

Abstract
We are developing a retroviral replicating vector (RRV) encoding cytosine deaminase as an anticancer agent for gliomas. Despite its demonstrated natural selectivity for tumors, and other safety features, such a virus could potentially cause off-target effects by productively infecting healthy tissues. Here, we investigated whether incorporation of a hematopoietic lineage-specific microRNA target sequence in RRV further restricts replication in hematopoietic lineage-derived human cells in vitro and in murine lymphoid tissues in vivo. One or four copies of a sequence perfectly complementary to the guide strand of microRNA 142-3p were inserted into the 3′ untranslated region of the RRV genome expressing the transgene encoding green fluorescent protein (GFP). Viral spread and GFP expression of these vectors in hematopoietic lineage cells in vitro and in vivo were measured by qPCR, qRT-PCR, and flow cytometry. In hematopoietic lineage-derived human cell lines and primary human stimulated peripheral blood mononuclear cells, vectors carrying the 142-3pT sequence showed a remarkable decrease in GFP expression relative to the parental vector, and viral spread was not observed over time. In a syngeneic subcutaneous mouse tumor model, RRVs with and without the 142-3pT sequences spread equally well in tumor cells; were strongly repressed in blood, bone marrow, and spleen; and generated antiviral immune responses. In an immune-deficient mouse model, RRVs with 142-3pT sequences were strongly repressed in blood, bone marrow, and spleen compared with unmodified RRV. Tissue-specific microRNA-based selective attenuation of RRV replication can maintain antiviral immunity, and if needed, provide an additional safeguard to this delivery platform for gene therapy applications.
Introduction
We are developing a replicating retroviral vector (RRV) for clinical use as an anticancer agent for high-grade glioma (https://clinicaltrials.gov/; NCT01156584, NCT01470794, and NCT01985256). This agent, Toca 511 (vocimagene amiretrorepvec), is derived from Moloney murine leukemia virus (MLV) with an amphotropic envelope gene and encodes a sequence-optimized yeast cytosine deaminase (yCD2) in conjunction with an encephalomyocarditis virus (EMCV)-derived internal ribosome entry site (IRES) (Perez et al., 2012). Toca 511 is cytocidal for infected cells only after administration of the antifungal drug 5-fluorocytosine (5-FC), converted in situ to the anticancer drug 5-fluorouracil (5-FU). This gammaretrovirus has natural specificity for tumors through its requirement for replicating cell targets, the partial inactivation of innate immunity in tumors, and the generally immune-suppressed tumor environment (Melcher et al., 2011; Ostertag et al., 2012). Anticancer efficacy has been demonstrated in a number of rodent models (Tai et al., 2005; Wang et al., 2006; Ostertag et al., 2012; Yin et al., 2013). Initial investigation of the spread of related RRVs has shown significant infection in lymphoid tissues in nude mice (Duerner et al., 2008), but restricted spread in immune-competent mice, rats (Wang et al., 2006; Hiraoka et al., 2007; Ostertag et al., 2012), and dogs (our unpublished data). Although amphotropic MLV can enter human lymphocytes and integrate into the host genome, it does not spread efficiently in primary lymphocytes in culture (Cornetta et al., 1993; Ebeling et al., 2003), likely in part because of the relatively high levels of the antiretroviral protein APOBEC3G (apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G) (Jin et al., 2005; Mous et al., 2011), the expression of which can be modulated by the interferon signaling pathway (Chen et al., 2006; Mous et al., 2011).
The natural attenuation of MLV replication in lymphoid cells of nonmurine origin is consistent with the published lack of both adverse sequelae and productive infection in healthy macaques over 2 years (Cornetta et al., 1990). While lymphomagenesis after transplantation of hematopoietic stem cells transduced with nonreplicative retroviral vectors into immune-ablated or immune-incompetent human patients has been reported, it is clear that this effect is dependent on the clinical status of patients and/or the transgene delivered (Müller et al., 2011). For example, to our knowledge, no instance of lymphomagenesis in clinical trial subjects has been linked to infusion of retroviral vector-transduced T cells. Persistent quantifiable lymphoid infection has not been observed so far in our clinical trials in more than 60 trial subjects (Aghi et al., 2013; Cloughesy et al., 2013). Nevertheless, it is conceivable that chronic active infection of lymphoid tissue in vivo could be observed in some situations, with possible risk of lymphomagenesis. To address this hypothetical outcome, we investigated whether further restriction of RRV in lymphoid tissue could be achieved by including targets for tissue-specific microRNAs (miRNAs) (Ebert and Sharp, 2010) in the RRV genome.
MiRNA142-3p, miRNA181, and miRNA223 are highly expressed in hematopoietic tissues in human and mouse (Chen et al., 2004; Baskerville and Bartel, 2005; Monticelli et al., 2005), whereas miRNA122 is expressed at high levels in the liver but not in other tissues (Lagos-Quintana et al., 2002). The incorporation of a target sequence, perfectly complementary to the guide strand of a specific miRNA of interest, in transcription-based vectors has been used as a way to control undesirable expression of transduced genes in off-target cells or tissue types (Brown et al., 2006, 2007). Incorporation of target sequences of tissue- or cell-enriched miRNAs into the viral genome can at least partially restrict off-target spread of replicating oncolytic viruses (Edge et al., 2008; Kelly et al., 2008; Ylosmaki et al., 2008; Cawood et al., 2009; Sakurai et al., 2012). Given the potential for the clinical use of RRVs, it was of interest to investigate the feasibility of this miRNA approach for RRV.
We designed RRVs each carrying one or four copies of the hematopoietic-specific miRNA142-3p target sequence (142-3pT and 142-3pT4X, respectively) in the 3′ untranslated region (UTR) of the viral genome and examined viral spread, gene expression, and genome stability of these RRVs in lymphoid and nonlymphoid cells, and in animal models. This miRNA-based approach can selectively and effectively repress RRV replication in human peripheral blood mononuclear cells (PBMCs), in human hematopoietic lineage-derived cell lines, and in whole blood cell lineages in two in vivo mouse models. This strategy has the potential to offer an additional safeguard in the RRV delivery platform for gene therapy applications.
Materials and Methods
Plasmid construction
The pAC3-GFP vector, also known as pAC3-emd or T5.0006, is an MLV-based RRV in which the yCD2 gene downstream of the IRES in the pAC3-yCD2 vector (Ostertag et al., 2012; Perez et al., 2012) has been replaced with an emerald green fluorescent protein (GFP)-encoding gene. Its structure and that of its derivatives pAC3-GFP-142-3pT and pAC3-GFP-142-3pT4X are shown in Fig. 1.
FIG. 1.

