

Abstract
Largazole is a potent and class I-selective histone deacetylase (HDAC) inhibitor purified from marine cyanobacteria and was demonstrated to possess antitumor activity. Largazole employs a unique prodrug strategy, via a thioester moiety, to liberate the bioactive species largazole thiol. Here we report alternate prodrug strategies to modulate the pharmacokinetic and pharmacodynamics profiles of new largazole-based compounds. The in vitro effects of largazole analogues on cancer cell proliferation and enzymatic activities of purified HDACs were comparable to the natural product. However, in vitro and in vivo histone hyperacetylation in HCT116 cells and implanted tumors, respectively, showed differences, particularly in the onset of action and oral bioavailability. These results indicate that, by employing a different approach to disguise the “warhead” moiety, the functional consequence of these prodrugs can be significantly modulated. Our data corroborate the role of the pharmacokinetic properties of this class of compounds to elicit the desired and timely functional response.
Keywords: Largazole, histone deacetylases, antitumor activity, natural products, prodrugs
Epigenetic changes have been recognized as hallmarks of cancers, with loss of acetylation in H4K16 and trimethylation in H4K20 and hypermethylated CpG islands being associated with malignancies.1 In addition, 75% of cancers were demonstrated to have high expression of class I histone deacetylases (HDACs).2,3 Canonical HDACs belonging to classes I, II, and IV utilize a Zn2+-dependent mechanism to modulate the deacetylation of histones and nonhistone proteins, leading to modulation of gene expression.4 Inhibition of HDACs is a validated strategy for cancer therapy with two small molecules, vorinostat and romidepsin, having gained approval for clinical use. Vorinostat (SAHA) represents a pan-selective HDAC inhibitor, with submicromolar potency at the enzyme level and micromolar cellular activity.5 However, romidepsin (FK228) possesses subnanomolar IC50s for selected HDACs and low nanomolar cellular activity and, because of its densely functionalized macrocycle, displays selectivity for class I HDACs.6 Aside from the use of HDAC inhibitors as monotherapy, these molecules are also being utilized in combination with other agents, with a distinct mechanism of action, to more efficiently modulate the growth of malignancies.4,7 Many other disorders appear to benefit from inhibition of certain HDAC class or isoform activity.4 Selective HDAC inhibitors are warranted primarily to probe the physiological and pathological roles of different HDAC isoforms and also minimize the side effects of treatment.4
Even though many promising HDAC inhibitors have been identified, two persistent problems are being encountered: the limited isoform selectivity and metabolic instability of these compounds.4 For example, the cyclic tetrapeptide HDAC inhibitors belonging to the trapoxin-class of compounds are among the most potent inhibitors in vitro.8 However, the relatively fast biotransformation of these inhibitors hindered further development of these compounds as therapeutics.4,9 SAHA and related HDAC inhibitors utilize a hydroxamic acid moiety, while FK228 and related structures are disulfide prodrugs that liberate the corresponding thiol as a Zn2+ complexing warhead. The latter has the advantage over hydroxamate-based inhibitors that a variety of prodrug strategies can be developed to modulate critical parameters to achieve the greatest possible desired effect. Introduction of the disulfide moiety was likewise utilized to improve the bioactivity of cyclotetrapeptide HDAC inhibitors.10−12
Largazole (1) (Figure 1) is a unique cyclodepsipeptide from the marine cyanobacterium Symploca sp.13,14 Among the unique structural features of largazole, the thioester moiety serves as a cryptic functional group for potent antiproliferative activity. Largazole, upon protein-assisted hydrolysis, rapidly liberates the bioactive species largazole thiol.15,16 On the basis of X-ray cocrystallization studies and molecular docking experiments, the thiol moiety serves as the “warhead” that complexes with the Zn2+ in the catalytic center of HDACs and the distinct and highly functionalized macrocycle facilitates the class I selectivity of largazole.14,16,17 In spite of the rapid biotransformation of largazole to largazole thiol, the latter possesses significant stability in vivo leading to pronounced biological activity in HCT116 mouse xenograft models.16 Analysis of the pharmacokinetic properties of largazole thiol in rats indicated a half-life of 30 min and presence in the systemic circulation for 2 h, after a 10 mg/kg bolus injection.18 Monitoring of the functional consequences of largazole thiol treatment in a mouse xenograft model showed tumor hyperacetylation after 4 h.16 Long-term studies over 14 days using daily treatment of 5 mg/kg largazole showed a significant decrease in tumor growth rate and tumor volume.16 Furthermore, a dosage of 10 mg/kg/day of largazole in combination with dexamethasone reduced the invasiveness of MDA-MB-231 breast cancer cells in orthotopic xenograft tumors by changing cell morphology and inducing E-cadherin relocalization to cell–cell junctions where E-cadherin can exert its anti-invasive properties.19
Figure 1.

