

Abstract
In order to quantify the interactions between molecules of biological interest, the determination of the dissociation constant (Kd) is essential. Estimation of the binding affinity in this way is routinely performed in “favorable” conditions for macromolecules. Crucial data for ligand–protein binding elucidation is mainly derived from techniques (e.g., macromolecular crystallography) that require the addition of high concentration of salts and/or other additives. In this study we have evaluated the effect of temperature, ionic strength, viscosity, and hydrophobicity on the Kd of three previously characterized protein–ligand systems, based on variation in their binding sites, in order to provide insight into how these often overlooked unconventional circumstances impact binding affinity. Our conclusions are as follows: (1) increasing solvent viscosity in general is detrimental to ligand binding, (2) moderate increases in temperature have marginal effects on the dissociation constant, and (3) the degree of hydrophobicity of the ligand and the binding site determines the extent of the influence of cosolvents and salt concentration on ligand binding affinity.
Keywords: Affinity, solvent, viscosity, ion strength, dissociation constant, fluorescence spectroscopy
The measurement of the dissociation equilibrium constant (Kd) is the corner stone of the drug discovery process since it encompasses the determination of properties of ligand binding to a receptor necessary for the elucidation of the structure-to-function correlations of potential drugs.1 For ligands that interact noncovalently with their biological target (in contrast to those through covalent binding), binding affinity of a protein–ligand complex (PL) in equilibrium with free ligand L and protein P is reflected in the values of this dissociation constant defined as Kd = [P][L]/[PL]. From this equation it is apparent that complexes with large Kd values have a low binding affinity and vice versa.
In general, experiments to determine Kd values are conducted in “favorable” conditions, e.g., in solutions containing low concentration of macromolecules, buffer salts, and required cofactors.2 In contrast, biochemical reactions in living systems take place in viscous media,3 containing high total concentrations of macromolecules and crowding from other molecular species.4,5 Even though significant efforts have been directed to monitor interactions (through kinetics, binding studies, biological assays, etc.) in conditions that emulate such biological phenomena, important information is derived from techniques that require extreme conditions, e.g., macromolecular X-ray crystallography necessitates high concentrations of salt and/or high viscosity solvents.6 Moreover, the addition of an organic solvent such as DMSO is often necessary to overcome the low aqueous solubility of potential inhibitors. Since the conditions described above affect the physicochemical properties of the solution, these will also in turn impact the binding affinity of ligands to target proteins.
In order to achieve a desired end point of biophysical characterization (biochemical assays, crystallization, etc.), the aforementioned additives and conditions are often varied assuming that such modifications would not influence ligand/protein interactions. Since the effects of solvent, solute, and temperature factors on ligand–protein binding affinity have to our knowledge not been studied extensively, we therefore attempt to quantitate their effect on ligand–protein affinity and examine their correlation to the hydrophilicity/hydrophobicity of the cognate binding pocket. The results of such a study will be essential for assay validation as well as for interpretation of crystallographic data.
We therefore investigate the effects of temperature, ionic strength (NaCl concentration), solvent viscosity (glycerol percentage), and cosolvent (DMSO) concentration on ligand–protein binding affinity. This was done by determining the Kd values of three protein–ligand systems, based on the differing hydrophobicity of the individual binding sites, using steady-state fluorescence. These include (1) TNF-α [binding of a hydrophobic ligand, (SPD304; CLogP = 8) to a hydrophobic binding pocket] monitored by tyrosine fluorescence; (2) cyclin A2(CA2) [binding of an amphiphilic and highly soluble peptide (RRLIF; CLogP = −0.96) to an amphipathic binding pocket] monitored by tryptophan-fluorescence; and (3) trypsin, a protein with no intrinsic fluorescence [binding of a hydrophilic fluorescent ligand (p-amino-benzamidine/PABA; CLogP = −0.48) to an amphipathic binding pocket].
