

Abstract
Exposing mice to a chronic hypoxic treatment (10% oxygen, 21 days) that promotes pulmonary hypertension was observed to attenuate the pulmonary vasoconstriction response to acute hypoxia (HPV) both in vivo and in isolated pulmonary arteries. Since catalase restored the HPV response in isolated arteries, it appeared to be attenuated by extracellular hydrogen peroxide. Chronic hypoxia promoted the detection of elevated lung superoxide, extracellular peroxide, extracellular SOD expression, and protein kinase G (PKG) activation [based on PKG dimerization and vasodilator-stimulated phosphoprotein (VASP) phosphorylation], suggesting increased generation of extracellular peroxide and PKG activation may contribute to the suppression of HPV. Aorta from mice exposed to 21 days of hypoxia also showed evidence for extracellular hydrogen peroxide, suppressing the relaxation response to acute hypoxia. Peroxide appeared to partially suppress contractions to phenylephrine used in the study of in vitro hypoxic responses. Treatment of mice with the heme precursor δ-aminolevulinic acid (ALA; 50 mg·kg−1·day−1) during exposure to chronic hypoxia was examined as a pulmonary hypertension therapy because it could potentially activate beneficial cGMP-mediated effects through promoting a prolonged protoporphyrin IX (PpIX)-elicited activation of soluble guanylate cyclase. ALA attenuated pulmonary hypertension, increases in both superoxide and peroxide, and the suppression of in vitro and in vivo HPV responses. ALA generated prolonged detectible increases in PpIX and PKG-associated phosphorylation of VASP, suggesting PKG activation may contribute to suppression of pulmonary hypertension and prevention of alterations in extracellular peroxide that appear to be attenuating HPV responses caused by chronic hypoxia.
Keywords: aminolevulinic acid, hypoxic pulmonary vasoconstriction, protein kinase G, pulmonary hypertension
chronic hypoxia is known to contribute to the development of pulmonary hypertension in humans with chronic obstructive pulmonary disease (7), and intermittent hypoxia associated with sleep apnea promotes both pulmonary and systemic hypertension (9). A loss of the hypoxic pulmonary vasoconstriction (HPV) response which maintains ventilation to perfusion ratios is seen in humans with chronic obstructive pulmonary disease (COPD) and in pulmonary arteries isolated from these individuals (25). Studies in animal models of chronic hypoxia have provided evidence for processes that are associated with increased generation of reactive oxygen species (ROS) from sources, including various Nox oxidases participating in the progression of hypoxia-induced pulmonary hypertension development (10, 12, 13, 15, 19, 21, 23). Previous studies in pulmonary arteries isolated from mice exposed to 21 days of hypoxia have detected an increased predominantly extracellular superoxide-mediated enhancement of vasoconstriction in arteries precontracted with agents, such as serotonin, which further increase superoxide generation (19). Rho kinase activation may participate in this action of superoxide (4). On the basis of studies in mice deficient in Nox2, this source of superoxide generation appears to be essential for both the development of pulmonary hypertension caused by exposure to chronic hypoxia and the increased extracellular superoxide seen in pulmonary arteries isolated from these animals (19). While less is known about the effects of chronic hypoxia on systemic vascular function, there is a report in rat mesenteric arteries, suggesting it depressed phenylephrine-induced contraction by a process that appears to be influenced by intracellular catalase activity (27). Chronic hypoxia has been reported to initially increase the expression of extracellular SOD (ecSOD) in lungs of mice after 1 day, and decreased ecSOD was detected after 35 days of chronic hypoxia (23). In addition, this study also demonstrated that mice with increased ecSOD expression were protected from the development of pulmonary hypertension on exposure to chronic hypoxia. While it is logical to assume hydrogen peroxide levels are altered by chronic hypoxia as a result of changes in ROS generation and the expression of SOD enzymes, minimal consideration has been given to the possibility that extracellular peroxide can influence vascular function in animals exposed to chronic hypoxia. Thus, this study focused on examining the hypothesis that exposure of mice to chronic hypoxia can alter vascular responses, such as HPV through increased extracellular peroxide generation under conditions that promote pulmonary hypertension.
Our laboratory recently detected evidence for the HPV response in isolated bovine pulmonary arteries being very sensitive to inhibition by increased extracellular peroxide that resulted from promoting an elevated expression of ecSOD (2). In this study, we employed the 21-day exposure of mice to chronic hypoxia (10% O2) model of pulmonary hypertension development (24) to examine the origins of a novel observation that pulmonary arteries isolated from these animals showed an attenuation of their contractile response to acute hypoxia, which are restored in the presence of the peroxide scavenger catalase. The response of the aorta to acute hypoxia from the mice studied was also examined to detect whether chronic hypoxia influenced vascular function through increasing extracellular peroxide in a systemic vascular segment. Because our laboratory has been studying the origins of how a chronic treatment of mice with the heme biosynthesis precursor δ-aminolevulinic acid (ALA) prevents the development of pulmonary hypertension (3), we examined whether treating mice with ALA during the 21-day period of exposure to hypoxia influenced changes caused by chronic hypoxia. We are interested in ALA since it can activate pulmonary vascular soluble guanylate cyclase (sGC) by promoting the accumulation of protoporphyrin IX (PpIX, 20) and because pharmacological activators of sGC that mimic the actions of PpIX (24) have been found to protect against pulmonary hypertension (11). Studies in lung slices were conducted to detect changes caused by chronic hypoxia on the levels of superoxide, extracellular peroxide, ecSOD expression, peroxide stimulation of a thiol oxidation-mediated dimerization of protein kinase G (PKG). Measurements of lung PpIX and increased PKG activity (based on its phosphorylation of VASP) were used to confirm that the in vivo treatment with ALA maintained the increases in these systems that were observed in previous studies exposing isolated pulmonary arteries to 1 day of organoid culture with ALA (20). Doppler flow echocardiography measurements of an indicator of pulmonary arterial pressure changes (28) were used to examine the effects of chronic hypoxia and ALA treatments on the acute in vivo hypoxic pulmonary vasoconstriction response elicited by acute exposure to 10% oxygen and to characterize the influence of these chronic treatments on the development of pulmonary hypertension.