Key HDAC inhibitor structures and synthesis of largazole disulfide analogues.
In this letter, we describe largazole disulfide analogues that present a different prodrug approach to modulate the pharmacological effects via changes in the pharmacokinetic properties. Here we synthesized homodimeric disulfides based on largazole thiol, and we furthermore “hetero-dimerized” largazole thiol with several Cys derivatives of different polarity to form mixed disulfides, in analogy to FK228. FK228, in contrast, however, forms an intramolecular disulfide with Cys of its macrocycle subunit and is not amenable to this tuning approach. By monitoring the level of histone hyperacetylation and largazole thiol in tumor cells in vitro and in vivo, we demonstrate significant changes in the functional consequences of largazole thiol, intricately related to the biotransformation of the largazole disulfide analogues.
Starting from the known trityl (Trt)-protected thiol 3,20 largazole disulfide analogues 4–6 were synthesized following standard iodine oxidation conditions (Figure 1).21 All three cases smoothly proceeded to provide the desired disulfide analogues 4–6 in excellent yields (88–93%). We expected that homodimer 4 of largazole thiol (2) would show very similar potency in cellular assays with possibly distinct bioavailability. Since 4 liberates two equivalents of largazole thiol (2) upon reduction, the compound would actually be twice as potent on a molar basis of prodrug. Simultaneously, we prepared a disulfide analogue 5 with cysteine tert-butyl ester. Another Cys-based disulfide analogue 6 with a free carboxylic acid was synthesized to improve water-solubility, and we aimed to interrogate the effects of these structural changes on the compounds’ activities in the cancer cell in vitro and at the target site in vivo.
The effects of largazole and largazole disulfide analogues on the enzymatic activity of the complete panel of canonical HDACs were assessed to determine the potency and isoform selectivity (Table 1). Compounds 4 and 5 showed comparable potency to largazole thiol in inhibiting HDACs, with subnanomolar IC50s against class I HDAC isoforms, particularly for HDACs 1–3. The potencies of largazole thiol and its disulfide analogues were slightly better than FK228 (Table 1). Since the active form is identical to that of 4 and 5, we expect that 6 would show a comparable profile, although we did not experimentally test its effect on the enzymatic activity of purified HDACs. Comparison of the antiproliferative effects of largazole and disulfide analogues on HCT116 human colorectal adenocarcinoma cells showed comparable IC50s, with only a 2-fold difference in activity. In contrast, 6 showed close to a 7-fold decrease in potency compared to largazole, with an IC50 of 34 ± 5 nM (48 h treatment), indicating that the free carboxylic acid moiety in 6 may significantly alter the properties of this compound, leading to changes in the antiproliferative activity.
Table 1. Inhibition of Purified HDACs and Growth of HCT116 Cancer Cells by Largazole and Disulfide Analogues (IC50 ± SD, nM).
| IC50 (nM)a | 2 | 4b | 5b | FK228b |
|---|---|---|---|---|
| HDAC1 | 0.4 ± 0.05 | 0.3 ± 0.01 | 0.4 ± 0.03 | 0.8 ± 0.03 |
| HDAC2 | 0.9 ± 0.09 | 0.5 ± 0.09 | 0.9 ± 0.01 | 1 ± 0.1 |
| HDAC3 | 0.7 ± 0.003 | 0.3 ± 0.02 | 0.6 ± 0.06 | 1.3 ± 0.1 |
| HDAC4 | NIc | NIc | NIc | 647 ± 20 |
| HDAC5 | NIc | NIc | NIc | 22%d |
| HDAC6 | 35 ± 10 | 61 ± 8 | 135 ± 10 | 45%d |
| HDAC7 | NIc | NIc | NIc | 34%d |
| HDAC8 | 102 ± 7 | 40%d | 35%d | 36%d |
| HDAC9 | NIc | NIc | NIc | 45%d |
| HDAC10 | 0.5 ± 0.02 | 0.2 ± 0.004 | 0.5 ± 0.04 | 0.9 ± 0.1 |
| HDAC11 | 3 ± 0.3 | 0.1 ± 0.0002 | 0.2 ± 0.03 | 0.3 ± 0.0003 |
| HCT116 cells antiproliferative activity | 5.9 ± 0.7 | 12 ± 2 | 13 ± 1 | 3.0 ± 0.2 |
IC50 values for enzymatic assays are averages from duplicate experiments. IC50 for HCT116 antiproliferative activity, n = 4.
