

Abstract
This study evaluated 18F-labeled IMPY [6-iodo-2-(4′-N,N-dimethylamino)phenylimidazo[1,2-a]pyridine] derivatives as agents for imaging β-amyloid plaque with positron emission tomography (PET). The precursor for radiolabeling and reference compounds was synthesized in up to five steps from commercially accessible starting materials. One of the two N-methyl groups of IMPY was substituted with either a 3-fluoropropyl (FPM-IMPY) or a 2-fluoroethyl (FEM-IMPY) group. FPM-IMPY and FEM-IMPY were found to have moderate affinity for Aβ- aggregates with Ki = 27 ± 8 and 40 ± 5 nM, respectively. A “one-pot” method for 18F-2-fluoroethylation and 18F-3-fluoropropylation of the precursor was developed. The overall decay-corrected radiochemical yields were 26–51%. In PET experiments with normal mouse, high uptake of activity was obtained in the brain after iv injection of each probe: 6.4% ID/g for [18F]FEM-IMPY at 1.2 min, and 5.7% ID/g for [18F]FPM-IMPY at 0.8 min. These values were similar to those of [123I/125I]IMPY (7.2% ID/g at 2 min). Polar and nonpolar radioactive metabolites were observed in both plasma and brain homogenates after injection of [18F]FEM or [18F]FPM-IMPY. In contrast to the single-exponential washout of [123I/125I]IMPY, the washouts of brain activity for the two fluorinated analogues were biphasic, with an initial rapid phase over 20 min and a subsequent much slower phase. Residual brain activity at 2 h, which may represent polar metabolites trapped in the brain, was 4.5% ID/g for [18F]FEM-IMPY and 2.1% ID/g for [18F]FPM-IMPY. Substantial skull uptake of [18F]fluoride was also clearly observed. With a view to slow the metabolism of [18F]FEM-IMPY, an analogue was prepared with deuteriums substituted for the four ethyl hydrogens. However, D4-[18F]FEM-IMPY showed the same brain uptake and clearance as the protio analogue. Metabolism of the [18F]FEM-IMPY was appreciably slower in rhesus monkey than in mouse. Autoradiography of postmortem brain sections of human Alzheimer’s disease patients with [18F]FEM-IMPY showed high displaceable uptake in gray matter and low nonspecific binding in the white matter. This study demonstrates that the IMPY derivatives have favorable in vivo brain pharmacokinetics and a moderate affinity for imaging β-amyloid plaques; however, further improvements are needed to reduce radioactive metabolites, increase binding affinity, and reduce lipophilicity.
1. Introduction
There are a number of reasons for the development of noninvasive biomarkers for Alzheimer’s disease (AD). Diagnosis and staging of the disease are of immediate interest for patients. In addition, quantitation of β-amyloid in brain would presumably allow presymptomatic identification of patients and monitoring of putative neuroprotective effects of novel treatments that are currently being investigated. Two of the characteristic pathological features for postmortem AD are the abundance of neuritic plaques composed of Aβ-amyloid and neurofibrillary tangles made of PHF-tau aggregates.1 Recent efforts to identify positron emission tomography (PET) Aβ-amyloid probes have generated a few leads, including [1C]-2-(4-(methylamino)phenyl)benzo[d]thiazol- 6-ol ([1C]6-OH-BTA-1) 2 and [18F]-2-(1-(2-(N-(2-fluoroethyl)- N-methylamino)naphthalen-6-yl)ethylidene)- malononitrile ([18F]FDDNP, Figure 1).3 Some derivatives of 6-iodo-2-(4′-N,N-dimethylamino)phenylimidazo[1,2- a]pyridine (IMPY, Figure 1) have also been identified as single-photon emission computed tomography (SPECT) probes for Aβ-amyloid.4 [123I/125I]IMPY has a high uptake in the normal mouse brain (peak: 7.2% dose/g at 2–5 min) and fast clearance (to <0.35% dose/g after 2 h of injection). Brain radioactivity in the Tg2576 AD transgenic mouse model is ~3.3 times higher than that in the age-matched control at 4 h after injection. This desirable pharmacokinetics makes [125I]IMPY an attractive agent for mapping brain Aβ plaques. Here, we report the synthesis and evaluation of two 18F-labeled analogues of IMPY, in which one of its N-methyl groups is replaced with [18F]3-fluoropropyl ([18F]FPM-IMPY) or [18F]2-fluoroethyl ([18F]FEM-IMPY), as prospective PET radioligands for β-amyloid.
Figure 1.
Structures of leading tracers for β-amyloid.
2. Materials and Methods
2.1. Preparation of Compounds. Chemicals, Reagents, and Solutions
[125I]-4-(6-iodobenzo[d]thiazol-2-yl)-N,N-dimethylbenzenamine ([125I]TZDM) was prepared as described previously.5 A solid form of the peptide Aβ40 monomer in the form of a trifluoroacetate salt was purchased from Biosource International Inc. (Camarillo, CA). Reagents used in the syntheses were purchased from Aldrich Chemical Co. or Fluka Chemical Company (Milwaukee, WI) and were used without further purification unless otherwise indicated. Water was purified through a Millipore water purification system, comprising a combination of two filters, one Rio, one reservoir, and one Milli-Q synthesis system (Bedford, MA). Acetonitrile (HPLC grade) was obtained from Fisher Scientific (Pittsburgh, PA). Phosphate buffer (pH 7.4) was made through mixing solutions of NaH2PO4 and Na2HPO4 (81:19 v/v). Brain tissue from deceased Alzheimer’s disease (AD) patients was obtained from Brain Bank of the Clinical Brain Disorders Branch, National Institute of Mental Health, National Institutes of Health. Both entorhinal cortex and hippocampal regions were used.
Instrument and General Conditions
Analytical HPLC except for radiosynthesis was performed using a reverse-phase column (Luna C18, 5 μm, 10.0 mm × 250 mm; Phenomenex) eluted with concentrated ammonia (0.25%) in acetonitrilewater at 6.2 mL/min. The chromatography system was fitted with a continuous wavelength UV–vis detector (Beckman System Gold 168 detector) and an autosampler (Beckman System Gold 508 autosampler). For semipreparative Beckman HPLC, a reverse-phase column (Luna C18, 5 μm, 21.2 mm × 250 mm; Phenomenex) was eluted at 20.0 mL/min. The HPLC system was fitted with a manual injector (5 mL injection loop). The purity of compounds was determined with HPLC monitored for UV absorbance at 280 nm (for IMPY derivatives) or 254 nm (for other aromatic compounds) and expressed as area percentage of all peaks. The 1H and 13C NMR spectra of all compounds were acquired on Bruker 400 MHz spectrometers using the chemical shifts of residual deuterated solvent as the internal standard; chemical shift (δ) data for the proton and carbon resonance were reported in parts per million (ppm) relative to the internal standard. Thin-layer chromatography (TLC) was performed using silica gel 60 F254 plates from EM Science, and compounds were visualized under UV light. Flash chromatography was carried out using a Biotage Horizon HPFC system (Charlottesville, VA, column sizes: 12 mm × 150 mm, 25 mm × 150 mm, 40 mm × 150 mm) with hexanes and ethyl acetate (EtOAc) as eluents with chromatographic solvent proportions expressed on a volume:volume basis. IR spectra were recorded using a Perkin-Elmer Spectrum One FT-IR spectrometer, and UV–vis spectra were recorded using a Lambda 40 UV–vis spectrometer. Mass spectra were acquired using either Thermo Finnigan LCQDECA LC–MS (MSHPLC column, Luna C18, 5 μm, 2.0 mm × 150 mm, Phenomenex; flow rate, 150 μL/min; eluent, MeOH and H2O mixture) or Thermo Finnigan PolarisQ GC–MS (GC column, capillary RTX-5ms 30 m × 0.25 mm; flow rate, 1 mL/min; carrier gas, He). High-resolution mass spectra were acquired under electron ionization (EI) conditions using a double-focusing high-resolution mass spectrometer (AUTOSPEC, Micromass Inc.) with samples introduced through a direct insertion probe. Elemental analyses of selected compounds were carried out by Midwest Microlab (Indianapolis, IN) or Galbraith Laboratories, Inc. (Knoxville, TN).
