Mice were intracerebroventricularly injected with AdCRE. One month later, they were intracardially perfused with 4% paraformaldehyde and 0.1% gluteraldehyde in PBS. The brains were removed, and those designated as controls were marked for identification by a nick in the cortex. Brains were then sectioned frontally at 24 μm on a sliding microtome, collected into alternating wells filled with PBS, and each well contained an experimental and control group. Immunohistochemistry was performed for phosphorylated-CREB (1:400; Cell Signaling) using a biotinylated goat anti-rabbit IgG antibody (Vector Laboratories; no. BA-1000) at a concentration of 1:200. Biotin detection occurred by treating the sections with Vectastain ABC Elite kit (Vector Laboratories), and then visualizing the immunoreactivity by 3,3′-diaminobeznidine (DAB) plus hydrogen peroxide (SIGMAfast 3,3′-diaminobeznidine; Sigma D4168). Sections were then rinsed in PBS and cover slipped. The number of immunopositive cell fragments was determined by counting cells greater in size than a 3.0-μm diameter circle.
Statistics.
We analyzed the data with one- or two-way ANOVA, with repeated measurements as appropriate. Bonferroni multiple-comparison procedures were used to further explore treatment effects. If equal variance or normality failed, we used nonparametric analysis of our data, such as Mann-Whitney U- or Wilcoxon tests. We considered significance at P < 0.05, and all data plotted are expressed as means ± SE.
RESULTS
Intracerebroventricular AdCRE induces human angiotensinogen production in the SFO of sRARed mice.
A construct with conditionally inducible human angiotensinogen expression was designed to test the hypothesis that production of ANG II in the SFO is sufficient on its own to increase fluid intake. Upon Cre-mediated recombination, the gene encoding dsRED and a stop sequence are removed, allowing the ubiquitous CAG promoter to drive expression of human angiotensinogen (ARed construct; Fig. 1A). The addition of Cre-recombinase to Cos-7 cells transfected with ARed resulted in decreased dsRED fluorescence (Fig. 1B), a 20-fold induction of human angiotensinogen mRNA, and an increase in hAGT protein (Fig. 1C).
To test whether we can specifically induce human angiotensinogen expression in the SFO of ARed mice, we used a method previously validated by us and others to selectively target the SFO (29, 59). As shown in CAG-LSL-tdTomato mice (Fig. 1D), a reporter model for Cre-recombination, intracerebroventricular injection of AdCRE induces robust expression of the tdTomato reporter in the SFO (Fig. 1E). There was no induction of tdTomato expression in other regions of the brain, including the MnPO, PVN, or rostral ventrolateral medulla in this experiment or in previously reported cohorts (13, 33). Consistent with the tdTomato reporter mice, intracerebroventricular injection of AdCRE into ARed transgenic mice significantly induced expression of human angiotensinogen mRNA selectively in the SFO (Fig. 1F). There was no induction of human angiotensinogen mRNA when AdGFP was injected, and the induction in the PVN was not significantly different from that obtained with nontransgenic control mice.
Expression of the human RAS selectively in the SFO is insufficient to increase BP.
We next bred mice expressing neuron-specific human renin (sR) with ARed mice (double transgenic: sRARed) to allow Cre-mediated induction of ANG-II production in the SFO. Unexpectedly, an analysis of multiple cohorts of sRARed mice revealed there was no significant difference in tail cuff systolic blood pressure (Fig. 2A) or heart rate (Fig. 2B) compared with control mice either before or after intracerebrobentricular injection of AdCRE. To ensure this was not due to a lack of sensitivity of the tail cuff measurement, we confirmed these results with radiotelemetry over a 5-wk period after injection of AdCRE (Fig. 2, C and D). Five weeks is sufficient time to mediate a blood pressure increase in the slow-pressor model of peripheral ANG II administration (11, 21). Importantly, the same cohort of AdCRE-treated sRARed mice used for the tail cuff measurements of blood pressure exhibited increased water intake (at the end of the blood pressure experiment) compared with control mice (Fig. 3A). This is important because it demonstrates that the lack of an increase in arterial pressure is not the result of a failed viral delivery or Cre-mediated recombination. The increase in water intake in AdCRE-treated sRARed mice was returned to normal by the AT1R blocker losartan (Fig. 3B). The maximum change in water intake from vehicle to losartan was significantly greater in sRARed compared with control mice (Fig. 3C). Thus, whereas increased expression of the human RAS specifically in the SFO of sRARed mice is sufficient to increase water intake, it is insufficient to raise blood pressure.
Fig. 2.
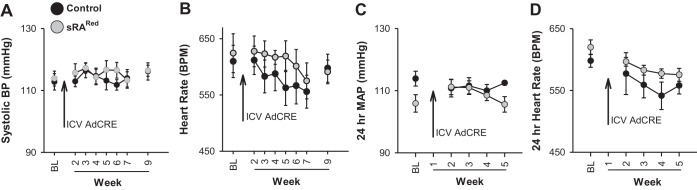
Blood pressure in sRARed mice. Tail-cuff was used to measure systolic blood pressure (BP; A) and heart rate (B) in control (n = 6) and sRARed mice (n = 6) before and up to 9 wk after intracerebroventricular injection of AdCRE. Radiotelemeters were used to measure 24-h mean arterial pressure (MAP; C) and heart rate (D) in control (n = 3–5) and sRARed mice (n = 4 or 5) before and 5 wk after intracerebroventricular injection of AdCRE.
Fig. 3.

Water intake in sRARed mice. A: water intake in one of the cohorts of control and sRARed mice used for the tail cuff measurements in Fig. 1 was taken at the end of the blood pressure experiment. B: 24-h water intake after either aCSF or increasing doses of intracerebroventricularly administered losartan in littermate control (n = 4) or sRARed (n = 5) mice previously injected with AdCRE intracerebroventricularly. C: three-parameter logarithmic regression was used to determine the maximum change in water intake from aCSF to intracerebroventricular losartan. *P < 0.05; †P = 0.056, Bonferroni post hoc comparison.