The binding of ligand to protein is generally dictated by a complex array of intermolecular interactions. The availability of structural information was an important prerequisite, and the systems studied were chosen on the basis of the different types of binding interactions described above (Figure 1). The X-ray structure (PDB ID: 2AZ5) of TNF-α with SPD304 revealed that hydrophobic interactions dominate the association of the ligand to the protein binging site. In contrast, CA2/RRLIF crystal structure (PDB ID: 1OKV) indicates that both hydrophobic and electrostatic interactions contribute to the affinity, although the charge–charge interactions provide the major contribution to binding.7 In the case of the trypsin/PABA system (PDB ID: 3GY4) the ligand amidinium group mimics the guanidinium side chain of an arginine residue, and as a result, the noncovalent complex is stabilized by a charge–charge interaction and reinforced by two hydrogen bonds, between the amidinium group and Asp189. In addition, three other hydrogen bonds are observed between the protein and the ligand. Furthermore, hydrophobic interactions contribute to the stability of the complex.8
Figure 1.
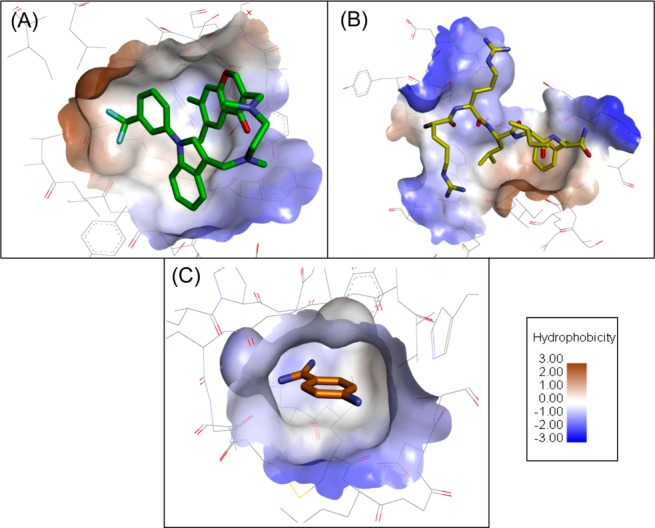
Binding site surfaces highlighting hydrophobicity degrees between (A) TNF-α dimer with SPD304 (PDB ID: 2AZ5); (B) CA2 with RRLIF peptide (PDB ID: 1OKV); and (C) trypsin with PABA (PDB ID: 3GY4). Hydrophobicity levels are visualized with brown-to-blue gradient.
TNF-α has been considered as an attractive therapeutic target for inhibiting inflammation and bone resorption.9 SPD304 is a recently discovered small-molecule antagonist that inhibits the trimerization of TNF-α.10 We have previously demonstrated that the dissociation constant of TNF-α/SPD304 (at 25 °C) complex is 5.36 ± 0.21 μM.11
Human CA2 participates in cell cycle regulation, DNA replication, and transcription. CA2 plays a critical role in cell-cycle regulation by binding and activating cyclin dependent kinase 2 (CDK2), thus promoting both G1/S and G2/M cell-cycle transitions. Inhibition of CDK2/CA2 activity through blocking of the substrate recruitment site in the cyclin A subunit has been demonstrated to be an effective method for inducing apoptosis in tumor cells.7,12 RRLIF is a peptide ligand, which inhibits (IC50 = 0.68 μM; in a competitive cyclin A-binding assay) the association of the CDK2/CA2 complex with its substrates.7 Fluorescence measurements of CA2 with RRLIF are illustrated in Figure 2A.1, and as shown, sequential additions of this ligand to CA2 resulted in a significant decrease of fluorescence intensity (∼15% quenching) compared to the blank experiment yielding a Kd of 24 ± 2.1 μM.
Figure 2.

