

Abstract

In a previous communication, our efforts leading from 1 to the identification of spiro[cyclohexane-dihydropyrano[3,4-b]indole]-amine 2a as analgesic NOP and opioid receptor agonist were disclosed and their favorable in vitro and in vivo pharmacological properties revealed. We herein report our efforts to further optimize lead 2a, toward trans-6′-fluoro-4′,9′-dihydro-N,N-dimethyl-4-phenyl-spiro[cyclohexane-1,1′(3′H)-pyrano[3,4-b]indol]-4-amine (cebranopadol, 3a), which is currently in clinical development for the treatment of severe chronic nociceptive and neuropathic pain.
Keywords: NOP receptor agonists, MOP receptor agonists, cebranopadol, analgesics
Recent publications indicate that small molecules activating both nociceptin/orphanin FQ peptide (NOP) and mu opioid peptide (MOP) receptors may potentiate opiate analgesia and at the same time display an improved side effects profile.1,2 We have recently reported the discovery of a series of small molecules, characterized by their high NOP and opioid receptor agonistic activity.3 This series included uncyclized (e.g., 1) as well as spirocyclic examples (e.g., 2a). The discovery of spirocyclic 2a originated from the respective uncyclized analogues, which were potent NOP and MOP receptor binders but sometimes hampered by only partial agonistic NOP and MOP receptor activity. In particular, the spiroindole derivates sparked our interest due to their structural novelty and favorable in vitro and in vivo properties. The leading spiroether 2a exhibited strong efficacy in preclinical models of acute (ED50 rat tail-flick: 3.63 nmol/kg i.v.) and neuropathic pain (ED50 rat spinal nerve ligation: 1.05 nmol/kg i.v.) but was hampered by poor pharmacokinetic (PK) properties in rats with high clearance, large volume of distribution, moderate half-life (Cl = 4.0 L/h·kg; Vss = 7.52 L/kg; t1/2 = 1.6 h), and a critically very low oral bioavailability (F = 4%).
We herein report our efforts to further optimize the spiroindole lead 2a, which eventually led to the discovery of trans-6′-fluoro-4′,9′-dihydro-N,N-dimethyl-4-phenyl-spiro[cyclohexane-1,1′(3′H)-pyrano[3,4-b]indol]-4-amine (3a, cebranopadol), a novel potent analgesic NOP and opioid receptor agonist, currently in clinical development for the treatment of severe chronic nociceptive and neuropathic pain.
The structure–activity relationship (SAR) established around the uncyclized scaffolds (e.g., 1) suggested that a broad variety of linkers such as alcohols, ethers, and amines are tolerated, showing high NOP and MOP receptor binding affinities.3 As a result, we applied this knowledge through analogous structural variations to lead structure 2a (region A, Figure 1). These changes were also combined with a targeted approach to improve the poor PK profile, in particular by addressing metabolically liable regions B and C.
Figure 1.
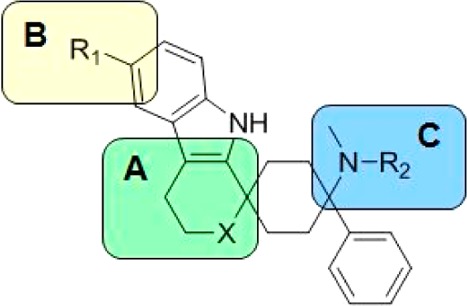
Sites of modification.