We previously showed that increased activity of the brain RAS increases resting metabolic rate (29, 30). Therefore, we sought to determine whether an SFO-selective increase in human RAS activity increased metabolic rate. Like blood pressure, there was no difference in resting metabolic rate, oxygen consumption (V̇o2), and heat production estimated by respirometry compared with controls after injection of AdCRE (Table 1).
Table 1.
Resting metabolism as measured by indirect calorimetry in littermate control (n = 6) and sRARed mice (n = 6) 68 days after intracerebroventricular injection of AdCRE
| Control | sRARed | |
|---|---|---|
| VO2, ml/min | 0.80 ± 0.02 | 0.71 ± 0.03 |
| VO2, ml·100 g−1·min−1 | 3.26 ± 0.17 | 2.95 ± 0.22 |
| RQ | 0.93 ± 0.01 | 1.02 ± 0.10 |
| Heat, kcal/h | 0.24 ± 0.01 | 0.22 ± 0.01 |
| Heat, kcal·kg−1·h−1 | 9.70 ± 0.52 | 8.90 ± 0.53 |
| TCO2 | 24.00 ± 0.91 | 24.33 ± 0.49 |
AdCRE, adenovirus expressing Cre-recombinase; RQ, respiratory quotient.
Expression of the human RAS selectively in the SFO is sufficient to increase fluid intake.
The data above suggest that increased activity of the human RAS in the SFO induces water intake when water is the only fluid offered. Consequently, we next examined fluid intake in mice given a two-bottle choice between water and isotonic saline (0.15 M NaCl). There was a significant increase in 24-h fluid intake in two separate cohorts of sRARed mice compared with controls starting 20 days after intracerebroventricular injection of AdCRE (Fig. 4A). This reflected an increase in 0.15 M NaCl (Fig. 4B) and total sodium uptake (Fig. 4C), but not an increase in water intake (Fig. 4D). Thus, when given a two-bottle choice with 0.15 M NaCl, the increase in fluid intake occurred because of an increased preference for saline (Fig. 4E). There was no change in food intake (Fig. 4F). Urine volume in AdCRE-treated sRARed mice was unchanged at baseline and at day 14 when there was no increase in fluid intake, but increased 1.9-fold at day 21 when the maximal increase in fluid intake was reached. We confirmed there was an increase in the expression of human angiotensinogen mRNA in the SFO of one of these cohorts similar to data in Fig. 1 (data not shown). There were no changes in serum chemistry or renal function (Table 2).
Fig. 4.

Fluid intake in two-bottle choice test. Intakes of total fluid (A), 0.15 M NaCl (B), total sodium (C), water (D), sodium preference (E), and food (F) in control (n = 6–13) and sRARed (n = 6–12) mice before and up to 24 days after intracerebroventricular injection of AdCRE. Mice were given a two-bottle comparator drink between 0.15 M NaCl and water. There are statistically significant interactions between genotype and period for total intakes of fluid and sodium. *P < 0.05, Bonferroni post hoc comparison.
Table 2.
Serum chemistry of littermate control (n = 5) and sRARed (n = 6) 32 days after intracerebroventricular injection of AdCRE
| Parameter | Control | sRARed |
|---|---|---|
| Na+, mmol/l | 148.50 ± 1.04 | 148.00 ± 0.97 |
| K+, mmol/l | 6.98 ± 0.39 | 6.87 ± 0.18 |
| Cl−, mmol/l | 116.25 ± 0.63 | 116.50 ± 0.89 |
| iCa2+, mmol/l | 1.20 ± 0.03 | 1.20 ± 0.05 |
| Anion gap, mmol/l | 15.75 ± 0.48 | 16.20 ± 0.89 |
| BUN, mg/dl | 21.00 ± 3.29 | 23.67 ± 2.04 |
| Creatinine, mg/dl | 0.23 ± 0.03 | 0.20 ± 0.00 |
| Hct, %PCV | 46.50 ± 1.04 | 46.00 ± 0.58 |
PCV, packed cell volume; BUN, blood urea nitrogen.
We next examined the intake and preferences for increased concentrations of saline. sRARed and control mice were placed into metabolic cages with a two-bottle choice between water and a comparator drink (0.15 M, 0.3 M, or 0.5 M NaCl) 20 days after intracerebroventricular injection of AdCRE (Fig. 5). When presented with 0.15 M NaCl as the comparator drink, total fluid intake in this cohort showed a trend toward being increased in sRARed mice (P = 0.056), and like the previous experiment and our previous studies in sRAFlox mice, this was a reflection of increased NaCl but not water intake (30). When presented with 0.3 M NaCl as the comparator drink, total fluid, water, and sodium intake was increased, but there was no increase in the preference for hypertonic saline. Urine sodium was also increased in AdCRE-treated sRARed mice given 0.3 M NaCl (Fig. 5F). There was no increase in fluid, water, or NaCl intake when 0.5 M NaCl was offered. Urine volume increased 2.13- and 2.26-fold in sRARed mice given 0.15 M and 0.3 M NaCl, respectively, but were not different from controls at 0.5 M NaCl.
Fig. 5.