4-Methylaminoacetophenone (2)
4-Aminoacetophenone (15.0 g, 111 mmol), sodium carbonate (35.3 g, 333 mmol), and iodomethane (13.8 mL, 31.5 g, 222 mmol) in water (400 mL) were stirred at gentle reflux for 5 h. [CAUTION! The iodomethane has a low boiling point (41–43 °C), so it refluxes energetically and tends to evaporate off.] The mixture was then cooled, and an NaOH solution (20% w/v, 80 mL) was added to quench the reaction. The mixture was stirred for another 15 min. Water was added to dissolve the inorganic salts. This mixture, containing the white organic solids insoluble in water, was extracted three times with CH2Cl2, and the collected organic phase was dried over Na2SO4. The solvent of the filtrate was removed by rotary evaporation to afford a crude solid (15 g), which was purified by flash chromatography (FCC) (silica gel, hexane–AcOEt = 3:1 v/v) to give the title compound as a slightly pale-yellow solid (4.1 g, 27 mmol, 25%) and 4-(N,N-dimethylamino)acetophenone (5.0 g, 30 mmol, 27%). 1H NMR (400 MHz, CDCl3): δ 7.82 (2H, d, 3JHH = 9.0 Hz), 6.54 (2H, d, 3JHH = 9.0 Hz), 4.26 (1H, bs, NH), 2.88 (3H, d, 3JHH = 4.5 Hz, N–Me), 2.49 (3H, s). 13C{1H} NMR (400 MHz, CDCl3): δ 196.4 (–CO–), 153.0 (Ph-1), 130.7 (Ph-2,6), 126.5 (Ph-4), 111.0 (Ph-3,5), 30.1 (CH3–N), 26.0 (CH3–CO). MS, m/z (CI): 167 [M + NH4]+, 150 [M + H]+. Anal. Calcd for C9H11-NO: C, 72.50; H, 7.40; N, 9.40. Found: C, 72.39; H, 7.27; N, 9.26.
N-(4-Acetylphenyl)-2,2,2-trifluoro-N-methylacetamide (3)
Compound 2 (1.25 g, 8.28 mmol) and triethylamine (TEA) (4.17 g, 41.4 mmol) were dissolved with stirring in anhydrous CH2Cl2 (45 mL) under dinitrogen. The mixture was cooled to −10 °C with an acetone–CO2 bath. A cooled (−10 °C) solution of trifluoroacetic anhydride (6.95 g, 33.1 mmol) in anhydrous CH2Cl2 (15 mL) was added via a syringe dropwise over 15 min. The mixture was allowed to warm to room temperature (RT), and the stirring was continued for 4 h. The reaction was quenched with NH4Cl solution (1 M, 30 mL). The mixture was stirred vigorously for 15 min. The organic phase was separated off, and the aqueous phase was extracted with CH2Cl2 (2 × 20 mL). The combined organic phase was washed with water and brine and dried over Na2-SO4. The solvent of the filtrate was removed by rotary evaporation. The crude solid (3.76 g) was purified by FCC [silica gel, hexane–CH2Cl2 (1:1 v/v) + 3% AcOEt] to yield the title compound as a transparent oil, which crystallized over 24 h to a pale-yellow solid (1.92 g, 7.84 mmol, 95%). 1HNMR (400 MHz, CDCl3): δ 8.01 (2H, d, 3JHH = 8.5 Hz), 7.34 (2H, d, 3JHH = 8.5 Hz), 3.37 (3H, s, N–CH3), 2.62 (3H, s). 13C{1H} NMR (400 MHz, CDCl3): δ 209.5 (–CO–), 156.6 (q, 3JCF = 36.4 Hz, –C(O)CF3), 149.1 (Ph-1), 144.7 (Ph-4), 129.7 (Ph-2,6), 127.6 (Ph-3,5), 116.2 (q, 2JCF = 288 Hz, CF3), 39.4 (N–CH3), 26.6 (CH3–CO). MS, m/z (CI): 263 [M + NH4]+, 246 [M + H]+. Anal. Calcd for C11H10F3NO2: C, 53.88; H, 4.11; N, 5.71. Found: C, 53.71; H, 4.20; N, 5.72.
N-[4-(2-Bromoacetyl)phenyl]-2,2,2-trifluoro-N-methylacetamide (4)
Compound 3 (1.9 g, 7.7 mmol) and tetra-n-butylammonium tribromide (4.45 g, 9.2 mmol) were dissolved in methanol (150 mL). The reaction was stirred at RT overnight. The solvent was then removed under reduced pressure, and the residue was dissolved in AcOEt (60 mL). The mixture was washed twice with water (2 × 30 mL) and once with brine. The organic solution was then dried over Na2-SO4, and the solvent from the filtrate was removed. The crude product was purified using FCC (hexane–CH2Cl2 = 1: 1 v/v/+ 2% AcOEt), yielding the title compound as a transparent oil (1.81 g, 5.58 mmol, 72%). 1H NMR (400 MHz, CDCl3): δ 8.06 (2H, d, 3JHH = 8.6 Hz), 7.38 (2H, d, 3JHH = 8.2 Hz), 4.43 (2H, s, CH2Br), 3.40 (3H, s, CH3N). 13C{1H} NMR (400 MHz, CDCl3): δ 190.1 (–CO–), 156.6 (q, 3JCF = 36.4, –C(O)CF3), 145.2 (Ph-1), 134.0 (Ph-4), 130.4 (Ph-2,6), 127.9 (Ph-3,5), 116.1 (q, 2JCF = 288 Hz, CF3), 39.4 (N–CH3), 30.4 (CH2–Br). MS, m/z (EI): 323–325 [M]+, 230 [M – CH2Br]+. Anal. Calcd for C11H9BrF3NO2: C, 40.77; H, 2.80; N, 4.32. Found: C, 40.87; H, 2.75; N, 4.18.
2,2,2-Trifluoro-N-[4-(6-iodoimidazo[1,2-a]pyridin-2-yl)- phenyl]-N-methylacetamide (5)
Compound 4 (1.8 g, 5.5 mmol) and 5-iodo-2-aminopyridine (1.26 g, 5.73 mmol) were dissolved in absolute ethanol (65 mL) and stirred under reflux for 2 h. The mixture was then cooled, and NaHCO3 (0.836 g, 9.94 mmol) was added. The reaction mixture was refluxed for another 4 h. The solvent was then removed under reduced pressure, and the residue was collected with AcOEt (70 mL) and washed with water (40 mL). The organic phase was then dried with Na2SO4 and the solvent was removed by rotary evaporation, affording a crude product (2.8 g). This was purified using FCC (silica gel, hexane–CH2Cl2–AcOEt = 2: 2: 1 by volume), yielding the title compound as a pale-yellow solid (1.51 g, 3.4 mmol, 62% yield). The product was recrystallized from Et2O–EtOH. 1H NMR (400 MHz, CDCl3): δ 8.40 (1H, s, H-5), 8.02 (2H, d, 3JHH = 8.4 Hz, H-2′,6′), 7.82 (1H, s, H-3), 7.42 (1H, d, 3JHH = 9.4 Hz, H-8), 7.35 (1H, dd, 3JHH = 9.5, 4JHH = 1.6 Hz, H-7), 7.29 (2H, d, 3JHH = 8.2 Hz, H-3′,5′), 3.37 (3H, s, CH3). 13C{1H} NMR (400 MHz, CDCl3): δ 156.9 (q, 3JCF = 35.2 Hz, –C(O)CF3), 144.8, 144.3, 140.4, 134.1, 133.1, 130.6, 127.8 (C–H, Ph), 127.2 (C–H, Ph), 118.7, 116.4 (q, 2JCF = 286 Hz, CF3), 108.2, 75.5 (C–I), 39.6 (N–CH3). MS, m/z (CI): 446 [M + H]+, 320 [M − I + H]+. Anal. Calcd for C16H11F3-IN3O: C, 43.17; H, 2.49; N, 9.44. Found: C, 43.25; H, 2.40; N, 9.32.
[4-(6-Iodoimidazo[1,2-a]pyridin-2-yl)phenyl]-N-methylamine (6, HM-IMPY)
Compound 5 (0.9 g, 2.0 mmol) and K2-CO3 (1.12 g, 8.1 mmol) were added to a solution of MeOH–H2O (20:1 v/v, 70 mL). This solution was refluxed under stirring for 1 h. The MeOH was then removed by rotary evaporation, and the organic residue with some water was extracted with Et2O (4 × 50 mL). The combined organic phase was washed with water and dried over Na2SO4. The volume of the filtrate was reduced to about 30 mL. The precipitate formed in the concentrated solution was decanted off and washed twice with Et2O (2 × 10 mL). The ethereal mother liquor was further concentrated until another batch of the precipitate appeared. The procedure above was repeated to afford collectively a pale-yellow solid (0.615 g, 1.7 mmol, 88%). 1H NMR (400 MHz, CDCl3): δ 8.30 (1H, s, H-5), 7.74 (2H, d, 3JHH = 8.6 Hz, H-2′,6′), 7.63 (1H, s, H-3), 7.35 (1H, d, 3JHH = 9.4 Hz, H-8), 7.26 (1H, dd, 3JHH = 9.5, 4JHH = 1.5 Hz, H-7), 6.64 (2H, d, 3JHH = 8.6 Hz, H-3′,5′), 3.85 (1H, s, N–H), 2.87 (3H, s, CH3). 13C{1H} NMR (400 MHz, CDCl3): δ 149.6, 147.1, 144.0, 132.0, 130.2, 127.3 (C–H, Ph), 122.2, 118.1, 112.5 (C–H, Ph), 106.3, 74.5 (C–I), 30.7 (N–CH3). MS, m/z (CI): 350 [M + H]+, 224 [M − I + H]+. HRMS Calcd, m/z (CI): C14H13-IN3 = 350.015 425. Found: 350.016039. Anal. Calcd for C14H12-IN3: C, 48.16; H, 3.46; N, 12.03. Found: C, 48.06; H, 3.26; N, 11.89.