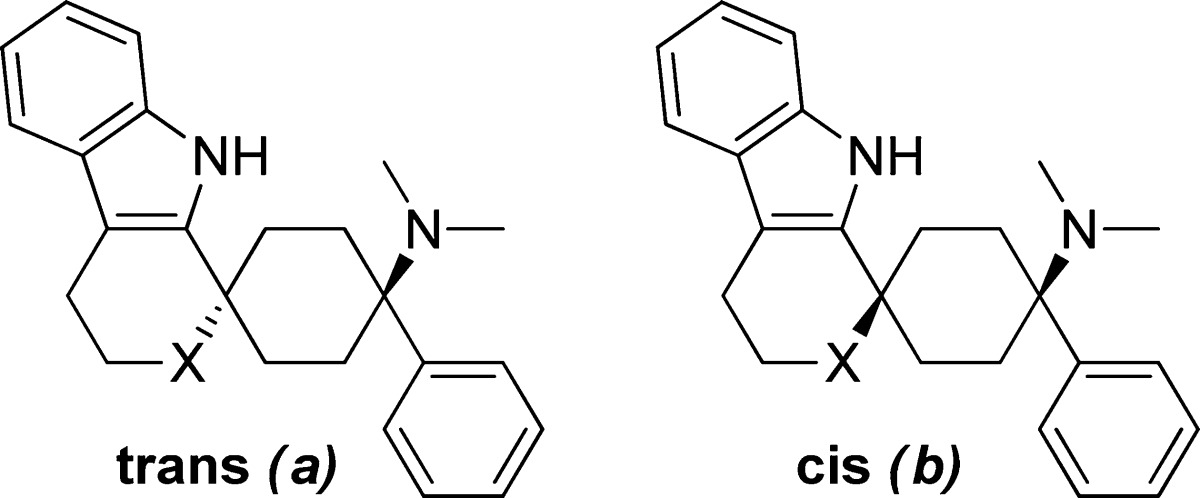
With this in mind, the transformation of the oxacyclic spiro moiety into a carba-, aza-, or thio-cyclic moiety was investigated. Advancing from the oxacyclic spiro 2a to the azacyclic moiety 5a led to equally potent NOP and MOP receptor binders, as well as the introduction of the N-methyl subunit 6a. Similarly, potency was retained when moving from the oxa-cycle 2a to the thio-cycle 7a and carbacycle 8a (Table 1).
Table 1. Modification of Spirocyclic Oxygen, Region A.
| % inhibition
@ 1 μM or Ki (nM) |
||||
|---|---|---|---|---|
| compd | X | config | hNOPr | hMOPr |
| 2ab | O | trans | 0.1 | 0.26 |
| 2bb | O | cis | 30% | 310 |
| 5ac | NH | trans | 0.19 | 0.39 |
| 6ac | NMe | trans | 0.55 | 0.24 |
| 7aa | S | trans | 3.7 | 3.1 |
| 8a | CH2 | trans | 1.26 | 1.95 |
Hemicitrate.
Hydrochloride.
Dihydrochloride.
The wide range of tolerated moieties suggests that, upon cyclization, the molecule has been brought into a preferred conformation at both receptors (Figure 2). As a consequence of these results and employing the most favored core from a lipophilicity point of view, we focused our efforts for further optimization on the oxacyclic moiety.
Figure 2.
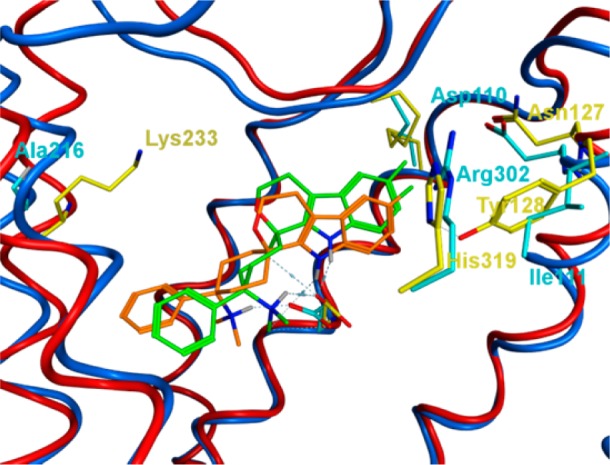
Three-dimensional structural overlay of the binding modes of 3a in the NOP and MOP receptors. NOP: Binding modes of 3a (orange) in the NOP receptor: NOP ribbons, blue; residues, cyan. MOP: Binding modes of 3a (green) in the MOP receptor: MOP ribbons, red; residues, yellow. MOP ribbons of helix 7, His319/Arg302, removed for clarity.
The synthesis of chemotypes described in this letter is shown in Scheme 1. Tryptophol 9 or its TMS ether 10, thiotryptophol 13, and commercially available tryptamines 14 and 15 were cyclized with 4-dimethylamino-4-phenyl-cyclohexanone 19 to give the corresponding spirocycles 2–7 under similar conditions to the ones reported previously.3
Scheme 1. Synthesis of Investigated Spirocycles.