Dissociation constant (Kd) determination of (A) CA2 with RRLIF at 25 °C in 50 mM Tris-HCl pH 8.0, 100 mM MgCl2; λex = 295 nm, and λem = 345 nm. Slits for excitation and emission were set at 5 and 20 nm, respectively, and (B) trypsin with PABA at 25 °C performed in 20 mM phosphate buffer (pH 7.4) containing 100 mM NaCl and 0.1% PEG6000: λex = 320 nm and λem = 370 nm. Slits for both excitation and emission were set at 10 nM. Left panels: Direct plot of fluorescence intensity against ligand total concentration. Sequential additions of ligand were made into a cuvette containing protein (CA2 or tryspin) (■) or Trp (or buffer) (●). The dissociation constant was calculated by fitting the fluorescence intensity corrected values (▲) to a quadric equation. Right panels: Saturation plot after calculation of free (L) and bound (PL) ligand concentrations. Insets: Scatchard plots.
A significant increase of fluorescence intensity of the ligand was observed during titration of trypsin with PABA, in previously characterized assay conditions13 (Figure 2B.1), as compared to the blank experiment, resulting in a Kd value of 4.1 ± 0.35 μM for this interaction. Using the raw data of Figure 2B.1 the saturation curve was generated (Figure 2B.2), and a Scatchard analysis subsequently was performed (Figure 2B.2; inset graph). The linearity of the Scatchard plot resulted in an n value of 1 suggesting one binding site. The resulted Kd of 4.1 μM is in good agreement with the previously reported value of 6 μM13 and the values of Ki of 713 and 8.25 μM14 determined kinetically.
To evaluate the effect of temperature, ionic strength (NaCl concentration) and viscosity (glycerol concentration) on the binding affinity of the three protein–ligand systems, we implemented a 23 full factorial design (Table 1) to quantify dissociation constants (Kd). As illustrated in Table 1, the effect of the three factors on Kd values revealed that glycerol concentration was a crucial effector of decreased binding affinity, followed by temperature (especially for CA2, which was unstable above 30 °C).
Table 1. Full Factorial Design Examined the Effect of Three Independent Variables on Kd of Equilibrium of Various Ligand–Protein Systemsa.
| factor |
Kd (μM) ligand–protein |
|||||
|---|---|---|---|---|---|---|
| run | temp (°C) | NaCl (mM) | glycerol (%v/v) | SPD304/TNF-α | PABA/trypsin | RRLIF/cyclinA2 |
| 1 | 20 | 100 | 5 | 6.14 ± 0.32 | 4.83 ± 0.29 | 21.50 ± 1.52 |
| 2 | 20 | 100 | 50 | 22.54 ± 1.17 | 25.21 ± 1.55 | 32.24 ± 2.18 |
| 3 | 37 | 100 | 5 | 6.46 ± 0.38 | 6.41 ± 0.44 | N.D. |
| 4 | 37 | 100 | 50 | 19.18 ± 1.02 | 22.53 ± 1.37 | N.D. |
| 5 | 20 | 500 | 5 | 6.38 ± 0.43 | 6.23 ± 0.52 | 9.06 ± 0.81 |
| 6 | 20 | 500 | 50 | 24.93 ± 1.95 | 27.52 ± 1.38 | 33.00 ± 2.21 |
| 7 | 37 | 500 | 5 | 7.17 ± 0.64 | 7.62 ± 0.57 | N.D. |
| 8 | 37 | 500 | 50 | 21.87 ± 1.72 | 23.62 ± 0.31 | N.D. |
The mean values of three independent measurements are presented. N.D.: not determined.
Titration of TNF-α with SPD304 at 20 °C (in the presence of 100 mM NaCl) in a solution containing 5% or 50% glycerol resulted in Kd values of 6.14 ± 0.32 μM (run 1) and 22.54 ± 1.17 μM (run 2), respectively. When the titration was performed at 37 °C in a solution containing 5% glycerol, a Kd of 6.46 ± 0.38 μM was obtained (run 3), while an increase of glycerol to 50% resulted in a 3-fold increase of the Kd value (run 4). A similar decrease in binding affinity of the trypsin/PABA system was observed in an increasingly viscous environment (runs 1–4). An increase of the Kd value for CA2/RRLIF binding from 24.5 to 32.24 μM was also observed by increasing glycerol concentration from 5 to 50% (runs 1 and 2, respectively). These results reveal that binding affinity is adversely affected by increasing solvent viscosity.
