

Abstract
We used positron emission tomography (PET) to measure the earliest change in dopaminergic synapses and glial cell markers in a chronic, low-dose MPTP non-human primate model of Parkinson’s disease (PD). In vivo levels of dopamine transporters (DAT), vesicular monoamine transporter-type 2 (VMAT2), amphetamine-induced dopamine release (AMPH-DAR), D2-dopamine receptors (D2R) and translocator protein 18 kDa (TSPO) were measured longitudinally in the striatum of MPTP-treated animals. We report an early (2 months) decrease (46%) of striatal VMAT2 in asymptomatic MPTP animals that preceded changes in DAT, D2R, and AMPH-DAR and was associated with increased TSPO levels indicative of a glial response. Subsequent PET studies showed progressive loss of all pre-synaptic dopamine markers in the striatum with expression of parkinsonism. However, glial cell activation did not track disease progression. These findings indicate that decreased VMAT2 is a key pathogenic event that precedes nigrostriatal dopamine neuron degeneration. The loss of VMAT2 may result from an association with α-synuclein aggregation induced by oxidative stress. Disruption of dopamine sequestration by reducing VMAT2 is an early pathogenic event in the dopamine neuron degeneration that occurs in the MPTP non-human primate model of PD. Genetic or environmental factors that decrease VMAT2 function may be important determinants of PD.
Keywords: MPTP, non-human primate, Parkinson’s disease, PET, translocator protein 18 kDa, VMAT2
Idiopathic Parkinson’s disease (PD) is the second most common neurodegenerative disorder and its prevalence increases with age (Dauer and Przedborski 2003). The clinical manifestation of PD includes resting tremor, rigidity, bradykinesia, and unsteady gait (Dauer and Przedborski 2003). These clinical symptoms result from the progressive loss of dopamine (DA) neurons in the substantia nigra pars compacta (SNpc) with prominent DAergic terminal loss in the caudate and putamen (Dauer and Przedborski 2003). Although the clinical manifestation of PD is associated with the loss of DA in the striatum, other neuronal systems and brain regions are also affected (Langston 2006).
Molecular imaging including positron emission tomography (PET) and single photon emission computer tomography with radiotracers selective for specific aspects of the DA synapse have been used to study nigrostriatal DA neuron terminal degeneration in PD patients. These include [18F]-fluoroDOPA to measure dopa decarboxylase activity, [123I]-β-CIT and [11C]-WIN 35,428 for dopamine transporters (DAT), and [11C]-dihydrotetrabenazine ([11C]-DTBZ) for vesicular monoamine transporter type 2 (VMAT2) (Wong et al. 1993; Pirker et al. 2002; Brooks 2003; Ravina et al. 2005; Bohnen et al. 2006). By necessity, imaging studies in PD patients are performed at a point when significant DA terminal degeneration has already occurred. Typically, PET studies in PD assess one aspect of the DA synapse (Pirker et al. 2002; Bohnen et al. 2006; Gerhard et al. 2006) and the use of a multi-tracer approach has been limited even in nonhuman primates models of PD (Lee et al. 2000; Doudet et al. 2006). Further, no multi-tracer PET study to date has included a marker of glial cell activation. Therefore, there is a paucity of knowledge on the in vivo dynamics of DA synapses and the role of glial cells in the slow and progressive nigrostriatal dopamine neuron degeneration that occurs at the early pre-clinical stages of PD. Thus, understanding the early changes in DA synapses and glial cell responses prior to clinical expression of disease may provide valuable insights on disease pathophysiology.
The goal of the present study was to use a multi-tracer PET imaging approach to investigate the earliest change(s) in striatal DA synapses in a chronic, low-dose MPTP nonhuman primate model of PD. We chose a chronic low-dose MPTP administration paradigm to mimic the slow and progressive degeneration that occurs in idiopathic PD. A second aim was to assess whether imaging the peripheral benzodiazepine receptor, renamed translocator protein 18 kDa (TSPO) (Papadopoulos et al. 2006) could serve as an in vivo marker to track glial cell activation.
Materials and methods
Animal husbandry and chronic MPTP administration
Male baboons (Papio anubis) (body weight ranging from 20–30 kg) were used. Nine animals were used in the study but one was excluded because it did not go through the entire experimental protocol. Baboons were singly housed in a colony room maintained at 22 ± 1°C, with free access to food and water. For MPTP–HCl (MPTP; Sigma, St Louis, MO, USA) administration, animals were sedated (8–15 mg/kg ketamine, i.m.) prior to treatment. MPTP was administered by intravenous injection once per week starting with a dose of 0.1 mg MPTP/kg body weight for the first 2 weeks then 0.2 mg/kg continuously for 7–8 months except the week when PET scans were performed. The MPTP dose was then increased to 0.4–0.6 mg/kg until the appearance of clinical symptoms. After the completion of the last PET imaging set, all animals were killed immediately by intravascular injection of 7–8 mL Buthanasia (each mL containing 390 mg pentobarbital sodium and 50 mg phenytoin sodium) (Schering-Plough, Kenilworth, NJ, USA), except one animal that was killed within 1 month. Brains were collected and embedded in warm 3% agarose. After the hardening of agarose, the embedded brains were sliced in a series of 0.4 cm coronal sections using a commercial meat slicer. For each brain slice, one of the hemispheres was immediately frozen by dry ice and the other hemisphere was post-fixed for immunohistochemistry. Brain sections were stored at −80°C until use. All animal studies were reviewed and approved by the Animal Care and Use Committee of the Johns Hopkins Medical Institutions, Baltimore, MD, USA.
Radioligand synthesis and PET image analysis
Synthesis of radioligands, performance of PET studies and PET image analysis were routinely performed by us at the Johns Hopkins Hospital Cyclotron and PET facilities. PET studies were performed at baseline (prior to MPTP administration) and at three different time points after initiation of weekly injections of MPTP. The following radioligands were synthesized using published methods. [11C]-(R)-PK11195: R-N-desmethyl-PK11195 [(R)-1-(2-chlorophenyl)-N-methyl-N-(1-methyl-propyl)-3-isoquinoline carboxamide] the precursor for the radioisotope labeled (R)-PK11195, was purchased from ABX (Advanced Biochemical Compounds, Dresden, Germany). [11C]-(R)-PK11195 was synthesized as described previously (Chen and Guilarte 2006) with a specific radioactivity of over 370 GBq/μmol (10 000 Ci/mmol). [11C]-WIN 35,428: [11C]-WIN 35,428 was synthesized as described previously (Wong et al. 1993). The average specific activity of the final product calculated at the end of synthesis was over 74 GBq/μmol (2000 Ci/mmol). [11C]-dihydrotetrabenazine (DTBZ): [11C]-DTBZ synthesis was accomplished by a modification of the method of Jewett et al. (1997) with specific activities over 74 GBq/μmol (2000 Ci/mmol). [11C]-raclopride: [11C]-raclopride was synthesized by a modification of the method of Ehrin et al. (1985) with specific activities over 296 GBq/μmol (8000 Ci/mmol).