(A) Relative microRNA 142-3p (miRNA142-3p) expression in peripheral blood mononuclear cells (PBMCs) of five healthy individuals and in hematopoietic lineage-derived cell lines. Total RNA was extracted from cells and reverse transcribed followed by qPCR with miRNA142-3p-specific reverse transcription primer and qPCR primers and probe to determine the expression level of miRNA142-3p relative to the endogenous small RNA RNU6B. The bar graph (in log10 scale) indicates the miRNA142-3p expression level relative to U87-MG cells by using the comparative (ΔΔCT) CT method. R, resting PBMCs; A, activated PBMCs. (B) Schematic diagram of plasmid DNA of retroviral replicating vector pAC3-GFP incorporating the 142-3pT sequence. (C) Identical sequences of mature human and murine miRNA142-3p (accession nos. MIMAT0000434 and MIMAT0000155, respectively). (D) 142-3pT sequence inserted into the pAC3-GFP vector to generate pAC3-GFP-142-3pT and pAC3-GFP-142-3pT4X vectors.
Cell culture
293T, U87-MG (HTB-14; American Type Culture Collection [ATCC], Manassas, VA), PC-3 (CRL-1435; ATCC), CEM (CCL-119; ATCC), U937 (CRL-1593.2; ATCC), and Tu2449 (Ostertag et al., 2012) cells were cultured as described in text and Materials and Methods in the online supplement (supplementary data are available online at www.liebertpub.com/hum).
MiRNA142-3p expression assay
miRNA-enriched RNA was extracted from cells with an Ambion mirVana miRNA isolation kit followed by DNase I treatment (AM1560 and AM1906; Life Technologies, Carlsbad, CA) according to the manufacturer's protocols. An Applied Biosystems TaqMan microRNA reverse transcription kit (4366596; Life Technologies) was used with RT primers for miRNA142-3p (assay 000464; Life Technologies) and RNU6B (assay 001093; Life Technologies) as endogenous controls to produce cDNA. Reverse transcription and quantitative PCRs were set up and carried out according to the manufacturer's protocols. 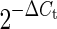 was calculated to obtain miR-142-3p expression relative to RNU6B in each sample. In U87-MG cells, the Ct value for miR-142-3p was at the lower limit of detection (Ct values between 38 and 40). When performing calculations for relative expression by
was calculated to obtain miR-142-3p expression relative to RNU6B in each sample. In U87-MG cells, the Ct value for miR-142-3p was at the lower limit of detection (Ct values between 38 and 40). When performing calculations for relative expression by 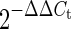 , the value of
, the value of 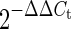 in U87-MG cells was assumed to be 1.
in U87-MG cells was assumed to be 1.
Virus production
Viral stock was produced by transient transfection of 293T cells by the calcium phosphate precipitation method and titered on PC-3 cells by qPCR to count integrated viral genomes, as described (Perez et al., 2012).
Flow cytometry
Viral replication was monitored by GFP expression. Cells were infected with GFP-encoding viruses at a multiplicity of infection (MOI) of 0.01 for U87-MG cells and at an MOI of 2 for U937 and CEM cells. Viral replication kinetic analyses were obtained by plotting the percentage of GFP-positive cells over time. For analysis of lineage-negative (lin−) bone marrow cells from infected, immune-deficient mice, see Materials and Methods in the online supplement. All flow cytometric data were acquired on a BD FACSCanto II cytometer running BD FACSDiva (BD Biosciences, San Jose, CA) and analyzed with FlowJo software (TreeStar, Ashland, OR).
Vector stability assay and amplification of IRES-GFP region
Vector stability was measured by serial passage on U87-MG cells as described previously (Perez et al., 2012).
Vector copy number of proviral DNA
Proviral vector copy numbers in genomic DNA were determined by qPCR as previously described (Perez et al., 2012).
Relative expression of cellular viral RNA by qRT-PCR
RNA was extracted from cells and quantitated by qRT-PCR. The average integrated vector copy number was used to determine the value of the normalization factor (ratio of parental vector copy number to142-3pT4X vector copy number) at each time point. The normalized relative expression of cellular viral RNA was determined by multiplying the normalization factor by the 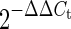 value.
value.
Preparation of PBMCs and RRV infection
Human T lymphocytes from healthy human donors were purified by density gradient centrifugation. Retrovirus supernatant was added at an MOI of 4 in microtiter plates, spun to increase infection efficiency, and incubated overnight at 37°C; the process was repeated after 24 hr.
RRV infection in vivo and biodistribution analysis
All animal experiments were conducted under protocols approved by the University of California, Los Angeles (Los Angeles, CA) Animal Research Committee. Studies for the syngeneic subcutaneous tumor model and the intravenous delivery to nude mice with no tumors were performed as described in Results, and in the online supplement Materials and Methods.
For biodistribution analysis, qPCR was performed to determine the vector copy number per microgram of tissue-derived genomic DNA. Ct values >38 were scored as nondetectable, and copy numbers less than 250 copies/μg were scored as below the lower limit of quantification (LLOQ). One-way analysis of variance was performed for statistical analysis, using GraphPad Prism (GraphPad Software, San Diego, CA). In the analysis, values from samples scored as LLOQ were included in the calculation. The value for a statistically significant difference was set at p<0.05.
Anti-MLV ELISA
The anti-MLV ELISA was performed as described in Materials and Methods in the online supplement. Test samples were determined as being anti-MLV positive or negative by comparing the mean optical density (OD) values with a given threshold value (mean value of OD of the negative control+2×standard deviation value).
Detection of viral protein
At the end of infection, cells were harvested and lysed for immunoblotting as previously described (Perez et al., 2012).
Results
MiRNA142-3p expression in hematopoietic lineage cells
Expression levels of miRNA142-3p are enriched in hematopoietic lineage cells compared with some monocytic and lymphoblastic cell lines (Chen et al., 2004; Merkerova et al., 2008). We confirmed this result in primary resting and activated PBMCs from five healthy individuals, and compared three established human cell lines (CEM, CEM-T4, and U937) of hematopoietic origin with two nonhematopoietic cell lines (U87-MG glioma and PC-3 prostate cancer; Fig. 1A). The expression of miRNA142-3p in both U87-MG and PC-3 cells was at the lower limit of detection (Ct values between 38 and 40) by qRT-PCR. MiRNA142-3p expression in primary PBMCs did not vary appreciably among individuals, or between resting and activated states. In lymphoid-derived cell lines (CEM, CEM-T4, and U937) miRNA142-3p expression levels were comparable to those in PBMCs. Thus, miRNA142-3p expression is enriched in hematopoietic lineage cells, and the state of activation appears to have little effect on miRNA142-3p levels in primary mononuclear cells.
pAC3-GFP vectors carrying 142-3pT sequences replicate efficiently in U87-MG cells
In pAC3-GFP the CD gene of pAC3-yCD2 was replaced with a GFP gene (Perez et al., 2012). We constructed pAC3-GFP vectors carrying either a single copy or four tandem repeats of 142-3pT (Fig. 1B–D). All three vectors generated similar transient transfection titers in the range of 1×106−2×106 transducing units (TU)/ml on PC-3 prostate cancer cells (Fig. 2A). The replication kinetics of pAC3-GFP-142-3pT and pAC3-GFP-142-3pT4X were comparable to that of pAC3-GFP (Fig. 2B and C) in U87-MG cells. As expected, neither repression of GFP expression nor restriction of viral replication was observed in U87-MG cells infected with vectors carrying the 142-3pT sequence, as miRNA142-3p expression was at the lower limit of detection in U87-MG cells.