MATERIALS AND METHODS
Materials.
All salts used for making physiological solutions were analyzed reagent grade from Baker Chemical. All gases were purchased from Tech Air (White Plains, NY). H2O2, ALA, and maleimide were obtained from Sigma Chemical (St. Louis, MO). cGMP-dependent protein kinase 1α (PKG 1α) antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). β-actin antibody was purchased from Sigma Chemical. ecSOD antibody was purchased from Upstate (Millipore, Billerica, MA). Vasodilator-stimulated phosphoprotein (VASP) and total-VASP antibodies were purchased from Cell Signaling (Beverly, MA).
Animal handling and exposure to chronic hypoxia.
Male C57BL/6J mice (8–10 wk) were purchased from Jackson Laboratories (Bar Harbor, ME). All animal protocols were approved by the Institutional Animal Care and Use Committee at New York Medical College. Mice were either exposed to normoxic (21% O2) or to normobaric hypoxic (10% O2) conditions for 21 days with or without ALA (50 mg·kg−1·day−1) in a hypoxic in vivo cabinet (Coy Laboratories, Grass Lake, MI) setup for measuring oxygen and recording time-dependent changes. ALA treatment was performed by adding ALA into the drinking water for 21 days. After 21 days, pulmonary arteries and aorta were isolated from each mouse after they were anesthetized with 50 mg/kg pentobarbital.
Detection of changes in pulmonary artery pressure by Doppler flow.
Transthoracic echocardiography (Vevo 770; Visual Sonics, Toronto, Ontario, Canada) was performed at the end of the 21-day protocol on mice, which were under light anesthesia, through a constant flow of isoflurane, using methods previously described (28). The pulmonary acceleration time-to-ejection time (PAT/ET) ratio was used to quantify relative changes in pulmonary arterial pressure, and this method was calibrated under a variety of conditions by direct measurements of right ventricular systolic pressure. After obtaining a baseline measurement with the mouse breathing room air, an acute hypoxic pulmonary vasoconstriction response was elicited by having the animal breathe 10% oxygen for a 6-min period, and responses at 2 min of exposure to hypoxia were evaluated and reported in the results.
Tissue preparation from mice.
Pulmonary arteries and the aorta were carefully isolated from each mouse in ice-cold buffer and maintained in cold Krebs-bicarbonate buffer until they were mounted for the experiment. After isolation of tissues, small rings were prepared from each aorta and pulmonary artery (∼180 μm internal diameter). These vascular segments were used for vascular reactivity studies.
Measurement of vascular reactivity in mouse pulmonary arteries and aorta.
Freshly prepared pulmonary arterial and aortic rings were used for studies measuring changes in isometric force, conducted initially under an atmosphere of 21% O2 and 5% CO2 in Krebs-bicarbonate buffer at 37°C. Pulmonary arterial rings were mounted on Danish Myograph Technology wire myographs, and aortic rings were mounted on World Precision Instruments force displacement transducers. Powerlab data acquisition systems from ADInstruments were used for the recording of time-dependent changes in force. Pulmonary rings and aortic rings were incubated at optimized passive tensions of 0.5 g and 2 g, respectively, for 1 h in Krebs-bicarbonate buffer containing 118 mM NaCl, 4.7 mM KCl, 1.5 mM CaCl2, 25 mM NaHCO3, 1.1 mM MgSO4, 1.2 mM KH2PO4, and 5.6 mM glucose gassed with 21% O2-5% CO2-74% N2 to maintain a pH of 7.4. Following this 1 h of incubation, the rings were depolarized with 123 mM KCl containing Krebs-bicarbonate buffer, and the rings were again reequilibrated with normal Krebs-bicarbonate buffer for another 30 min before being exposed to the protocols used in the results section.
Western blot analysis.
PKG dimer expression was detected in mouse lung tissue by running a Western blot under a protein-denaturing condition, as published previously by our laboratory (22). Changes in thiol-redox were avoided in all samples by adding 100 mM malemide during sample preparation. Relative changes in PKG1α monomer and dimer forms are reported as the percent of the total PKG1α. Changes in PKG1α monomer and dimer expression were quantified after normalization to β-actin, and phosphorylated VASP was normalized to total VASP. Total PKG levels and VASP levels were not altered as a result of chronic hypoxic treatment. The molecular masses of the PKG monomer and dimer are 75 and 150 kDa, respectively. ecSOD expression was also detected in mouse lung tissue. Molecular weight of ecSOD is 45 kDa. The molecular weight of phosphorylated VASP and total VASP is 50 kDa.
Measurement of protoporphyrin IX fluorescence.