Required pretreatment with DTT to reduce the disulfide bond.
NI denotes no significant inhibition if inhibition percent is <20% at the maximum tested concentration (1 μM for all compounds).
Percent inhibition at the highest concentration tested (1 μM).
Cellular HDAC inhibition by largazole and largazole disulfide analogues was assessed by monitoring hyperacetylation of histone H3 (Lys9/14) via immunoblot analysis. The antiproliferative effects of largazole, 4, and 5 paralleled the effects on histone hyperacetylation at 8 and 24 h post-treatment and showed a dose-dependent increase (Figure 2A,B). In contrast, the slightly lower potency of 6 in the antiproliferative assay can be inferred from the late onset of histone hyperacetylation, where changes in histone hyperacetylation can only be observed after 24 h of treatment (Figure 2B).
Figure 2.
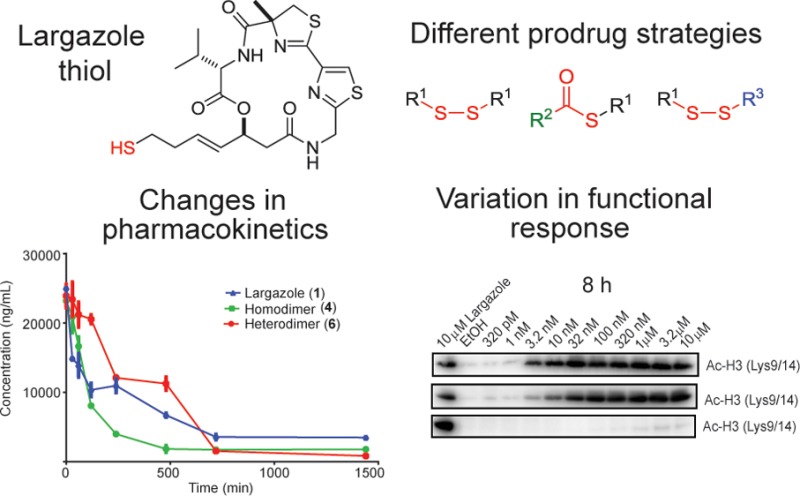
In vitro monitoring of histone hyperacetylation and metabolic stability of largazole and largazole disulfide analogues. Time-course monitoring of histone hyperacetylation in HCT116 cells: (A) 8 and (B) 24 h. (C) Cellular stability of prodrugs in HCT116 cell lysates. (D) Amount of generated largazole thiol from each prodrug (measured as NEM adduct).
Reduced cell permeability of 6 is presumably one major contributing factor to the differences noticed (see below). In order to assess the contribution of biotransformation to the functional responses, the cellular stability of largazole and disulfide analogues was monitored by incubating the compounds with whole cell lysates of HCT116 and monitoring the level of the parent compounds and largazole thiol using HPLC–MS. The liberated largazole thiol was derivatized using N-ethylmaleimide (NEM) prior to HPLC–MS to prevent autoxidation and electrophilic addition, to permit more accurate quantitation. Largazole thiol (2) was quickly liberated from largazole (1), with a rapid increase observed at 30 min (Figure 2C,D). In contrast, an appreciable change in the largazole thiol level following treatment with 6 was observed later, after approximately 4 h of incubation (Figure 2D).
This suggests that the free carboxylic acid moiety of 6 may have contributed to the slower biotransformation of the prodrug. The possible change in the rate of biotransformation of largazole disulfide analogues is compensated in 4 by delivery of two molecules of largazole thiol from one molecule of the prodrug. Hence, significant increase in largazole thiol level is observed after 1 h of treatment with 4. The cellular stability of 5 was not monitored due to lability of the tert-butyl group under ionizing condition in HPLC–MS, preventing the accurate quantitation of 5.