PET imaging acquisition and analysis
Baseline and a series of PET studies during MPTP administration including [11C]-(R)-PK11195 for TSPO, [11C]-WIN 35,428 for DAT, and [11C]-DTBZ for VMAT2 were performed. In a subset (n = 3) of the MPTP exposed baboons, PET studies were also performed to assess the early effect of MPTP administration on amphetamine-induced dopamine release (AMPH-DAR) and D2R using the D2-dopamine receptor ligand [11C]-raclopride. These studies were performed using a bolus plus continuous infusion protocol recently described by us (Guilarte et al. 2006; Zhou et al. 2006) at baseline and at the MPTP-1 and MPTP-2 time points and within 2 weeks of the DAT, VMAT2, and TSPO PET studies. On the day of the PET studies, two intravenous catheters and an arterial catheter were placed for infusion of anesthesia, injection of radiotracer, and arterial blood sampling, respectively. Baboons were initially anesthetized intramuscularly with 8–10 mg/kg alfadolone and alfaxolone acetate (Saffan) (Arnolds Veterinary Products, Shropshire, UK) and intubated. Anesthesia was maintained throughout the study by a continuous intravenous infusion drip of 6–9 mg/kg/h Saffan. To ensure reproducibility of positioning during the multiple PET studies, a custom-made facemask was fitted to the baboon’s head, which was then attached to the head holder of the PET bed during the PET study. A laser matched with markings on the facemask for ensuring the exact head position in each study. After the animal was positioned in the PET scanner, a transmission scan was obtained with twin 370-MBq (10 mCi) 68Ge pin sources for 10 min and used for attenuation correction of the subsequent emission PET scans. PET scanning started immediately after intravenous injection of 740 MBq (20 mCi) of high specific activity [11C]-labeled tracers. A GE Advance PET camera (General Electric, Milwaukee, WI, USA) with an axial resolution (full-width at half-maximum) of 5.8 mm and an in-plane resolution of 5.4 mm was used in 3D mode for image acquisition. This scanner acquires 35 simultaneous slices of 4.25-mm thickness, enclosing a total longitudinal field of view of 15 cm. Thirty serial dynamic PET images were acquired during the 90 min after injection of radioligand using the following image sequence: 4 × 15 s frames, 4 × 30 s frames, 3 × 1 min frames, 2 × 2 min frames, 5 × 4 min frames, and 12 × 5 min frames. In all cases, over 32 arterial samples/PET study (for radioactivity) were obtained during the 90 min acquisition time. Arterial samples were also obtained at 5, 10, 15, 30, 60, and 90 min after the injection of the radiotracer for analysis by means of HPLC for the correction of the metabolism of tracers. PET scans were reconstructed using ramp-filtered back-projection in a 128 × 128 matrix; with a transaxial pixel size of 2 × 2 mm. Images were corrected for attenuation and decay.
Image analysis
Volume of interests (VOIs) was defined on individual animals’ spoiled gradient MRIs for the caudate nucleus, putamen, cerebellum, and the whole gray matter using a 3D interactive segmentation approach. Briefly, the upper and lower MRI intensities were selected for each working structure using MRI intensity plots to display between-the-threshold voxels in a color scale and others in a gray scale. MRI voxels shown in color were included to the working VOI after eliminating unwanted continuations to other structures manually. Finally, VOIs were manually edited according to visual inspections in three orthogonal views. Additional VOIs (frontal cortex, temporal lobe, parietal lobe, occipital lobe, and thalamus) were obtained by transferring standard VOIs to individual subjects’ MRI spaces according to the standard-to-individual MRI spatial normalization parameters. The gray matter VOI was used to define outer (vs. CSF space) and inner (vs. white matter) boundaries of individual VOIs. VOIs were transferred from MRI to PET space according to MRI-to-PET co-registration parameters which were defined on each PET using mutual information theory (Maes et al. 1997; Kuwabara et al. 2004): Voxel values of a VOI ranged from 0.5 (a cut-off at the periphery) to 1 (at the center). Time-radioactivity curves (TACs) of VOIs were obtained by applying individual VOIs to PET frames.
[11C]-(R)-PK-11195 PET analysis
We employed the plasma input graphical analysis of the reversible ligand (Logan et al. 1990) to obtain regional distribution volumes of [11C]-(R)-PK-11195. The plot reached an asymptote at 40 min in all cases and regions where distribution volume is given by the slope. In a preliminary study, we also employed one-tissue, one-vascular compartmental model, two-tissue, one-vascular compartmental models with reversible (i.e., with the dissociation constant (k4), the k4-model) and irreversible (k4 = 0, the k3-model) binding (Kropholler et al. 2005). All tested compartmental models showed systematic deviations of normalized residues (i.e., sum across all subjects and regions of predicted TAC minus measured TAC over-predicted TAC). Thus, we selected the model-free approach. The method used by Gerhard et al. (2006) that ‘reference’ voxels (i.e., no specific binding of PK11195) extracted by cluster analysis of the whole brain voxels was not applicable to our study because we found that MPTP neurotoxicity affected cortical and subcortical regions (Supplementary material Fig. S1).
[11C]-WIN 35,428 and [11C]-DTBZ PET analysis
We employed a linear version of the simplified reference tissue method (MRTM2) (Lammertsma and Hume 1996) for WIN 35,428 and DTBZ using the cerebellum as the reference region. One common value was estimated for the brain-to-blood clearance rate constant of the reference region (k2R) per scan according to the method proposed by Wu and Carson with a modification (Wu and Carson 2002). The primary outcome variable of MRTM2 was binding potential (BP) while R1, the striatum-to-cerebellum blood-to-brain clearance rate constant (K1) ratio was also estimated regionally. In addition, [11C]-WIN 35,428 and [11C]-DTBZ PET scans were analyzed by the bolus-plus-infusion transformation (Kuwabara et al. 2004) that was shown to be more robust against statistical errors (i.e., noises) of PET measurements.
[11C]-raclopride with amphetamine challenge to assess in vivo dopamine release and D2-dopamine receptors
Extended simplified reference tissue model was used for modeling tracer kinetics in the pre- and post-amphetamine challenge phases (Zhou et al. 2006). Using this method, we are able to obtain D2R BP from the pre-amphetamine phase and AMPH-DAR from the post-amphetamine phase. Briefly, animals were studied with a bolus (B) plus continuous infusion (I) method (B/I = 75) using the D2-DAR ligand [11C]-raclopride. Animals received a continuous infusion of [11C]-raclopride over a 90-min scanning period with AMPH (2.0 mg/kg i.v.) delivered at 40 min from the onset of the [11C]-raclopride infusion. The change in [11C]-raclopride binding following AMPH was measured and used to infer the magnitude of DA release (Zhou et al. 2006).
Ex vivo methods
All methods used in post-mortem tissue are standard in our laboratory and are described below.
Quantitative receptor autoradiography in post-mortem tissue
These fresh frozen slabs at the level of the caudate/putamen from the frozen brain hemispheres were sectioned at 20 μm on a freezing cryostat (Leica, Nussloch, Germany) in the coronal plane, thaw-mounted on 50 × 75 × 1.0 mm adhesion superfrost plus slides (Brain Research Laboratories, Newton, MA, USA) and stored at −80°C until use.
TSPO autoradiography
[3H]-(R)-PK11195 were used to assess TSPO levels in the caudate/putamen and SN. Adjacent brain sections were used with both radioligands as follows: Slides were dried for 30 min at 37°C and pre-washed for 5 min at 22–24°C in 50 mmol/L Tris–HCl buffer (pH 7.4). Slides were incubated at 22–24°C for 30 min in buffer containing 1 nmol/L [3H]-(R)-PK11195 (specific activity: 3164 GBq/mmol or 85.5 Ci/mmol; custom synthesized by NEN Life Science Products, Boston, MA, USA) to determine total binding. Non-specific binding was determined in adjacent brain slices in the presence of 10 μmol/L racemic PK11195. Slides were washed twice for 3 min each in 4°C buffer and dipped twice in dH2O at 4°C. Slides were dried under a stream of cool air and apposed to Kodak Bio-Max MR films (Eastman Kodak, Rochester, NY, USA) for 5 weeks at 22–24°C.
DAT autoradiography
The selective DAT radioligand [125I]-RTI-121 was used to assess DAT levels in caudate/putamen and SN. Slides were thawed and pre-incubated for 30 min at 22–24°C in 1X phosphate-buffered saline (PBS). This was followed with a 60 min, 22–24°C incubation with 30 pmol/L [125I]-RTI-121 (Perkin-Elmer, Waltham, MA, USA) in 1X PBS with 10 mmol/L NaI. Specific binding was determined in the presence of 10 μmol/L GBR-12909 (Sigma). The slides were then washed twice in 1X PBS at 4°C for 20 min each, followed by two 5 s dips in 4°C dH2O. Sections were air-dried and apposed to Kodak Bio-Max MR film for 20 h at 22–24°C.