6-Iodo-2-[4′-N-(2-fluoroethyl)methylamino]phenylimidazo[ 1,2-a]pyridine (7, FEM-IMPY)
Compound 6 (HM-IMPY) (87.0 mg, 0.249 mmol) and 2-fluoroethyl triflate (143.3 mg, 0.731 mmol) were dissolved in 2.5 mL of dry CH3CN in a microwave tube (10 mL volume). The mixture was subjected to CEM microwave irradiation (200 W, 100 °C, 10 min). A yellowish-orange solution was generated. The mixture was injected onto an HPLC column (21 mm × 250 mm; Phenomenex), eluted with an aqueous ammonia solution (0.25% v/v of concentrated ammonia and water)–MeCN ammonia solution (0.25% v/v of concentrated ammonia and MeCN) gradient (20–100% MeCN ammonia solution over 15 min) at 20 mL/min. The retention time of the compound was 13.0 min. The solvent of the collected portion was removed under reduced pressure to generate a white powder (32.2 mg, 33%). 1H NMR (400 MHz, CDCl3): δ 8.60 (s, 1H), 8.05 (d, 3JHH = 8.9 Hz, 2H), 7.92 (s, 1H), 7.62 (d, 3JHH = 9.4 Hz, 1H), 7.52 (d, 3JHH = 9.4 Hz, 1H), 7.0 (d, 3JHH = 8.9 Hz, 2H), 4.86 (dt, 2JFH = 48 Hz, 3JHH = 5.6 Hz, 2H, CH2F), 3.93 (dt, 3JFH = 24 Hz, 3JHH = 5.6 Hz, 2H, CH2N), 3.30 (s, 3H, NCH3). 19F NMR (400 MHz, CDCl3): δ (relative to CFCl3), −222.5 (hept, 2JHF = 45.2 Hz, 3JHF = 22.6 Hz). UV–vis (EtOH, nm): 355 (ε = 2.65 × 104), 285 (ε = 3.63 × 104), 234 (ε = 4.49 × 104). LC–MS, m/z: 396.2 [M + H]+. HRMS Calcd, m/z (FAB+): C16H16IFN3 = 396.0373. Found: 396.0378.
6-Iodo-2-[4′-N-(2-fluoropropyl)methylamino]phenylimidazo[ 1,2-a]pyridine (8, FPM-IMPY)
The same synthetic procedure as that used for FEM-IMPY was used except that 3-fluoropropyl triflate replaced 2-fluoroethyl triflate. HM-IMPY (57.4 mg, 0.164 mmol) and 3-fluoropropyl triflate (129.1 mg, 0.614 mmol) were used to generate a yellow solution. The HPLC retention time of the compound was 13.8 min. A pale-yellow product was obtained (26.1 mg, 39%). 1H NMR (400 MHz, CDCl3): δ 8.15 (s, 1H), 7.61 (d, 3JHH = 8.9 Hz, 2H), 7.48 (s, 1H), 7.21 (d, 3JHH = 9.4 Hz, 1H), 7.11 (dd, 3JHH = 9.4 Hz, 4JHH = 1.6 Hz, 1H), 4.34 (dt, 2JFH = 47.2 Hz, 3JHH = 5.6 Hz, 2H), 3.36 (t, 3JHH = 7.0 Hz, 2H, CH2–N), 1.81 (dpent, 3JFH = 33.4 Hz, 3JHH = 6.7 Hz, 2H, CH2), 2.83 (s, 3H, NCH3). 19F NMR (400 MHz, CDCl3): δ (relative to CFCl3), −221.4 (2JHF = 48.9 Hz, 3JHF = 30.1 Hz). UV–vis, in ethanol (nm): 355 (ε = 2.08 × 104), 285 (ε = 3.30 × 104), 234 (ε = 3.99 × 104). LC–MS, 410.2 [M + H]+. HRMS Calcd, m/z (FAB+): C17H18IFN3 = 410.0529. Found: 410.0534.
D4-Ethylene Glycol Bis-tosylate (9)6
To a mixture of anhydrous ethylene-d4 glycol-d2 (1.0 g, 15 mmol) and dry pyridine (4.0 mL) was added p-toluenesulfonyl chloride (5.9 g, 31 mmol) portionwise, keeping the temperature at 0 °C. The mixture was stored in a refrigerator overnight. The mixture was then poured into ice–water (60 mL), and the resulting white precipitate was collected by filtration and washed with cold water (2 × 25 mL). The solid was recrystallized from EtOH to give 4.2 g (76%) of the desired product as white crystals. Mp: 125 °C. 1H NMR (400 MHz, CDCl3): δ 2.46 (s, 6H), 7.34 (dm, 4H, 3JHH = 8.0 Hz), 7.74 (dm, 4H, 3JHH = 8.0 Hz). 13C NMR (400 MHz, CDCl3): δ 21.67, 127.96, 129.95, 132.42, 145.25. GC–MS: 373.86. IR (KBr): 553, 564, 576, 661, 693, 709, 819, 860, 908, 972, 1020, 1067, 1082, 1101, 1179, 1194, 1299, 1311, 1371, 1598 cm−1. HRMS Calcd, m/z (FAB+): C16H15D4O6S2 = 375.0875. Found: 375.0865.
Production of [18F]fluoride
No carrier added (NCA) [18F]-fluoride in 18O-enriched water was provided by the PET Center, National Institutes of Health. Briefly, it was obtained from the 18O(p,n)18F reaction by the proton irradiation of 18O-enriched water (>95 atom %). This is the most effective method of generating NCA [18F]fluoride.7
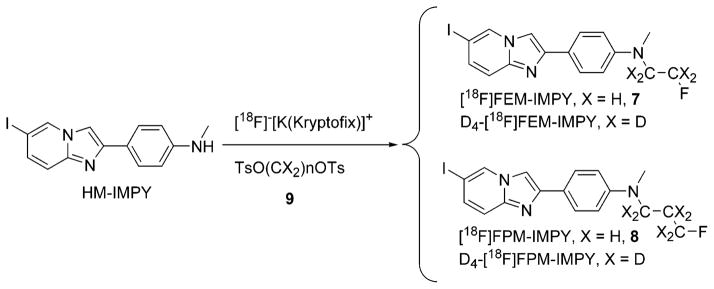
2.2. Preparation of Radioligands
NCA [18F]fluoride (100–250 mCi in 0.25 mL of H218O) was added with Kryptofix 222 (5 mg, 13.3 μmol) and K2CO3 (0.5 mg, 3.6 μmol) in MeCN–water (95:5 v/v, 0.1 mL) and dried azeotropically by addition–evaporation cycles (4 × 2 mL of MeCN). Ethylene glycol bistosylate or 1,3-propandiol bis-tosylate (2.0 mg) in MeCN (250 μL) was then added, and the closed glass reaction vessel was heated at 110 °C for 10 min to generate [18F]2-fluoroethyl tosylate ([18F]F(CH2)2OTs) or [18F]3-fluoropropyl tosylate ([18F]F(CH2)3OTs), respectively.8 The above solution was transferred to HM-IMPY (2–5 mg) in a V-vial, and the reaction mixture was concentrated to ~50 μL. This residue was heated at 135 °C for 45 min and then diluted with MeCN–water (3: 1 v/v, 1.5 mL) for direct injection onto an HPLC column (Luna C-18, 10 mm × 250 mm, Phenomenex) eluted with MeCN + 0.25% triethylamine (TEA) and H2O + 0.25% TEA (isocratic 50:50, v/v) at 6.0 mL/min, with eluate monitored for absorbance at 365 nm and radioactivity. For detection of radioactive compounds, a γ-ray detector (Bioscan Flow Count fitted with a NaI(Tl) detector) was used in series with the UV absorbance detector. The radioactive fraction with the same retention time as the respective reference ligand (tR: FEM-IMPY, 18 min; FPM-IMPY, 21 min) was collected. H2O (~100 mL) was added to the collected portion of the product. The solution was passed through a C-18 Sep-Pak cartridge (Waters, Milford, MA), which was washed at least twice with H2O. The radioactive product was washed out of the cartridge by absolute ethanol (1.0 mL) into a V-vial with Tween 80 (~25 μL). The solvent was evaporated under a stream of nitrogen at RT. Formulation of [18F]FEM-IMPY or [18F]FPM-IMPY in saline (0.9% w/v, 0.5 mL) provided a concentrated solution (2–18.4 mCi/mL) for mouse, rat, and monkey PET experiments.