FIG. 2.

Replication kinetics and green fluorescent protein (GFP) expression levels of pAC3-GFP, pAC3-GFP-142-3pT, and pAC3-GFP-142-3pT4X vectors in U87-MG cells. (A) Viral titer of pAC3-GFP, pAC3-GFP-142-3pT, and pAC3-GFP-142-3pT4X vectors in PC-3 cells. (B) Replication kinetics of pAC3-GFP, pAC3-GFP-142-3pT, and pAC3-GFP-142-3pT4X vectors. U87-MG cells were infected with each vector at a multiplicity of infection (MOI) of 0.01 on day 0 and passaged on days 3, 6, and 9 postinfection. The percentage of GFP-positive cells was determined by flow cytometry with proper gating to exclude GFP-negative cells. The replication kinetics of each vector was obtained by plotting the percentage of GFP-positive cells versus time. (C) Comparison of MFI of GFP expression at the indicated time points postinfection. All experiments were performed in triplicate, and the data shown represent one of the three independent experiments (means+SD). (D) Schematic diagram of integrated proviral DNA and locations of the primer sets. 5-MLV-U3-B and 3-MLV-Psi primers and probe were used to determine the average copy number of vector per cell and viral titer by qPCR. UCLA5-127 and UCLA3-37 primers were used to assess the integrity of the IRES-GFP transgene of integrated proviral DNA spanning the IRES-GFP region by end-point PCR. (E) Stability of IRES-GFP transgene in pAC3-GFP, pAC3-GFP-142-3pT, and pAC3-GFP-142-3pT4X proviral DNA over multiple serial infections in U87-MG cells. DNA molecular marker (1 Kb Plus marker; Life Technologies) was included in the first lane of each gel. The numbers above each lane indicate the number of infection cycles for each vector. Arrowheads indicate size of the PCR product expected for the undeleted IRES-GFP region (1445 bp for pAC3-GFP, 1492 bp for pAC3-GFP-142-3pT, and 1575 bp for pAC3-GFP-142-3pT4X vector). NTC, no-template control; +, positive control using plasmid DNA corresponding to each vector as a template in PCR.
pAC3-GFP vectors carrying 142-3pT sequences are stable through multiple rounds of infection in U87-MG cells
We examined the stability of these vectors over serial infection cycles, by collecting viral supernatant from fully infected U87-MG cells, infecting fresh U87-MG cells for 12 cycles, harvesting the genomic DNA after each infection, and amplifying a 1.4-kb PCR product to assess the integrity of the integrated viral genome (Fig. 2D), as previously described (Logg et al., 2002; Perez et al., 2012). Results are shown in Fig. 2E; PCR products <1.4 kb represent partial or complete deletion of viral genome in the IRES-GFP region. The pAC3-GFP-142-3pT and pAC3-GFP-142-3pT4X vectors showed complete stability up to infection cycle 6, similar to the parental vector. After infection cycle 6, emergence of deletion mutants, indicated by PCR products<1.4 kb, were observed for all three vectors. However, the 1.4-kb band carrying the intact IRES-GFP region could still be detected up to infection cycle 12, consistent with previous data (Logg et al., 2001; Wang et al., 2006; Perez et al., 2012). Our findings indicate that the pAC3-GFP vector can tolerate insertion of miRNA target sequences in the 3′ UTR without any significant impairment of viral replication and vector stability.
Vectors carrying 142-3pT sequences show repression of transgene expression in PBMCs and are stable
Activated PBMCs from one of the five donors were infected with pAC3-GFP, pAC3-GFP-142-3pT, or pAC3-GFP-142-3pT4X vectors at an MOI of 4, and viral spread was monitored by flow cytometry. Because of the short-term viability of PBMCs in culture and their low infectivity for amphotropic MLV (Sabatino et al., 1997; Ebeling et al., 2003), viral replication kinetics could be assessed in vitro only up to day 10 postactivation with OKT-3 and IL-2. On day 3 postinfection, there was little difference in the percentage of GFP-positive cells among the three vectors. The parental vector continued to spread on day 6 in culture, whereas cells infected with pAC3-GFP-142-3pT vector or pAC3-GFP-142-3pT4X vector remained static or showed a slight decrease in the percentage of GFP-positive cells over time. By day 10 postinfection, a significant difference in viral spread was observed among the three vectors (Fig. 3A). Despite small differences in viral spread on day 3, remarkable differences in the level of GFP expression were observed at early time points postinfection, as indicated by comparison of the mean fluorescence intensity (MFI) between the parental vector and 142-3pT-restricted vectors (Fig. 3B). Notably, the pAC3-GFP-142-3pT4X vector appeared to be more effective in repressing GFP expression than the pAC3-GFP-142-3pT vector (Fig. 3B). Furthermore, Fig. 3C shows that the IRES-GFP region from the genomic DNA of infected PBMCs remained stable over the entire course of infection for all three vectors.
FIG. 3.

Repression of viral spread in PBMCs infected with pAC3-GFP vector carrying the 142-3pT sequence. (A) Replication kinetics of pAC3-GFP, pAC3-GFP-142-3pT, and pAC3-GFP-142-3pT4X vectors in PBMCs. Activated PBMCs were infected with each vector at an MOI of 4 on day 0 and passaged on days 3, 6, and 10 postinfection. The percentage of GFP-positive cells was determined by flow cytometry, using proper gating to exclude CD3- and GFP-negative cells. The replication kinetics of each vector was obtained by plotting the percentage of GFP-positive cells versus time. (B) Comparison of mean fluorescence intensity of GFP expression at the indicated time points postinfection. (C) Stability of pAC3-GFP, pAC3-GFP-142-3pT, and pAC3-GFP-142-3pT4X proviral DNA in PBMCs. DNA molecular marker (1 Kb Plus marker) was included in the first lane of the gel. The numbers above each lane indicate the day postinfection. The arrowhead indicates the size of the PCR product expected for the undeleted IRES-GFP region (1445 bp for pAC3-GFP, 1492 bp for pAC3-GFP-142-3pT, and 1575 bp for pAC3-GFP-142-3pT4X). NC, naive cells, negative control. (D) Schematic diagram of cellular viral RNA isoforms. 5′ GFP, 3′ GFP primers and probe, and 5′ env2, 3′ env2 primers and probe, which recognize both unspliced and spliced cellular viral RNA, were used, respectively, to measure the level of cellular viral RNA by qRT-PCR. (E) Normalized expression level of cellular viral RNA in PBMCs infected with pAC3-GFP, pAC3-GFP-142-3pT, and pAC3-GFP-142-3pT4X, using the env2 primer set versus the GFP primer set. The expression level is presented relative to the parental vector, which is set to 1.