It is evident that a disulfide prodrug strategy also affects the cellular permeability of the prodrug and, consequently, the uptake and biotransformation, particularly if charged or ionizable groups are incorporated. In order to achieve early functional response, utilizing either a more easily (but intracellularly) converted prodrug such as the thioester of largazole or liberating two molecules of the bioactive species such as in 4 would be effective strategies. The stability profiles of largazole and disulfide analogues in mouse serum were also evaluated (Figure S1, Supporting Information). Largazole showed rapid conversion to largazole thiol, while 4 and 6 did not show any largazole thiol formed during in vitro incubations and based on HPLC–MS monitoring. This suggests that the concentration of reducing agents, primarily glutathione, present in mouse serum during in vitro incubations, may be limiting and hence will fail to activate the disulfide prodrugs, which would result in preferred intracellular activation.
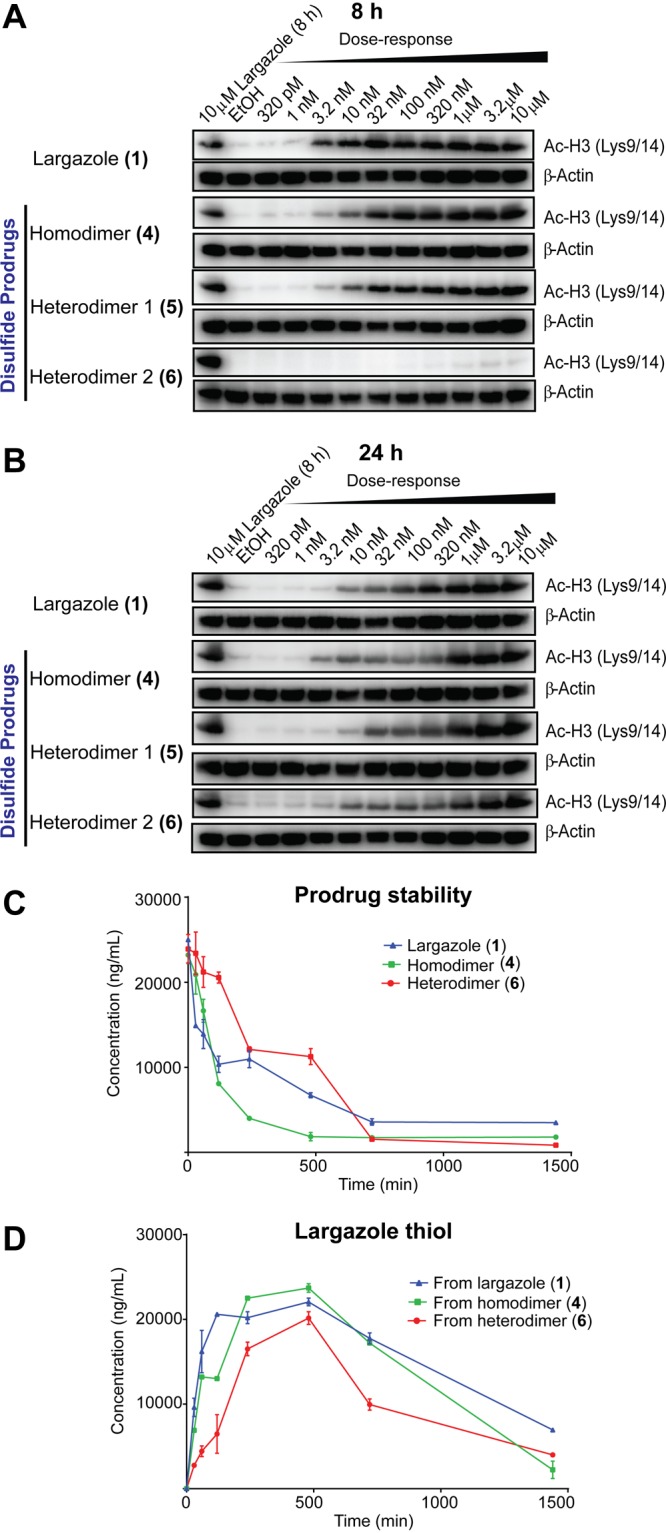
Next, we assessed the in vivo activity of largazole and largazole disulfide analogues using a HCT116 mouse xenograft model. Varying dosages (10 mg/kg, 50 mg/kg) and routes of administration (intraperitoneal and oral) were utilized. Tumors were excised after 4, 12, and 24 h, and the levels of histone hyperacetylation and largazole thiol in tumors were assessed by immunoblotting and HPLC–MS, respectively. Using a dosage of 10 mg/kg and i.p. administration, 4 showed the highest