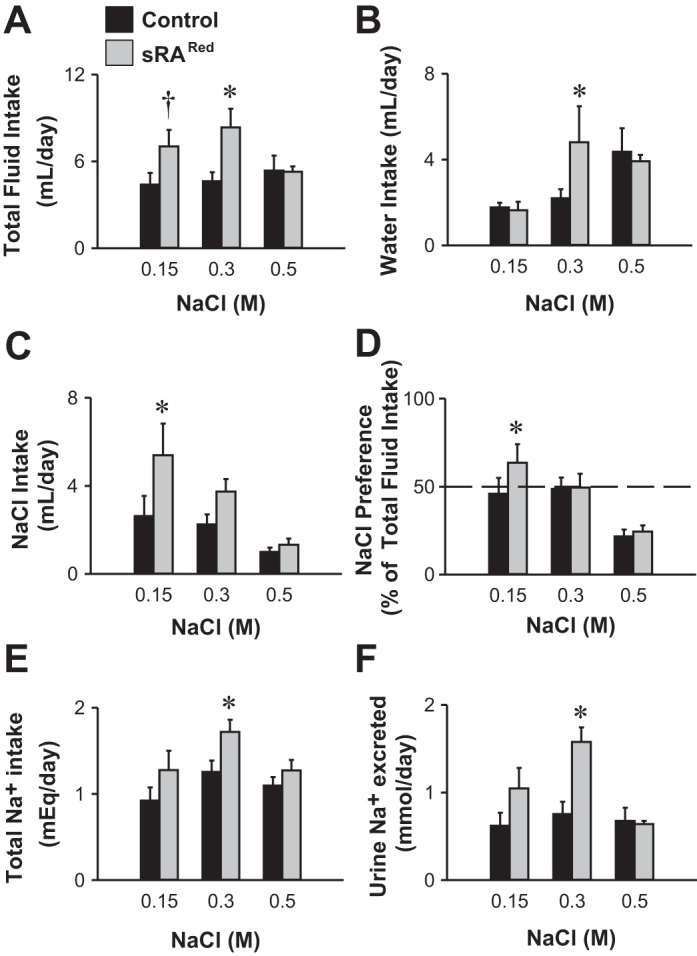
Fluid intake in two-bottle choice test with different comparator drinks. Total fluid (A), water (B), saline (C), and sodium (E) intakes in littermate control (n = 9) and sRARed (n = 6) mice after intracerebroventricular injection of AdCRE and given sequential two-bottle comparator drink tests of water vs. NaCl (0.15 M, 0.3 M, or 0.5 M). Preference for drinking NaCl (D) and excretion of sodium in the urine (F) of control and sRARed mice. There are statistically significant interactions between genotype and molarity for total fluid intake and urine sodium. *P < 0.05, †P = 0.056, Bonferroni post hoc comparison.
Increased fluid intake in AdCRE-treated sRARed is mediated by PKC.
In addition to the evidence for SFO-specific expression of human angiotensinogen mRNA obtained above, we sought to obtain direct evidence of increased human angiotensinogen protein or ANG II in the SFO of AdCRE-treated sRARed mice. However, the level of both products was below the level of detection by immunohistochemistry, perhaps explaining why there was no increase in blood pressure. Therefore, we sought to assay for surrogate markers downstream of AT1R activation. We have previously shown that elevated fluid intake due to hyperactivity of the brain RAS is mediated through PKC-α (13). We injected the pan-active PKC inhibitor BIM into AdCRE-treated sRARed. PKC inhibition corrected the increased water intake in AdCRE-treated sRARed, lowering it to the level observed in control mice (Fig. 6, A and B). There was no change in water intake when the ERK pathway was inhibited in AdCRE-treated sRARed mice with FR180204 (Fig. 6, C and D). Phosphorylation of CREB has been shown to be a downstream effector of ANG-II AT1R action in the rostral ventrolateral medulla and vascular smooth muscle cells (12, 67). We observed both an increase in the intensity (Fig. 7) and number of phospho-CREB-labeled cells (Fig. 8) within the SFO and MnPO of sRARed compared with control mice. There was no increase in the number of phospho-CREB-positive cells in the PVN, supraoptic nuclei, nucleus accumbens, or lateral hypothalamus, nor in the cortex and hippocampus (data not shown).
Fig. 6.
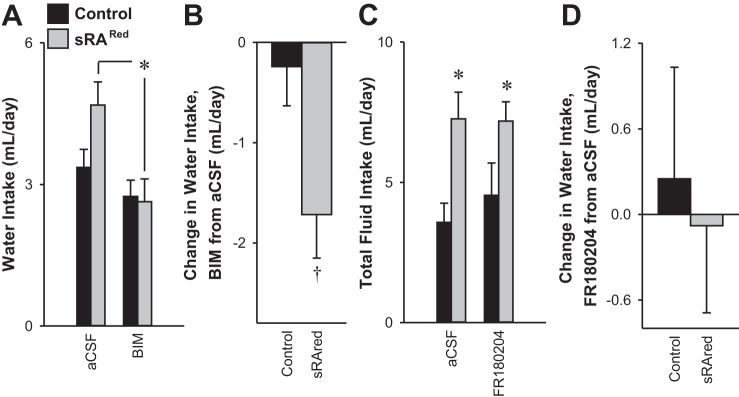
Water intake in response to PKC and ERK inhibition. Water intake (A) and the change in water intake (B) of control (n = 8) and sRARed (n = 11) mice previously injected intracerebroventricularly with AdCRE and treated with either artificial cerebrospinal fluid (aCSF) or bisindolylmaleimide I (BIM). Total fluid intake (C) and the change in total fluid intake (D) of littermate control (n = 5) and sRARed mice (n = 7) previously injected intracerebroventricularly with AdCRE, given a two-bottle comparator drink of water vs. 0.3 M NaCl and treated with either aCSF or FR180204 intracerebroventricularly. *P < 0.05, Bonferroni post hoc comparison. †P < 0.05, independent t-test.
Fig. 7.

Expression of phosphorylated CREB. Representative images of p-CREB within the SFO and MnPO of littermate control and sRARed mice previously injected intracerebroventricularly with AdCRE. Scale is 100 μm for the SFO and 200 μm for the MnPO. *, choroid plexus; 3V, third ventricle; ac, anterior commissure.
Fig. 8.