Reagents and conditions: (a) (i) HN(TMS)2, TMSCl, THF; (ii) NaHCO3, Et2O, 88–100%; (b) (i) Na2S2O3, H2O, EtOH reflux; (ii) i-PrOH reflux, 75%; (c) 50 v/v H3PO4/H2O, Et2O, reflux, 96%; (d) (i) HY or 40 w/w HNMe2/H2O, MeOH, 4 N HCl, KCN, 0 °C to rt; (ii) H2O, Et2O, DCM, 84–97%; (e) (i) PhMgCl, THF, 0 °C to rt; (ii) NH4Cl, H2O, 0 °C to rt, Et2O; (f) 2-butanone, TMSCl, 0 °C to rt, 35–58% over steps (e) and (f); (g) (i) 7.5 N HCl, Et2O; (ii) NaOH, 78–97%; (h) 5-(TES)-4-pentin-1-ol, MeCN, Ph3PI2, imidazole, 70%; (j) (i) t-BuLi/pentane, Et2O, −75 to −70 °C; (ii) 13, Et2O, 28%; (k) (i) iodoaniline, [1,3-bis(2,6-i-Pr2Ph)-imidazol-2-ylidene]-(3-Cl-Py)Pd(II)-chloride, Na2CO3, DMF, 100 °C; (ii) Na2S2O3, 66%; (l) TBAF, THF, reflux, 50 °C, 36%; (m) TMSOTf, DCM, rt, 33–99%; (n) (i) H3PO4, conc. AcOH; (ii) NaOH, 96%; (o) TfOH, DCM, rt; (ii) NaOH; (iii) MeOH, 65–67%; (p) TFA, DCE, 3–31%; (q) (i) TMSOMs, DCM, rt; (ii) filtration; (iii) evaporation filtrate, recryst EtOH; (iii) citric acid, EtOH, 65 °C to rt, 11% 7a; (r) (i) MsOH, DCM; (ii) filtration, 63–99%; (s) citric acid, hot ethanol, 5 °C 12 h, 53–99%.
To achieve carbacycle 8, another route was necessary: 5-triethylsilyl-4-pentyn-1-ol 20 was converted into the corresponding iodide 21, which after metal–halogen exchange with tBuLi was added to previously described319 to give 22. Larock cyclization4 of the triethylsilyl alkyne moiety with ortho-iodoaniline gave the indole-3-yl-propyl moiety in 23.
After silyl deprotection with TBAF, spirocyclization of 24 was achieved with TMSOTf as promoter to give 8a after separation from the accompanying cis diastereomer by HPLC.5
The spirocyclization of 4-methylamino-cyclohexanone 27 gave the corresponding N-monomethyl analogue 28a.
Also cyclic amino heads, such as 4-phenyl-4-pyrrolidin-1-yl-cyclohexanone, were successfully synthesized and spirocyclized in analogous manner to give 38b–40b.
As illustrated in our previous publication, compound 2a, following intravenous administration, showed excellent efficacy in the rat tail-flick and spinal nerve ligation model while having a promising side effects profile. Nevertheless, it suffered from poor oral bioavailability and high clearance. To understand this further we subjected 2a to metabolite ID. These investigations identified N-demethyl compound 28a (Table 2) and 6′-hydroxy compound 48a (Table 3) as metabolites in humans and rodents. Our optimization efforts therefore aimed at targeting both sites of metabolism to improve the PK profile.
Table 2. Variation of the Amine Head, Region C.

| % inhibition
@ 1 μM or Ki (nM) |
||||
|---|---|---|---|---|
| compd | Y | config | hNOPr | hMOPr |
| 2aa | NMe2 | trans | 0.1 | 0.26 |
| 28ab | NHMe | trans | 1.1 | 0.36 |
| 38bc | piperidin-1-yl | cis | <10% | 43% |
| 39bd | 4-Me-piperazin-1-yl | cis | <10% | <10% |
| 40bc | morpholin-4-yl | cis | <10% | <10% |
Hydrochloride.
Hemicitrate.
Methanesulfonate.
Dimethanesulfonate.