Repression of viral spread in PBMCs is mediated by selective reduction of viral mRNA
Cellular viral RNA levels in PBMCs were measured and first normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and subsequently further normalized to the average copy number of integrated proviral DNA per cell with env2 and GFP amplicons (Fig. 3D). Reductions in normalized cellular viral RNA were observed at all time points for both 142-3pT-restricted vectors, as compared with the parental vector (Fig. 3E), with day 10 levels appearing qualitatively to be most markedly suppressed (about 25% of control or less). Therefore, our data indicate selective repression of transcripts from the pAC3-GFP-142-3pT and pAC3-GFP-142-3pT4X vectors, consistent with the proposed RNA interference (RNAi) mechanism of action.
To examine the possibility that 142-3pT-carrying vectors might accumulate mutations after infection, we isolated and cloned IRES-GFP PCR products from genomic DNA (day 10 postinfection) of PBMCs infected with the pAC3-GFP-142-3pT or pAC3-GFP-142-3pT4X vector. Ten clones were analyzed for each vector, and the sequencing data revealed that 2 of 10 clones for the pAC3-GFP-142-3pT vector had A-to-G mutations. Similarly, 1 of 10 clones for the pAC3-GFP-142-3pT4X vector had C-to-T and G-to-A mutations within and proximal to the seed recognition sequence, respectively (Supplementary Fig. S1). Our results indicate that the spread of RRV incorporating the 142-3pT sequence can be restricted in cultured PBMCs by reduction of viral RNA levels, and the majority of these vectors appear to be stable during the entire course of infection.
Vectors carrying 142-3pT sequences show repression of transgene expression in hematopoietic lineage-derived cell lines
To examine longer term repression than is possible in PBMC experiments, we examined established cell lines of myeloid (U937) and lymphoid (CEM) origin. Cells were infected with pAC3-GFP, pAC3-GFP-142-3pT, or pAC3-GFP-142-3pT4X vector at an MOI of 2. Both lymphoid cell lines supported viral replication of the parental pAC3-GFP vector, as indicated by a gradual increase in the percentage of GFP-positive cells over time (Fig. 4 shows results from U937 cells; see the online supplement for the CEM cell line). Some GFP expression was observed in U937 cells infected with pAC3-GFP-142pT vector during the entire course of infection, whereas the level of GFP expression was fully repressed in cells infected with pAC3-GFP-142-3pT4X vector (Fig. 4A). In terms of vector stability, initial deletion of the IRES-GFP region occurred at about the same time for both pAC3-GFP and pAC3-GFP-142-3pT but became almost complete for pAC3-GFP-142-3pT, whereas the majority of the parental pAC3-GFP vector remained full length. In contrast, the pAC3-GFP-142-3pT4X vector remained stable throughout the entire course of infection (Fig. 4B). The presence of the intact full-length 1.4-kb product, and of a low-abundance product arising from the small percentage of cells transduced by the initial viral inoculum, is consistent with suppression of replication both at the proviral DNA level (vector copy number; Fig. 4C) and at the RNA level after initial infection (Fig. 4D). Furthermore, qRT-PCR results showed effective repression of viral replication of pAC3-GFP-142-3pT vector up to day 21, after which emergence of deletion mutants was observed, and proviral and viral RNA levels increased. In contrast, the pAC3-GFP-142-3pT4X proviral vector remained intact and viral RNA levels showed sustained suppression through day 28 (Fig. 4B–D). In correlation, there was no detectable titer from U937 cells infected with pAC3-GFP-142-3pT4X (Fig. 4E), and sustained repression of viral capsid and envelope gene expression was observed in U937 cells infected with pAC3-GFP-142-3pT4X vector (Fig. 4F). Thus, the lower level of cellular viral RNA observed in U937 cells infected with pAC3-GFP-142-3pT4X correlates with low proviral DNA level, undetectable level of viral protein, and infectious particle production, leading to sustained repression of viral spread.
FIG. 4.

Repression of viral spread in U937 cells infected with pAC3-GFP vector carrying the 142-3pT sequence. (A) Replication kinetics of pAC3-GFP, pAC3-GFP-142-3pT, and pAC3-GFP-142-3pT4X vectors in U937 cells. Cells were infected with each vector at an MOI of 2 on day 0 and passaged at the indicated time points. The percentage of GFP-positive cells was determined by flow cytometry, with proper gating to exclude GFP-negative cells. The replication kinetics of each vector was obtained by plotting the percentage of GFP-positive cells versus time. (B) Stability of pAC3-GFP, pAC3-GFP-142-3pT, and pAC3-GFP-142-3pT4X proviral DNA in U937 cells. DNA molecular marker (1 Kb Plus marker) was included in the first lane of the gel. The numbers above each lane indicate the days postinfection. The arrowheads indicate the size of the PCR product expected for the undeleted IRES-GFP region (1445 bp for pAC3-GFP, 1492 bp for pAC3-GFP-142-3pT, and 1575 bp for pAC3-GFP-142-3pT4X vectors). NC, naive cells, negative control. The 17-day lane for pAC3-GFP-1423pT appears under-amplified for unknown reasons, but shows a full-length band. (C) Vector copy number of proviral DNA in U937 cells infected with pAC3-GFP, pAC3-GFP-142-3pT, and pAC3-GFP-142-3pT4X vector. (D) Normalized expression level of cellular viral RNA in U937 cells infected with pAC3-GFP, pAC3-GFP-142-3pT, and pAC3-GFP-142-3pT4X vector. Expression levels are presented relative to the parental vector, which is set to 1. (E) Viral titers produced by infected U937 cells. At the end of infection, cells were seeded at 1×106 in 5 ml of culture medium. At 48 hr postseeding, viral supernatant from each sample was collected for titration in PC-3 cells by qPCR. (F) Viral proteins produced by U937 cells infected with pAC3-GFP, pAC3-GFP-142-3pT, and pAC3-GFP-142-3pT4X vectors. At the end of infection, cells were seeded at 1×106 in 5 ml of culture medium. At 48 hr postseeding, cells were harvested and lysed for immunoblotting. Twenty micrograms of cell lysate was loaded onto each lane as indicated by the loading control, GAPDH. Lanes 1–4: noninfected cells and cells infected with pAC3-GFP, pAC3-GFP-142-3pT, or pAC3-GFP-142-3pT4X vector, respectively. Asterisk (*) indicates deletion of the IRES-GFP cassette in vectors carrying the 142-3pT sequences.