VMAT2 autoradiography
[3H]-α-Dihydrotetrabenazine ([3H]-DTBZ) was used as a selective radioligand for determination of VMAT2 levels in brain slices. Slides were pre-washed in 20 mmol/L HEPES, 300 mmol/L sucrose buffer (pH 8.0) for 15 min at 22–24°C. Slides were incubated at 22–24°C for 60 min in buffer with 3.0 nmol/L [3H]-DTBZ (specific activity: 740 GBq/mmol or 20 Ci/mmol) (ARC; American Radio-labeled Chemicals Inc., St Louis, MO, USA) to determine total binding. Non-specific binding was determined in adjacent brain slices with 2 μmol/L non-radioactive DTBZ (ARC). Slides were then rinsed three times at 22–24°C in 40 mmol/L HEPES, 32 mmol/L sucrose buffer (pH 8.0) for 5 min each followed by a rinse in dH2O at 4°C and dried under a stream of cool air. Slides were apposed to Kodak Bio-Max MR films for 8 weeks at 22–24°C.
In all quantitative receptor autoradiography studies, reference [3H] or [125I]-microscales standards (Amersham, Arlington Heights, IL, USA) were included with each film to ensure linearity of optical density and to allow quantitative analysis of the images. Images from autoradiograms were captured using an image analysis system (Loats Associates Inc., Westminster, MD, USA) and densitometric analysis was performed using NIH Image v1.62.
Immunohistochemistry
Brain slabs post-fixed in 4% paraformaldehyde were sectioned (50 μm thickness) using a freezing microtome (Leica). Free floating sections were pre-treated with 3.3% H2O2 and 10% methanol in Tris-buffer saline to reduce endogenous peroxidase level and increase the permeability of tissue for 20 min following by incubation in blocking solution containing 5% normal serum, 0.2% Triton X-100 for 1 h at 22–24°C. Sections were incubated with antibodies against Glial fibrillary acidic protein (1 : 5000, rabbit, Dako, Carprinteria, CA, USA), LN3 antibody (1 : 500, mouse, Novocastra, Newcastle, UK), tyrosine hydroxylase (TH) (1 : 2000, rabbit, Chemicon, Temecula, CA, USA), nitrotyrosine (NT; 1 : 500, rabbit, Chemicon), and α-synuclein (1 : 5000, Chemicon; with or without pre-treatment with 20% formic acid) at 4°C for 48 h. After rinsing in TBS, sections were incubated with biotinylated secondary antibodies (1 : 250, Vector, Burlingame, CA, USA) for 60 min and the avidin–biotin peroxidase complex solution (1 : 1600, ABC Elite, Vector) for 60 min at 22–24°C. The reaction product was visualized with 3, 3′-diam-inobenzidine as the chromogen and 0.03% H2O2. Immunolabeled brain sections were mounted on 50 × 75 × 1.0 mm adhesion superfrost plus slides (Brain Research Laboratories) and covers-lipped. Immunolabeled brain sections for NT were counter-stained with 0.5% Cresyl Violet (Chroma-Gesellschaft, Köngen, Germany). TH images in the striatum were acquired using the Inquiry system (Loats Associate) and optical densities measured using NIH Image v1.62. Brain sections were analyzed using Olympus BX51 microscope with an attached Olympus DP70 video camera and motorized stage (Prior Scientific Instruments Ltd, Cambridge, England) and the CAST-GRID software (Olympus, Denmark). Counting of TH-positive cells in SNpc was performed independently by two investigators.
Immunofluorescense
For combined labeling, sections were incubated in a cocktail of primary antibodies followed by incubation in corresponding fluorophore-conjugated secondary antibodies (overnight, at 4°C). The following combinations of markers were used for co-localization analysis: TH (1 : 2000, mouse, Chemicon) and α-synuclein (1 : 2500, rabbit, Chemicon); VMAT2 (1 : 2000, rabbit, Phoenix Pharm Inc., St Joseph, MO, USA) and α-synuclein (1 : 2500, mouse, Transduction Labs, Lexington, KY, USA); VMAT2 (1 : 2000, rabbit, Phoenix Pharm Inc.) and TH (1 : 2000, mouse, Chemicon); NT (1 : 500, rabbit, Chemicon) and TH (1 : 2000, mouse, Chemicon). Sections were mounted on slides and coverslipped using mounting media containing 4′-diamidino-2-phenylindole (Vector). Sections were examined using a Zeiss 510 Meta (Carl Zeiss, Jena, Germany) confocal microscope.
HPLC-electrochemical analysis of dopamine, homovallinic acid and dihydroxyphenylacetic acid levels in brain tissue
Caudate and putamen samples were assayed for DA and its metabolites homovallinic acid and dihydroxyphenylacetic acid using a HPLC (Shimadzu North America, Columbia, MD, USA) with electrochemical detection system (ESA Coulochem III ECD, ESA Inc., Chelmsford, MA, USA). Each sample was assayed in duplicate according to a previously described protocol (Guilarte et al. 2006) with the modification that a commercially available mobile phase was used (MDTM-70-1332; ESA Labs.). Briefly, tissue was sonicated in 10 volumes of 0.1 mol/L perchloric acid and the homogenate was centrifuged at 7400 g for 15 min and the resulting supernatant was filtered through Phenex 4 mm non-sterile 0.2 μm nylon syringe filters. 20 μL of the filtered supernatant (1 : 1 dilution for controls) was injected into a 5 μm C18 (4.6 × 150 mm) reverse-phase column with a flow rate of 0.6 mL/min. Monoamine detection occurred with a guard potential of +0.35 V, channel one potential of the analytical cell set to −0.15 V and channel two potential set to +0.22 V, each channel with a sensitivity of 1 μA. Integration of the peaks was performed using LC Solution software (Shimadzu). The concentration of individual samples was calculated using a line of best fit (R2 > 0.99) from the injected standards (0.1–8 μg/mL).
Neurological assessment
A non-human primate behavioral assessment battery was used to evaluate animals at baseline and in follow-up studies during MPTP treatment, typically prior to PET studies. Behavioral assessment was performed with the animals in their cages in the alert awake state. All sessions were rated live by a neurologist (JRB) and videotaped for blind rating. The non-human primate behavioral assessment battery includes items of the Unified Parkinson’s Disease Rating Scale suitable for non-human primates. These included salivation, swallowing, falling, facial expression, tremor at rest, action or postural tremor of hands, posture and body bradykinesia and hypokinesia.
Statistical analysis
Statistical analysis of PET studies was performed using regression with clustering on animal to account for repeated measures on the same animal over time. Differences between baseline and post-MPTP behavioral rating data were compared using a non-parametric Wilcoxon signed rank test. A linear regression model was used for the comparison between PET results (the last time point) and receptor autoradiography. Concentrations of DA and metabolites in caudate and putamen were analyzed using Student’s t-test. The counting of TH positive cells in the SNc were also analyzed using Student’s t-test for MPTP treatment effect. Post-mortem receptor autoradiography studies were analyzed using a regression model with random intercept from treatment effect (MPTP vs. control) and regions with sub-regions with consideration of individual animals and interaction. Statistical significance was set at p < 0.05. All values are expressed as mean ± SEM.
Results
MPTP dosing paradigm and general characteristics of animals
Cumulative MPTP doses and time (in days) from the first MPTP dose to the different imaging time points were as follows (n = 5 animals): From the start of MPTP administration to the first imaging set (MPTP-1) = 1.2 ± 0.0 mg/kg and 56.8 ± 2.8 days. From the start of MPTP administration to the second imaging set (MPTP-2) = 5.4 ± 0.8 mg/kg and 231.6 ± 18.7 days. From the start of MPTP administration to the third imaging set (MPTP-3) = 7.1 ± 1.0 mg/kg and 297.2 ± 19.4 days. No significant differences in body weight or brain weight between naïve control (n = 3) and MPTP-treated animals (n = 5) were measured at the end of the study. The body weight (kg) of animals was: naïve control = 28.0 ± 1.6 and MPTP-treated = 26.6 ± 1.1. The brain weight (g) was: naïve control = 183.7 ± 8.0 and MPTP-treated = 169.2 ± 7.1.