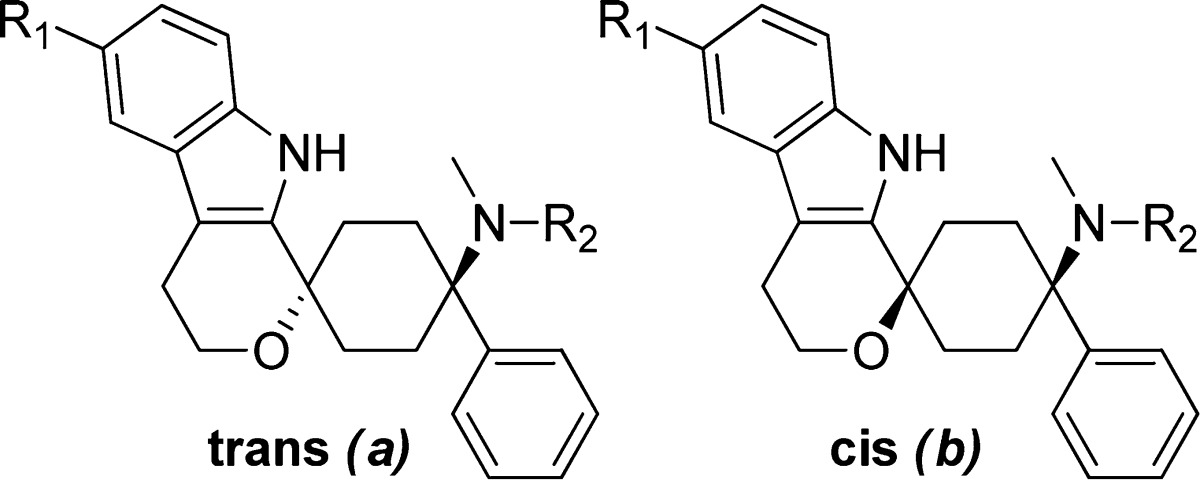
Table 3. Variation of Substituents at Major Metabolic Positions, Region B.
| % inhibition
@ 1 μM or Ki (nM) |
|||||
|---|---|---|---|---|---|
| compd | R1 | R2 | config | hNOPr | hMOPr |
| 3aa | F | Me | trans | 0.9 | 0.7 |
| 3ba | F | Me | cis | 97.4 | 46 |
| 4aa | F | H | trans | 11 | 2 |
| 47aa | Me | Me | trans | 4.5 | 2.7 |
| 48aa | OH | Me | trans | 1 | 0.4 |
| 49ab | OMe | Me | trans | 9.4 | 1.1 |
Hemicitrate.
Hydrochloride.
The dimethyl amine head was tackled first. Synthesis of the N-demethyl metabolite, analogue 28a, resulted in a compound with potent NOP and MOP receptor binding activity. For the cyclic amine heads piperidine 38b, N-methyl piperazine 39b, and morpholine 40b, we only obtained the generally less active cis stereoisomer. However, because the introduction of cyclic amine heads resulted in complete loss of binding affinities, we concluded that there is little room for optimization here and retained the dimethylamino moiety for further optimization. We therefore turned our interest to the 6′-position of the indole spiro scaffold with the aim to block this point of metabolic instability.
As previously reported, the relative stereochemistry remained crucial for high NOP and MOP receptor binding affinities3 with the trans stereoisomer being the favored stereoisomer with respect to binding affinity (2a vs 2b; 3a vs 3b). This has been rationalized, through a bidentate, chelating binding mode to Asp130, which is only feasible from the trans configuration.3
Pleasingly, a variety of small substituents such as Me (47a), F (3a, 3b, and 4a), OH (48a), and OMe (49a) could be obtained by use of trytophols 41–43 and TMS ether 45 and were well tolerated by both receptors. Indeed, as described in Table 3, all trans stereoisomers demonstrated binding affinities below 20 nM at both the NOP and MOP receptor.
Despite the good receptor binding properties, we decided only to progress compounds with a NOP/MOP selectivity factor of <5, as our focus was for equipotent NOP and MOP receptor binders. On this basis, compounds 3a, 4a, 5a, and 48a were advanced into in vitro functional testing.
Equipotent binding at the MOP and NOP receptor was rationalized by overlaying the recently published crystal structures of the MOP and NOP receptors.6,7 Major differences of the MOP and NOP receptor binding sites are Lys233 (MOP)/Ala216 (NOP), Asn127 (MOP)/Asp110 (NOP), His319 (MOP)/Arg302 (NOP), and Tyr128 (MOP)/Ile111 (NOP) switches (Figure 2).
This should allow for similar pi–pi stacking interactions of aromatic moieties at the NOP and MOP receptors. Hydrophobic cavities are conserved in both receptors. We have subsequently docked compound 3a in both receptors and observed only slightly different binding modes;8 therefore, similar binding affinities at the NOP and MOP receptors are to be expected.