The concentration of NaCl had a minimal impact on the Kd values of TNF-α/SPD304 system (Table 1) as expected for a majorly hydrophobic interaction. In contrast, elevated NaCl concentration seems to inhibit binding of PABA to trypsin (runs 1 and 5), whereas it promotes cyclin A2/RRLIF binding. Titration of CA2 with RRLIF in buffer containing 100 mM NaCl resulted in a Kd of 21.5 ± 1.52 μM (run 1). When NaCl was increased to 500 mM, binding affinity was significantly increased, resulting in a 2-fold decrease of Kd to 9.06 ± 0.81 μM (run 4). It has been previously demonstrated that CA2 is stable in higher (>500 mM) NaCl concentration.12
To further investigate the effects of solvent viscosity on dissociation constants of the TNF-α/SPD304 and trypsin/PABA systems, we performed a series of experiments increasing the glycerol concentration (up to 50%), at various temperatures (25, 30, or 37 °C) (Figure 3). For glycerol concentrations in the range of 0–10%, similar binding curves (Figure S1, Supporting Information) were obtained when TNF-α was titrated with SPD304 at (25 and 30 °C), resulting in Kd values of ∼5 μM. In contrast, the addition of 25% or 50% glycerol (Figure S1, Supporting Information) resulted in 3- and 4-fold respective increases of Kd values. As illustrated in Figure 3, Kd values obtained at 25 °C in the presence of 25% and 50% glycerol were slightly higher (lower affinity) than those obtained at 30 °C. When experiments were performed at 37 °C in buffers without glycerol, a Kd of 6.18 ± 0.26 μM was obtained. In contrast, when the titration was performed in buffer containing 10% glycerol (at the same temperature), a Kd of 5.45 ± 0.26 μM was obtained. The increase in glycerol concentration to 25 and 50% resulted in a 2.5- and 4-fold increase of Kd value. Notably, Kd values obtained at 37 °C in viscous solutions (25 and 50% glycerol) were lower than those obtained at 25 or 30 °C.
Figure 3.
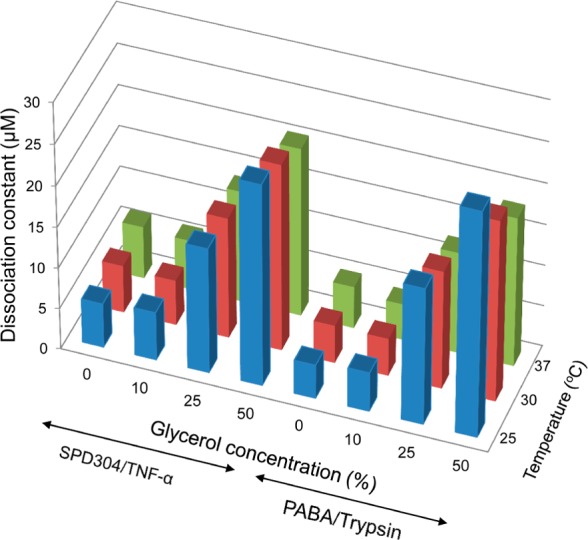
Effect of temperature and glycerol concentration on SPD304/TNF-α and PABA/trypsin binding affinity. The mean values of three independent measurements are presented.
A similar impact of solvent viscosity on the binding affinity of PABA to trypsin was observed (Figure 3). When titrations were performed in the absence of glycerol, the dissociation constant was increased by approximately 12% and 25%, when increasing the temperature from 25 to 30 and 37 °C, respectively. The addition of glycerol to the reaction mixture to a ratio of 10% had minimal impact on the dissociation constant of the trypsin/PABA complex at any tested temperature. In contrast, a decrease of binding affinity (therefore an increase of Kd) of the complex was observed at high glycerol concentrations (>25%). Interestingly, Kd values obtained at 37 °C in viscous solutions (25 and 50% glycerol) were lower than those obtained at 25 or 30 °C (Figure 3).