The final product was analyzed on a Luna column (4.6 mm × 250 mm; Phenomenex) eluted with MeCN + 0.25% TEA and H2O + 0.25% TEA (isocratic at 60: 40 v/v) at 2.0 mL/min (tR: FEM-IMPY, 6 min; FPM-IMPY, 8 min). Radioactivity and absorbance (365 nm) were monitored to confirm the radiochemical and chemical purity of the [18F]FEM-IMPY or [18F]-FPM-IMPY and to measure its specific radioactivity.
2.3. In Vitro Binding Assay and Nonspecific Binding in Human AD Brain Homogenate
The Aβ40 monomer was aggregated by dissolving this peptide (0.5 mg/mL) in a buffer [sodium phosphate (pH 7.4, 10 mM) containing EDTA (1 mM)]. The solution was incubated at 37 °C for 46 h with gentle and constant shaking at 30 strokes per minute. After formation of the Aβ40 aggregates, the solution was dispensed into separate vials and stored at −80 °C for future use. Binding was assayed in 12 mm × 75 mm borosilicate glass tubes as described in ref 4. For inhibition studies, the reaction mixture contained Aβ40 aggregates (10–20 nM in the final mixture, 50 μL) or human AD brain homogenate (100 μg in the final mixture, 100 μL), inhibitors (10−5–10−10 M in 10% ethanol, 50 μL), [125I]TZDM (in 40% EtOH, 0.05 nM in the final mixture, 50 μL, 100 000 dpm/tube), and aqueous EtOH (10% v/v) in a final volume of 1.0 mL. Nonspecific binding was defined by adding thioflavin-T (10 μM) for [125I]TZDM binding in the same assay tubes. The mixture was incubated at RT for 3 h for 125I-labeling or 40 min for 18F-labeling, and the bound and the free radioactivity was separated by a vacuum filtration through Whatman GF/B filters using a Brandel M-24R cell harvester followed by washes with aqueous ethanol (10% v/v, 2 × 3 mL) at RT. Filters containing the bound 125I-labeled ligand were counted in a γ-counter (Packard 5000) with 70% counting efficiency. Under the assay conditions, the nonspecifically bound fraction was less than 15% of the total radioactivity. The results of inhibition experiments of synthetic amyloid aggregates were subjected to nonlinear regression analysis using software EBDA9 by which Ki values were calculated.
Binding was also assayed in the same way with [18F]FEM-IMPY as radioligand and incubation for 40 min. The inhibition curve of [18F]FEM-IMPY by FEM-IMPY in AD brain tissue homogenates was fitted [KaleidaGraph 3.5] to the following equation, which is derived from the modified Hill equation:10
where F(x) is the radioactivity bound relative to control, M is the maximal percentage of [18F]FEM-IMPY bound specifically, H is the Hill coefficient (close to 1), [L] is the concentration of inhibitor, and N is the percentage of nonspecific binding.
2.4. Autoradiography
Coronal sections of cerebrum from a confirmed case of Alzheimer’s disease (female, 59-years old, postmortem interval (PMI) of 47.5 h) and a normal control (female, 64-years old, PMI of 28.5 h) were frozen rapidly in a 50:50 mixture of dry ice and isopentane, sealed in a plastic bag, and stored at −76 °C before sectioning. Frozen blocks of the medial temporal lobe (hippocampal region) were sectioned at a thickness of 14 μm, mounted on gelatin-coated glass slides, dried, and stored under desiccant at −76 °C. Slide-mounted tissue sections were removed from the freezer, thawed at room temperature for 20 min, and air-dried. They were then immersed in a 10% neutral phosphate-buffered formalin solution at room temperature for 1 h, delipidized in xylene for 40 min, and rehydrated through ethanols to water.
FEM-IMPY, 1.0 mg, was dissolved in 1.265 mL of ethanol, resulting in a stock solution of 2.00 × 10−3 M, which was then further diluted to 1 × 10−3 M with 10% ethanol. One of the Alzheimer’s disease tissue slides (Figure 3b) was pretreated with FEM-IMPY stock solution for 60 min at room temperature and washed with water for 30 s. All slides were then placed flat at the bottom of a glass dish and were incubated for 25 min at RT in [18F]-FEM-IMPY solution (made up with 0.1 mCi in about 0.1 mL of ethanol added to 10 mL of 0.9% saline). They were then dipped in distilled water for 30 s, then immersed in ethanol for 15 min and lastly in water for 30 s. The slides were dried on a hot plate with a stream of cold air and placed in a cassette with the exposed sides facing up. The phosphor imaging plate was held with the blue side down. The entire cassette was placed in the dark at RT overnight for 18F labeling. Digital autoradiography was acquired using a FUJI BAS 5000 phosphorimager (FUJI, Tokyo, Japan) with a resolution of 25 μm.
Figure 3.
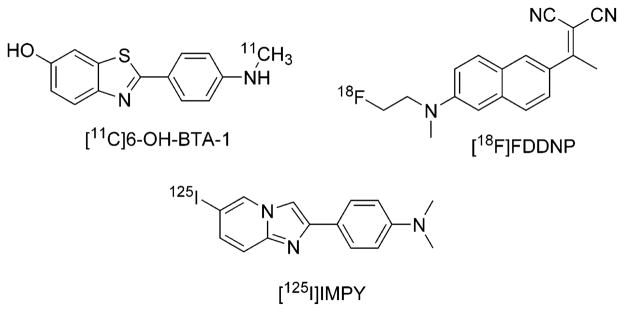
Autoradiography of [18F]FEM-IMPY and immuno-fluorescent (IF) detection of β-amyloid plaques in postmortem brain tissue sections of medial temporal lobe from an AD patient and an age-matched control: (a) AD tissue + [18F]FEM-IMPY; (b) AD tissue + IF protocol; (c) AD tissue pretreated with FEM-IMPY + [18F]FEM-IMPY; (d) control tissue + [18F]-FEM-IMPY.
2.5. Partition Coefficient Determination
Ligand (3–5 mg) was dissolved in sodium phosphate buffer (0.1 M, pH 7.4, 6.0 mL) and n-octanol (1.0 mL). The septum-sealed mixture was vortexed for 1 min, shaken vigorously for another min, filtered, stood at RT for 10 min, and centrifuged at 5000g for 10 min. An aliquot of organic or aqueous layer was taken through the septum by syringe. An aliquot of octanol layer (5.0 μL) was added to MeCN (5.0 mL) to prepare a stock solution. Both aqueous and organic phases (1.0 mL each) were analyzed by HPLC using MeCN and water with 0.25 vol % of concentrated ammonia solution in each as a gradient mixture (5–95% of the MeCN solution in 15 min) at 2.0 mL/min. Partition coefficients (PC) were determined by the concentration ratio of the the organic phase to the aqueous phase.
2.6. In Vivo Biodistribution in Normal Mice
For radioligand injection, a 30 gauge needle was attached to a polyethylene catheter (PE 10) and inserted into the tail vein of male ICR mice (2–3 months old, average of 20–30 g). The needle and catheter were secured with tissue adhesive. For arterial blood sampling, femoral artery catheter (PE 10) insertion with 1% isoflurane anesthesia was performed. Subcutaneous tunneling and tethering at the nape secured the arterial catheter. When fully anesthetized in the induction chamber, the mice were placed on a heating pad. For dynamic scanning, heads and bodies of mice and rats were fixed with tape and kept with 1% isoflurane anesthesia via a nose cone. Body temperature was monitored with a rectal probe and maintained at 36–37 °C. Then NCA radioligand (400–450 μCi) in saline (0.9% w/v, 0.03–0.15 mL) with Tween solution (0.1%) was injected and flushed with saline (0.070 mL).