Protoporphyrin IX fluorescence from lung tissue was determined by a previously published method used by our laboratory (20). Increases in protoporphyrin IX fluorescence were measured using an excitation wavelength of 485 ± 20 and emission of 620 ± 40 nm in lung tissues of control and ALA-treated mice. Lung tissues were placed on the bottom of the ∼6-mm diameter wells of a sterile 96-well microplate with 200 μl of Krebs containing 10 mM HEPES buffer (pH 7.4). Protoporphyrin IX fluorescence was measured from the bottom surface of the plate using BIOTEK fluorescent microplate reader (model FLx800i), as published previously (20). Data are reported in the arbitrary fluorescence units measured (AU) after subtraction of the low levels of background fluorescence observed in the absence of lung tissue.
Superoxide and peroxide measurement using chemiluminescence.
As published previously by our laboratory (2), changes in superoxide were measured from quantifying the chemiluminescence of 5 μM lucigenin in a liquid scintillation counter (LS6000IC; Beckman Instruments, San Diego, CA) with a single active photomultiplier tube in a dark room. Initial background chemiluminescence was measured in plastic scintillation minivials containing 5 μM lucigenin in 1 ml of Krebs solution buffered with 10 mM HEPES-NaOH (pH 7.4) in the absence of tissue. This measurement was subtracted from subsequent measurements made in the presence of lung tissue. The tissue was weighed at the end of the experiment, and the counts were divided by weight to give the readings in counts per minute per gram of tissue. Peroxide released from lung tissue was detected by substituting 10 μM luminol and 1 nM horseradish peroxidase for lucigenin in the chemiluminescence methods (2) used for superoxide.
RESULTS
Exposure of mice to 21 days of hypoxia promotes pulmonary hypertension and an attenuation of the in vivo HPV response.
The PAT/ET ratio obtained from Doppler flow measurements with heart rates maintained at ∼500 beats/min was used to detect changes in pulmonary arterial pressure elicited by the 21-day exposure to hypoxia. Data in Fig. 1 indicate this treatment significantly decreases the PAT/ET ratio. Calibration of these measurements based on right ventricular systolic pressure as an indicator of pulmonary arterial pressure suggested that pulmonary arterial pressure increased from 19.0 mmHg to 31.8 mmHg as a result of the exposure to 21 days of hypoxia. Data in Fig. 1 also show that an acute exposure to 10% oxygen elicited a decrease in PAT/ET ratios consistent with a HPV response increasing in pulmonary artery systolic pressure by 6.1 mmHg. The acute exposure to hypoxia did not appear to elicit a HPV response in mice exposed to 21 days of hypoxia.
Fig. 1.
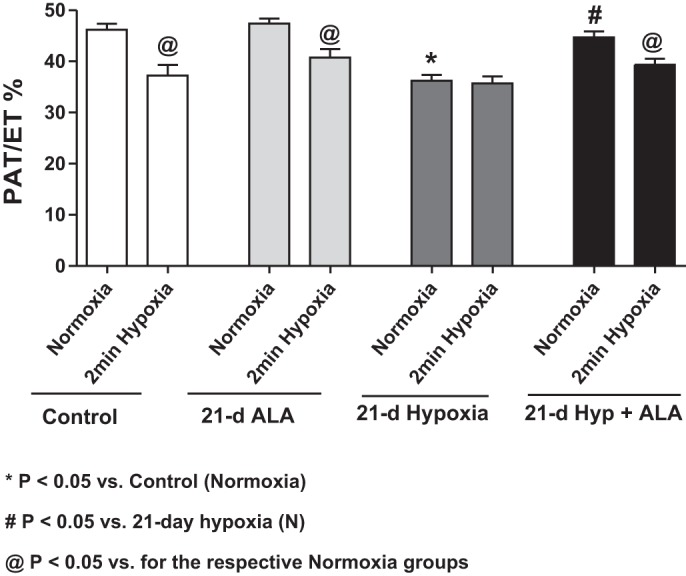
Data from Doppler flow echocardiography detection of relative increases in pulmonary arterial pressure based on decreased acceleration time (PAT)-to-ejection time (ET) ratios showing that 21 days of exposure hypoxia (10% oxygen) causes pulmonary hypertension and an attenuation of the acute hypoxic pulmonary vasoconstriction response (2-min hypoxia) in a manner that is prevented by treating mice with δ-aminolevulinic acid (ALA) (50 mg·kg−1·day−1) during the exposure to chronic hypoxia. *P < 0.05 vs. control (normoxia). #P < 0.05 vs. 21-day hypoxia (N). @P < 0.05 vs. for the respective normoxia groups (n = 5 or 6).
Treatment of mice with ALA prevents pulmonary hypertension development and the loss of HPV caused by exposure of mice to 21 days of hypoxia.
The PAT/ET ratio data in Fig. 1 show that a 21-day treatment with ALA did not significantly alter pulmonary arterial pressure (20.0 mmHg) or the increase in pressure caused by acute hypoxia (7.1 mmHg). In contrast, treatment with ALA during the 21-day period of exposure to hypoxia resulted in an estimated pressure of 22.8 mmHg, suggesting ALA prevented the increased pulmonary arterial pressure caused by the chronic hypoxic exposure, and it restored the increase in pressure elicited by acute hypoxia (5.7 mmHg). Thus, the ALA treatment used prevented both the development of pulmonary hypertension and loss of HPV elicited by 21 days of exposure to hypoxia.
ALA increases protoporphyrin IX fluorescence and promotes activation of soluble guanylate cyclase based on increased PKG-mediated phosphorylation of VASP.