To further elucidate the effect of solvent viscosity on binding affinity, we monitored shifts in the emission spectra of PABA during titration with trypsin. PABA is weakly fluorescent in neutral aqueous buffers and has an emission maximum at 374 nm. Binding to trypsin results in a blue shift of the emission peak to 362 nm and significant enhancement of fluorescence intensity.13 Therefore, we monitored emission maximum shifts during titration of trypsin with PABA at 37 °C in 20 mM phosphate buffer (pH 7.4) containing 0% or 50% glycerol (Figure 4). Figure 4A.1 illustrates indicative emission spectra during sequential additions of PABA in a trypsin solution (1.5 μM) at 37 °C, in the absence of glycerol. A progressive shift of the emission maximum from 362 to 374 nm was observed as the ligand concentration was increased from 0.33 to 28 μM. In contrast, sequential additions of PABA in buffer without protein (control experiment) resulted in emission maxima of ∼375 nm (Figure 4A.1; inset). Binding curves (Figure 4A.2) were generated by plotting the average fluorescence intensity values from 362 to 375 nm (for each addition) versus the ligand concentration, and curve fitting analysis resulted in a Kd of 5.3 ± 0.31 μM. Subsequent to these experiments, we monitored changes in emission maximum in the presence of 50% glycerol (Figure 4B) to determine solvent viscosity effects. We observed a significant enhancement of emission intensity of PABA as the glycerol proportion in the buffer increased from 0 to 50%. Interestingly, titration of trypsin with PABA at 37 °C in buffer containing 50% glycerol also resulted in a progressive shift of emission maximum from 375 nm to ∼365 nm, after the ligand concentration reached 10.8 μM (Figure 4B.1). As expected, a shift in the emission spectrum was not observed in the blank experiment (Figure 4B.1; inset). A saturation plot (Figure 4B.2, inset) was obtained after subtraction of values of blank experiment (titration of PABA in phosphate buffer, from raw data), and a Kd of 18.21 ± 0.98 μM was calculated, which is 3-fold higher than that obtained at 37 °C, in buffer containing 5% glycerol (Table 1; run 3).
Figure 4.

Fluorescence titration of trypsin with PABA in buffer containing 0% (A) or 50% (B) glycerol. During titration, changes of a ligand’s emission spectrum due to its binding with the protein were recorded. Experiments were performed at 37 °C in 20 mM phosphate buffer (pH 7.4) containing 100 mM NaCl, 0.1% PEG6000, and 0% or 50% glycerol (A and B, respectively). Left panels: emission spectra of PABA during its titration in trypsin (1.5 μM) solution at various conditions. Inset: emission spectra of PABA during its titration in buffer (blank experiment). Right panels: Direct plot of fluorescence raw data versus total ligand concentration during sequential additions of PABA to trypsin solution. Inset: Fitting of corrected fluorescence intensity values (after subtraction of values of blank experiments) to a quadric equation for Kd determination. Arrows indicate the points (concentrations) where the emission spectrum was recorded. (C) Monitoring of emission spectra changes at various time intervals during interaction of PABA with trypsin at 37 °C in buffer containing 0% (left) or 50% (right) glycerol. F.I.: fluorescence intensity.
To investigate whether protein–ligand binding is diffusion limited, we monitored emission spectral alterations during binding of trypsin with PABA (10 μM) in buffers containing 0 or 50% glycerol, at various time intervals. For this, PABA was incubated for 10 min at 37 °C. Trypsin (5 μM) was injected, and perturbations in fluorescence emission spectra were recorded. As illustrated in Figure 4C.1, an immediate shift of emission maximum from 374 to 362 nm and a 5-fold fluorescence enhancement were observed, directly after the addition of protein in the ligand’s solution without glycerol (approximately 2 s are required to record the spectrum). Prolonged incubation (600 s) of trypsin with PABA had no further impact on the emission spectrum. In stark contrast, interaction of PABA with trypsin in the viscous medium took place at a slower pace as illustrated in Figure 4C.2, whereas a progressive shift of emission maximum from 374 to 362 nm followed by a significant enhancement of fluorescence intensity were observed in binding time.