We used a small animal PET camera [Advanced Technology Laboratory Animal Scanner (ATLAS)] with an 11.8 cm diameter aperture, 2 cm axial field-of-view ring type research scanner surrounding the animal with 15 mm deep, 18 LGSO (7 mm)/GSO (8 mm) phoswich detector modules.11 This phoswich design achieved good resolution [1.1–1.8 mm full width at half-maximum (fwhm)] at the center with 3D OSEM (ordered subset expectation maximization) and 2D OSEM/single slice rebinning (SSRB) reconstruction methods, while preserving a sensitivity of 2.7% (100–650 keV).
For dynamic scanning, sequential brain scanning was performed in the same bed position immediately after iv injection of the radioligand; the scan lasted 1–2 h. Sequential time-activity curve (TAC) data were acquired immediately after radioligand injection to evaluate the brain uptake and washout in each mouse. The %ID/brain, %ID/g, and %ID-kg/g were calculated by both TAC of the PET scan and in vitro tissue counts (determined under section 2.7).
2.7. Metabolism of Radioligands in Normal Mice
Consecutive blood samples (each 10–35 μL for mouse) that were taken during PET scanning and dissected homogenized brain taken after scanning were analyzed to evaluate the metabolites and parent radioligand in the blood and brain. (As a general rule, 10% of the total blood volume can be collected in one experiment every 2–4 weeks. Total blood volume can be calculated as approximately 7.5% of body weight.)
The metabolites were determined with reverse-phase HPLC analysis on a column (Nova-Pak C18 Radial-Pak Cartridge, 4 μm, 8 mm × 10 mm; Waters Corp., Milford, MA) and with radial compression module RCM-100 and eluted with a mixture of MeOH, H2O, and TEA (70:30:0.1) at 1.0 mL/min. The HPLC was equipped with a Beckman System Gold 126 solvent module and a flow-through NaI(Tl) scintillation detector (model FC-4000) and rate meter (Bioscan, Washington, DC).
2.8. In Vivo Biodistribution in Rhesus Monkey
A male rhesus monkey (7.12 kg) was initially immobilized with ketamine (15 mg/kg) and subsequently anesthetized with isoflurane (1.5%) for the duration of the PET scan. The monkey was placed prone in the PET camera (GE Advance tomograph; GE Medical Systems, Waukesha, WI). A urinary catheter was inserted and clamped so that the activity overlaying the bladder represented the total urinary excretion during the scanning interval. ECG (Electrocardiogram), body temperature, and heart and respiration rates were measured throughout the experiment. Before injection of the radioligand, each animal received a brain transmission scan (32 min) with two rotating rods containing 68Ge for subsequent attenuation correction of the emission image. Brain emission scans were performed following the intravenous administration of [18F]-FEM-IMPY (2.9 mCi). Serial dynamic transaxial images were acquired for a total of 120 min for the head in 3D mode (33 frames with scan duration ranging from 30 s to 5 min). The tomographic images were analyzed with PMOD 2.4 (pixelwise modeling computer software; PMOD Group, Zurich, Switzerland). 12 All frames of the original reconstructed PET data were summed, and this summed image was coregistered to a T1- weighted magnetic resonance (MR) image acquired separately on a GE 1.5 T Signa MR scanner (GE Medical Systems, Waukesha, WI), using image analysis software MEDx (Sensor Systems Inc., Sterling, VA). The summed PET image was then fused with the coregistered MR image using an image fusion tool in PMOD. Several regions of interest (ROIs) for the source organs were then manually defined on this fused image with anatomical structures identified on the MR image.
3. Results
3.1. Chemical Synthesis
The nucleus of imidazo- [1,2-a]pyridines is generally made by condensation between a 2-aminopyridine and an α-halogenoketone or aldehyde in the presence of a mild base such as sodium bicarbonate according to Scheme 1. The pyridine nitrogen attacks the α-carbon of the ketone, displacing the halide and forming a pyridinium salt. The salt is deprotonated by the weak base. The imine group generated attacks the carbonyl group intramolecularly to give an amino alcohol intermediate, which dehydrates spontaneously to form the imidazole.
Scheme 1.

The synthesis of HM-IMPY is shown in Scheme 2. The secondary amine 2 was protected using trifluoroacetic anhydride.13 The trifluoroacetyl group gives multiple advantages. The bromination can be done in one step and under mild conditions, using tetra-n-butylammonium tribromide in MeOH according to a modified procedure.14 This bromination is regioselective, occurring only on the methyl group, thus avoiding byproducts from bromination on the ring. This occurs because the trifluoroacetyl group depresses the ER (electron resonance) effect of the N-methylamino group. The reaction solvent appears to be critical for this reaction. The trifluoroacetyl group as a protecting group also provides for easy removal at the end of synthesis by weak bases under mild conditions. The condensation reaction between the aminopyridine derivative and bromoketone was conducted under mild basic conditions.15 2-Amino- 5-iodopyridine was available either commercially or through electrophilic iodination at the 5-position of 2-aminopyridine using I2 and periodic acid in acid media (Scheme 3).16 Ideally, HM-IMPY might be derived from the condensation reaction between 5-iodo-2-aminopyridine and 2-bromo-4′-methylaminoacetophenone, but the attempt to synthesize the latter by the usual process (Scheme 4) was frustrated by the poor yield and complex mixture of products at the dibromination step. The monoiodination of 4-methylaminoacetophenone with I2 and Selectfluor17 in MeOH gave a mixture of compounds with many ring-iodinated by products.
Scheme 2.

Scheme 3.
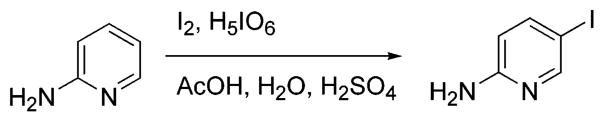
Scheme 4.

An alternative synthesis of HM-IMPY is the mono-demethylation of the imidazopyridine, IMPY.15 This reaction was attempted in different ways according to methods reported in the literature for the demethylation of N,N-dimethylanilines (Scheme 5). In the first approach TiCl4/CH2Cl218 gave only polymerized products. In the second, tetra-n-butylammonium periodate, a potent oxygen donor, was used to hydroxylate one of the methyl groups in the presence of a metalloporphyrin catalyst.19 The α-amino alcohol was then expected to decompose easily to give the secondary amine and formaldehyde. However, this method had to be abandoned because the reaction was low-yielding with many side reactions. The third approach20 was more effective and allowed us to obtain the desired imidazopyridine in 30% yield. The proposed mechanism consists of a double single-electron-transfer oxidation of the nitrogen center by the iodosobenzene, giving iodobenzene and an iminium ion. The iminium ion should be attacked by the N3− ion, forming an aminomethylene azide derivative. Quenching the reaction mixture with an aqueous solution of NaHCO3 allows the azide to be displaced by H2O to form the α-amino alcohol, which dissociates quickly to form formaldehyde and the secondary amine. The problem with this procedure is that the reaction is not reproducible enough on scale-up. Moreover, the product is quite labile under the oxidative reaction conditions.
Scheme 5.
3.2. Radiolabeling
A “one-pot” radiosynthesis of [18F]FEM-IMPY or [18F]FPM-IMPY was developed experimentally and gave radiochemical yields ranging from 26% to 51% (mean, 35%), corrected to the start of radiosynthesis, and in >93% radiochemical purity (Scheme 6). Specific radioactivity was determined to be 2.3–17.9 Ci/μmol at EOS (end of synthesis).
Scheme 6.
3.3. Binding to Aβ40 Aggregates in Solution and in AD Brain Homogenates
Both FEM-IMPY and FPM-IMPY compete well with [125I]TZDM for binding to Aβ-40 aggregates, a previously established selective probe.5 This is one of three binding sites reported in the literature for thioflavin-type ligand series. The other two are for a Congo red series5 and an FDDNP series.21 The Ki values decrease and the experimentally determined log D values (log P at pH 7.4) increase with increasing length of the side chain on amino group (Table 1).
Table 1.
The log D and Ki Values of IMPY Derivatives against [125I]TZDM for Binding to Aβ(1–40) Aggregates
| ligand | logD | Ki [nM] |
|---|---|---|
| IMPY | 3.58 ± 0.44 | 15.0 ± 5.0 |
| FEM-IMPY | 4.41 ± 0.10 | 27 ± 8 |
| FPM-IMPY | 4.60 ± 0.19 | 40 ± 5 |
With increasing concentrations of FEM-IMPY, [18F]-FEM-IMPY binding to Aβ-amyloid plaques of the human AD brain homogenates should decrease and level off after all specific binding sites have been displaced. We observed that about half of all binding was displaceable (or specific, Figure 2). The curve was fitted to a modified Hill equation to accommodate the nonspecific binding of the ligand, yielding Ki = 83 ± 69 nM (with this and subsequent data expressed as the mean ± SD). This binding constant compares well with that determined from the synthetic amyloid aggregates (Ki = 27 ± 8 nM).