More detailed characterization of 3a hemicitrate revealed binding affinities of 2.6 nM (N = 7, ± SD = 1.4) and 18.0 nM (N = 11, ± SD = 20.0 nM) at the hKOP and hDOP receptors.9
Functional investigations on 3a, 4a, 5a, and 48a revealed that all compounds acted as full agonists at both the MOP and NOP receptor and could therefore be progressed (Table 4). We nevertheless decided to drop 5a because of concerns with regards to hERG (data not shown).
Table 4. Functional Assay Results for Selected Compounds.
| EC50 GTPγS (nM) |
rel. eff.a GTPγS |
|||
|---|---|---|---|---|
| compd | hNOPr | hMOPr | hNOPr | hMOPr |
| 3ab | 13 | 1.2 | 89% | 104% |
| 4ab | 60 | 2.6 | 101% | 110% |
| 5ac | 1.3 | 0.2 | 108% | 101% |
| 48ab | 120 | 1.2 | 75% | 96% |
Efficacy of 100% is defined as maximum [35S]GTPγS binding induced by stimulation with nociceptin (NOP receptor) and DAMGO (MOP receptor).
Hemicitrate.
Dihydrochloride.
After these promising in vitro results we progressed 3a, 4a, and 48a into in vivo PK profiling. Compound 3a showed good mouse PK properties with low clearance (Cl = 0.96 L/h·kg), moderate half-life (t1/2 = 2.57 h), medium volume of distribution (Vc = 2.96 L/kg), and good oral bioavailability (F = 44%). In rats, 3a achieved a significant improvement in oral bioavailability (F = 23%) as compared to 2a (F = 4%).3 The mouse PK of 4a is characterized by a very high clearance (Cl = 15.3 L/h·kg), very high volume of distribution (Vz = 72.5 L/kg), longer half-life (t1/2 = 3.50 h), and good oral bioavailability (F = 44%).
Compounds 3a, 4a, and 48a were initially compared with regards to their activity in a mouse tail-flick model of acute nociceptive pain using a method modified from D’Amour and Smith.10 Antinociceptive activity of the compounds was quantified after i.v. and p.o. dosing by measuring the increase in the latency for tail withdrawal from a radiant heat source in male NMRI mice11 (Table 5). Compound 3a induced dose-dependent inhibition of heat nociception with ED50 values of 40.1 nmol/kg i.v. and 77.0 nmol/kg p.o. (Figure 3 and Table 5). With both routes of administration, the maximum attainable antinociceptive response was obtained, indicating full antinociceptive efficacy.
Table 5. In Vivo Results for Selected Compounds in a Mouse Tail-Flick Model of Acute Nociceptive Pain.
| tail-flick
mouse |
|||
|---|---|---|---|
| compd | ED50 (95% CI) [nmol/kg] | Emax (%MPE) | duration of action (min)e |
| 3ad | i.v.: 40.1 (30.4–48.5) | >95 | 180 |
| p.o.: 77.0 (57.8–98.1) | 100 | >480 | |
| 4ad | i.v.: 434 (347–499) | 100 | 300 |
| p.o.: 1432 (1172–1758) | >95 | >600 | |
| 48ac | i.v.: 22.2 (18.0–27.6) | >95 | n.d. |
| p.o.: 3062 (2622–3538) | 100 | n.d. | |
| fentanyl citrate | i.v.: 11.2 (8.5–15.1) | 100 | 40 |
| p.o.: 1956 (1378–2797) | 100 | 90 | |
| hydromorphone HCl | i.v.: 622 (529–778) | 100 | n.d. |
| p.o.: 18753 (13466–25284) | >95 | n.d. | |
Hydrochloride.
Dihydrochloride.
Citrate.
Hemicitrate.
Duration of action was determined for a dose that produces approximately 90% MPE; it describes the duration for which the antinociceptive efficacy is >20% MPE. n.d. = not determined.
Figure 3.

Mouse tail-flick test: Dose–response curves of cebranopadol (3a) hemicitrate (A) and 4a hemicitrate (B) obtained after oral administration. Each point of the graph represents the mean ± SEM of the maximum possible effect (MPE); n = 10 animals per group. *p < 0.05 versus vehicle.