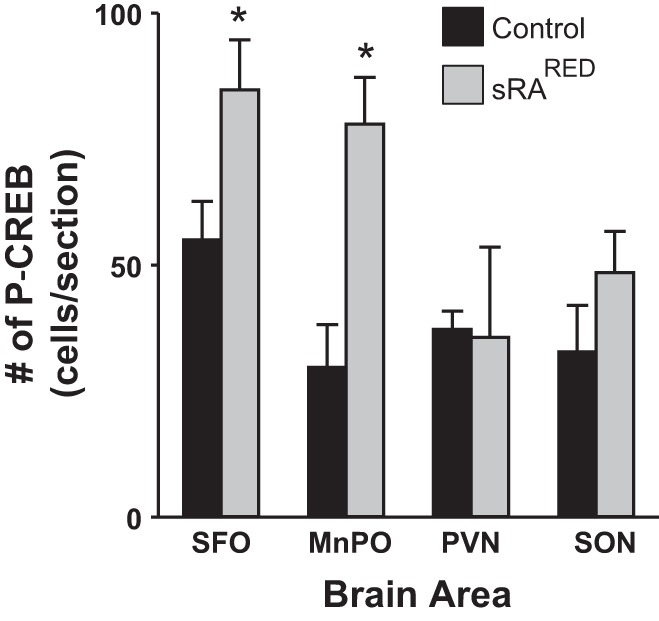
Quantification of p-CREB. The number of immunopositive cells within the SFO, MnPO, PVN, and SON of littermate control (n = 5) or sRARed (n = 5–8) mice previously injected intracerebroventricularly with AdCRE (C). *P < 0.05 vs. control.
DISCUSSION
There are several important findings in this study. First, the induction of human RAS expression selectively in the SFO of transgenic mice causes increased water intake that is mediated by an AT1R- and PKC-dependent mechanism. Second, increased fluid intake, when given as a choice between 0.15 M NaCl and water, is due to increased consumption of 0.15 M NaCl but not water, suggestive of an increased preference for saline under nonaversive conditions. Third, when the mice are provided a choice between 0.3 M NaCl and water, their increased fluid intake was largely due to a preference for water, although they exhibited increased sodium intake as well. There was no increase in fluid intake when 0.5 M NaCl was provided, suggesting it was aversive. From this, we can conclude that localized production of ANG II in the SFO in this model induces drinking but does not alter sodium appetite, formally defined as increased preference for NaCl under aversive conditions. Finally, SFO-selective expression of the human RAS is insufficient to increase arterial pressure or resting metabolic rate, at least at the level of expression observed in the AdCRE-treated sRARed model. Thus, it is possible that the neural pathways controlling drinking may be more sensitive to ANG II than the pathways controlling blood pressure or metabolism.
It has long been known that direct administration of ANG II to the SFO induces drinking and that the induction of drinking in response to centrally or peripherally administered ANG II can be blocked by lesions of the SFO (49, 57, 58). We previously showed that overexpression of the human renin and angiotensinogen genes throughout the brain (the sRAFlox model) results in a marked elevation of both water intake and blood pressure that is dependent upon expression of the ANG II substrate in the SFO (59). In a recent study, we showed that the increase in ANG II-mediated drinking is due to activation of PKCα in the SFO (13). In the current study, we generated a new model to address whether SFO-selective induction of ANG II production is sufficient on its own to mediate increased drinking and blood pressure. Unlike the sRAFlox model in which Cre-recombinase causes an ablation of human angiotensinogen expression, in the sRARed model, angiotensinogen expression, and consequently, ANG II production, is induced by Cre-recombinase. The strength of this model is that the ARed construct allows for temporal and spatial control over the production of ANG II dictated by the site of Cre-recombinase expression in the presence of human renin.
We showed that intracerebroventricular injection of AdCRE into ARed mice significantly induces the production of human angiotensinogen mRNA in the SFO, but not the MnPO. While the increase in human angiotensinogen mRNA in the PVN was not significant, there was a trend for its induction, which may be due to interanimal variability. There is substantial evidence supporting an angiotensinergic neural connection between the SFO and the PVN, which uses ANG II as the neurotransmitter (reviewed in Ref. 24). Moreover, ANG II directly injected into the PVN increases blood pressure (3). Thus, we cannot rule out that in some mice, increased ANG II production in the PVN may have contributed to the phenotypes that we observed. We also cannot rule out the possibility of increased production of human angiotensinogen in other relevant nuclei. This may be important as nuclei in the AV3V region, which have extensive interconnectivity with the SFO, have been implicated in mediating drinking responses to ANG II (7). For example, although there was no evidence for induction of human angiotensinogen in the MnPO and no evidence that AdCRE caused recombination in the MnPO, we found elevated phosphorylated CREB in the MnPO of AdCRE-treated sRARed mice. The increase in phosphorylated CREB could be an indicator of ANG II-mediated neural activity in the MnPO. This would be consistent with extensive angiotensinergic interconnectivity between the SFO and MnPO, as well as the role of the MnPO in drinking (14, 25, 35, 43).
There were several surprising results of our study. First, the increase in fluid intake in AdCRE-treated sRARed mice occurred over a period of 3 wk. This is surprising because direct injection of ANG II centrally or directly into the SFO elicits a rapid increase in drinking behavior (56, 58). The increase in water intake that we observed in sRAFlox mice was quite robust, but because transgenes in these mice were not inducible and were expressed lifelong, there was no way to determine when the dipsogenic response started. We know that the production of human angiotensinogen mRNA in AdCRE-treated sRARed mice was an order of magnitude lower than observed in sRAFlox mice, and human angiotensinogen protein and angiotensin peptides were both undetectable in sRARed mice. Thus, the delayed drinking response may result from a slow accumulation of ANG II over time. We are certain that the increased drinking is ANG II-dependent because single transgenic sR or ARed mice treated with AdCRE had no response, and is AT1R-dependent because it was normalized with losartan. The delayed response may also be due to the observation from some studies that compared with rats, higher doses of ANG II have been reported to be required to mediate drinking responses in mice (54). Other studies reported similar drinking responses to central ANG II between mice and rats (19, 36).