In CEM cells, deletion of the IRES-GFP region in cells infected with parental vector was observed in the early stages of infection. In this case, the lack of full viral spread, as monitored by GFP expression in the CEM cell line, is partly due to the emergence of deletion mutations. However, CEM cells infected with pAC3-GFP-142-3pT4X vector showed similar prolonged repression of viral replication with stable vector, reduction in cellular viral RNA, and undetectable titer (Supplementary Figs. S2 and S3). Together, our results indicate that in cells in which viral replication was effectively repressed by miRNA142-3p-mediated RNA interference, the integrated viral genome remained stable and further virus spread did not occur.
Data from hematopoietic lineage cell lines suggest that the lack of GFP expression in cells infected with vectors carrying the 142-3pT sequence was mediated by miRNA142-3p regulation at least during the early time points after infection, and that cells infected with vector carrying four copies of the 142-3pT sequence showed more efficient reduction of viral gene expression, which in turn correlated with more durable repression of viral spread.
Vectors carrying 142-3pT sequences spread efficiently in tumors of immune-competent mice
Humans and mice share an identical mature miRNA142-3p sequence. Therefore, the effect of the 142-3pT sequence could be evaluated in vivo by monitoring the biodistribution of the vectors in immune-competent, tumor-bearing mice. Tu2449 mouse glioma cells, of which 0.01% of the cells were fully transduced with pAC3-GFP, pAC3-GFP-142-3pT, or pAC3-GFP-142-3pT4X vector, were implanted subcutaneously into the right flank of each mouse and the proviral vector copy number in tumor, whole blood, bone marrow, and spleen was determined on approximately day 20 after tumor engraftment. In addition, sera from mice of the control and experimental groups were also collected to measure the anti-MLV immune response by ELISA, as suppression of expression of xenoantigens in lymphoid tissue has been associated with lack of immune response to these xenoantigens in other systems (Brown et al., 2006). Tumor growth rates among the control and experimental groups were comparable (Fig. 5A) and viral spread was almost completely restricted to tumors for all three vectors (from 60,000 to 750,000 copies in tumors and mostly below the LLOQ or not detected in blood, spleen, or bone marrow) (Supplementary Table S1). In this experiment, vectors carrying the 142-3pT4X sequence appeared to spread slightly more efficiently than the parental vector (Fig. 5B). There was no significant difference in ELISA-detected immune response among the experimental groups bearing the RRV-infected tumor (Fig. 5C). Together, the data suggest that incorporation of the 142-3pT sequence into the RRV does not appreciably affect viral replication or the host anti-MLV immune response.
FIG. 5.

Biodistribution of retroviral replicating vector (RRV) in immune-competent mice engrafted with tumor infected with vector carrying the 142-3pT sequences. (A) Tumor growth in mice engrafted with tumor infected with RRV carrying the 142-3pT sequences. (B) Scatter plot of the vector copy number of proviral DNA in tumor infected with pAC3-GFP, pAC3-GFP-142-3pT, or pAC3-GFP-142-3pT4X vector on approximately day 20 after tumor engraftment. Each symbol represents one mouse. (C) Scatter plot of anti-MLV immune response measured on approximately day 20 after tumor engraftment. Control group consisted of mice engrafted with subcutaneous tumor without RRV infection. Mean values and standard deviations are shown. One-way analysis of variance (ANOVA) was performed for statistical analysis, and values from samples scored as lower limit of quantification (LLOQ) (fewer than 250 copies/μg) were included in the analysis. *Significant difference (p<0.05). OD, optical density; ns, not significant.
Vectors carrying 142-3pT sequences show repression of viral spread in lymphoid tissues of immune-deficient nude mice
Replication of RRV with or without the 142-3pT sequence in lymphoid tissues of immune-competent mice described previously was not robust enough to demonstrate suppression of viral spread. Therefore, we evaluated the effectiveness of viral suppression in lymphoid tissues in vivo, using immune-deficient nude mice to remove antiviral adaptive immune responses as conflating issues in data interpretation. In the immune-deficient mouse model, viral suppression was evaluated by monitoring the biodistribution of the vectors in blood, bone marrow, and spleen on days 15 and 30 after intravenous administration of RRV. Mice were infected with 4×105 TU of pAC3-GFP, pAC3-GFP-142-3pT, or pAC3-GFP-142-3pT4X vector by tail vein injection. Vector levels in genomic DNA from whole blood, bone marrow, and spleen were measured on days 15 and 30 postinfection. On day 15 postinfection with the parental vector, viral infection was detected in blood and bone marrow, but not in spleen. In contrast, viral spread was mostly below the lower limit of quantification in mice infected with the pAC3-GFP-142-3pT or pAC3-GFP-142-3pT4X vector (Supplementary Tables S2 and S3), an effect that was statistically significant (p<0.05). By day 30, viral spread was observed in all of these tissues among the three groups. However, mice infected with pAC3-GFP-142-3pT or pAC3-GFP-142-3pT4X vector continued to show marked sustained repression of viral spread in all of these tissues, which was statistically significant compared with the parental vector (Fig. 6A–C; Supplementary Tables S2 and S3). When data were analyzed to differentiate dose effect of the target sequence between the pAC3-GFP-142-3pT and pAC3-GFP-142-3pT4X vectors, a statistically significant difference between the two vectors was observed only in blood, despite trends in other tissues (Supplementary Table S2). Intriguingly, the median vector copy number of all three vectors appeared to be higher in bone marrow than in blood and spleen by day 30 postinfection. Therefore, we determined whether the lineage-negative (lin−) stem progenitor cells derived from bone marrow (BM) could be infected with RRV, and if so, whether RRV incorporating the 142-3pT sequence restricted viral spread in this particular cell population. We repeated the experiment and harvested BM samples from three sets of three randomly pooled mice in each group in order to obtain sufficient numbers of lin− cells to measure GFP expression levels and vector copy number. Figure 6D and E, and Supplementary Table S4, show that the pAC3-GFP vector does infect lin− stem progenitor cells in immune-deficient nude mice, but that vectors incorporating 142-3pT sequences were effectively suppressed in this population as analyzed either by GFP expression or vector copy number. Similar results were obtained in the lin+ cell population (Supplementary Table S4). All together, the results indicate that the spread of RRV incorporating the 142-3pT sequence can be effectively restricted in lymphoid tissues in vivo, and that the restriction is more effective with incorporation of multiple 142-3pT sequences. This in vivo effect is presumably by the same mechanisms observed in primary human PBMCs and hematopoietic lineage-derived cell lines.
FIG. 6.