Lung tissues from 21-day ALA-treated mice were used to determine whether changes in protoporphyrin IX (PpIX) fluorescence could be detected. The 21-day treatment with ALA caused an increase in PpIX fluorescence (Fig. 2A). Western blotting was used to detect changes in PKG activation based on phosphorylation of the serine-239 site on VASP (18). The 21-day treatment with ALA also caused an increase in VASP phosphorylation associated with activation of PKG (Fig. 2B), suggesting the detected increase in PpIX was stimulating cGMP generation by sGC.
Fig. 2.

A comparison of lung tissue from 21-day control and 21-day ALA-treated mice showing that ALA increased the levels of protoporphyrin IX (PpIX) fluorescence (A) and VASP phosphorylation (B). Summary data showing that superoxide (C) and extracellular peroxide (D) levels are significantly increased by chronic hypoxia and that ALA treatment reversed these increases in lung tissues of chronic hypoxic mice. *P < 0.05 vs. control; #P < 0.05 vs. 21-day hypoxia; n = 6.
Chronic hypoxia increases superoxide and peroxide levels, while ALA treatment during chronic hypoxia reduces superoxide and peroxide levels.
Lung tissue from chronic hypoxic mice showed an increase in the detection of superoxide (Fig. 2C) and extracellular peroxide (Fig. 2D). ALA treatment restored the detection of superoxide (Fig. 2C) and peroxide (Fig. 2D) in lung tissue from mice exposed to chronic hypoxia to the levels seen in control mice exposed to 21 days of normoxia in the absence or presence of ALA.
Expression of PKG dimer, phosphorylated VASP and ecSOD was increased in 21-day chronic hypoxic mouse lung tissue.
Lung tissue from mice exposed to 21 days of normoxia and hypoxia were used to examine the effect of chronic hypoxia on PKG dimerization (Fig. 3A), VASP phosphorylation (Fig. 3B) and ecSOD expression (Fig. 3C) because peroxide can stimulate pulmonary vasodilation through activation of PKG by dimerization (5, 22) and because ecSOD promotes extracellular peroxide generation (2). Lungs from 21-day hypoxic mice showed both increased PKG dimerization, PKG-associated VASP phosphorylation, and increased expression of ecSOD.
Fig. 3.

Western blot analysis showing the effects of exposure of mice to 21 days of hypoxia compared with a 21-day exposure to normoxia control on expression of PKG-dimerization (A) (n = 8). *P < 0.05 vs. control monomer. #P < 0.05 vs. control dimer. PKG monomer and dimer were analyzed as the % of total PKG after the total PKG was normalized to actin. B: VASP-phosphorylation (n = 8); *P < 0.05 vs. control and ecSOD (C) (n = 6); *P < 0.05 vs. control in mouse lung tissue.
Exposure of mice to 21 days of hypoxia elicits in isolated pulmonary arteries a peroxide-mediated attenuation of the HPV contractile response.
When contracted with 10 nM phenylephrine, pulmonary arteries from 21-day hypoxic mice showed inhibition of the contraction (Fig. 4A). These same pulmonary arteries when treated with 1 μM catalase for 15 min before the addition of 10 nM phenylephrine showed an improved contraction to phenylephrine, suggesting extracellular peroxide had a role in suppressing force generation. Catalase did not alter contractions to 10 nM phenylephrine in pulmonary arteries from mice exposed to 21 days of normoxic conditions. Pulmonary arteries were precontracted with 10 nM phenylephrine (PE) under aerobic conditions and then were exposed to acute hypoxia by changing the gassing in the myograph chambers from 21% O2, 5% CO2, and 74% N2 to 5% CO2 and 95% N2 (Po2 ∼8–10 Torr) for 20 min to elicit an in vitro HPV-type response. Pulmonary arteries isolated from 21-day hypoxic mice showed attenuation of the HPV response when they were exposed to acute hypoxia (Fig. 4B). Catalase (1 μM) had a significant effect on restoring the impairment of contraction to acute hypoxia seen in pulmonary arteries (Fig. 3B). Pulmonary arteries from both groups showed a comparable level of contraction to 100 nM phenylephrine [21-day normoxia = 0.17 ± 0.02; 21-day hypoxia = 0.18 ± 0.03 (n = 9–10)] and to 123 mM KCl [21 day normoxia = 0.30 ± 0.04; 21 day hypoxia = 0.32 ± 0.03 (n = 12–13)], suggesting that contractions to HPV and to phenylephrine that are similar in magnitude to HPV responses appeared to be very sensitive inhibition by extracellular peroxide.
Fig. 4.

Effects of the absence and presence of scavenging extracellular peroxide with catalase on contractile responses of pulmonary arteries isolated from mice exposed to 21 days of normoxia or hypoxia in the absence and presence of 21-day treatment with ALA (50 mg·kg−1·day−1) to 10 nM phenylephrine (A) and the contractile response (B) elicited by a subsequent exposure to 20 min of acute hypoxia. *P < 0.05 vs. control; #P < 0.05 vs. 21-day hypoxia; n = 8–10. @P < 0.05 for significant effects of catalase for respective PE/acute hypoxia groups.
Treatment of mice with ALA restores the HPV response in pulmonary arteries, which was attenuated by exposure to 21 days of hypoxia.