The second surprising finding was the lack of an increase in baseline blood pressure. Lesions of the SFO block the pressor action of peripheral ANG II, AT1R in the SFO are thought to mediate responses to some psychological stressors, and hypertension can be induced by electrically stimulating the SFO (34, 38, 48, 56). The SFO richly expresses AT1Rs, which are required to mediate changes in blood pressure in response to central sodium and DOCA-salt (33, 65). On the contrary, lesions of the SFO did not attenuate experimental renal hypertension nor the hypertension caused by short-term ANG II infusion, and, in fact, augmented blood pressure in the latter (4, 37). We have to consider the possibility that whereas the level of expression of human renin and angiotensinogen may have been sufficient to stimulate the neural pathways involved in drinking, it may have been insufficient to stimulate the pathways required to mediate an increase in blood pressure. Additional studies would be needed to test the hypothesis that there is a differential sensitivity to the dipsogenic and pressor actions of ANG II in the SFO. Lesion studies suggest that other brain regions in addition to the SFO mediate blood pressure increases to ANG II in a model of ANG II/high-salt hypertension (52). Differential effects of ANG II on blood pressure and drinking were also observed in Sprague-Dawley rats in response to AT2R inhibition (66).
AdCRE-treated sRARed mice increase their water intake when water is the only fluid offered. This is likely due to the direct effects of ANG II within the brain because it is blocked by intracerebroventricular administration of either losartan or an inhibitor of PKC and because there is no evidence for an alteration in renal function and serum chemistry. We recently showed that drinking in the sRAFlox model is due to PKCα activation in the SFO (13). In rats, ANG II stimulates central PKCα activity, and PKC blockers prevent the dipsogenic response to intracerebroventricular ANG II (27). On the contrary, there was no effect on ANG II-stimulated water intake when the ERK pathway was blocked. This is consistent with studies showing that ANG II-induced water and salt intakes are mediated by divergent intracellular signaling cascades downstream of the AT1R, PKC for water intake, and MAP kinase for NaCl intake (15, 16, 22). This divergence in AT1R signaling pathways controlling water and salt intake was recently supported by the identification of a novel small peptide encoded by the AT1R gene, which selectively blocks the ANG II-ERK1/2 pathway both in vitro and in vivo (45). As we did not measure the effect of ERK inhibition when mice were provided saline, we are unable to determine whether the ERK pathway is selectively activated in response to SFO ANG II under those conditions.
It is well established that central ANG II causes increased intake of dilute saline solutions (5, 6, 22, and reviewed by Fitzsimons; see reference 26). Consistent with this, when given a choice between 0.15 M NaCl and water, sRARed mice prefer saline over water. On the contrary, when offered a choice between 0.3 M NaCl or water, the preference is for water, and thus, sRARed mice do not exhibit an increase in salt appetite despite the preference for palatable saline. Increased sodium appetite has been reported in sodium-depleted rats caused by coadministration of furosemide and captopril, which was attenuated by lesion of the SFO (46, 51). Rats given peripheral injections of either renin or ANG II, central injections of ANG II, or DOCA-salt also exhibit a sodium appetite (1, 5). Sodium appetite is also increased in central ANG II-treated transgenic mice overexpressing AT1R receptors selectively in neurons (41). It is possible that we did not observe an increase in sodium appetite in sRARed mice because the mice were sodium replete on a standard rather than low-sodium diet or that the low level of ANG II produced in this model did not gain access to the MnPO, although this is not consistent with our data showing increased phosphorylated CREB in the MnPO (25). It should be noted that the mice used to measure phosphorylated CREB were not provided saline. It is notable that compared with rats, mice are much more resistant to ANG II-induced drinking despite a similar increase in c-Fos staining in the SFO (54). Also, sRAFlox mice exhibit the same pattern of increased sodium intake when palatable, but not when aversive, and, therefore, exhibit increased “intake” but not “appetite” (30). Thus, our data using intracerebroventricular AdCRE-treated sRARed mice suggest that SFO-specific ANG II hyperactivity is sufficient to recapitulate the fluid intake patterns that are present with whole brain RAS hyperactivity in mice.
Perspectives and Significance
We show that production of ANG II selectively in the SFO is sufficient on its own to increase sodium and fluid intakes via a preference for palatable saline, although there is no increase in sodium appetite or change in blood pressure. This suggests that the mechanisms controlling drinking and blood pressure in response to local production and action of ANG II in the SFO may differ, either in terms of its dose response, or more likely the cellular specificity of where ANG II acts. Whether ANG II acts on neurons projecting to nuclei controlling water and electrolyte homeostasis or to nuclei controlling the preganglionic neurons of the sympathetic nervous system likely dictates the response.
GRANTS
This work was supported through research grants from the National Institutes of Health (NIH) to C. D. Sigmund (HL-048058, HL-061446, HL-062984), to C. D. Sigmund and J. L. Grobe (HL-084207), and to J. L. Grobe (HL-098276). Transgenic mice were generated at the University of Iowa Transgenic Animal Facility supported, in part, by grants from the NIH and from the Roy J. and Lucille A. Carver College of Medicine. The authors also gratefully acknowledge the generous research support of the Roy J. Carver Trust.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: J.P.C., J.L.G., and C.D.S. conception and design of research; J.P.C., M.D.C., and D.R.D. performed experiments; J.P.C., M.D.C., D.R.D., J.L.G., and C.D.S. analyzed data; J.P.C., M.D.C., J.L.G., and C.D.S. interpreted results of experiments; J.P.C. and C.D.S. prepared figures; J.P.C. and C.D.S. drafted manuscript; J.P.C., M.D.C., J.L.G., and C.D.S. edited and revised manuscript; J.P.C., M.D.C., D.R.D., J.L.G., and C.D.S. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors would like to thank Xin Tian for generation of the ARed construct. We also thank Dr. L. Philip Sanford, Norma Sinclair, JoAnne Schwarting, and Patricia Yarolem for generating and genotyping mice. The University of Iowa Vector Core generated the adenoviruses.