In vivo response of dopaminergic synapses and glial cell markers to MPTP-induced neurotoxicity in the non-human primate striatum
PET imaging was used to evaluate DAT, VMAT2, AMPH-DAR, D2R, and TSPO in the baboon striatum at baseline and at the three different time points during MPTP administration. At the earliest time point examined after initiation of MPTP administration (MPTP-1; 2 months), there was a significant decrease in VMAT2 BP in the caudate (46%) and putamen (35%) relative to baseline (Fig. 1). The decrease in VMAT2 was associated with a significant increase (40%) in TSPO levels, indicative of glial cell activation (Fig. 1), and near significant reductions in AMPH-DAR (29%, p = 0.056) (Fig. 2). No significant effect of MPTP treatment was measured on D2R or DAT BP at this time (Figs 1 and 2). Continued administration of the same low-dose (0.2 mg/kg) MPTP regimen for an additional 4–5 months (MPTP-2) resulted in significant reductions in DAT levels in the caudate (22%) and putamen (17%) relative to baseline, while VMAT2 levels remained significantly decreased from baseline, but at the same level relative to MPTP-1 (Fig. 1). A significant reduction in AMPH-DAR (52%; p < 0.05) at MPTP2 with no change in D2R levels was also observed (Fig. 2). TSPO levels in the striatum remained elevated but the differences did not reach statistical significance relative to baseline (Fig. 1).
Fig. 1.
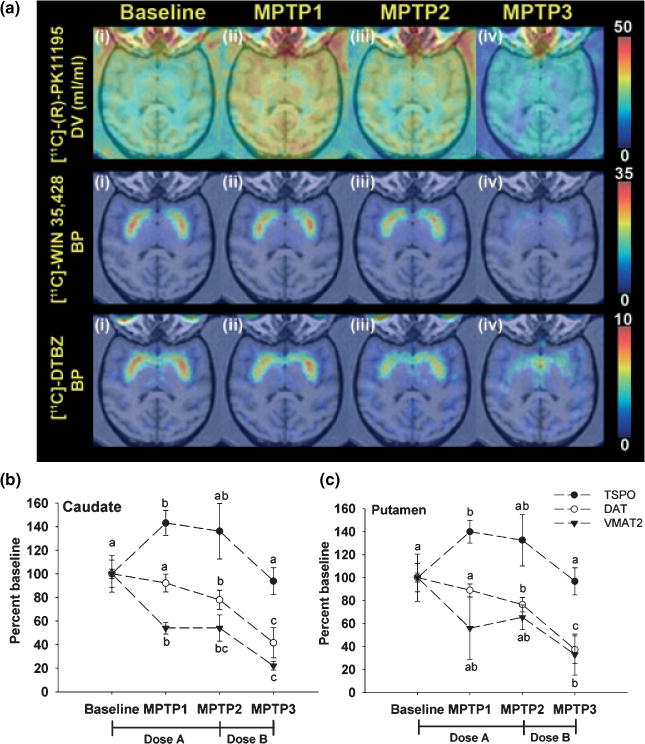
Effect of chronic MPTP treatment on striatal DAT, VMAT2, and TSPO measured by PET (a) Representative trans-axial parametric brain images of [11C]-(R)PK11195 PET for TSPO (upper row), [11C]-WIN 35,428 for DAT (middle row) and [11C]-DTBZ for VMAT2 (lower row) at (i) baseline, (ii) MPTP-1, (iii) MPTP-2, and (iv) MPTP-3. The pseudo-color represents the level of binding based on the scale on the right side of each series of PET scans. The PET images are co-registered with SPGR MRI imaging at the level of the putamen. PET imaging results for TSPO, DAT, and VMAT2 in the caudate (b) and putamen (c). Values were transformed to percentage of baseline with error bars representing standard error of the mean (SEM) (n = 5). For a particular marker, different letters represent statistical significance at p < 0.05. The MPTP dosing paradigms as follows, Dose a: 0.1 mg/kg per week was injected for the first 2 weeks, then 0.2 mg/kg per week for 5 weeks (MPTP-1) and continuing for another 5–6 months (MPTP-2); Dose b: 0.4–0.6 mg/kg per week until the appearance of clinical symptoms (MPTP-3). Binding potential values at baseline are VMAT2: 0.82 ± 0.13 (caudate) and 0.91 ± 0.10 (putamen); baseline DAT: 2.05 ± 0.12 (caudate) and 2.11 ± 0.08 (putamen). The distribution volume (DV) of baseline TSPO is 1.77 ± 0.21 ml/per mL tissue (caudate) and 1.91 ± 0.24 mL/per mL tissue (putamen) (value = mean ± SEM, n = 5).
Fig. 2.
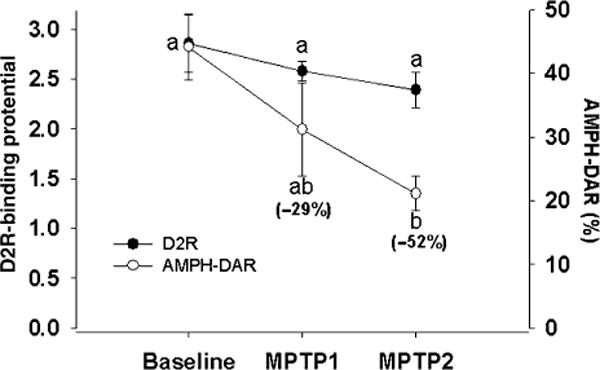
PET imaging of amphetamine-induced dopamine release (AMPH-DAR) and D2 dopamine receptor (D2R) binding potential in the striatum at baseline, MPTP-1 and MPTP-2. Values are mean ± SEM (n = 3). Different letters represent statistical significance at p < 0.05.
After the PET studies at MPTP-2 were performed, the weekly MPTP dose was increased (0.4–0.6 mg/kg) in order to induce a parkinsonian state. Increased MPTP administration resulted in a rapid and marked loss of DAT in the caudate (58%) and putamen (61%) at MPTP-3 relative to baseline with further reductions in VMAT2 levels in the caudate (78%) and putamen (69%) (Fig. 1). TSPO levels at the MPTP-3 time point were not elevated either in the caudate or putamen and did not differ from baseline (Fig. 1).
Effect of chronic low-dose MPTP administration on in vivo TSPO expression in extra-striatal brain regions
We assessed the TSPO response to MPTP-induced neurodegeneration in other brain regions besides the striatum. The PET data indicates a similar pattern of TSPO changes as in the striatum in cortical and subcortical brain regions including the temporal, parietal, and occipital cortices and in the thalamus and pons (Supplementary material Fig. S1). These findings suggest that glial cell activation is not restricted to the striatum but is also present in other brain areas in this non-human primate model of PD.
Behavioral assessment before and during chronic MPTP administration
Behavioral assessment was performed at approximately the same time points as the PET studies in order to directly correlate in vivo levels of DAergic and glial cell markers with neurological disease. The results indicate that there were no significant alterations in behavior at the MPTP-1 and MPTP-2 time points relative to baseline. However, a significant increase in salivation, tearing, resting tremor and rating scales for bradykinesia and hypokinesia emerged after the MPTP dose was increased following MPTP-2. The average score for bradykinesia and hypokinesia was 2.8 ± 0.6 (mean ± SEM) (n = 5, p < 0.05) in a 0 (normal) to 4 (severe) scale.
Confirmation of in vivo PET findings in post-mortem brain tissue using quantitative receptor autoradiography
We performed DAT, VMAT2, and TSPO autoradiography in brain specimens from MPTP-treated animals and naïve controls. We observed a significant decrease in DAT and VMAT2 levels in all subregions of the caudate and putamen but not in the nucleus accumbens (Fig. 3). TSPO levels in the caudate and putamen were slightly increased but were not significantly different from controls except in the lateral and medial aspects of the caudate (Fig. 3). We also found that TSPO levels were not significantly different in the substantia nigra (SN) of MPTP-treated animals relative to controls (data not shown).