Figure 2.

Determination of specific vs nonspecific binding of [18F]FEM-IMPY in postmortem AD brain tissue homogenate. Each data point came from the average of four measurements, and the bar represents the standard deviation (SD).
3.4. Labeling AD Brain Sections
The pattern of radioactivity in the adjacent coronal AD brain specimen without competitor showed specific binding of [18F]FEM-IMPY to cortical areas containing neuritic plaques (NP) (Figure 3a). Specific binding of [18F]FEM-IMPY to regions of cortical gray matter with NPs was eliminated in the AD specimen pretreated with nonradioactive FEM-IMPY, compared to autoradiography without competitor (Figure 3c). No specific binding was observed for a control brain specimen (Figure 3d).
3.5. Studies in Normal Mice
In the averaged TAC for four individual mice administered with [18F]FEM-IMPY (Figure 4), the brain uptake of the activity (%ID-kg/g) peaks between 0.5 and 2 min after injection. The elimination of the activity follows an exponential decay but is near zero after about 20 min. The individual TAC data were well fitted to the double-exponential formula
Figure 4.

Time–activity curve in brain and skull after iv injection of [18F]FEM-IMPY or D4-[18F]FEM-IMPY in normal mice. Each point represents the average of four mice. The standard error of each point for the brain is within 3–12% and that for the skull is 5–25% after 1.2 min. For clarity, the error bar is not shown. D4-skull represents data from D4-[18F]FEM-IMPY.
where y is the activity, a is the residual radioactivity trapped inside the brain, b and d are constants, c is the rate of uptake, e is the rate of elimination, and t is the time after injection. The first exponential portion in the equation represents the uptake of the activity in brain, and the second exponential portion represents the elimination of the activity.
The average TAC from four individual mice injected with [18F]FPM-IMPY is shown in Figure 5. Assuming an average brain weight of 0.40 and 25 g body weight, analogous fitting was performed for [125I]IMPY data from Kung et al.4 The constants from the individual curve fittings are summarized in Table 2.
Figure 5.

Time–activity curve in brain and skull after iv injection of [18F]FPM-IMPY in normal mice. Each point represents the average of four mice. The standard error of each point for the brain is within 2–12% and that for the skull is 7–38% after 1.2 min. For clarity, the error bar is not shown.
Table 2.
Curve-Fitting Constants of TAC Obtained after Intravenous Administration of Radiolabeled IMPY or Derivatives to Mice
| administered radioligand | max activity [(ID%-kg)/g] | time at max activity [min] | washout rate [min−1] | residual activity at 120 min, 102 × [(ID%-kg)/g] | rate of bone uptake, 103 × [min−1] |
|---|---|---|---|---|---|
| [125I]IMPY4 | 0.216 ± 0.010 | 2 | 0.122 ± 0.024 | 1.08 ± 0.22 | NA |
| [18F]FEM-IMPY | 0.161 ± 0.019 | 0.50 | 0.334 ± 0.021 | 11.1 ± 0.05 | 1.60 ± 0.294 |
| [18F]FPM-IMPY | 0.157 ± 0.018 | 1.83 | 0.127 ± 0.006 | 4.79 ± 0.11 | 7.28 ± 2.85 |
The residual activity in brain at 2 h was much greater for [18F]FEM-IMPY than for [125I]IMPY.4 Much less residual radioactivity was left for [125I]IMPY itself during the time course of the measurement, while a substantial amount of radioactivity was trapped inside the brain for [18F]FEM-IMPY. Values for curve-fitting constants for [18F]FPM-IMPY fall between [18F]IMPY and [18F]FEM-IMPY with respect to residual brain activity and clearance.
Substantial bone uptake of radioactivity was observed in the skull after the administration of [18F]FEM-IMPY or [18F]FPM-IMPY to normal mice. The averaged bone uptake TACs for each radioligand in four individual mice are shown in Figures 4 and 5. The bone uptake TAC of [18F]FEM-IMPY can be fitted to a linear function except for a few initial points (less than 5 min), while that of [18F]FPM-IMPY is fitted more accurately with an exponential curve. The average rate for bone uptake of radioactivity is faster for [18F]FPM-IMPY than for [18F]FEM-IMPY (Table 2). A significant reduction in skull bone uptake of radioactivity was observed for [18F]-D4-FEM-IMPY (Figure 4 and Table 2). The radioactivity in the skull bone was reduced by ~34% over the span of the experiment upon substitution of the 4-ethylene protons by deuteriums. However, there was no effect of deuterium substitution on brain uptake or clearance of radioactivity.
3.6. Studies in Monkey
The TACs of [18F]FEM-IMPY in a rhesus monkey are shown in Figure 6 and Table 3. Brain activity increased quickly and reached substantial levels, with peak values of 0.26–0.35 %ID-kg/g at 3–6 min after injection. Washout of brain activity was similar in all brain regions with a mono-exponetial washout rate of 0.05–0.09 min−1 (corresponding to a half-life T1/2 = 8–14 min). Activity in bone increased in an exponential fashion and reached levels of ~0.1 %ID-kg/g, similar to residual activity in brain (Figure 6).
Figure 6.

Time–activity curves of different regions of brain after iv injection of [18F]FEM-IMPY in a rhesus monkey.
Table 3.
Uptake and Elimination Rate Constants of Radioactivity in Various Brain Regions of a Rhesus Monkey after Intravenous Administration of [18F]FEM-IMPY
| component | elimination [min−1] | uptake [min−1] |
|---|---|---|
| frontal cortex | 0.0606 ± 0.0024 | 0.859 ± 0.030 |
| striatum | 0.0859 ± 0.0037 | 0.972 ± 0.040 |
| brainstem | 0.0488 ± 0.0020 | 1.00 ± 0.040 |
| cerebellum | 0.0854 ± 0.0040 | 1.09 ± 0.050 |
| jaw bone | NA | 0.373 ± 0.058 |
3.7. Metabolite Analysis. 3.7.1. Metabolism in Blood in Vivo
The distribution of radioactivity among chemical species following injection of the radioligand was monitored by HPLC. The TACs for several components of activity in whole blood are shown in Figure 7. Curves for parent, lipophilic metabolite (L-metabolite), and total radioactivity were fitted to a single-exponential decay, and the curve for polar metabolite (P-metabolite) was fitted to a double-exponential function. The half-lives for all radioactive components were less than 2 min. This is much shorter than those within the brain of the same animal. The rate constants are summarized in Table 4. No significant difference was found for [18F]D4-FEM-IMPY (data not shown), compared with [18F]FEM-IMPY. Only a single polar radioactive metabolite was seen in plasma at <30 min after injection of [18F]FPM-IMPY.
Figure 7.

Time–activity curves of different radioactive components after iv injection of [18F]FEM-IMPY in the in vivo whole blood of normal mice.
Table 4.
Elimination Rate Constants for [18F]FEM-IMPY and Radioactive Derivatives in Vivo in Whole Blood
| component | uptake [min−1] | elimination [min−1] |
|---|---|---|
| parent | NA | 1.14 ± 0.16 |
| P-metabolite | 0.576 ± 0.980 | 0.575 ± 2.21 |
| L-metabolite | NA | 0.722 ± 0.347 |
| total radioactivity | NA | 0.801 ± 0.158 |
3.7.2. Metabolism in Brain in Vivo
HPLC analysis of brain homogenates obtained 2 h after injection of [18F]-FEM-IMPY showed virtually no parent compound. The only radioactive species observable were polar (P-metabolite), presumably fluoride ion or a related small-molecule ionic species such as [18F]FCH2CO2−. Analogous metabolites were observed for [18F]FPM-IMPY.
3.7.3. Metabolism in Brain Tissue in Vitro
Incubation of mouse brain homogenates with each radioligand at 37 °C for various lengths of times resulted in radioactive metabolites. HPLC analysis showed the same pattern of metabolism as described for whole blood in vivo. The TACs for [18F]FEM-IMPY are displayed in Figure 8. For unknown reasons, degradation of the radiolabeled ligand did not go to completion but only to about 10% conversion. The curves were fitted to mono-and biexponential functions (to account for the accumulation and consumption). No significant difference was observed for the disappearance of [18F]FEM-IMPY (t1/2 = 15 min) compared to that of [18F]D4-FEM-IMPY (t1/2 = 14 min). However, [18F]FPM-IMPY (t1/2 = 11 min) degraded more quickly than [18F]FEM-IMPY.
Figure 8.

Time–activity curves of different radioactive components after incubation of [18F]FEM-IMPY in the in vitro brain homogenate of normal mice.