REFERENCES
- 1.Acerbo MJ, Johnson AK. Behavioral cross-sensitization between DOCA-induced sodium appetite and cocaine-induced locomotor behavior. Pharmacol Biochem Behav 98: 440–448, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen AM, O'Callaghan EL, Hazelwood L, Germain S, Castrop H, Schnermann J, Bassi JK. Distribution of cells expressing human renin-promoter activity in the brain of a transgenic mouse. Brain Res 1243: 78–85, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Bains JS, Potyok A, Ferguson AV. Angiotensin II actions in paraventricular nucleus: functional evidence for neurotransmitter role in efferents originating in subfornical organ. Brain Res 599: 223–229, 1992 [DOI] [PubMed] [Google Scholar]
- 4.Bruner CA, Mangiapane ML, Fink GD. Subfornical organ. Does it protect against angiotensin II-induced hypertension in the rat? Circ Res 56: 462–466, 1985 [DOI] [PubMed] [Google Scholar]
- 5.Bryant RW, Epstein AN, Fitzsimons JT, Fluharty SJ. Arousal of a specific and persistent sodium appetite in the rat with continuous intracerebroventricular infusion of angiotensin II. J Physiol 301: 365–382, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buggy J, Fisher AE. Evidence for a dual central role for angiotensin in water and sodium intake. Nature 250: 733–735, 1974 [DOI] [PubMed] [Google Scholar]
- 7.Buggy J, Fisher AE. Anteroventral third ventricle site of action for angiotensin-induced thirst. Pharmacol Biochem Behav 4: 651–660, 1976 [DOI] [PubMed] [Google Scholar]
- 8.Buggy J, Fisher AE, Hoffman WE, Johnson AL, Phillips MI. Ventricular obstruction: effect on drinking induced by intracranial injection of angiotensin. Science 190: 72–74, 1975 [DOI] [PubMed] [Google Scholar]
- 9.Bunnemann B, Iwai N, Metzger R, Fuxe K, Inagami T, Ganten D. The distribution of angiotensin II AT1 receptor subtype mRNA in the rat brain. Neurosci Lett 142: 155–158, 1992 [DOI] [PubMed] [Google Scholar]
- 10.Burnett CM, Grobe JL. Direct calorimetry identifies deficiencies in respirometry for the determination of resting metabolic rate in C57BL/6 and FVB mice. Am J Physiol Endocrinol Metab 305: E916–E924, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao X, Peterson JR, Wang G, Anrather J, Young CN, Guruju MR, Burmeister MA, Iadecola C, Davisson RL. Angiotensin II-dependent hypertension requires cyclooxygenase 1-derived prostaglandin E2 and EP1 receptor signaling in the subfornical organ of the brain. Hypertension 59: 869–876, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan SH, Wang LL, Tseng HL, Chan JY. Upregulation of AT1 receptor gene on activation of protein kinase Cβ/nicotinamide adenine dinucleotide diphosphate oxidase/ERK1/2/c-fos signaling cascade mediates long-term pressor effect of angiotensin II in rostral ventrolateral medulla. J Hypertens 25: 1845–1861, 2007 [DOI] [PubMed] [Google Scholar]
- 13.Coble JP, Johnson RF, Cassell MD, Johnson AK, Grobe JL, Sigmund CD. Activity of PKC-α within the subfornical organ is necessary for fluid intake due to brain angiotensin. Hypertension 64: 141–148, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.da Silva RK, Saad WA, Renzi A, Menani JV, Camargo LA. Effect of lateral hypothalamus lesions on the water and salt intake, and sodium and urine excretion induced by activation of the median preoptic nucleus in conscious rats. J Auton Nerv Syst 53: 195–204, 1995 [DOI] [PubMed] [Google Scholar]
- 15.Daniels D, Mietlicki EG, Nowak EL, Fluharty SJ. Angiotensin II stimulates water and NaCl intake through separate cell signalling pathways in rats. Exp Physiol 94: 130–137, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daniels D, Yee DK, Faulconbridge LF, Fluharty SJ. Divergent behavioral roles of angiotensin receptor intracellular signaling cascades. Endocrinology 146: 5552–5560, 2005 [DOI] [PubMed] [Google Scholar]
- 17.Davisson RL, Oliverio MI, Coffman TM, Sigmund CD. Divergent functions of angiotensin II receptor isoforms in brain. J Clin Invest 106: 103–106, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davisson RL, Yang G, Beltz TG, Cassell MD, Johnson AK, Sigmund CD. The brain renin-angiotensin system contributes to the hypertension in mice containing both the human renin and human angiotensinogen transgenes. Circ Res 83: 1047–1058, 1998 [DOI] [PubMed] [Google Scholar]
- 19.Denton DA, Blair-West JR, McBurnie M, Osborne PG, Tarjan E, Williams RM, Weisinger RS. Angiotensin and salt appetite of BALB/c mice. Am J Physiol Regul Integr Comp Physiol 259: R729–R735, 1990 [DOI] [PubMed] [Google Scholar]
- 20.Deschepper CF, Bouhnik J, Ganong WF. Colocalization of angiotensinogen and glial fibrillary acidic protein in astrocytes in rat brain. Brain Res 374: 195–198, 1986 [DOI] [PubMed] [Google Scholar]
- 21.Edgley A, Kett M, Anderson W. ‘Slow pressor’ hypertension from low-dose chronic angiotensin II infusion. Clin Exp Pharmacol Physiol 28: 1035–1039, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Felgendreger LA, Fluharty SJ, Yee DK, Flanagan-Cato LM. Endogenous angiotensin II-induced p44/42 mitogen-activated protein kinase activation mediates sodium appetite but not thirst or neurohypophysial secretion in male rats. J Neuroendocrinol 25: 97–106, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferguson AV. Neurophysiological analysis of mechanisms for subfornical organ and area postrema involvement in autonomic control. Progr Brain Res 91: 413–421, 1992 [DOI] [PubMed] [Google Scholar]