Fig. 3.
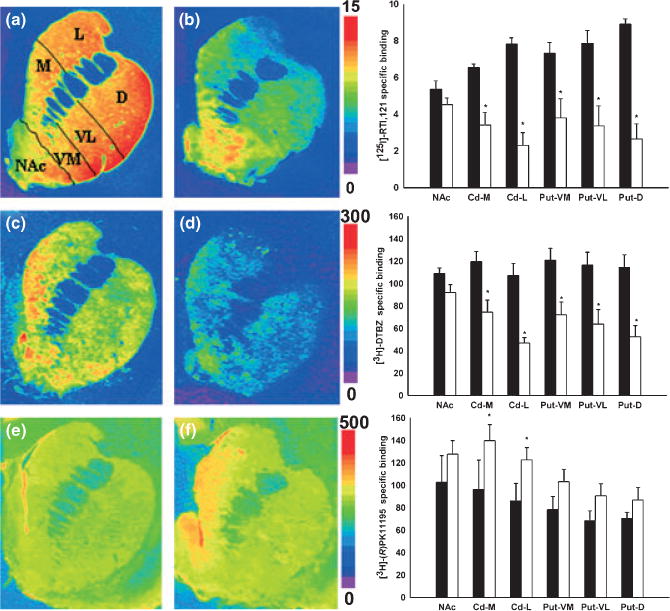
Representative brain autoradiograms (coronal plane) of [125I]-RTI 121 binding to DAT (a and b), [3H]-DTBZ binding to VMAT2 (c and d) and [3H]-(R)-PK11195 binding to TSPO (e and f) at the level of the caudate and putamen in control (a, c and e) and MPTP-treated baboons (b, d, and f). Pseudo-color bar on the right side of each image set represents the level of receptor binding (fmol/mg tissue). Specific binding of DAT, VMAT2, and TSPO measured in subregions of the caudate, putamen and nucleus accumbens in control (black bar, n = 3) and MPTP animals (white bar, n = 5) (corresponding graph next to each image). Each value represents the mean ± SEM. (*p < 0.05). NAc = nucleus accumbens, Cd-M = caudate-medial, Cd-L = caudate-lateral, Put-VM = putamen-ventromedial, put-VL = ventrolat-eral, D = putamen-dorsal.
Tyrosine hydroxylase (TH) immunohistochemistry and analysis of DA and metabolite concentrations in brain tissue
We performed immunohistochemistry for TH in DAergic terminals in the striatum and cell bodies in the SNpc. Figure 4(a) and (b) shows that the loss of TH-positive terminals in the striatum was consistent with the pattern of [125I]-RTI-121 binding to DAT and [3H]-DTBZ binding to VMAT2 observed in MPTP-treated animals (Fig. 3). Counting of SNpc TH-positive cells obtained independently by two investigators (M-KC and TV) showed a significant loss (45% reduction, p < 0.05) in MPTP-treated animals relative to naïve controls (Figs 4c and d). The loss of TH-positive terminals was associated with significant reductions (>80%) in the concentrations of DA and metabolites in the striatum of MPTP-treated animals relative to naïve controls (Fig 4e).
Fig. 4.
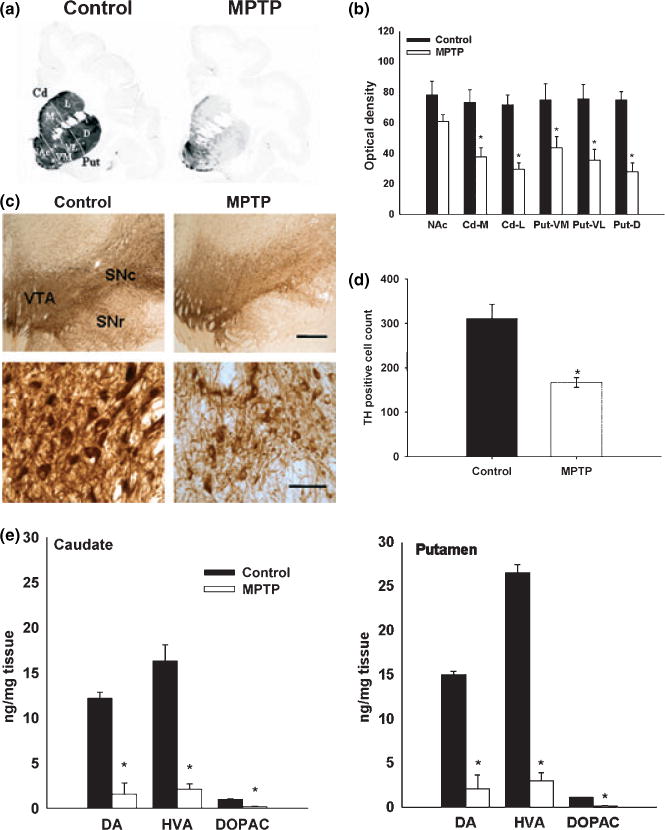
(a) Representative coronal section of TH immunohistochemistry in control and MPTP-treated baboon brains at the level of the caudate and putamen. The intensity of gray scale represents the level of TH immunostaining. (b) TH optical density in the subregions of caudate, putamen and nucleus accumbens in control (n = 3) and MPTP animals (n = 5). Each value represents the mean ± SEM. (*p < 0.05). (c) Representative coronal section of TH immunohistochemistry in control and MPTP baboon brains at the level of the substantia nigra. (Scale bar = 1000 μm in upper row and 100 μm in lower row). There is a significant loss of TH immunostaining in the substantia nigra pars compacta (SNc) and pars reticulata (SNr) in the MPTP-treated animals compared to control (upper row). Higher magnification images of TH positive neurons in the SNc of control and MPTP-treated animals (lower row). (d) Analysis of TH positive cell numbers in the SNc in control (n = 3) and MPTP-treated animals (n = 3). Each value represents the mean ± SEM (*p < 0.05). (e) Dopamine and metabolite concentrations in the caudate and putamen of control and MPTP-treated animals. Values are mean ± SEM of controls (n = 3) and MPTP animals (n = 5) (*p < 0.01).
Glial cell markers immunohistochemistry
Glial fibrillary acidic protein immunohistochemistry showed reactive astrocytosis in all regions of the caudate and putamen of MPTP-treated animals compared to controls (Fig. 5). LN3 immunohistochemistry also showed activated microglia in the putamen of MPTP-treated animals, especially in the dorsal and ventrolateral aspects, with essentially no immunoreactivity in the caudate when compared to control tissue (Fig. 5). Activation of microglia and astrocytes were observed in the SNpc of MPTP-treated animals but the degree of activation varied between animals (data not shown).
Fig. 5.
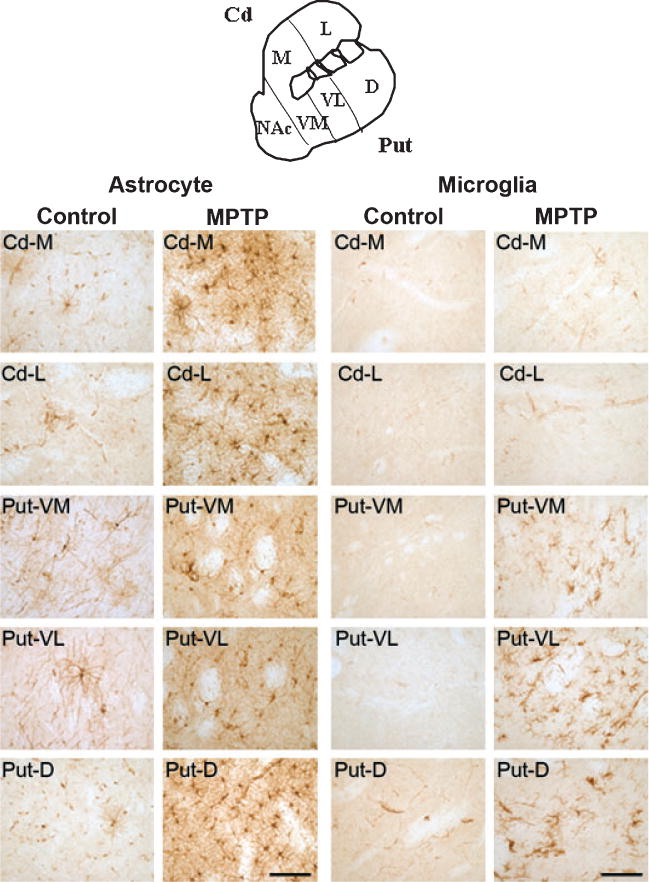
Glial fibrillary acidic protein (GFAP) immunohistochemistry for astrocytes and LN3 immunohistochemistry for microglia in baboon caudate and putamen (see sketch) of control and MPTP-treated animals. (Scale bar = 100 μm). There are morphological changes in astrocytes from a resting state (control) to the activated state (MPTP) with an increase of GFAP immunostaining per cell (hypertrophy) and the total number of GFAP positive cells in subregions of the caudate and putamen. There are also morphological changes of microglia from a resting state with small cell bodies and highly ramified processes (control) to activated microglia with ‘bushy’ appearing cells as well as round phagocytotic cells (but only in some areas of the putamen from MPTP animals). We observed no microglial staining in the caudate of MPTP-treated animals.