Biodistribution of RRV in immune-deficient mice infected with vectors carrying the 142-3pT sequence. Viral spread on day 30 postadministration in (A) blood, (B) bone marrow, and (C) spleen of mice infected with pAC3-GFP, pAC3-GFP-142-3pT, or pAC3-GFP-142-3pT4X vector was determined by qPCR using tissue-derived genomic DNA. (D) Lineage-negative (lin−) bone marrow cells from infected, immune-deficient mice were analyzed by flow cytometry for GFP expression. (E) Viral spread in the lin− cell population infected with pAC3-GFP, pAC3-GFP-142-3pT, or pAC3-GFP-142-3pT4X vector. The control group consisted of mice injected with PBS. Mean values and standard deviations are shown. One-way ANOVA was performed for statistical analysis, and values from samples scored as LLOQ (fewer than 250 copies/μg) were included in the analysis. *Significant difference (p<0.05). ns, not significant.
Discussion
RRVs efficiently infect malignant tumor target cells in vivo in animal models, and are tumor selective in immune-competent animals (Wang et al., 2006; Ostertag et al., 2012). However, implantation of immune-ablated monkeys with autologous hematopoietic stem cells, transduced with a mixture of replication-competent amphotropic MLV, contaminating MCF viruses, and other infectious particles derived from “ping-pong” amplification of a nonreplicative retroviral vector in mouse producer cells, gave rise to lymphoma in 3 of 10 immune-ablated monkeys in 6–7 months (Vanin et al., 1994; Purcell et al., 1996). Unlike the others, the three affected monkeys did not have a detectable antiviral immune response and allowed extensive viral replication to occur in vivo. In similar experiments in immune-ablated monkeys implanted with autologous hematopoietic stem cells transduced with vector and amphotropic MLV (made in a more controlled fashion, without the ping-pong process), four of four monkeys survived without clinical sequelae or detectable virus, and remained seropositive for 3 years (Cornetta et al., 1991). Therefore, published data in mouse and rat tumor models and in monkeys (Cornetta et al., 1990; Wang et al., 2006; Ostertag et al., 2012) and our unpublished data in dogs all suggest that MLV-based RRV replication is well controlled in blood cells of immune-competent animals without the need for the microRNA target strategy used here. Also, Toca 511-encoded CD activates the prodrug 5-FC into the anticancer drug 5-FU, which removes cells that have been infected by the virus. However, a small theoretical risk of uncontrolled viral replication in hematopoietic lineage cells remains in settings in which RRVs do not elicit an antiviral immune response, or are not otherwise destroyed. Suppression of infection in hematopoietic lineage tissues is one strategy that could further reduce the possibility of such an event in human subjects. Various strategies for tissue-specific targeting of RRV have been explored (Logg et al., 2001; Schneider et al., 2003; Tai et al., 2003; Metzl et al., 2006; Duerner et al., 2008). However, lymphohematopoietic cells were not specifically detargeted, and the potential to improve the safety of MLV-based replicating vectors by such approaches is unclear. In this study, we examined whether repression of viral replication could be achieved by the interaction between miRNA142-3p and its target sequence in the 3′ UTR of RRV in primary human PBMCs, hematopoietic lineage-derived cell lines, in an immune-competent tumor-bearing mouse model, and an immune-deficient mouse model.
Our RRVs can tolerate the insertion of 142-3pT sequences, replicate efficiently, and remain stable over multiple rounds of viral infection in U87-MG glioma cells. The tolerability of sequence insertion within the 3′ UTR in our study is consistent with data previously reported (Logg et al., 2001; Wang et al., 2006). Furthermore, the vectors incorporating the 142-3pT sequence replicated as efficiently as the parental control vector in tumors in vivo. After the initial infection event, viral replication was effectively repressed in PBMCs infected with vectors carrying the 142-3pT sequence relative to the control vector, and both single-copy and 4X copy types of vectors remained stable and repressed GFP protein expression during the course of infection. This was also true in longer experiments with U937 and CEM cell lines. Although the use of GFP expression as one of the readouts for repression of viral spread was complicated by the emergence of GFP deletion mutants in the cell lines, the repression of viral replication was confirmed by additional methods including assessment of vector stability, measurement of the relative expression level of cellular viral RNA, and detection of viral proteins.
In the syngeneic tumor model, the absence of viral spread in lymphoid tissue for all three vectors did not affect viral spread within the tumor, growth of the tumor, or the anti-MLV immune response mounted by the host. These results suggest that active viral replication in hematopoietic lineage cells is not required for efficient infection of tumor cells in vivo. In addition, the absence of viral spread in lymphoid tissue, regardless of the presence or absence of miRNA142-3pT sequences, shows that lack of infection of lymphoid cells does not lead to a delay in onset of an anti-MLV immune response beyond the expected time frame of 10–20 days in mice. The opposite effect (inhibition of anti-xenoantigen immune responses) has been reported for nonreplicating lentiviral vectors incorporating miRNA142-3p target sequences (Brown et al., 2006, 2007; Brown and Naldini, 2009). The difference may be due to different routes of administration or potency of immune stimulation between RRV and nonreplicating lentiviral vectors, as the latter completely lack viral protein expression. The nonreplicating MLV-based vectors, which led to the development of leukemia in some patients with X-linked severe combined immunodeficiency (SCID) receiving ex vivo gene therapy (Hacein-Bey-Abina et al., 2010) also express no viral proteins, and it is conceivable that, because RRVs express viral proteins that robustly elicit antiviral immune responses, RRVs may paradoxically present less risk than nonreplicating retroviral vectors in certain clinical settings. Our observations showing lack of interference of antiviral immune response by miRNA-based detargeting with RRV are novel and have positive clinical safety implications.
Repression of viral replication in lymphoid tissue, particularly in bone marrow, via miRNA142-3p was demonstrated in the nude mouse model in the course of a 30-day infection, showing this repression is independent of an antiviral immune response. In general, the vector carrying four copies of the 142-3pT sequence was repressed more effectively than the vector carrying a single copy.
Relative cellular viral RNA levels of vector with 142-3pT sequences in cell lines were significantly reduced compared with those produced by the control vector, in PBMCs and cell lines. However, when the relative cellular viral RNA expression levels were normalized to average vector copy number per cell, we did not observe 100% RNA degradation even in the early time period after initial infection in all cases. Incomplete repression of viral spread was most pronounced in PBMCs, in which low vector copy number due to low infectivity occurred. The potential discrepancy between strong repression of GFP fluorescence and moderate repression of viral RNA levels observed in PBMCs suggests that additional mechanisms besides RNA degradation are involved. Possible additional mechanisms are translational inhibition of GFP protein synthesis or diversion of RNA to viral particle assembly, away from the RNAi cellular compartment. Both mechanisms could occur concomitantly, as the emergence of deletion mutants observed in the later stage of infection presumably could arise only from new infections via the pool of viral RNA directed to viral particle assembly.