- 24.Ferguson AV, Washburn DL, Latchford KJ. Hormonal and neurotransmitter roles for angiotensin in the regulation of central autonomic function. Exp Biol Med (Maywood) 226: 85–96, 2001 [DOI] [PubMed] [Google Scholar]
- 25.Fitts DA, Freece JA, Van Bebber JE, Zierath DK, Bassett JE. Effects of forebrain circumventricular organ ablation on drinking or salt appetite after sodium depletion or hypernatremia. Am J Physiol Regul Integr Comp Physiol 287: R1325–R1334, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Fitzsimons JT. Angiotensin, thirst, and sodium appetite. Physiol Rev 78: 583–686, 1998 [DOI] [PubMed] [Google Scholar]
- 27.Fleegal MA, Sumners C. Drinking behavior elicited by central injection of angiotensin II: roles for protein kinase C and Ca2+/calmodulin-dependent protein kinase II. Am J Physiol Regul Integr Comp Physiol 285: R632–R640, 2003 [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez AD, Wang G, Waters EM, Gonzales KL, Speth RC, Van Kempen TA, Marques-Lopes J, Young CN, Butler SD, Davisson RL, Iadecola C, Pickel VM, Pierce JP, Milner TA. Distribution of angiotensin type 1a receptor-containing cells in the brains of bacterial artificial chromosome transgenic mice. Neuroscience 226: 489–509, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grobe JL, Buehrer BA, Hilzendeger AM, Liu X, Davis DR, Xu D, Sigmund CD. Angiotensinergic signaling in the brain mediates metabolic effects of deoxycorticosterone (DOCA)-salt in C57 mice. Hypertension 57: 600–607, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grobe JL, Grobe CL, Beltz TG, Westphal SG, Morgan DA, Xu D, de Lange WJ, Li H, Sakai K, Thedans DR, Cassis LA, Rahmouni K, Mark AL, Johnson AK, Sigmund CD. The Brain renin-angiotensin system controls divergent efferent mechanisms to regulate fluid and energy balance. Cell Metab 12: 431–442, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grobe JL, Xu D, Sigmund CD. An intracellular renin-angiotensin system in neurons: fact, hypothesis, or fantasy. Physiology 23: 187–193, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hermann K, Raizada MK, Sumners C, Phillips MI. Presence of renin in primary neuronal and glial cells from rat brain. Brain Res 437: 205–213, 1987 [DOI] [PubMed] [Google Scholar]
- 33.Hilzendeger AM, Cassell MD, Davis DR, Stauss HM, Mark AL, Grobe JL, Sigmund CD. Angiotensin type 1a receptors in the subfornical organ are required for deoxycorticosterone acetate-salt hypertension. Hypertension 61: 716–722, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishibashi S, Nicolaidis S. Hypertension induced by electrical stimulation of the subfornical organ (SFO). Brain Res Bull 6: 135–139, 1981 [DOI] [PubMed] [Google Scholar]
- 35.Johnson AK, Cunningham JT, Thunhorst RL. Integrative role of the lamina terminalis in the regulation of cardiovascular and body fluid homeostasis. Clin Exp Pharmacol Physiol 23: 183–191, 1996 [DOI] [PubMed] [Google Scholar]
- 36.Johnson RF, Beltz TG, Thunhorst RL, Johnson AK. Investigations on the physiological controls of water and saline intake in C57BL/6 mice. Am J Physiol Regul Integr Comp Physiol 285: R394–R403, 2003 [DOI] [PubMed] [Google Scholar]
- 37.Knuepfer MM, Johnson AK, Brody MJ. Effect of subfornical organ ablation on the development of renal hypertension. Clin Exp Hypertens A 6: 1027–1034, 1984 [DOI] [PubMed] [Google Scholar]
- 38.Krause EG, de Kloet AD, Scott KA, Flak JN, Jones K, Smeltzer MD, Ulrich-Lai YM, Woods SC, Wilson SP, Reagan LP, Herman JP, Sakai RR. Blood-borne angiotensin II acts in the brain to influence behavioral and endocrine responses to psychogenic stress. J Neurosci 31: 15009–15015, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lavoie JL, Cassell MD, Gross KW, Sigmund CD. Adjacent expression of renin and angiotensinogen in the rostral ventrolateral medulla using a dual-reporter transgenic model. Hypertension 43: 1116–1119, 2004 [DOI] [PubMed] [Google Scholar]
- 40.Lavoie JL, Cassell MD, Gross KW, Sigmund CD. Localization of renin expressing cells in the brain using a REN-eGFP transgenic model. Physiol Genomics 16: 240–246, 2004 [DOI] [PubMed] [Google Scholar]
- 41.Lazartigues E, Sinnayah P, Augoyard G, Gharib C, Johnson AK, Davisson RL. Enhanced water and salt intake in transgenic mice with brain-restricted overexpression of angiotensin (AT1) receptors. Am J Physiol Regul Integr Comp Physiol 295: R1539–R1545, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lind RW, Johnson AK. Subfornical organ-median preoptic connections and drinking and pressor responses to angiotensin II. J Neurosci 2: 1043–1051, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lind RW, Swanson LW, Ganten D. Angiotensin II immunoreactivity in the neural afferents and efferents of the subfornical organ of the rat. Brain Res 321: 209–215, 1984 [DOI] [PubMed] [Google Scholar]
- 44.Lind RW, Swanson LW, Ganten D. Organization of angiotensin II immunoreactive cells and fibers in the rat central nervous system. An immunohistochemical study. Neuroendocrinology 40: 2–24, 1985 [DOI] [PubMed] [Google Scholar]