Pulmonary arteries isolated from mice simultaneously treated with hypoxia and ALA showed an attenuation of the inhibition of contraction to 10 nM phenylephrine that was seen in pulmonary arteries isolated from mice treated with hypoxia for 21 days in the absence of ALA (Fig. 4A). Pulmonary arteries isolated from mice treated with ALA during the exposure to 21 days of chronic hypoxia also showed a restoration of the attenuated contraction to acute hypoxia seen in mice exposed to 21 days of hypoxia in the absence of ALA (Fig. 4B). On the other hand, 1 μM of catalase treatment did not significantly improve the contraction to acute hypoxia in pulmonary arteries isolated from mice exposed to 21 days of hypoxia together with ALA (Fig. 4B).
Exposure of mice to 21 days of hypoxia elicits a peroxide-mediated attenuation of the aortic relaxation response to acute hypoxia.
Aortas from hypoxic mice contracted with 100 nM phenylephrine (PE) showed a lower level of force generation compared with the 21-day normoxic control aortas (Fig. 5A). When incubated with 1 μM catalase for 15 min before contraction, there was restoration of PE contraction in the hypoxic mouse. Aortas from mice exposed for 21 days to normoxic and hypoxic conditions were precontracted with 100 nM PE under aerobic conditions and then were exposed to acute hypoxia by changing the gassing in the tissue baths from 21% O2, 5% CO2, and 74% N2 to 5% CO2 and 95% N2 (Po2 ∼8–10 Torr) for 20 min. Aorta isolated from mice exposed to normoxic conditions relaxed on exposure to acute hypoxia. However, aorta isolated from hypoxic mice showed a complete attenuation of relaxation when they were exposed to acute hypoxia (See Fig. 5B). Note that aortas isolated from the 21-day aerobic control mice showed a 43 ± 5% relaxation of the 100 nM phenylephrine force on exposure to acute hypoxia, and aortas isolated from the 21-day hypoxic mice actually showed a small contraction (−24 ± 10% relaxation) to acute hypoxia. Acute treatment with catalase had a significant effect in reversing the attenuation of the relaxation to acute hypoxia seen in aortas isolated from chronic hypoxia-exposed mice (Fig. 5B). Aortas from both groups showed a comparable level of contraction to 123 mM KCl [21 day normoxia = 0.37 ± 0.03; 21 day hypoxia = 0.38 ± 0.04 (n = 12)], suggesting that contractions to phenylephrine appeared to be more sensitive to inhibition by extracellular peroxide. While catalase appeared to cause an increase in force in the normoxic control animal aortas in a manner that was not statistically significant (Fig. 5A, P = 0.22), this trend might be related to previous documentation of peroxide-associated endothelium-derived hyperpolarizing factor responses in this vascular segment (6).
Fig. 5.

Effects of the absence and presence of scavenging extracellular peroxide with catalase on contractile responses of aorta isolated from mice exposed to 21 days of normoxia or hypoxia in the absence and presence of 21-day treatment of mice with ALA (50 mg·kg−1·day−1) to 100 nM phenylephrine (A) and the relaxation response (B) elicited by a subsequent exposure to 20 min of acute hypoxia. *P < 0.05 vs. control; @P < 0.05 for significant effects of catalase for respective PE/acute hypoxia groups; n = 6–8.
Treatment of mice with ALA does not restore in the isolated aorta the relaxation response elicited by acute hypoxia that was attenuated by exposure of mice to 21 days of hypoxia.
Aorta isolated from mice simultaneously treated with ALA and chronic hypoxia showed a similar attenuation of the contraction to 100 nM phenylephrine, as seen in 21-day hypoxic mouse aorta (Fig. 5A). In addition, aorta from mice treated with both ALA and chronic hypoxia showed a similar attenuation of the relaxation to hypoxia that was observed in aorta isolated from mice treated with chronic hypoxia in the absence of ALA (Fig. 5B), suggesting that treatment with ALA was not preventing alterations in aortic responses caused by chronic hypoxia. While 1 μM catalase treatment appeared to improve these attenuated responses in 21-day ALA-treated hypoxic mouse aorta (Fig. 5B), the effects of catalase were not statistically significant.
DISCUSSION
The present study identified evidence for 21 days of chronic hypoxia causing an extracellular hydrogen peroxide-mediated marked suppression of the acute contractile response of pulmonary arteries to hypoxia based on a reversal of these effects by catalase. Peroxide also inhibited contraction to lower, but not higher levels of force generation by phenylephrine. This was associated with detection of elevated levels of superoxide, extracellular peroxide, ecSOD, and PKG activation by thiol oxidation-mediated subunit dimerization associated with increased VASP phosphorylation at the PKG site in lung tissue. These observations are consistent with the model shown in Fig. 6, illustrating how increased generation of superoxide and its conversion by enzymes, such as ecSOD, to levels of extracellular peroxide that could activate PKG were potentially contributing to the catalase-attenuated suppression of contraction to hypoxia and phenylephrine. Interestingly, the heme biosynthetic precursor ALA attenuated the detection of increases in both superoxide and peroxide, and the peroxide-associated suppression of contraction to both phenylephrine and acute exposure to hypoxia. ALA generated detectable increases in PpIX, an activator of sGC (16, 20), and it increased PKG-associated phosphorylation of VASP. Since pharmacological activators of sGC, which bind at the PpIX site (26), attenuate and/or reverse pulmonary hypertension development (11), activation of PKG by increased cGMP and the inhibition of pulmonary hypertension could be a prominent factor in how ALA is potentially preventing increases in superoxide and/or peroxide generation that caused the observed alterations in vascular function, resulting from the 21 days of exposure to hypoxia.
Fig. 6.