In order to characterize the duration of analgesic action, equi-effective dosages of the high dose range (>90% of the maximum possible effect (MPE)) were chosen. After oral administration, significant antinociceptive activity was observed for more than 8 h (last test point measured), where 85% MPE was still reached. The duration of action after intravenous administration lasted up to 3 h, where 38% MPE was measured. These investigations demonstrate that 3a is highly effective in an animal model of acute nociceptive pain. It is about as equi-potent to the strong opioid fentanyl citrate but shows a significantly longer duration of action and, in contrast to fentanyl, displays high potency and long lasting efficacy as well after oral administration (see Table 5).
Compound 4a also exerted dose-dependent activity in the mouse tail-flick test (Figure 3), with ED50 values of 434 nmol/kg i.v. and 1432 nmol/kg p.o. This is about 10-fold less potent than 3a after both i.v. and p.o. dosing, which can be clearly explained by the observed higher volume of distribution and clearance. The potency of 48a after oral dosing was surprisingly low as compared to cebranopadol 3a and could only be partially explained by its lower oral bioavailability (F = 14%).
A model of streptozotocin (STZ)-induced diabetic polyneuropathy was used to explore the efficacy of 3a in a neuropathic pain state (see Supporting Information for details). A dose of 2.1 nmol/kg i.v. of 3a was tested in STZ-treated diabetic CD-1 mice and, for comparison, in nondiabetic control animals. While 3a induced a significant reduction of thermal hyperalgesia assessed in a hot plate test, it was without an antinociceptive effect in the control animals. The antihyperalgesic efficacy of 2.1 nmol/kg i.v. of 3a was calculated to be 54.1% MPE (n = 10, SEM ± 4.6% MPE; p < 0.001 vs vehicle control) (Figure 4). Thus, in the neuropathic pain state, 3a showed about half-maximum antihyperalgesic activity at a dose being 20 times lower than the ED50 value in the tail-flick model of acute nociception.
Figure 4.

Dose-dependent effects of 3a (cebranopadol) hemicitrate on heat nociception (tail-flick and hot plate in STZ control animals), on thermal hyperalgesia (hot plate in STZ diabetic animals), and on motor coordination (rota rod) after i.v. administration in mice. #, Relative effect refers to percentage of maximum possible effect (% MPE) for tail-flick and STZ models, or relative decrease in performance time on the rotating rod (rota rod). Error bars indicate SEM (n = 10). *, p < 0.05 versus vehicle control.
We decided to progress 3a, due to its high efficacy and potency after oral and intravenous administration, for in-depth profiling. The cytochrom-P450 inhibition potential of 3a was estimated after incubation with human CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4/5 and probe substrates selectively metabolized by these enzymes. None of the tested cytochrome P450 enzymes were inhibited in vitro by 3a up to 250 nM.
The interaction of 3a with safety-relevant cardiovascular targets was characterized: L-type Ca2+-channel (benzothiazepine site) rat brain cortex Ki (μM) = 0.8 (N = 2); L-type Ca2+-channel (phenylalkylamine site) rat brain cortex Ki (μM) = 0.48 (N = 2); L-type Ca2+-channel (dihydropyridine site) rat whole brain Ki (μM) > 100 (N = 2); and hERG channel human recombinant Ki (μM) > 100 (N = 2). Taking into account the high affinity of 3a to interact with the primary targets (Ki < 1 nM), the affinities toward these safety relevant targets are expected to be without biological relevance. More detailed electrophysiological investigations using the whole-cell patch-clamp technique12 revealed that 3a did not affect hERG-mediated potassium currents in transfected HEK293 cells up to 10 μM.