histone hyperacetylation at 4 and 12 h (Figure 3A). Largazole (1) samples showed rapid loss of histone hyperacetylation after 4 h, the levels at later time points were, however, sustained (Figure 3A). In contrast, 6 caused a late onset of histone hyperacetylation, with the maximum level observed at later time points (Figure 3A). These in vivo results paralleled the in vitro assessment of histone hyperacetylation (Figure 2A,B). In order to further verify the results, the dosage was increased to 50 mg/kg. Analogous results were obtained in both dosages using i.p. administration (Figure 3C). Largazole caused maximum histone hyperacetylation at 4 h, with a rapid decrease but sustained level of histone hyperacetylation at later time points (Figure 3C). Compound 4 showed greater effects on histone hyperacetylation, and because it represents the delivery of two molecules of largazole thiol, sufficient amounts of the bioactive species can be obtained even at later time points, sufficient to yield a functional response (Figure 3C). A decrease in histone hyperacetylation was observed only after 12 h. A strong effect of 6 on histone hyperacetylation was also consistently observed at 24 h at least at the higher dose (Figure 3C).
Figure 3.
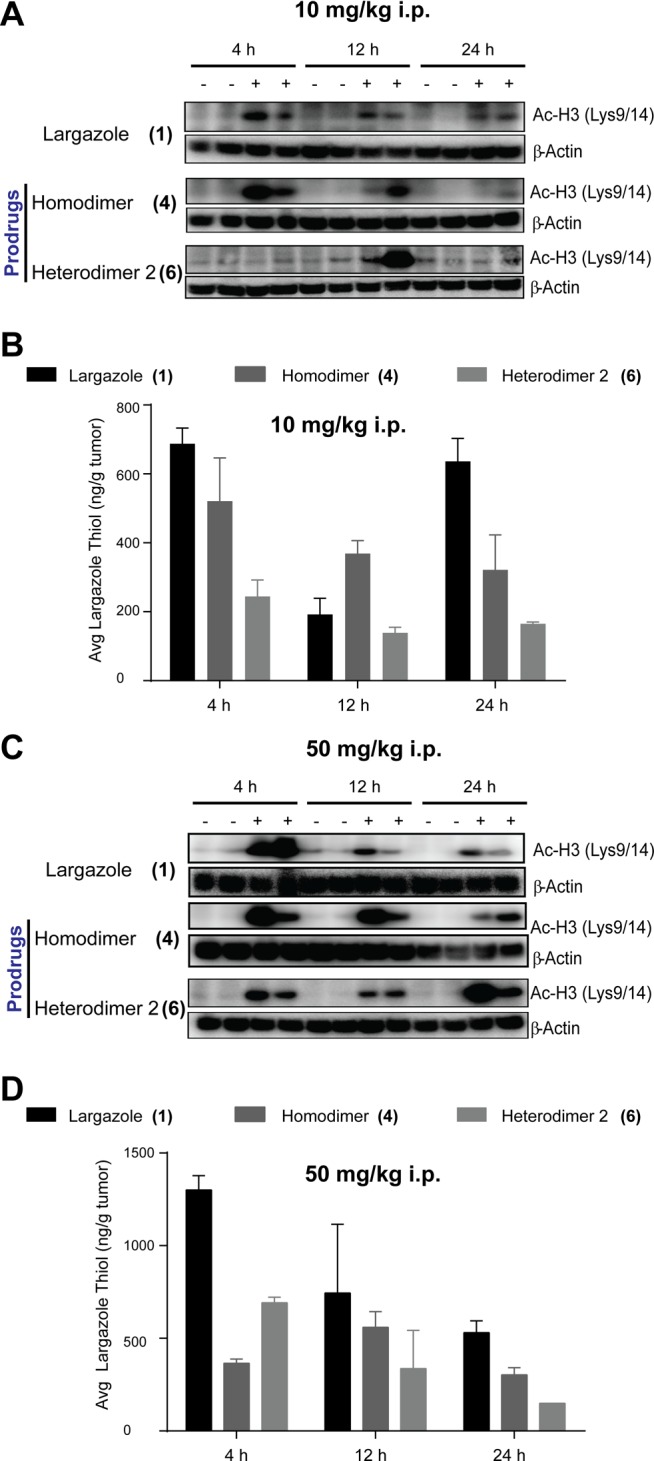
In vivo monitoring of histone hyperacetylation and largazole thiol levels in excised tumors (n = 2). Histone hyperacetylation (A,B) and largazole thiol level (C,D) in excised tumors from mice treated with 10 mg/kg (A,C) and 50 mg/kg (B,D) intraperitoneal administration.