The results obtained by studying the effect of temperature and viscosity on the binding affinity of TNF-α/SPD304 and trypsin/PABA systems suggest that (i) binding is affected by a decrease of the solution’s viscosity as a result of the temperature increase (Figure S2, Supporting Information); therefore, the ligand can “reach” the protein. It has been previously demonstrated that the diffusion controlled rate for the binding of a small ligand to a protein will be inversely proportional to the viscosity of the medium,15 and (ii) glycerol both stabilizes and preserves protein conformation at high temperatures. Our results revealed that Kd values obtained at 37 °C in the presence of 10% glycerol were lower compare to those obtained in the absence of glycerol (Figures 3 and S1, Supporting Information). However, ionic strength modulates the magnitude of electrostatic interactions. The direct dependence of Kd on ionic strength has been recognized as a common feature of protein–protein interactions that are electrostatically rate enhanced and diffusion limited.16 For a protein complex that has charge complementarity in the interface, the association rate constant Kd is expected to be enhanced by electrostatic interactions. To assess the effect of ionic strength on the tested protein–ligand interactions, fluorescence spectroscopy experiments were conducted at salt concentrations varying from 0 to 500 mM.
As illustrated in Table 2, the dissociation constant of TNF-α/SPD304 revealed a weak linear dependence on ionic strength (Figure S3.A, Supporting Information). Contrary to the results observed with the TNF-α/SPD304 complex, the binding affinity of PABA to trypsin appears to be negatively affected by elevated salt concentration (Figure S3.B, Supporting Information). Titration of trypsin with PABA in the presence of 50 or 100 mM NaCl resulted in Kd values of 4.32 ± 0.31 and 4.12 ± 0.42 μM, respectively. The addition of 250 or 500 mM of NaCl in the reaction buffer resulted in a 1.5- and 2-fold increase of Kd (Table 2). However, an altogether reverse effect of salt concentration on CA2/RRLIF binding affinity was observed. When titration of CA2 was performed in the presence of 100 mM NaCl, a Kd value of approximately 24 μM was obtained by curve-fitting analysis (Figure S3.C, Supporting Information). In a protein solution containing 250 mM NaCl, a Kd value of 17.45 ± 0.98 μM was obtained. Interestingly, in the presence of 500 mM NaCl, a 3-fold decrease of Kd (8.47 ± 0.62 μM) was observed (Table 2).
Table 2. Dissociation Constant Values for Three Different Complexes Calculated by Fluorescence Spectroscopy at Different Salt Concentrationsa.
|
Kd (μM) ligand–protein |
|||
|---|---|---|---|
| NaCl (mM) | SPD304/TNF-α | PABA/tryspin | RRLIF/cyclinA2 |
| 50 | 5.42 ± 0.35 | 4.32 ± 0.31 | N.D. |
| 100 | 5.53 ± 0.51 | 4.12 ± 0.42 | 24.36 ± 1.25 |
| 250 | 5.71 ± 0.32 | 6.64 ± 0.35 | 17.45 ± 0.98 |
| 500 | 5.92 ± 0.42 | 8.58 ± 0.49 | 8.47 ± 0.62 |
Experiments performed in 10 mM citrate phosphate (pH 6.5) containing 5% DMSO, 50 mM Tris-HCl pH 8.0 containing 0.01% monothioglycerol, and 20 mM phosphate buffer (pH 7.4) containing 0.1% PEG6000, respectively, at 25 °C. The mean values of three independent measurements are presented. N.D.: not determined.
Because of the amphipathic nature of both protein (CA2) and ligand (RRLIF), one of the key questions is whether RRLIF binding to CA2 is primarily governed by electrostatic or hydrophobic interactions. We have previously suggested that the apparent RRLIF affinity for CA2 is in fact primarily driven by electrostatic interactions.7 Current data further corroborate the above findings for the salt concentrations implemented.