Having ensured a low potential to induce cardiac side effects by 3a we embarked on assessing its side effects profile. Treatment with opioid drugs can be associated with severe side effects on the central nervous system (CNS) and gastrointestinal functions.13
In order to investigate its potential to affect the CNS, 3a was investigated in a rota-rod test.14 In male CD-1 mice, intravenous administration of 3a at doses up to 376 nmol/kg did not significantly influence motor coordination in the rota-rod test. At a dose of 454 nmol/kg, 3a induced a significant impairment of the rota-rod performance. The time that animals were able to remain on the rotating rod was shortened transiently for about 30–60 min after administration. The maximum effect was observed 15 min after administration, when the time on the rod was shortened from the maximum performance time of 120 s to 72 ± 17 s (mean ± SEM). Concurrently, the number of failures increased significantly up to 2.6 ± 0.9 (mean ± SEM). The no observed effect level (NOEL) for 3a was therefore considered to be 376 nmol/kg. An ED50 value could not be calculated but is estimated to be >454 nmol/kg. Compound 3a is, thus, characterized by a safety margin ≥10-fold between the ED50 value in the tail-flick test and the NOEL for effects on motor coordination (Figure 4). This is in clear contrast to standard opioids such as morphine or oxycodone that impair motor coordination within the antinociceptive dose range.15
Effects of 3a on the gastrointestinal system were investigated in the charcoal transit test in male NMRI mice.16 In this assay, effects on gut motility were measured as changes in transit rate of an orally administered charcoal suspension. Intravenous administration of 3a at doses between 10 and 454 nmol/kg induced a dose-dependent inhibition of intestinal charcoal transit. NOEL was 21 nmol/kg i.v., and the ED50 was 87 nmol/kg i.v. A comparison of this ED50 value to the ED50 value in the mouse tail-flick assay points toward a separation of the analgesic dose range from doses inhibiting gastrointestinal motility. In contrast to standard opiates like morphine, which inhibits intestinal transit in mice already at doses below the ED50 in the tail-flick assay,16,173a clearly showed effects on gut motility at doses higher than those required to induce antinociception.
In summary, 3a (cebranopadol) exhibited high efficacy in a mouse model of acute pain with a potency comparable to the strong opioid fentanyl. In a model of polyneuropathic pain, 3a (cebranopadol) showed an about 20 times higher potency as compared to the acute pain model. In contrast to standard opioids, which induce marked CNS side effects and inhibition of gastrointestinal transit at doses below or within the half-maximum effective analgesic dose range,17,183a (cebranopadol) exhibits distinctly improved margins between the analgesic dose range and doses inducing significant gastrointestinal or CNS side effects, which point towards a broader therapeutic window (Figure 4).
A more detailed investigation of cebranopadol’s pharmacological profile has been published elsewhere.9 Currently, cebranopadol is in clinical development for the treatment of severe chronic nociceptive and neuropathic pain.
Acknowledgments
We would like to thank H. Steinhagen, P. Ratcliffe, S. Frosch, and E. Hoppe for very helpful discussions, F. Theil (ASCA GmbH) for conception and realization of compounds, T. Koch for obtaining NOP and MOP receptor binding data,and S. Brenner, P. Günther, B. Liebenhoff, M. Mülfarth, and R. Woloszczak for technical assistance with the in vivo experiments. Detailed 1H and NOE NMR analysis for determination of stereochemical configuration by P. Jonas and M. Schade, high-resolution mass by J. Bergstreiser and M. Fuhr, and eADME characterization by S. Steufmehl are greatly acknowledged.
Glossary
Abbreviations
- CHO
Chinese hamster ovary cells
- CI
confidence interval
- CNS
central nervous system
- DOP
delta opioid peptide
- EC50
concentration with half-maximum inducible [35S]GTPγS binding
- ED50
half-maximum effective dose
- Emax
maximum possible effect for the agonist
- GTPγS
guanosine-5′-[γ-thio]triphosphate
- hNOPr
Human nociceptin/orphanin FQ receptor
- hMOPr
Human μ opioid receptor
- HCl
hydrochloride
- IC50
half-maximum inhibitory concentration
- i.v.
intravenous
- Ki
dissociation constant for inhibitor binding
- KOP
kappa opioid peptide
- MeCN
acetonitrile
- MOP
mu opioid peptide
- MPE
maximum possible effect
- NOEL
no observed effect level
- NOP
nociceptin/orphanin FQ peptide
- SNL
spinal nerve ligation
- TES
triethylsilyl
Supporting Information Available
Assay description and experimental procedures for the synthesis and characterization of selected compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Cremeans C. M.; Gruley E.; Kyle D. J.; Ko M.-C. Roles of μ-opioid receptors and nociceptin/orphanin FQ peptide receptors in buprenorphine-induced physiological responses in primates. J. Pharmacol. Exp. Ther. 2012, 343, 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll L. The use of bifunctional NOP/Mu and NOP receptor selective compounds for the treatment of pain, drug abuse, and psychiatric disorders. Curr. Pharm. Des. 2013, 19, 7451–60. [PubMed] [Google Scholar]
- Schunk S.; Linz K.; Hinze C.; Frormann S.; Oberbörsch S.; Sundermann B.; Zemolka S.; Englberger W.; Germann T.; Kless A.; Christoph T.; Kögel B. Y.; Schröder W.; Harlfinger S.; Saunders D.; Sonnenschein H. Discovery of spiro[cyclohexane-dihydropyrano[3,4-b]indole]-amines as potent NOP and opioid receptor agonists. ACS Med. Chem. Lett. 2014, 10.1021/ml500116x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larock R. C.; Yum E. K. Synthesis of indoles via palladium-catalyzed heteroannulation of internal alkynes. J. Am. Chem. Soc. 1991, 113, 6689–90. [Google Scholar]
- Hinze C.; Aulenbacher O.; Sundermann B.; Oberboersch S.; Friderichs E.; Englberger W.; Koegel B.-Y.; Linz K.; Schick H.; Sonnenschein H.; Henkel B.; Rose V. S.; Lipkin M. J.. Preparation of spirocyclic indoles as ORL-1 receptor ligands for the treatment of pain. U.S. Patent 8,053,576, Nov 8, 2011.