The highest concentration of largazole thiol in the tumor was obtained with largazole treatment after 4 h, based on HPLC–MS monitoring (Figure 3B,D). Largazole thiol generated rapidly decreased after 4 h (Figure 3B,D). In contrast to our initial observation with incubations using HCT116 cell lysates (Figure 2D), 4 did not generate greater amounts of largazole thiol (2) compared to largazole (Figure 3B,D). The amount of largazole thiol (2) generated from 4, however, was almost constant over time. Compound 6, however, gave mostly the lowest concentration of largazole thiol (Figure 3B,D).
Obviously the cell permeability of 6 is expected to be lower because of the carboxylic acid moiety. The late onset of histone hyperacetylation from treatment with 6 and the intense level of histone hyperacetylation arising from 4 cannot be solely accounted for by cell permeability and the levels of largazole thiol at the tumor site in vivo. One explanation could be the reduced permeability into the nucleus once the intact compound reached the cytoplasm; however, intracellular reduction would probably be faster. It is likely that largazole disulfide analogues undergo biotransformation in the macrocyle and independent of the disulfide bonds, to generate largazole thiol analogues with an intact thiol moiety but with minor modification to the macrocycle. Since the method used in the HPLC–MS profiling utilized multiple reaction monitoring (MRM), this detection method is highly specific for largazole thiol and cannot detect generated biotransformation products.
Oral bioavailability of largazole thiol was evident from the observed histone hyperacetylation at 4 and 12 h following 50 mg/kg largazole treatment (Figure 4A). However, no functional response from the oral treatment using 4 and 6 was observed (Figure 4A). The results of the histone hyperacetylation paralleled the results from HPLC–MS monitoring of largazole thiol, where a level in the range as seen for 10 mg/kg i.p. administration was only observed with largazole (1) treatment (Figure 4B). This suggested that oral bioavailability of largazole is reduced approximately 5-fold compared with the i.p. route. The low levels of largazole thiol from 4 and 6 and the low functional response observed with the oral route of administration for these two compounds indicate poor oral bioavailability. This may be due to the increased polar surface area and number of hydrogen bonding donors and acceptors in 4 and 6 compared to largazole. Increased oral bioavailability has been correlated with low polar surface area and low number of rotatable bonds.22
Figure 4.
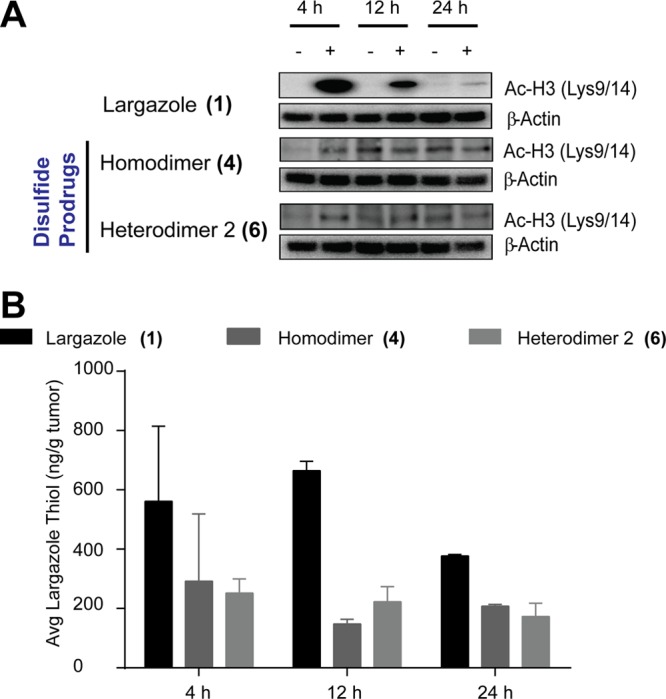
Oral bioavailability studies. (A) Histone hyperacetylation. (B) Largazole thiol level in excised tumors from mice treated with 50 mg/kg, oral administration (n = 2).