Overall, our findings reveal that an interaction of hydrophobic nature (SPD304/TNF-α) is not influenced by the variation of ionic strength. In contrast, binding of soluble and polar ligands (RRLIF and PABA) to proteins exhibiting partial hydrophilic binding sites (CA2 and trypsin) is profoundly influenced by the ionic strength of the solution. Notably, in the case of the ligand with the highest hydrophilicity (RRLIF; CLogP = −0.96), a 3-fold Kd decrease was observed. On the contrary, in the case of PABA exhibiting a CLogP value of −0.48 (half compared to that of RRLIF), a 2-fold Kd increase was reordered. The respective increase or decrease of binding affinity might be attributed to the nature (positive or negative) of the specific charges of both the protein binding site and the docked ligand.
A major concern during development and validation of a biochemical assay is the potential impact of the low aqueous solubility of potential ligands. Because of the strong relationship between solubility and Kd, bioassay development should properly assess low-solubility compounds.17 DMSO is the organic cosolvent most widely implemented to solubilize compounds in aqueous buffers. However, in several cases, DMSO interference can vary depending on the protein.18 We have previously demonstrated that TNF-α can tolerate up to 10% DMSO; however, Kd values increased 1.5-fold when cosolvent concentration increased from 5 to 10%.11 This indicates that, in the case of a protein with a hydrophobic binding pocket and a hydrophobic ligand, DMSO concentration must be fine-tuned since high levels might prove inhibiting. Therefore, the minimum cosolvent volume, in which the ligand remains soluble, must be determined.
To further examine this preliminary data obtained for TNF-α, we determined the stability of CA2 and trypsin in aqueous solutions containing various concentrations of DMSO (2.5, 5, and 10%) by monitoring the changes of absorbance at 280 nm (A280nm) as previously described.11 The results revealed that both proteins are remarkably stable in the presence of a high (10%) concentration of DMSO since the decrease of A280nm was less than 7% (data not shown). We subsequently tested the effect of cosolvent concentration on CA2/RRLIF and trypsin/PABA binding interaction. In the presence of 2.5, 5, and 10% DMSO, similar binding curves were obtained for the titration of CA2 with RRLIF resulting in each case in a Kd of approximately 24 μM (Figure S4.A, Supporting Information), indicating that DMSO has negligible effect on CA2/RRLIF binding affinity. Varying the DMSO concentration does, however, have a slight effect on trypsin/PABA affinity since in the presence of 2.5, 5, and 10% DMSO the obtained Kd values were 4.4 ± 0.35, 5.1 ± 0.47, and 5.8 ± 0.57 μM, respectively (Figure S4.B, Supporting Information). In accordance with our working hypothesis, interactions of predominantly hydrophilic nature (CA2/RRLIF and trypsin/PABA) are not significantly influenced by the presence of a hydrophobic cosolvent (DMSO) in concentrations up to 10%, whereas binding interfaces driven by primarily hydrophobic interactions (SPD304/TNF-α) are.
In conclusion, the findings of this work highlight the factors influencing protein–ligand interactions that should be taken into account during assay development and validation. If higher temperatures are required, e.g., in the case of conditions emulating biological systems, elevated glycerol concentration might preserve protein stability. Nevertheless, high solvent viscosity has a negative impact on protein–ligand affinity in general, and therefore the optimum polyol concentration should be defined accordingly. Moreover, our results revealed a diverse dependence of the protein–ligand complex formation on ionic strength. To this end, while determining salt concentration in assay buffers, one should take under consideration information regarding the hydrophobic or hydrophilic nature of the binding site and ligand. In addition, although the addition of organic solvents is mandatory for insoluble ligands, it should not exceed a critical concentration, ensuring that the interactions of a ligand within the hydrophobic environment of the protein’s binding pocket remain favorable.