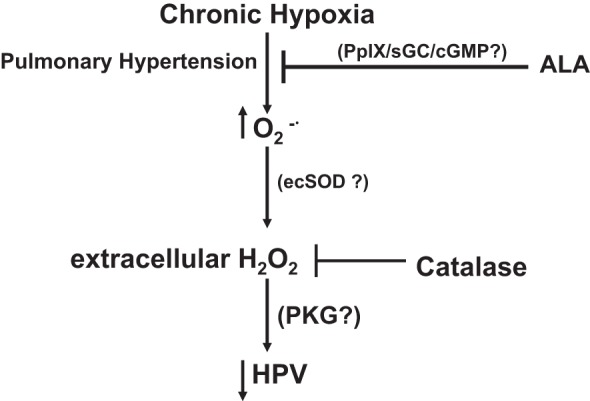
Model showing systems hypothesized to be potentially contributing to how exposing mice to 21 days of chronic hypoxia attenuates the acute pulmonary arterial hypoxic pulmonary vasoconstriction (HPV) response through increasing hydrogen peroxide. The beneficial effects of 21 days of ALA treatment functioning through attenuating pulmonary hypertension and the generation of vasodilator levels of extracellular hydrogen peroxide potentially through its attenuation of the generation of superoxide are shown. PKG, cGMP protein kinase; PpIX, protoporphyrin IX.
Catalase did not alter contraction to phenylephrine or responses to hypoxia in pulmonary arteries from mice, which were not exposed to hypoxia, suggesting that extracellular peroxide is not normally present in amounts that influence these responses. Although aortas from control mice not exposed to chronic hypoxia may show some evidence of peroxide-suppressing contraction to phenylephrine, it did not alter the relaxation response to hypoxia in vitro. While there is much evidence for chronic hypoxia-generating increases in ROS, there has been minimal consideration in previous studies for chronic hypoxia suppressing vascular contractile function or acute responses to hypoxia by increasing the generation of hydrogen peroxide.
In the present study, we observed that ecSOD remained elevated after 21 days of hypoxia, and this was associated with the detection of increased superoxide and extracellular peroxide in lung tissue. These observations are consistent with both an increased generation of superoxide and its conversion by SOD enzymes to extracellular peroxide. Recent studies from our laboratory suggest that increased ecSOD activity causes attenuation of the acute contraction of isolated bovine pulmonary arteries to hypoxia through a mechanism that appears to involve the conversion of a secreted source of superoxide that seems to be derived from Nox2 oxidase (1, 29) into levels of extracellular hydrogen peroxide that suppressed the HPV response (2). Since increased ecSOD was observed in lung tissue from mice exposed to 21 days of hypoxia, it could be a contributing factor in generating levels of peroxide that suppress HPV. Our previous studies have also detected evidence for peroxide functioning to attenuate the relaxation of bovine coronary arteries to hypoxia through stimulating an ERK MAP kinase-mediated force enhancing mechanism (14). On the basis of the restoration of the attenuated relaxation to acute hypoxia by catalase observed in aorta from mice exposed to 21 days of hypoxia, increased extracellular peroxide appeared to be the cause of the attenuation of this response. Thus, exposure of mice to 21 days of hypoxia appears to suppress contraction to phenylephrine and responses to acute hypoxia by elevating the levels of extracellular peroxide in both the pulmonary and systemic vasculature. However, other factors, such as the generation of vasoconstrictor levels of superoxide from elevated levels of pulmonary hypertension mediators, such as serotonin, endothelin, and prostaglandins (e.g., prostaglandin H2 and thromboxane A2) may also be important in the in vivo expression of pulmonary hypertension (19). In addition, the remodeling effects of changes in ROS could dominate over potential beneficial vasodilator effects of increased peroxide.
We investigated whether increased extracellular peroxide under chronic hypoxia alters PKG dimerization as PKG plays an important role in vascular relaxation activated by peroxide and other mechanisms (5, 18, 22). The detected increase in PKG dimerization caused by 21-day exposure of mice to hypoxia was also associated with an increase in PKG activity based on the detection of increased VASP phosphorylation. Overexpression of ecSOD in knock-in mice, or in animals with lung-targeted transfection methods, and perhaps as a result of increased heme oxygenase expression (2, 8), prevents and/or reverses the development of pulmonary hypertension caused by chronic hypoxia or monocrotaline (17, 23), suggesting that upregulation of ecSOD has beneficial effects in chronic hypoxic tissue. These observations together with our data suggest that chronic hypoxia elevates extracellular peroxide in a manner that could be hypothesized to function as a protective feedback mechanism to attenuate pulmonary arterial contraction to vasoconstrictors and hypoxia.
Data in Fig. 1 and our ongoing recent studies suggest that ALA prevents the hypoxia-induced pulmonary hypertension in the chronic hypoxic mouse model (3). As shown in Fig. 2, A and B, 21-day ALA treatment leads to the generation of PpIX, which can promote activation of sGC/PKG (16, 20). However, data in Figs. 1 and 4A did not detect evidence in the absence of chronic hypoxia for the prolonged treatment with ALA promoting decreases in pulmonary arterial pressure or a depression of contraction in pulmonary arteries or aortas. A prominent effect of treating mice with ALA during the 21-day period of hypoxia was improving pulmonary artery contractile function associated with an attenuation of peroxide generation and its suppression of both contraction to 10 nM phenylephrine and the acute HPV response. ALA appeared also to function by attenuating the increases in superoxide caused by exposure to 21 days of hypoxia, and this could potentially have beneficial effects, such as preventing Rho kinase activation (4). Interestingly, the ALA treatment did not appear to reverse the effects of chronic hypoxia in the aorta, suggesting some of its pulmonary associated actions on processes elicited by chronic hypoxia may be linked to it preventing the development of pulmonary hypertension. Although the level of systemic hypoxia was not measured in the current study, any changes elicited by treatment with ALA did not influence the functional changes in aortic reactivity caused by exposure to chronic hypoxia. Because it appears that cGMP-mediated activation of PKG seems to be beneficial in preventing the development and promoting reversal of pulmonary hypertension (11), the stimulation of PKG by peroxide may also function as a beneficial mechanism in pulmonary hypertension. However, the high sensitivity of the HPV response to impairment by extracellular peroxide may limit the beneficial nature of vasodilator effects of extracellular peroxide. In addition, other mechanisms activated by peroxide may contribute to the progression of hypoxia-associated disease processes.