In summary, the pharmacokinetic profiles of largazole analogues are intricately related to their pharmacological effects, and the modifications to the prodrug strategy employed serve as a cryptic method to accomplish this. While the compounds showed a similar biological activity profile in enzyme assays, differences were observed in cultured cells and in mouse xenograft models. Depending on the prodrug strategy employed, the pharmacodynamics profile can be modulated primarily through changes in the absorption, distribution, metabolism, and excretion (ADME) of the compounds. In the case of 6, there is a late onset of functional response, which may indicate poor absorption of the less permeable carboxylic acid prodrug and/or conversion of the prodrug to generate a largazole thiol analogue. The oral bioavailability of largazole highlights the advantage of the thioester functionality as a prodrug strategy. This additional feature of largazole circumvents the disadvantages of the pan-selective but orally available SAHA and the class I-isoform selective but low orally available FK228, providing a potential avenue for improvements of HDAC inhibitors.23,24 Additional mixed disulfides by heterodimerization with non-Cys thiols will expand opportunities to modulate selected parameters and tune overall compound properties, which is an advantage of the largazole scaffold over FK228-like compounds that form an intramolecular disulfide bond with Cys present in their core structures. It is also evident that histone hyperacetylation is not fully correlated to largazole thiol levels in vivo and highlights the potential role of alternate biotransformation products. Future experiments are then aimed at rigorously characterizing the in vivo biotransformation products of largazole and largazole disulfide analogues.
Supporting Information Available
Synthetic procedures, in vitro and in vivo assays, HPLC–MS monitoring methods, NMR and MS data, and additional figure and schemes. This material is available free of charge via the Internet at http://pubs.acs.org.
This research was supported by the National Institutes of Health National Cancer Institute (Grant R01CA138544). We are grateful to the North Carolina Biotechnology Center (NCBC; Grant No. 2008-IDG-1010) for funding of NMR instrumentation and to the National Science Foundation (NSF) MRI Program (Award ID No. 0923097) for funding of mass spectrometry instrumentation.
The authors declare the following competing financial interest(s): HL is a co-founder of Oceanyx Pharmaceuticals, Inc., which is negotiating licenses for largazole-related patents and patent applications.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sharma S.; Kelly T. K.; Jones P. A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M.; Oda Y.; Eguchi T.; Aishima S.-I.; Yao T.; Hosoi F.; Basaki Y.; Ono M.; Kuwano M.; Tanaka M.; Tsuneyoshi M. Expression profile of class I histone deacetylases in human cancer tissues. Oncol. Rep. 2007, 18, 769–774. [PubMed] [Google Scholar]
- Song J.; Noh J. H.; Lee J. H.; Eun J. W.; Ahn Y. M.; Kim S. Y.; Lee S. H.; Park W. S.; Yoo N. J.; Lee J. Y.; Nam S. W. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS 2005, 113, 264–268. [DOI] [PubMed] [Google Scholar]
- Salvador L.; Luesch H.. HDAC inhibitors and other histone modifying natural products as emerging anticancer agents. In Natural Products and Cancer Drug Discovery; Koehn F. E., Ed.; Springer: New York, 2013; pp 59–95. [Google Scholar]
- Marks P. A.; Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [DOI] [PubMed] [Google Scholar]
- Harrison S. J.; Bishton M.; Bates S. E.; Grant S.; Piekarz R. L.; Johnstone R. W.; Dai Y.; Lee B.; Araujo M. E.; Prince H. M. A focus on the preclinical development and clinical status of the histone deacetylase inhibitor, romidepsin (depsipeptide, Istodax). Epigenomics 2012, 4, 571–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolden J.; Peart M. J.; Johnstone R. W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discovery 2006, 5, 769–784. [DOI] [PubMed] [Google Scholar]