Supporting Information Available
Protein expression and purification; determination of dissociation constant (Kd) from fluorescence measurements and viscosity measurements. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was partly funded by National Institute of Health through the research project grant 5R01CA131368.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Gohlke H.; Klebe G. Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors. Angew. Chem., Int. Ed. 2002, 41, 2645–2676. [DOI] [PubMed] [Google Scholar]
- Minton A. P. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J. Biol. Chem. 2001, 276, 10577–10580. [DOI] [PubMed] [Google Scholar]
- Minton A. P.; Wilf J. Effect of macromolecular crowding upon the structure and function of an enzyme: glyceraldehyde-3-phosphate dehydrogenase. Biochemistry 1981, 20, 4821–4826. [DOI] [PubMed] [Google Scholar]
- Fulton A. B. How crowded is the cytoplasm. Cell 1982, 30, 345–347. [DOI] [PubMed] [Google Scholar]
- Zimmerman S. B.; Trach S. O. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J. Mol. Biol. 1991, 222, 599–620. [DOI] [PubMed] [Google Scholar]
- Carvalho A. L.; Trincao J.; Romao M. J. X-ray crystallography in drug discovery. Methods Mol. Biol. 2009, 572, 31–56. [DOI] [PubMed] [Google Scholar]
- Kontopidis G.; Andrews M. J. I.; McInnes C.; Cowan A.; Powers H.; Innes L.; Plater A.; Griffiths G.; Paterson D.; Zheleva D. I.; Lane D. P.; Green S.; Walkinshaw M. D.; Fischer P. M. Insights into cyclin groove recognition: Complex crystal structures and inhibitor design through ligand exchange. Structure 2003, 11, 1537–1546. [DOI] [PubMed] [Google Scholar]
- Clark S. M.; Konermann L. Determination of ligand-protein dissociation constants by electrospray mass spectrometry-based diffusion measurements. Anal. Chem. 2004, 76, 7077–7083. [DOI] [PubMed] [Google Scholar]
- Yuan H.; Qian H.; Liu S.; Zhang X.; Li S.; Wang W.; Li Z.; Jia J.; Zhao W. Therapeutic role of a vaccine targeting RANKL and TNF-alpha on collagen-induced arthritis. Biomaterials 2012, 33, 8177–8185. [DOI] [PubMed] [Google Scholar]
- Sun H.; Yost G. S. Metabolic activation of a novel 3-substituted indole-containing TNF-alpha inhibitor: Dehydrogenation and inactivation of CYP3A4. Chem. Res. Toxicol. 2008, 21, 374–385. [DOI] [PubMed] [Google Scholar]
- Papaneophytou C. P.; Mettou A. K.; Rinotas V.; Douni E.; Kontopidis G. A. Solvent selection for insoluble ligands, a challenge for biological assay development: a TNF-alpha/SPD304 study. ACS Med. Chem. Lett. 2013, 4, 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Ren J.; Qu X. Biophysical studies on the full-length human cyclin A2: protein stability and folding/unfolding thermodynamics. J. Phys. Chem. B 2008, 112, 8346–8353. [DOI] [PubMed] [Google Scholar]
- Evans S. A.; Olson S. T.; Shore J. D. p-Aminobenzamidine as a fluorescent probe for the active site of serine proteases. J. Biol. Chem. 1982, 257, 3014–3017. [PubMed] [Google Scholar]
- Mares-Guia M.; Shaw E. Studies on the active center of trypsin. The binding of amidines and guanidines as models of the substrate side chain. J. Biol. Chem. 1965, 240, 1579–1585. [PubMed] [Google Scholar]
- van Holde K. E. A hypothesis concerning diffusion-limited protein-ligand interactions. Biophys. Chem. 2002, 101, 249–254. [DOI] [PubMed] [Google Scholar]
- Zhou H. X. Disparate ionic-strength dependencies of on and off rates in protein-protein association. Biopolymers 2001, 59, 427–433. [DOI] [PubMed] [Google Scholar]
- Di L.; Kerns E. H. Biological assay challenges from compound solubility: strategies for bioassay optimization. Drug Discovery Today 2006, 11, 446–451. [DOI] [PubMed] [Google Scholar]
- Johnston P. A.; Johnston P. A. Cellular platforms for HTS: three case studies. Drug Discovery Today 2002, 7, 353–363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.