Perspectives and Significance
Diseases exposing lungs to chronic hypoxia such as chronic obstructive pulmonary disease are known to both contribute to the development of pulmonary hypertension in humans (7) and to be associated with a loss of the HPV response, which maintains ventilation to perfusion ratios (25). The results of this study detected evidence that increased extracellular hydrogen peroxide could be a factor in how chronic hypoxia can promote an impairment of HPV under conditions where pulmonary hypertension develops. Data in this study also provided evidence for how treatment with the amino acid ALA used for the biosynthesis of heme and stimulation of cGMP by PpIX could function to prevent the development of both hypoxia-induced pulmonary hypertension and the extracellular peroxide-associated impairment of HPV. These studies raise new questions in areas, such as determining whether the vasodilator and/or PKG-activating actions of extracellular peroxide are beneficial in preventing the development of pulmonary hypertension or do these processes contribute to enhancing hypoxemia by impairing gas exchange in diseases such as COPD? If the actions of extracellular peroxide contribute to disease progression, therapeutic approaches could also be developed on the basis of enhancing the scavenging of this source of peroxide.
DISCLOSURES
Michael S. Wolin is the inventor on a patent held by New York Medical College on the beneficial effects of promoting smooth muscle relaxation by generation of protoporphyrin IX. No other conflicts of interest are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: D.P. and M.S.W. conception and design of research; D.P. and R.A. performed experiments; D.P. and M.S.W. analyzed data; D.P. and M.S.W. interpreted results of experiments; D.P. and M.S.W. prepared figures; D.P. and M.S.W. drafted manuscript; D.P., R.A., and M.S.W. edited and revised manuscript; D.P., R.A., and M.S.W. approved final version of manuscript.
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health Grants R01HL-031069, P01HL-043023, R01HL-066331, and R01HL-115124. Portions of this study were presented at the 2011 Experimental Biology Meeting in Washington, D. C. (24).
REFERENCES
- 1.Ahmad M, Kelly MR, Malpani S, Kelly J, Wolin MS. Nox2 derived extracellular superoxide attenuates hypoxic pulmonary vasoconstriction through a novel mechanism in the presence of increased extracellular superoxide dismutase (Abstract). FASEB J 25: 1102.–4., 2011 [Google Scholar]
- 2.Ahmad M, Zhao X, Kelly MR, Kandhi S, Perez O, Abraham NG, Wolin MS. Heme oxygenase-1 induction modulates hypoxic pulmonary vasoconstriction through upregulation of ecSOD. Am J Physiol Heart Circ Physiol 297: H1453–H1461, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alhawaj R, Patel D, Ahmad M, Rémond MC, Leonard Eisenberg LMM, Wolin MS. Treatment of mice with delta-aminolevulinic acid, a generator of the guanylate cyclase activator protoporphyrin IX, prevents the development of hypoxia-induced pulmonary hypertension (Abstract). FASEB J 26: 873.–20., 2012 [Google Scholar]
- 4.Broughton BR, Jernigan NL, Norton CE, Walker BR, Resta TC. Chronic hypoxia augments depolarization-induced Ca2+ sensitization in pulmonary vascular smooth muscle through superoxide-dependent stimulation of RhoA. Am J Physiol Lung Cell Mol Physiol 298: L232–L242, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burgoyne JR, Madhani M, Cuello F, Charles RL, Brennan JP, Schröder E, Browning DD, Eaton P. Cysteine redox sensor in PKGIα enables oxidant-induced activation. Science 317: 1393–1397, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Capettini LSA, Cortes SF, Gomes MA, Silva GAB, Pesquero JL, Lopes MJ, Teixeira MM, Lemos VS. Neuronal nitric oxide synthase-derived hydrogen peroxide is a major endothelium-dependent relaxing factor. Am J Physiol Heart Circ Physiol 295: H2503–H2511, 2008 [DOI] [PubMed] [Google Scholar]
- 7.Chaouat A, Naeije R, Weitzenblum E. Pulmonary hypertension in COPD. Eur Respir J 32: 1371–1385, 2008 [DOI] [PubMed] [Google Scholar]
- 8.Christou H, Morita T, Hsieh CM, Koike H, Arkonac B, Perrella MA, Kourembanas S. Prevention of hypoxia-induced pulmonary hypertension by enhancement of endogenous heme oxygenase-1 in the rat. Circ Res 86: 1224–1229, 2000 [DOI] [PubMed] [Google Scholar]
- 9.Dempsey JA, Veasley SC, Morgan BJ, O'Donnell CP. Pathophysiology of sleep apnea. Physiol Rev 90: 47–112, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dennis KE, Aschner JL, Milatovic D, Schmidt JW, Aschner M, Kaplowitz MR, Zhang Y, Fike CD. NADPH oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol 297: L596–L607, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dumitrascu R, Weissmann N, Ghofrani HA, Dony E, Beuerlein K, Schmidt H, Stasch JP, Gnoth MJ, Seeger W, Grimminger F, Schermuly RT. Activation of soluble guanylate cyclase reverses experimental pulmonary hypertension and vascular remodeling. Circulation 113: 286–295, 2006 [DOI] [PubMed] [Google Scholar]