Sustained repression of viral replication in PBMCs, U937 cells, and CEM cells infected with the pAC3-GFP-142-3pT4X vector (demonstrated by repression of GFP expression, vector stability, and reduction in cellular viral RNA and viral titer), and the marked in vivo repression of viral spread in hematological tissue in vivo, suggest that tissue-specific miRNA-based detargeting approaches can be effective in restricting retrovirus replication. Previously, analyses of insertions of miRNA target sequence in a replicating oncolytic picornavirus (coxsackievirus) up to day 45 after virus administration showed that nearly 50% of the virus from viremic mice have mutated sequences, and hence represent escape mutants (Kelly et al., 2008). In contrast, the finding that derepression of GFP expression, or an increase in cellular viral RNA and titer, was not observed in U937 cells infected with the pAC3-GFP-142-3pT4X vector, suggests that even if there were mutations in one or more copies of the miRNA target sequence within the 142-3pT4X vector, the viral genome was still susceptible to suppression by the RNAi pathway with remaining copies. However, it appears likely that in the absence of complete suppression, the RRVs carrying 142-3pT sequences will produce escape mutants on further extensive replication in vivo.
We have shown here repression of both gene expression and viral replication of our RRV system in primary human PBMCs, in hematopoietic lineage-derived cell lines, and in lymphoid tissues in vivo. In contrast, an engineered adenoviral vector carrying the miRNA122 target sequence in the 3′ UTR of the E1A gene has been shown to be less affected by the same approach, with only a mild reduction in viral titer (Ylosmaki et al., 2008; Cawood et al., 2009). In general, single-stranded RNA viruses are considered more prone to accumulate mutations than at least double-stranded DNA viruses. However, the immediate and direct availability of RNA target(s) for miRNA-mediated suppression of gene expression in single-stranded RNA viruses may render the miRNA target-mediated suppression of replication most feasible with viruses with these genomes, provided near complete repression of replication can be achieved. This is what is observed when replication is efficiently repressed using the miRNA target strategy described here (e.g., 142-3pT4X in U397 cells), and mutations appear to accumulate slowly (Supplementary Fig. S4). This also suggests that inclusion of other target sequences in compatible insertion sites in the viral genome might provide further suppression of viral replication. Further improvements in this kind of vector design may be achieved by placing multiple different miRNA target sequences in the viral genome, by engineering RRV carrying a tissue-specific promoter in place of the U3 region in the LTR (Logg et al., 2002; Metzl et al., 2006), or both.
The biolocalization and virulence of viruses or viral vectors with miRNA targets intended to affect changes in tissue tropism should be fully investigated before widespread use. For example, one report showed that a naturally occurring virus that does not express in lymphoid tissue due to miRNA repression may result in some reduction in antiviral immune function, with changes in biological activity that are not beneficial (Trobaugh et al., 2014). Also, the presence of conventional anti-retroviral antibody responses and other antiviral innate immunity seen in syngeneic animals, with or without the target sequences, also suggests that in immune-competent animals the miRNA target strategy may be unnecessary, and may not lead to any difference in the already low levels of virus distribution in lymphoid tissues.
Supplementary Material
Acknowledgments
The authors thank Nicholas A. Boyle, PhD, Alessandro Lobbia, PhD, and Debra Gessner, MS, for critical reading of the manuscript. The original RRV system was developed with support from NIH grants R01 CA85908, CA105171, and CA121258 to N.K. This work was also supported in part by Accelerate Brain Cancer Cure, the American Brain Tumor Association, the Musella Foundation, the National Brain Tumor Society, Voices Against Brain Cancer, and the U.S. Department of Health and Human Services.
Author Disclosure Statement
A.H.L., O.D., K.W., D.K.G., D.O., D.J.J., and H.G. are fulltime employees of Tocagen. C.R.L. and N.K. are paid consultants to Tocagen and Tocagen provided research contract support to UCLA for some of the experiments described here. Y.L. is a former paid consultant of Tocagen. N.T., C.R.L., S.K., and E.C.Y. are supported in part by NIH grant U01 NS059821 and California Institute for Regenerative Medicine grant TR2-01791 to N.K. N.T. is also supported by NIH training grant T32 AI060567.
References
- Aghi M., Vogelbaum M.A., Jolly D.J., et al. (2013). Phase 1 trial of a retroviral replicating vector (Toca 511) in recurrent high grade glioma patients demonstrates the importance of real-time MRI-guided delivery for dose-related evaluation of safety and efficacy [Abstract ST-001]. Neuro Oncol. 15(Suppl. 3), iii217–iii225 [Google Scholar]
- Baskerville S., and Bartel D.P. (2005). Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 11, 241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown B.D., and Naldini L. (2009). Exploiting and antagonizing microRNA regulation for therapeutic and experimental applications. Nat. Rev. Genet. 10, 578–585 [DOI] [PubMed] [Google Scholar]
- Brown B.D., Venneri M.A., Zingale A., et al. (2006). Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat. Med. 12, 585–591 [DOI] [PubMed] [Google Scholar]
- Brown B.D., Gentner B., Cantore A., et al. (2007). Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat. Biotechnol. 25, 1457–1467 [DOI] [PubMed] [Google Scholar]
- Cawood R., Chen H.H., Carroll F., et al. (2009). Use of tissue-specific microRNA to control pathology of wild-type adenovirus without attenuation of its ability to kill cancer cells. PLoS Pathog. 5, e1000440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.Z., Li L., Lodish H.F., and Bartel D.P. (2004). MicroRNAs modulate hematopoietic lineage differentiation. Science 303, 83–86 [DOI] [PubMed] [Google Scholar]
- Chen K., Huang J., Zhang C., et al. (2006). Alpha interferon potently enhances the anti-human immunodeficiency virus type 1 activity of APOBEC3G in resting primary CD4 T cells. J. Virol. 80, 7645–7657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloughesy T.F., Liau L.M., Chiocca E.A., et al. (2013). Evidence supporting mechanism of action and drug activity in phase I trial of patients with recurrent high grade glioma administered Toca 511 at surgical resection followed by Toca FC [Abstract IT-006]. Neuro Oncol. 15(Suppl. 3), iii68–iii74 [Google Scholar]
- Cornetta K., Moen R.C., Culver K., et al. (1990). Amphotropic murine leukemia retrovirus is not an acute pathogen for primates. Hum. Gene Ther. 1, 15–30 [DOI] [PubMed] [Google Scholar]
- Cornetta K., Morgan R.A., Gillio A., et al. (1991). No retroviremia or pathology in long-term follow-up of monkeys exposed to a murine amphotropic retrovirus. Hum. Gene Ther. 2, 215–219 [DOI] [PubMed] [Google Scholar]