3.7.4. LC–MS Analysis of Intermediates
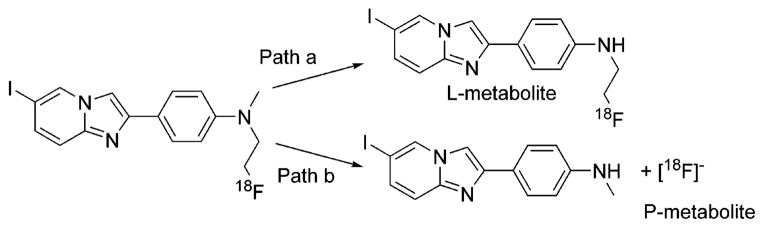
Incubation of a [18F]FEM-IMPY and FEM-IMPY (carrier, 15 μg) mixture with mouse brain homogenate provided sufficient material for the isolation of the radioactive metabolites by HPLC in three fractions: polar metabolite (P-metabolite), lipophilic metabolite (L-metabolite), and parent [18F]FEM-IMPY. The retention time of P-metabolite was very close to that of added [18F]fluoride under the same conditions. Each fraction was collected, allowed to decay, and analyzed by LC–MS (electrospray). The potential metabolites of FEM-IMPY were examined in each fraction according to the proposed loss or addition of fragment(s) from the FEM-IMPY molecule, namely, (1) hydroxylation by either the addition of an oxygen atom at one of the nitrogen atoms or the insertion of an oxygen atom into one of the C–H bonds (m/z 412), (2) N-demethylation at the tertiary amino group (m/z 382), (3) N-defluoroethylation at the tertiary amino group (m/z 350), and (4) simultaneous N-demethylation and N-defluoroethylation at the tertiary amino group (m/z 336). The chromatographic separation followed by electrospray analysis generatedm/z 382 and m/z 350 ions specific for metabolites of N-demethylated and N-defluoroethylated FEM-IMPY, respectively, but not for hydroxylated metabolites in the HPLC fractions. No significant amount of primary amine, resulting from complete N-dealkylation of FEM-IMPY, was observed. The metabolic pathways consistent with the MS analysis are shown in Scheme 7.
Scheme 7.
4. Discussion
An efficient chemical synthesis for the common precursor, HM-IMPY, has been developed among several available synthetic routes to the compound. This allows the syntheses of FEM-IMPY and FPM-IMPY, either as reference compounds or as 18F-labeled ligands. HM-IMPY is soluble in acetone, CH2Cl2, and CHCl3 but is insoluble in MeCN and water. It is stable in solid form but decomposes gradually in solution when exposed to air. The most reactive sites of the molecule toward electrophiles such as alkyl halides and triflates are the aromatic amino group and the aromatic imino group, with the reactivity of the latter rarely observed. It also reacts with acid, but the exact location is unclear at this time; it is probably the imino site. Both FEM-IMPY and FPM-IMPY are more lipophilic than HM-IMPY or IMPY. The order of the retention times on reverse-phase HPLC is HM-IMPY < IMPY < FEM < FPM.
HM-IMPY reacts slowly with ethylene glycol bistosylate at 130–150 °C. For example, the reaction is less than 5% complete after 10 min under microwave conditions (300 W and 150 °C). However, addition of [K- (Kryptofix 222)]+[F]− accelerates the alkylation to generate FEM- or FPM-IMPY in a 70–80% yield under anhydrous conditions with the same microwave conditions. Alkylation also occurs if 2-fluoroethyl triflate or 3-fluoropropyl triflate or the corresponding tosylate is used. This may explain the success of the “one-pot” synthesis of [18F]FEM-IMPY and [18F]FPM-IMPY, where excess ethylene glycol bis-tosylate or 1,3-propanediol bistosylate does not interfere with the radiosynthesis. Indeed, premixing of HM-IMPY, ethylene glycol bistosylate or 1,3-propanediol bis-tosylate, and [K-(Kryptofix 222)]+[18F]− generated the same [18F]FEM- and [18F]FPM-IMPY in a truly “one-pot” reaction. This suggests that FCH2CH2OTs, not the N-tosylate of IMPY, is the reactive intermediate in the radiosynthesis.
The binding affinities of IMPY, FEM-IMPY, and FPM-IMPY to synthetic Aβ aggregates are comparable but decrease gradually in the same order. This suggests that there is increasingly unfavorable steric interaction between IMPY derivatives and the protein pocket with an increase in alkyl chain length. However, removing one of the alkyl groups on the amino group dramatically reduces the binding affinity. The Ki values for HM-IMPY and FEH-IMPY (6-iodo-2-[4′-N-(2-fluoroethyl)- amino]phenylimidazo[1,2-a]pyridine) are 140 ± 20 and 161 ± 30 nM (our present work), a 5-fold lower affinity than those of FEM-IMPY and FPM-IMPY. This suggests that the binding pocket around the amino group is highly hydrophobic. If the amino group is replaced by a hydroxy group, the Ki is greater than 1000 nM (this work). The Ki is 451 ± 59 nM if the amino group is substituted by a methoxy group (this work). Both the number of alkyl groups and their electron-donating ability contribute to the binding affinity of IMPY derivatives to Aβ40 aggregates. In conclusion, the binding pocket around the amino group is a small hydrophobic pocket.
All of the IMPY derivatives reported here have quite high lipophilicity, with log D values between 3.6 and 4.6 at pH 7.4. This high lipophilicity may explain the relatively high nonspecific binding of [18F]FEM-IMPY to human AD brain homogenates, which is approximately 50% of the total binding.
The results of in vivo biodistribution studies in normal mice showed that both FEM-IMPY and FPM-IMPY easily penetrate the blood–brain barrier after intravenous injection. In PET experiments, high uptake of activity was obtained for both probes (6.4% ID/g for [18F]-FEM-IMPY and 5.7% ID/g for [18F]FPM-IMPY at 0.5–1 min). These values compare well with [123I/125I]IMPY (7.2% ID/g in 2 min).4 However, in contrast to the single-exponential washout of [123I/125I]IMPY, the washout of radioactivity from each probe examined here was biphasic, being rapid over the first 20 min and much slower thereafter. Residual brain radioactivity was 4.5% ID/g for [18F]FEM-IMPY and 2.1% ID/g for [18F]FPMIMPY at 2 h after injection.
Homogenates of ex vivo mouse brain after injection of the radioligand were analyzed by HPLC. The parent compounds of both ligands disappeared rapidly. The residual activity represented polar metabolite(s), one of which is probably the [18F]fluoride ion. This means that the residual radioactivity observed for both [18F]FEM-IMPY and [18F]FPM-IMPY came from the polar metabolite and not from nonspecific binding. Lipophilicity of the ligands does not play any role in this case. Detailed analysis of the in vivo whole blood of mice showed that the metabolism in blood is even faster than that in the brain. The contribution of the lipophilic intermediate (L-intermediate) is unclear at this time. Given its low concentration observed both in vivo and in vitro, it may not contribute significantly to the TACs.
The in vivo instability of the ligands within mouse brain is also observed in vitro. Identical metabolites for [18F]FEM-IMPY and [18F]FPM-IMPY were observed with both in vivo and in vitro experiments. However, the in vitro metabolism ended after about a 10% consumption of the radioligand, which may have been caused by instability of the enzymes or depletion of cofactors responsible for the metabolism of the radioligand. In the brain, the enzyme may be monoamine oxidase, which has been shown to be involved in the metabolism of dopamine and other amine compounds.22 In the periphery, the enzyme responsible could be MAO or a P-450 subtype.
The in vitro metabolites were isolated with the help of added carrier. Mass spectrum analysis of the metabolites demonstrated the presence of a dealkylated aromatic amino group, consistent with the assumption of the instability of tertiary amino groups in an oxidative environment. Indeed, an analogous oxidative reaction was used in the synthesis of HM-IMPY (Scheme 5).
Substantial uptake of [18F]fluoride in the skull was also clearly observed for both [18F]FEM-IMPY and [18F]-FPM-IMPY. A deuterium isotope effect is common for both chemical and biological reactions and has been observed for the pharmacokinetics of certain PET radiotracers.23 [18F]D4-FEM-IMPY was synthesized and evaluated for its effect on metabolism in both brain and periphery. No deuterium effect was observed for the brain uptake of the radioligand, and no clear reduction of residual brain radioactivity was found. However, a significant reduction of the bone uptake of radioactivity in the skull was clearly identified. This suggests that the metabolism for the IMPY derivative in the periphery but not in the brain is sensitive to small structural changes in the molecule.
In vivo distribution studies of the radioligands in rhesus monkeys showed species differences in the rate of metabolism of these radioligands. Both residual brain activity and bone uptake were substantially less in rhesus monkey compared to mice. No significant difference in activity uptake among regions of rhesus brain was observed, consistent with the absence of amyloid plaques in these normal monkeys.