- Taunton J.; Collins J. L.; Schreiber S. L. Synthesis of natural and modified trapoxins, useful reagents for exploring histone deacetylase function. J. Am. Chem. Soc. 1996, 118, 10412–10422. [Google Scholar]
- Masuoka Y.; Shindoh N.; Inamura N.. Histone deacetylase inhibitors from microorganisms: The Astellas experience. In Progress in Drug Research; Amstutz R., Petersen F., Eds.; Birkhauser Verlag: Basel, Switzerland, 2008; pp 337–359. [DOI] [PubMed] [Google Scholar]
- Nishino N.; Jose B.; Okamura S.; Ebisusaki S.; Kato T.; Sumida Y.; Yoshida M. Cyclic tetrapeptides bearing a sulfhydryl group potently inhibit histone deacetylases. Org. Lett. 2003, 5, 5079–5082. [DOI] [PubMed] [Google Scholar]
- Shivashimpi G. M.; Amagai S.; Kato T.; Nishino N.; Maeda S.; Nishino T. G.; Yoshida M. Molecular design of histone deacetylase inhibitors by aromatic ring shifting in chlamydocin framework. Bioorg. Med. Chem. 2007, 15, 7830–7839. [DOI] [PubMed] [Google Scholar]
- Sasaki K.; Ito T.; Nishino N.; Khochbin S.; Yoshida M. Real-time imaging of histone H4 hyperacetylation in living cells. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 16257–16262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taori K.; Paul V. J.; Luesch H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J. Am. Chem. Soc. 2008, 130, 1806–1807. [DOI] [PubMed] [Google Scholar]
- Hong J.; Luesch H. Largazole: from discovery to broad-spectrum therapy. Nat. Prod. Rep. 2012, 29, 449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Y.; Taori K.; Kim H.; Hong J.; Luesch H. Total synthesis and molecular target of largazole, a histone deacetylase inhibitor. J. Am. Chem. Soc. 2008, 130, 8455–8459. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Salvador L. A.; Byeon S.; Ying Y.; Kwan J. C.; Law B. K.; Hong J.; Luesch H. Anticolon cancer activity of largazole, a marine-derived tunable histone deacetylase inhibitor. J. Pharmacol. Exp. Ther. 2010, 335, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole K. E.; Dowling D. P.; Boone M. A.; Phillips A. J.; Christianson D. W. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J. Am. Chem. Soc. 2011, 133, 12474–12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M.; Salvador L. A.; Sy S. K. B.; Tang Y.; Singh R. S. P.; Chen Q.-Y.; Liu Y.; Hong J.; Derendorf H.; Luesch H. Largazole pharmacokinetics in rats by LC-MS/MS. Mar. Drugs 2014, 12, 1623–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law M. E.; Corsino P. E.; Jahn S. C.; Davis B. J.; Chen S.; Patel B.; Pham K.; Lu J.; Sheppard B.; Nørgaard P.; Hong J.; Higgins P.; Kim J.-S.; Luesch H.; Law B. K. Glucocorticoids and histone deacetylase inhibitors cooperate to block the invasiveness of basal-like breast cancer cells through novel mechanisms. Oncogene 2013, 32, 1316–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers A.; West N.; Taunton J.; Schreiber S. L.; Bradner J. E.; Williams R. M. Total synthesis and biological mode of action of largazole: a potent class I histone deacetylase inhibitor. J. Am. Chem. Soc. 2008, 130, 11219–11222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamber B.; Hartmann A.; Eisler K.; Riniker B.; Rink H.; Sieber P.; Rittel W. The synthesis of cystine peptides by iodine oxidation of S-trityl-cysteine and S-acetamidomethyl-cysteine peptides. Helv. Chim. Acta 1980, 63, 899–915. [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H. Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [DOI] [PubMed] [Google Scholar]
- Li Z.; Chan K. K. A subnanogram API LC/MS/MS quantitation method for depsipeptide FR901228 and its preclinical pharmacokinetics. J. Pharm. Biomed. Anal. 2000, 22, 33–44. [DOI] [PubMed] [Google Scholar]
- Kantharaj E.; Jayaraman R.. Histone deacetylase inhibitors as therapeutic agents for cancer therapy: Drug metabolism and pharmacokinetic properties. In Drug Development—A Case Study Based Insight into Modern Strategies; Rundfeldt C., Ed.; InTech: Rijeka, Croatia, 2011; pp 101–111. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.