Effect of MPTP administration on α-synuclein and VMAT2 in TH-positive neurons of the substantia nigra
TH and α-synuclein double-label confocal microscopy showed that in MPTP-treated animals there was increased expression and aggregation of α-synuclein in the cytoplasm of TH-positive neurons that appeared to be undergoing degeneration (Fig. 6a). The pattern of α-synuclein staining was diffused aggregation like what has been described in nigral neurons from PD patients (Kuusisto et al. 2003; Mori et al. 2006) and in cells exposed to mitochondrial inhibitors (Lee et al. 2002). To confirm that α-synuclein levels were increased in DA neurons in the SNpc of MPTP-treated animals and the increase fluorescence was not due to autofluorescence, we also performed α-synuclein immunohistochemistry. We observed large neurons with increased α-synuclein staining in the SNpc and in the ventral tegmental area of MPTP-treated animals relative to those in control animals (Supplementary material Fig. S2).
Fig. 6.
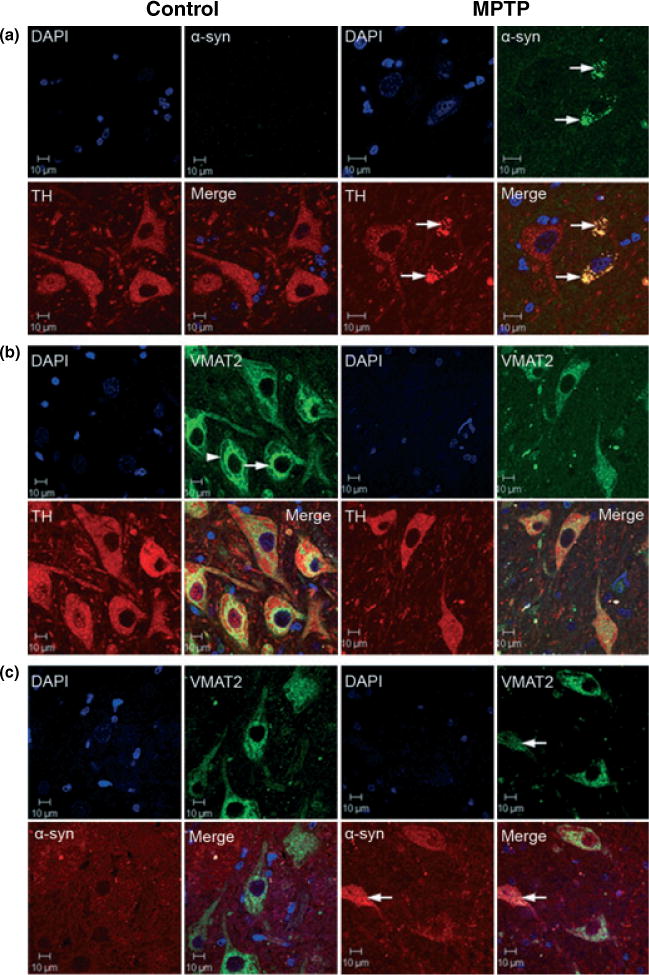
Confocal microscopy images of α-synuclein, TH and VMAT2 immunofluorescense labeling in control and MPTP-treated brains at the level of the substantia nigra pars compacta (coronal section) (Scale bar = 10 μm). (a) TH (red), α-synuclein (green), DAPI (blue) and merged triple labeling. There is a significant amount of α-synuclein punctate staining (or aggregation) co-localized with TH immunostaining in the cytosol of neurons from MPTP-treated animals that appear to be undergoing degeneration (see arrows). The insert in the α-synuclein panel from MPTP-treated animals is representative of the occasional α-synuclein-positive Lewy body-like spherical aggregates observed in MPTP-treated animals. (b) DAPI (blue), VMAT2 (green), TH (red) and merged triple labeling. In neurons from control animals, VMAT2 staining expressed a distinct subcellular pattern with a band labeling pattern in perinuclear regions (see arrow) and adjacent to the plasma membrane (see arrow head); the latter may be representative of a releasable pool of secretory vesicles. In nigral neurons from MPTP-treated animals, VMAT2 labeling expressed a more homogeneous distribution throughout the cytoplasm with disruption of the distinct labeling pattern observed in neurons from control animals. (c) DAPI (blue), VMAT2 (green), α-synuclein (red) and merged triple labeling. α-synuclein co-localized with VMAT2 as aggregates in the cytoplasm of degenerating neurons in MPTP-treated animals (see arrows).
We also examined the pattern of VMAT2 staining in TH-positive neurons in the SNpc of control and MPTP-treated animals. VMAT2 staining in control neurons had a distinct pattern with perinuclear and plasma membrane labeling (Fig. 6b). This normal pattern of cellular VMAT2 expression was dramatically changed in TH-positive neurons from MPTP-treated animals in which there was a more homogeneous distribution throughout the cytoplasm and a reduced level of staining (Fig. 6b). Importantly, VMAT2 co-labeled with α-synuclein aggregates in the cytoplasm of SNpc dopaminergic neurons from MPTP-treated animals suggestive of an interaction of these two proteins in the degenerative process (Fig. 6c).
Nitrotyrosine immunostaining in TH-positive neurons
In the SNpc of MPTP-treated animals there was NT-positive staining in large neurons that were Nissl negative suggestive of NT localization in degenerating neurons (Fig. 7b). This was in contrast to the lack of NT staining in Nissl positive large neurons in the SNpc of control animals (Fig. 7a). In MPTP animals, we also observed NT-positive cellular process and cell bodies that resemble microglia in close proximity to NT-positive and Nissl negative large neurons (Fig. 7b). This finding suggests that oxidative stress is present in neurons and possibly in microglia in the SNpc of MPTP-treated animals. To confirm that NT-positive and Nissl negative large neurons are DA neurons in the SNpc, we performed double-label immunofluorescense with confocal microscopy for NT and TH. Fig. 7c shows that NT was not present in TH-positive neurons in the SNpc from control animals but NT co-localized with TH-positive neurons from MPTP-treated animals (Fig. 7d) confirming that dopamine-containing neurons were in an active state of oxidative stress. These observations support increased levels of NT in microglia and TH-positive neurons in MTPT animals.
Fig. 7.