- 45.Liu J, Yosten GL, Ji H, Zhang D, Zheng W, Speth RC, Samson WK, Sandberg K. Selective inhibition of angiotensin receptor signaling through Erk1/2 pathway by a novel peptide. Am J Physiol Regul Integr Comp Physiol 306: R619–R626, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu B, Yan J, Yang X, Li J, Chen K. Involvement of brain ANG II in acute sodium depletion induced salty taste changes. Regul Pept 179: 15–22, 2012 [DOI] [PubMed] [Google Scholar]
- 47.MacGregor DP, Murone C, Song K, Allen AM, Paxinos G, Mendelsohn FA. Angiotensin II receptor subtypes in the human central nervous system. Brain Res 675: 231–240, 1995 [DOI] [PubMed] [Google Scholar]
- 48.Mangiapane ML, Simpson JB. Subfornical organ lesions reduce the pressor effect of systemic angiotensin II. Neuroendocrinology 31: 380–384, 1980 [DOI] [PubMed] [Google Scholar]
- 49.Mangiapane ML, Simpson JB. Subfornical organ: forebrain site of pressor and dipsogenic action of angiotensin II. Am J Physiol Regul Integr Comp Physiol 239: R382–R389, 1980 [DOI] [PubMed] [Google Scholar]
- 50.Morimoto S, Cassell MD, Sigmund CD. Glial- and neuronal-specific expression of the renin-angiotensin system in brain alters blood pressure, water intake, and salt preference. J Biol Chem 277: 33235–33241, 2002 [DOI] [PubMed] [Google Scholar]
- 51.Morris MJ, Wilson WL, Starbuck EM, Fitts DA. Forebrain circumventricular organs mediate salt appetite induced by intravenous angiotensin II in rats. Brain Res 949: 42–50, 2002 [DOI] [PubMed] [Google Scholar]
- 52.Osborn JW, Hendel MD, Collister JP, Ariza-Guzman PA, Fink GD. The role of the subfornical organ in angiotensin II-salt hypertension in the rat. Exp Physiol 97: 80–88, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paxinos G, Watson CS. The mouse brain in stereotaxic coordinates. New York: Academic, 2012 [Google Scholar]
- 54.Rowland NE, Goldstein BE, Robertson KL. Role of angiotensin in body fluid homeostasis of mice: fluid intake, plasma hormones, and brain Fos. Am J Physiol Regul Integr Comp Physiol 284: R1586–R1594, 2003 [DOI] [PubMed] [Google Scholar]
- 55.Sakai K, Agassandian K, Morimoto S, Sinnayah P, Cassell MD, Davisson RL, Sigmund CD. Local production of angiotensin II in the subfornical organ causes elevated drinking. J Clin Invest 117: 1088–1095, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simpson JB, Epstein AN, Camardo JS., Jr Localization of receptors for the dipsogenic action of angiotensin II in the subfornical organ of rat. J Comp Physiol Psychol 92: 581–601, 1978 [DOI] [PubMed] [Google Scholar]
- 57.Simpson JB, Routtenberg A. Subfornical organ lesions reduce intravenous angiotensin-induced drinking. Brain Res 88: 154–161, 1975 [DOI] [PubMed] [Google Scholar]
- 58.Simpson JB, Routtenberg A. Subfornical organ: a dipsogenic site of action of angiotensin II. Science 201: 379–381, 1978 [DOI] [PubMed] [Google Scholar]
- 59.Sinnayah P, Lazartigues E, Sakai K, Sharma RV, Sigmund CD, Davisson RL. Genetic ablation of angiotensinogen in the subfornical organ of the brain prevents the central angiotensinergic pressor response. Circ Res 99: 1125–1131, 2006 [DOI] [PubMed] [Google Scholar]
- 60.Sinnayah P, Lindley TE, Staber PD, Cassell MD, Davidson BL, Davisson RL. Selective gene transfer to key cardiovascular regions of the brain: comparison of two viral vector systems. Hypertension 39: 603–608, 2002 [DOI] [PubMed] [Google Scholar]
- 61.Sinnayah P, Lindley TE, Staber PD, Davidson BL, Cassell MD, Davisson RL. Targeted viral delivery of Cre recombinase induces conditional gene deletion in cardiovascular circuits of the mouse brain. Physiol Genomics 18: 25–32, 2004 [DOI] [PubMed] [Google Scholar]
- 62.Smith PM, Ferguson AV. Circulating signals as critical regulators of autonomic state—central roles for the subfornical organ. Am J Physiol Regul Integr Comp Physiol 299: R405–R415, 2010 [DOI] [PubMed] [Google Scholar]
- 63.Stec DE, Morimoto S, Sigmund CD. Vectors for high-level expression of cDNAs controlled by tissue-specific promoters in transgenic mice. Biotechniques 31: 256–260, 2001 [DOI] [PubMed] [Google Scholar]
- 64.Stornetta RL, Hawelu Johnson CL, Guyenet PG, Lynch KR. Astrocytes synthesize angiotensinogen in brain. Science 242: 1444–1446, 1988 [DOI] [PubMed] [Google Scholar]
- 65.Tiruneh MA, Huang BS, Leenen FH. Role of angiotensin II type 1 receptors in the subfornical organ in the pressor responses to central sodium in rats. Brain Res 1527: 79–86, 2013 [DOI] [PubMed] [Google Scholar]
- 66.Wang H, Gallinat S, Li HW, Sumners C, Raizada MK, Katovich MJ. Elevated blood pressure in normotensive rats produced by ‘knockdown’ of the angiotensin type 2 receptor. Exp Physiol 89: 313–322, 2004 [DOI] [PubMed] [Google Scholar]
- 67.Xie Z, Liu D, Liu S, Calderon L, Zhao G, Turk J, Guo Z. Identification of a cAMP-response element in the regulator of G-protein signaling-2 (RGS2) promoter as a key cis-regulatory element for RGS2 transcriptional regulation by angiotensin II in cultured vascular smooth muscles. J Biol Chem 286: 44646–44658, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang G, Gray TS, Sigmund CD, Cassell MD. The angiotensinogen gene is expressed in both astrocytes and neurons in murine central nervous system. Brain Res 817: 123–131, 1999 [DOI] [PubMed] [Google Scholar]
- 69.Yang YS, Hughes TE. Cre stoplight: a red/green fluorescent reporter of Cre recombinase expression in living cells. Biotechniques 31: 1036, 1038, 1040–1036, 1038, 1041, 2001 [DOI] [PubMed] [Google Scholar]