- 12.Frazziano G, Champion HC, Pagano PJ. NADPH oxidase-derived ROS and the regulation of pulmonary vessel tone. Am J Physiol Heart Circ Physiol 302: H2166–H2177, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fresquet F, Pourageaud F, Leblais V, Brandes RP, Savineau JP, Marthan R, Muller B. Role of reactive oxygen species and gp91phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. Br J Pharmacol 148: 714–723, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao Q, Zhao X, Ahmad M, Wolin MS. Mitochondrial-derived hydrogen peroxide inhibits relaxation of bovine coronary arterial smooth muscle to hypoxia through stimulation of ERK MAP kinase. Am J Physiol Heart Circ Physiol 297: H2262–H2269, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupte SA, Wolin MS. Relationships between vascular oxygen sensing mechanisms and hypertensive disease processes. Hypertension 60: 269–275, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ignarro LJ, Wood KS, Wolin MS. Activation of purified soluble guanylate cyclase by protoporphyrin IX. Proc Natl Acad Sci USA 79: 2870–2873, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamezaki F, Tasaki H, Yamashita K, Tsutsui M, Koide S, Nakata S, Tanimoto A, Okazaki M, Sasaguri Y, Adachi T, Otsuji Y. Gene transfer of extracellular superoxide dismutase ameliorates pulmonary hypertension in rats. Am J Respir Crit Care Med 177: 219–226, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Lincoln TM, Dey N, Sellak H. cGMP-dependent protein kinase signaling mechanisms in smooth muscle: from the regulation of tone to gene expression. J Appl Physiol 91: 1421–1430, 2001 [DOI] [PubMed] [Google Scholar]
- 19.Liu JQ, Zelko IN, Erbynn EM, Sham JS, Folz RJ. Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox). Am J Physiol Lung Cell Mol Physiol 290: L2–L10, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Mingone CJ, Gupte SA, Chow JL, Ahmad M, Abraham NG, Wolin MS. Protoporphyrin IX generation from delta-aminolevulinic acid elicits pulmonary artery relaxation and soluble guanylate cyclase activation. Am J Physiol Lung Cell Mol Physiol 291: L337–L344, 2006 [DOI] [PubMed] [Google Scholar]
- 21.Mittal M, Roth M, Konig P, Hofmann S, Dony E, Goyal P, Selbitz AC, Schermuly RT, Ghofrani HA, Kwapiszewska G, Kummer W, Klepetko W, Hoda MA, Fink L, Hanze J, Seeger W, Grimminger F, Schmidt HH, Weissmann N. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ Res 101: 258–267, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Neo BH, Kandhi S, Wolin MS. Roles for soluble guanylate cyclase and a thiol oxidation-elicited subunit dimerization of protein kinase G in pulmonary artery relaxation to hydrogen peroxide. Am J Physiol Heart Circ Physiol 299: H1235–H1241, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nozik-Grayck E, Suliman HB, Majka S, Albietz J, Van Rheen Z, Roush K, Stenmark KR. Lung EC-SOD overexpression attenuates hypoxic induction of Egr-1 and chronic hypoxic pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol 295: L422–L430, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patel D, Ahmad M, Alhawaj R, Wolin M. Exposure of mice to chronic hypoxia promotes the development of a hydrogen peroxide-mediated relaxation and attenuation of pulmonary and aortic responses to acute hypoxia (Abstract). FASEB J 25: 1102.–3., 2011 [Google Scholar]
- 25.Peinado VI, Santos S, Ramírez J, Roca J, Rodriguez-Roisin R, Barberà JA. Response to hypoxia of pulmonary arteries in chronic obstructive pulmonary disease: an in vitro study. Eur Respir J 20: 332–338, 2002 [DOI] [PubMed] [Google Scholar]
- 26.Stasch JP, Schmidt PM, Nedvetsky PI, Nedvetskaya TY, SAKH, Meurer S, Deile M, Taye A, Knorr A, Lapp H, Muller H, Turgay Y, Rothkegel C, Tersteegen A, Kemp-Harper B, Muller-Esterl W, Schmidt HH. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest 116: 2552–2561, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sweazea K, Walker BR. Antioxidant and vasodilatory effects of heme oxygenase on mesenteric vasoreactivity following chronic hypoxia. Microcirculation 16: 131–141, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thibault HB, Kurtz B, Raher MJ, Shaik RS, Waxman A, Derumeaux G, Halpern EF, Bloch KD, Scherrer-Crosbie M. Noninvasive assessment of murine pulmonary arterial pressure: validation and application to models of pulmonary hypertension. Circ Cardiovasc Imaging 3: 157–163, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang G, Zhang F, Muh R, Yi F, Chalupsky K, Cai H, Li PL. Autocrine/paracrine pattern of superoxide production through NAD(P)H oxidase in coronary arterial myocytes. Am J Physiol Heart Circ Physiol 292: H483–H495, 2007 [DOI] [PubMed] [Google Scholar]