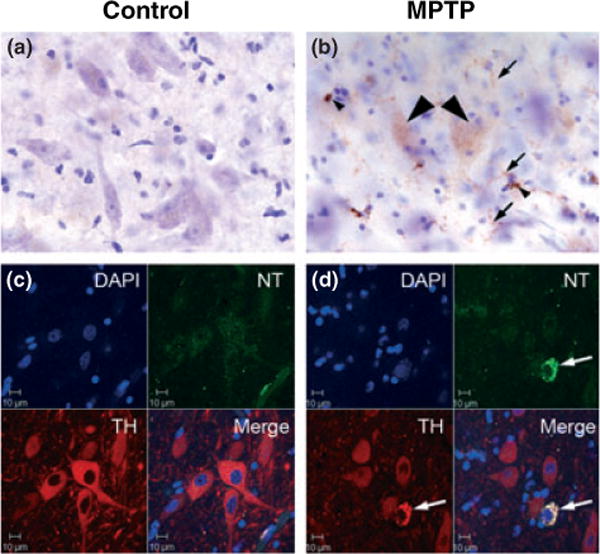
Immunostaining for nitrotyrosine (NT) in the SNpc of control and MPTP-treated animals. (a) The SNpc from controls lacked NT immunostaining (b) in MPTP-treated SNpc there was increased NT staining in large, Nissl negative cells that appear to be degenerating (arrowhead). The small arrows indicate a widespread increase in NT in cells with a morphology resembling microglia. (c) NT and TH double labeling immunofluorescence confocal microscopy in the SNpc from a control animal and (d) a MPTP-treated animal. No NT labeling was observed in TH-positive neurons from control animals but increased NT label was present in TH-positive neurons in the SNpc from MPTP-treated animals. Scale bar = 10 μm.
Discussion
We report in vivo evidence that the earliest response of the DAergic synapse in the striatum of asymptomatic MPTP-treated non-human primates is the selective loss of the vesicular protein VMAT2. We found a nearly 50% reduction in VMAT2 levels prior to changes in DAT levels in the striatum at the earliest time point measured, i.e., 2 months after initiation of low-dose MPTP administration (MPTP-1). The decrease in VMAT2 at this early time point was associated with a significant up-regulation of striatal TSPO indicative of a glial response to injury. These in vivo changes in DA synapse dynamics and glial cell activation were observed when animals were asymptomatic. These findings indicate that a reduction in VMAT2 and DA storage capacity is a key pathogenic event in the retrograde degeneration of nigrostriatal DA neurons in the MPTP non-human primate model of PD. The marked loss of VMAT2 function could set in motion a chain of events resulting in DA terminal degeneration and cell loss.
Lotharius and Brundin (2002) have noted the possibility that a reduction in DA storage sites may be a key pathogenic factor of PD since PET studies in PD patients have found a greater reduction in VMAT2 than fluoroDOPA PET suggesting that VMAT2 is reduced beyond what could be explained by a loss of nigrostriatal DA neurons. Our present study is the first to directly demonstrate in vivo, in a non-human primate model that recapitulates many of the features of idiopathic PD that, VMAT2 is the first pre-synaptic DA terminal marker that is reduced in the striatum prior to terminal degeneration.
The significance of maintaining vesicular DA storage is accentuated in a recent study published during the preparation of our manuscript. Using a VMAT2 hypomorph mouse expressing 5% of VMAT2 levels relative to wild-type, Caudle et al. (2007) found that these animals express a progressive age-dependent nigrostriatal DA system degeneration indicating that mishandling of DA from reduced VMAT2 is sufficient to cause progressive DA neuron degeneration. Similar to our present findings, the VMAT2 hypomorph mice displayed an increase in protein carbonyl and 3-NT formation, decreased striatal DA, decreased expression of DAT and TH, increased levels and aggregation of α-synuclein as well as loss of DA neurons in the SNpc (Caudle et al. 2007). Together our results and the work by Caudle and colleagues provide direct evidence that mishandling of DA storage by reduced VMAT2 expression is sufficient to cause DA-mediated oxidative stress and nigrostriatal DA system degeneration.
The function of VMAT2 in DA terminals is to transport DA from de novo synthesis and DAT re-uptake from the synapse into vesicles for storage and subsequent release (Kanner and Schuldiner 1987). This function has been postulated as being protective by preventing generation of reactive oxygen species in the cytoplasm from the oxidation of free, unprotected DA (Miller et al. 1999; Dauer and Przedborski 2003; Fleckenstein and Hanson 2003). Consistent with this notion, VMAT2 over-expression has been shown to decrease MPTP neurotoxicity (Chen et al. 2005) and VMAT2 heterozygous knockout mice have increased vulnerability to MPTP and methamphetamine-induced neurotoxicity (Takahashi et al. 1997; Gainetdinov et al. 1998; Fumagalli et al. 1999). Loss of VMAT2 function in secretory vesicles of DAergic terminals has been associated with the degeneration caused by over-expression of mutant forms of α-synuclein and parkin in familial forms of PD (Lotharius and Brundin 2002). Synaptic vesicle permeabilization by α-synuclein protofibrils and leakage of DA to the cytoplasm has been proposed as a potential mechanism for synaptic vesicle loss and neurodegeneration in familial forms of PD (Volles et al. 2001; Volles and Lansbury 2002). An association of α-synuclein and VMAT2 has been observed in Lewy bodies staining positive for VMAT2 in PD (Yamamoto et al. 2006).
We now show increased α-synuclein staining in TH-positive neurons in the SNpc of MPTP-treated animals with morphological features similar to early stages of α-synuclein aggregation in PD (Kuusisto et al. 2003), and in cells exposed to mitochondrial inhibitors (Lee et al. 2002) (Fig. 6a). VMAT2 immunolabeling in DA neuron cell bodies from MPTP-treated animals demonstrates disruption of the normal pattern of cellular VMAT2 distribution (Fig. 6b). Further, in MPTP-treated SNpc DA neurons, VMAT2 co-localized with increased levels of α-synuclein aggregates suggesting that these two proteins may associate in the degenerative process (Fig. 6c). Experimental support for an interaction of α-synuclein with VMAT2 comes from a recent study by Dean et al. (2007). These investigators report that using the yeast-two hybrid assay, α-synuclein interacts with VMAT2 to regulate VMAT2 activity. Therefore, the early and selective loss of VMAT2 in this non-human primate model of PD may be due to an association of VMAT2 with α-synuclein aggregation induced by an active state of oxidative stress.
The in vivo loss of VMAT2 prior to changes in DAT levels in the early asymptomatic phase of our study supports a change in the dynamics of DA uptake and storage. The PET results indicate an increase in the cellular DAT/VMAT2 ratio and this change may lead to increased intra-neuronal free DA concentrations overwhelming vesicular storage capacity. The accumulation of free, unprotected DA in the cytoplasm promotes auto-oxidation, generation of reactive oxygen species and initiation of degenerative events (Miller et al. 1999; Lotharius and Brundin 2002). Dysregulation of α-synuclein function by MPP+ or reactive oxygen species has been shown to decrease the synthesis and recycling of synaptic vesicles (Lotharius and Brundin 2002) and to enhance DAT trafficking to the plasma membrane (Sidhu et al. 2004), an effect that would increase the cellular DAT/VMAT2 ratio as observed in the early stage of our study. An active state of oxidative stress in DA neurons from MPTP-treated animals is supported by increased NT labeling in TH-positive neurons and in microglia (Fig. 7).
A unique aspect of our study was to assess the longitudinal in vivo response of the glial marker TSPO by PET in conjunction with DAergic markers. We found that while TSPO levels were increased shortly after MPTP administration, it lacked correlation with disease progression. This pattern of TSPO expression is reminiscent of the early appearance of a peak microglia response that precedes DA neuron degeneration in MPTP-treated mice (Liberatore et al. 1999) and may represent an early inflammatory response by microglia (Liberatore et al. 1999) that could set in motion a self-perpetuating cycle of injury (Block et al. 2007).