- Cornetta K., Nguyen N., Morgan R.A., et al. (1993). Infection of human cells with murine amphotropic replication-competent retroviruses. Hum. Gene Ther. 4, 579–588 [DOI] [PubMed] [Google Scholar]
- Duerner L.J., Schwantes A., Schneider I.C., et al. (2008). Cell entry targeting restricts biodistribution of replication-competent retroviruses to tumour tissue. Gene Ther. 15, 1500–1510 [DOI] [PubMed] [Google Scholar]
- Ebeling S.B., Simonetti E.R., Borst H.P., et al. (2003). Human primary T lymphocytes have a low capacity to amplify MLV-based amphotropic RCR and the virions produced are largely noninfectious. Gene Ther. 10, 1800–1806 [DOI] [PubMed] [Google Scholar]
- Ebert M.S., and Sharp P.A. (2010). Emerging roles for natural microRNA sponges. Curr. Biol. 20, R858–R861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edge R.E., Falls T.J., Brown C.W., et al. (2008). A let-7 microRNA-sensitive vesicular stomatitis virus demonstrates tumor-specific replication. Mol. Ther. 16, 1437–1443 [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S., Hauer J., Lim A., et al. (2010). Efficacy of gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 363, 355–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka K., Kimura T., Logg C.R., et al. (2007). Therapeutic efficacy of replication-competent retrovirus vector-mediated suicide gene therapy in a multifocal colorectal cancer metastasis model. Cancer Res. 67, 5345–5353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X., Brooks A., Chen H., et al. (2005). APOBEC3G/CEM15 (hA3G) mRNA levels associate inversely with human immunodeficiency virus viremia. J. Virol. 79, 11513–11516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly E.J., Hadac E.M., Greiner S., and Russell S.J. (2008). Engineering microRNA responsiveness to decrease virus pathogenicity. Nat. Med. 14, 1278–1283 [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M., Rauhut R., Yalcin A., et al. (2002). Identification of tissue-specific microRNAs from mouse. Curr. Biol. 12, 735–739 [DOI] [PubMed] [Google Scholar]
- Logg C.R., Logg A., Tai C.K., et al. (2001). Genomic stability of murine leukemia viruses containing insertions at the Env-3′ untranslated region boundary. J. Virol. 75, 6989–6998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logg C.R., Logg A., Matusik R.J., et al. (2002). Tissue-specific transcriptional targeting of a replication-competent retroviral vector. J. Virol. 76, 12783–12791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcher A., Parato K., Rooney C.M., and Bell J.C. (2011). Thunder and lightning: Immunotherapy and oncolytic viruses collide. Mol. Ther. 19, 1008–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkerova M., Belickova M., and Bruchova H. (2008). Differential expression of microRNAs in hematopoietic cell lineages. Eur. J. Haematol. 81, 304–310 [DOI] [PubMed] [Google Scholar]
- Metzl C., Mischek D., Salmons B., et al. (2006). Tissue- and tumor-specific targeting of murine leukemia virus-based replication-competent retroviral vectors. J. Virol. 80, 7070–7078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monticelli S., Ansel K.M., Xiao C., et al. (2005). MicroRNA profiling of the murine hematopoietic system. Genome Biol. 6, R71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mous K., Jennes W., De Roo A., et al. (2011). Intracellular detection of differential APOBEC3G, TRIM5α, and LEDGF/p75 protein expression in peripheral blood by flow cytometry. J. Immunol. Methods 372, 52–64 [DOI] [PubMed] [Google Scholar]
- Müller L., Milsom M., and Williams D. (2011). Insertional mutagenesis in hematopoietic cells: Lessons learned from adverse events in clinical gene therapy trials. In: Dupuy A.J. and Largaespada D.A., eds. Insertional Mutagenesis Strategies in Cancer Genetics (Springer, New York: ), pp 131–165 [Google Scholar]
- Ostertag D., Amundson K.K., Lopez Espinoza F., et al. (2012). Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-fluorouracil using a nonlytic retroviral replicating vector. Neuro Oncol. 14, 145–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez O.D., Logg C.R., Hiraoka K., et al. (2012). Design and selection of Toca 511 for clinical use: Modified retroviral replicating vector with improved stability and gene expression. Mol. Ther. 20, 1689–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell D.F., Broscius C.M., Vanin E.F., et al. (1996). An array of murine leukemia virus-related elements is transmitted and expressed in a primate recipient of retroviral gene transfer. J. Virol. 70, 887–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatino D.E., Do B.Q., Pyle L.C., et al. (1997). Amphotropic or gibbon ape leukemia virus retrovirus binding and transduction correlates with the level of receptor mRNA in human hematopoietic cell lines. Blood Cells Mol. Dis. 23, 422–433 [DOI] [PubMed] [Google Scholar]
- Sakurai F., Katayama K., and Mizuguchi H. (2012). MicroRNA-regulated transgene expression systems for gene therapy and virotherapy. Front. Biosci. 17, 2389–2401 [DOI] [PubMed] [Google Scholar]
- Schneider R.M., Medvedovska Y., Hartl I., et al. (2003). Directed evolution of retroviruses activatable by tumour-associated matrix metalloproteases. Gene Ther. 10, 1370–1380 [DOI] [PubMed] [Google Scholar]
- Tai C.K., Logg C.R., Park J.M., et al. (2003). Antibody-mediated targeting of replication-competent retroviral vectors. Hum. Gene Ther. 14, 789–802 [DOI] [PubMed] [Google Scholar]
- Tai C.K., Wang W.J., Chen T.C., and Kasahara N. (2005). Single-shot, multicycle suicide gene therapy by replication-competent retrovirus vectors achieves long-term survival benefit in experimental glioma. Mol. Ther. 12, 842–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trobaugh D.W., Gardner C.L., Sun C., et al. (2014). RNA viruses can hijack vertebrate microRNAs to suppress innate immunity. Nature 506, 245–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanin E.F., Kaloss M., Broscius C., and Nienhuis A.W. (1994). Characterization of replication-competent retroviruses from nonhuman primates with virus-induced T-cell lymphomas and observations regarding the mechanism of oncogenesis. J. Virol. 68, 4241–4250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Tai C.K., Kershaw A.D., et al. (2006). Use of replication-competent retroviral vectors in an immunocompetent intracranial glioma model. Neurosurg. Focus 20, E25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin D., Zhai Y., Gruber H.E., et al. (2013). Convection-enhanced delivery improves distribution and efficacy of tumor-selective retroviral replicating vectors in a rodent brain tumor model. Cancer Gene Ther. 20, 336–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylosmaki E., Hakkarainen T., Hemminki A., et al. (2008). Generation of a conditionally replicating adenovirus based on targeted destruction of E1A mRNA by a cell type-specific microRNA. J. Virol. 82, 11009–11015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.