In a preliminary manner, we double-labeled individual tissue sections with [18F]FEM-IMPY and with fluorescent-labeled antibody (DAKO) to amyloid (Figure 3b). The sections showed a general correspondence of colabeling. However, the phosphorimager plate demonstrated clumps of activity that were enriched in the gray matter, with individual spots about 100 μm in size and roughly spherical in shape. Thus, the resolution of the plate was degraded relative to the primary distribution of radioligand perhaps because of the distance traveled by the energetic β+ positron before annihilation by an electron. In any case, the general pattern of the autoradiographic labeling (each area of activity measuring about 100 μm in diameter) showed a strong visual correlation with higher densities of fluorescently labeled amyloid plaques (each area about 10 μm in diameter). The staining in gray matter was displaceable with cold FEM-IMPY. The nonspecific binding of [18F]FEM-IMPY in AD brain sections is much less compared with that determined from in vitro binding assays (50%). This may come from the xylene defatting step used for the AD brain sections pretreatment.
Conclusions
The two 18F-labeled IMPY derivatives enter the brain of normal mice readily and quickly. However, the washout shows a biphasic behavior. The first phase is similar to the behavior of [125I]IMPY. The brain retention of activity may be due to trapping of polar metabolite( s). The probes are quickly metabolized in plasma, with substantial defluorination and bone uptake. Rhesus monkey showed much slower metabolism for these derivatives than mouse. These results suggest that IMPY derivatives are promising candidates as PET imaging agents for Alzheimer’s disease, based on their favorable pharmacokinetics and their high specific labeling of β-amyloid plaques in vitro. However, more work is required to fine-tune these structures in order to reduce metabolism, increase affinity, and decrease lipophilicity. Radiolabeling beyond the N-alkyl chains in these structures is likely to be preferred.
Acknowledgments
We gratefully thank Dr. Shuiyu Lu (MIB, NIMH) for his help in the microwave reactions, Mr. Jinsoo Hong (MIB, NIMH) for assistance in determining the specific radioactivity of the radioligands, Mr. Kun Park (MIB, NIMH) for determination of log D, Dr. Senda Beltaifa (CDBD, NIMH) for the initial brain tissue processing, and Dr. Joel E. Kleinman (CDBD, NIMH) for access and discussion of postmortem brain tissues.
References
- 1.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 2.Mathis CA, Wang Y, Holt DP, Huang GF, Debnath ML, Klunk WE. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem. 2003;46:2740–2754. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- 3.Shoghi-Jadid K, Small GW, Agdeppa ED, Kepe V, Ercoli LM, Siddarth P, Read S, Satyamurthy N, Petric A, Huang SC, Barrio JR. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry. 2002;10:24–35. [PubMed] [Google Scholar]
- 4.Kung MP, Hou C, Zhuang ZP, Zhang B, Skovronsky D, Trojanowski JQ, Lee VM, Kung HF. IMPY: an improved thioflavin-T derivative for in vivo labeling of beta-amyloid plaques. Brain Res. 2002;956:202–210. doi: 10.1016/s0006-8993(02)03436-4. [DOI] [PubMed] [Google Scholar]
- 5.Zhuang ZP, Kung MP, Hou C, Skovronsky DM, Gur TL, Plossl K, Trojanowski JQ, Lee VM, Kung HF. Radioiodinated styrylbenzenes and thioflavins as probes for amyloid aggregates. J Med Chem. 2001;44:1905–1914. doi: 10.1021/jm010045q. [DOI] [PubMed] [Google Scholar]
- 6.Hwang KO, Lightner DA. Synthesis and spectroscopic properties of N,N-bridged dipyrrinones. Tetrahedron. 1994;50:1955–1966. [Google Scholar]
- 7.Stocklin G, Pike VW. Radiopharmaceuticals for Positron Emission Tomography. Kluwer Academic Publishers; Dordrecht, The Netherlands: 1993. [Google Scholar]
- 8.Studenov AR, Berridge MS. Synthesis and properties of 18F-labeled potential myocardial blood flow tracers. Nucl Med Biol. 2001;28:683–693. doi: 10.1016/s0969-8051(01)00233-5. [DOI] [PubMed] [Google Scholar]
- 9.Munson PJ, Rodbard D. Ligand: a versatile computerized approach for characterization of ligand-binding systems. Anal Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- 10.Bennett JP. Methods in binding studies. In: Yamamura HI, Enna SJ, Kuhar MJ, editors. Neurotransmitter Receptor Binding. Raven Press; New York: 1978. pp. 57–90. [Google Scholar]
- 11.Green MV, Seidel J, Vaquero JJ, Jagoda E, Lee I, Eckelman WC. High resolution PET, SPECT and projection imaging in small animals. Comput Med Imaging Graphics. 2001;25:79–86. doi: 10.1016/s0895-6111(00)00057-4. [DOI] [PubMed] [Google Scholar]
- 12.Mikolajczyk K, Szabatin M, Rudnicki P, Grodzki M, Burger C. A JAVA environment for medical image data analysis: initial application for brain PET quantitation. Med Inf (London) 1998;23:207–214. doi: 10.3109/14639239809001400. [DOI] [PubMed] [Google Scholar]
- 13.Baker R, Castro JL. Total synthesis of (+)-macbecin-I. J Chem Soc, Perkin Trans. 1990;1:47–65. [Google Scholar]
- 14.Kajigaeshi S, Kakinami T, Okamoto T, Fujisaki S. Synthesis of bromoacetyl derivatives by use of tetrabutylammonium tribromide. Bull Chem Soc Jpn. 1987;60:1159–1160. [Google Scholar]
- 15.Zhuang ZP, Kung MP, Wilson A, Lee CW, Plossl K, Hou C, Holtzman DM, Kung HF. Structure–activity relationship of imidazo[1,2-a]pyridines as ligands for detecting β-amyloid plaques in the brain. J Med Chem. 2003;46:237–243. doi: 10.1021/jm020351j. [DOI] [PubMed] [Google Scholar]
- 16.Hsieh HP, Mclaughlin LW. Syntheses of 2 pyridine Cnucleosides as deletion-modified analogs of DT and DC. J Org Chem. 1995;60:5356–5359. [Google Scholar]
- 17.Stavber S, Jereb M, Zupan M. Selectfluor F-TEDA-BF4 mediated and solvent directed iodination of aryl alkyl ketones using elemental iodine. Chem Commun. 2002:488–489. doi: 10.1039/b200240j. [DOI] [PubMed] [Google Scholar]
- 18.Periasamy M, Jayakumar KN, Bharathi P. Aryltitanium species through the reaction of N,N-dialkylarylamines with TiCl4: Oxidative coupling, N-dealkylation, and reaction with electrophiles. J Org Chem. 2000;65:3548–3550. doi: 10.1021/jo991864+. [DOI] [PubMed] [Google Scholar]
- 19.Takata T, Ando W. Mild and selective oxygen atom transfer–normal-Bu4NIO4 with metalloporphyrins. Tetrahedron Lett. 1983;24:3631–3634. [Google Scholar]
- 20.Magnus P, Lacour J, Weber W. N-Methyl azidonation of N,N-dimethylarylamines with the iodosylbenzene/trimethylsilylazide reagent combination and some reactions of α-azidomethylamines. Synthesis. 1998:547–551. [Google Scholar]
- 21.Agdeppa ED, Kepe V, Petri A, Satyamurthy N, Liu J, Huang SC, Small GW, Cole GM, Barrio JR. In vitro detection of (S)-naproxen and ibuprofen binding to plaques in the Alzheimer’s brain using the positron emission tomography molecular imaging probe 2-(1-[6-[(2-[(18)F]fluoroethyl)(methyl)- amino]-2-naphthyl]ethylidene)malono nitrile. Neuroscience. 2003;117:723–730. doi: 10.1016/s0306-4522(02)00907-7. [DOI] [PubMed] [Google Scholar]
- 22.Binda C, Li M, Hubalek F, Restelli N, Edmondson DE, Mattevi A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc Natl Acad Sci USA. 2003;100:9750–9755. doi: 10.1073/pnas.1633804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fowler JS, Wolf AP, MacGregor RR, Dewey SL, Logan J, Schlyer DJ, Langstrom B. Mechanistic positron emission tomography studies: demonstration of a deuterium isotope effect in the monoamine oxidase-catalyzed binding of [11C]L-deprenyl in living baboon brain. J Neurochem. 1988;51:1524–1534. doi: 10.1111/j.1471-4159.1988.tb01121.x. [DOI] [PubMed] [Google Scholar]