- Thompson A. A.; Liu W.; Chun E.; Katritch V.; Wu H.; Vardy E.; Huang X.-P.; Trapella C.; Guerrini R.; Calo G.; Roth B. L.; Cherezov V.; Stevens R. C. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 2012, 485, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Mathiesen J. M.; Sunahara R. K.; Pardo L.; Weis W. I.; Kobilka B. K.; Granier S. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standard modeling techniques have been applied to dock, optimize, and visualize the binding modes of our compounds as implemented in MOE2012.10 (Molecular Operating Environment 2012, CCG, Montreal, Canada).
- Linz K.; Christoph T.; Tzschentke T. M.; Koch T.; Schiene K.; Gautrois M.; Schröder W.; Kögel B. Y.; Beier H.; Englberger W.; Schunk S.; De Vry J.; Jahnel U.; Frosch S. Cebranopadol: A novel potent analgesic nociceptin/orphanin FQ peptide and opioid receptor agonist. J. Pharmacol. Exp. Ther. 2014, 349, 535–548. [DOI] [PubMed] [Google Scholar]
- D’Amour F. E.; Smith D. L. A method for determining loss of pain sensation. J. Pharmacol. Exp. Ther. 1941, 72, 74–79. [Google Scholar]
- Terlinden R.; Kogel B. Y.; Englberger W.; Tzschentke T.M. In vitro and in vivo characterization of tapentadol metabolites. Methods Find. Exp. Clin. Pharmacol. 2010, 32, 31–38. [DOI] [PubMed] [Google Scholar]
- Hamill O. P.; Marty A.; Neher E.; Sakmann B.; Sigworth F. J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981, 391, 85–100. [DOI] [PubMed] [Google Scholar]
- Zöllner C.; Stein C. Opioids. Handb. Exp. Pharmacol. 2007, 31–63. [DOI] [PubMed] [Google Scholar]
- Dunham N. W.; Miya T. S. A note on a simple apparatus for detecting neurological deficits in rats and mice. J. Am. Pharm. Assoc. 1957, 46, 208–209. [DOI] [PubMed] [Google Scholar]
- Winter L.; Nadeson R.; Tucker A. P.; Goodchild C. S. Antinociceptive properties of neurosteroids: A comparison of alphadolone and alphaxalone in potentiation of opioid antinociception. Anesth. Analg. 2003, 97, 798–805. [DOI] [PubMed] [Google Scholar]
- Niemegeers C. J. E.; Lenaerts F. M.; Janssen P. A. J. Loperamide (R 18553), a novel type of antidiarrheal agent. Part 2: In vivo parenteral pharmacology and acute toxicity in mice. Comparison with morphine, codeine and diphenoxylate. Arzneimittelforschung 1974, 24, 1636–1641. [PubMed] [Google Scholar]
- Tzschentke T. M.; de Vry J.; Terlinden R.; Hennies H. H.; Lange C.; Strassburger W.; Haurand M.; Kolb J.; Schneider J.; Buschmann H.; Finkam M.; Jahnel U.; Friderichs E. Tapentadol hydrochloride. Analgesic, mu-opioid receptor agonist, noradrenaline reuptake inhibitor. Drugs Future 2006, 31, 1053–1061. [Google Scholar]
- Meert T. F.; Vermeirsch H. A. A preclinical comparison between different opioids: antinociceptive versus adverse effects. Pharmacol., Biochem. Behav. 2005, 80, 309–326. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.