Studies by Ouchi et al. (2005) in subjects in the early stages of PD found no significant increase in [11C]-(R)-PK11195 binding to TSPO in the striatum. Based on the behavioral expression of parkinsonism in our animals, a comparable time point to the early stages of PD would be at the MPTP-3 imaging time point where we also found no significant increase in [11C]-(R)-PK11195 binding to TSPO in the striatum (Fig. 1). Consistent with our results, Ouchi et al. (2005) also found a lack of correlation between glial activation measured by TSPO-PET and disease progression. On the other hand, Gerhard et al. (2006) reported a small but significant increase in [11C]-(R)-PK11195 binding to TSPO in the striatum in a larger population of PD patients in the early and late stages of the disease. Taken together, these studies indicate that the glial/TSPO response in the striatum resulting from DA terminal degeneration is not very robust. Hurley et al. (2003) using a similar chronic, low-dose MPTP dosing paradigm as in our study also showed a relative lack of a microglial response in the striatum of neurologically affected non-human primates. Thus, there is a relative lack of a TSPO response in the symptomatic phase of the disease that is not readily detected by the resolution of current PET scanners. This explanation is consistent with the finding that in Huntington’s disease, a neurodegenerative condition in which there is frank loss of neuronal cell bodies intrinsic to the striatum, there is a very robust TSPO response that can be measured by PET (Pavese et al. 2006). It should be noted that in the PD study by Gerhard et al. (2006), they measured increased TSPO levels in cortical and subcortical structures besides the caudate and putamen. This finding is similar to our results in non-human primates (Supplementary material Fig. S1) and is consistent with the known degeneration that occurs in other brain nuclei in PD (Langston 2006).
In summary, our findings suggest that the early loss of VMAT2 may be a precursor to PD. Environmental factor and/or genetic mutations that reduce VMAT2 may increase susceptibility to PD. Therapeutic strategies to prevent degradation of VMAT2 or restore its function may be fruitful areas of investigation in PD research.
Supplementary Material
Acknowledgments
This work was supported by an award from the Michael J. Fox Foundation for Parkinson’s Research and NIEHS grant number ES07062 to TRG. The authors would like to thank Dr Robert F. Dannals and Dr Hayden T. Ravert for the synthesis of PET tracers. Dr Michael Griswold, Dr Brian Caffo and Ms. Shuchih Su provided statistical consultation. We especially thank Paige A. Finley for her assistance in the care and handling of animals during the PET studies. We also wish to give our thanks to Ted M. Dawson MD, PhD for reading the manuscript and providing constructive comments. This work was performed in partial fulfillment of doctoral degree requirements for M-KC.
Abbreviations used
- [11C]-DTBZ
[11C]-dihydrotetrabenazine
- AMPH-DAR
amphetamine-induced dopamine release
- BP
binding potential
- DAT
dopamine transporters
- PBS
phosphate-buffered saline
- PD
Parkinson’s disease
- PET
positron emission tomography
- TH
tyrosine hydroxylase
- VMAT2
vesicular monoamine transporter-type 2
- VOIs
volume of interests
Footnotes
Supplementary material
The following supplementary material is available for this article online:
This material is available as part of the online article from http://www.blackwell-synergy.com.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Bohnen NI, Albin RL, Koeppe RA, Wernette KA, Kilbourn MR, Minoshima S, Frey KA. Positron emission tomography of monoaminergic vesicular binding in aging and parkinson disease. J Cereb Blood Flow Metab. 2006;26:1198–1212. doi: 10.1038/sj.jcbfm.9600276. [DOI] [PubMed] [Google Scholar]
- Brooks DJ. Imaging end points for monitoring neuroprotection in parkinson’s disease. Ann Neurol. 2003;53(Suppl 3):S110–8. doi: 10.1002/ana.10480. discussion S118–119. [DOI] [PubMed] [Google Scholar]
- Caudle WM, Richardson JR, Shepherd KR, et al. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci. 2007;27:8138–8148. doi: 10.1523/JNEUROSCI.0319-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MK, Guilarte TR. Imaging the peripheral benzodiazepine receptor response in central nervous system demyelination and remyelination. Toxicol Sci. 2006;91:532–539. doi: 10.1093/toxsci/kfj172. [DOI] [PubMed] [Google Scholar]
- Chen CX, Huang SY, Zhang L, Liu YJ. Synaptophysin enhances the neuroprotection of VMAT2 in MPP+-induced toxicity in MN9D cells. Neurobiol Dis. 2005;19:419–426. doi: 10.1016/j.nbd.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson’s disease: Mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dean ED, Torres GE, Miller GW. Alpha-synuclein interacts with VMAT2 to regulate VMAT2 activity. Toxicol Sci. 2007;96(1S):45. [Google Scholar]
- Doudet DJ, Rosa-Neto P, Munk OL, Ruth TJ, Jivan S, Cumming P. Effect of age on markers for monoaminergic neurons of normal and MPTP-lesioned rhesus monkeys: A multi-tracer PET study. Neuroimage. 2006;30:26–35. doi: 10.1016/j.neuroimage.2005.09.044. [DOI] [PubMed] [Google Scholar]
- Ehrin E, Farde L, de Paulis T, et al. Preparation of 11C-labelled raclopride, a new potent dopamine receptor antagonist: Preliminary PET studies of cerebral dopamine receptors in the monkey. Int J Appl Radiat Isot. 1985;36:269–273. doi: 10.1016/0020-708x(85)90083-3. [DOI] [PubMed] [Google Scholar]
- Fleckenstein AE, Hanson GR. Impact of psychostimulants on vesicular monoamine transporter function. Eur J Pharmacol. 2003;479:283–289. doi: 10.1016/j.ejphar.2003.08.077. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Gainetdinov RR, Wang YM, Valenzano KJ, Miller GW, Caron MG. Increased methamphetamine neurotoxicity in heterozygous vesicular monoamine transporter 2 knock-out mice. J Neurosci. 1999;19:2424–2431. doi: 10.1523/JNEUROSCI.19-07-02424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Fumagalli F, Wang YM, Jones SR, Levey AI, Miller GW, Caron MG. Increased MPTP neurotoxicity in vesicular monoamine transporter 2 knock-out mice. J Neurochem. 1998;70:1973–1978. doi: 10.1046/j.1471-4159.1998.70051973.x. [DOI] [PubMed] [Google Scholar]
- Gerhard A, Pavese N, Hotton G, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic parkinson’s disease. Neurobiol Dis. 2006;21:404–412. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Guilarte TR, Chen MK, McGlothan JL, et al. Nigrostriatal dopamine system dysfunction and subtle motor deficits in manganese-exposed non-human primates. Exp Neurol. 2006;202:381–390. doi: 10.1016/j.expneurol.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Hurley SD, O’Banion MK, Song DD, Arana FS, Olschowka JA, Haber SN. Microglial response is poorly correlated with neurodegeneration following chronic, low-dose MPTP administration in monkeys. Exp Neurol. 2003;184:659–668. doi: 10.1016/S0014-4886(03)00273-5. [DOI] [PubMed] [Google Scholar]
- Jewett DM, Kilbourn MR, Lee LC. A simple synthesis of [11C]-dihydrotetrabenazine (DTBZ) Nucl Med Biol. 1997;24:197–199. doi: 10.1016/s0969-8051(96)00213-2. [DOI] [PubMed] [Google Scholar]
- Kanner BI, Schuldiner S. Mechanism of transport and storage of neurotransmitters. CRC Crit Rev Biochem. 1987;22:1–38. doi: 10.3109/10409238709082546. [DOI] [PubMed] [Google Scholar]
- Kropholler MA, Boellaard R, Schuitemaker A, van Berckel BN, Luurtsema G, Windhorst AD, Lammertsma AA. Development of a tracer kinetic plasma input model for (R)-[11C]-PK11195 brain studies. J Cereb Blood Flow Metab. 2005;25:842–851. doi: 10.1038/sj.jcbfm.9600092. [DOI] [PubMed] [Google Scholar]
- Kuusisto E, Parkkinen L, Alafuzoff I. Morphogenesis of lewy bodies: Dissimilar incorporation of alpha-synuclein, ubiquitin, and p62. J Neuropathol Exp Neurol. 2003;62:1241–1253. doi: 10.1093/jnen/62.12.1241. [DOI] [PubMed] [Google Scholar]
- Kuwabara H, Lee JS, Wong DF. Measurements of dopamine D2 receptor density with bolus-plus-infusion transformation of bolus-injection [11C]raclopride PET. NeuroImage. 2004;22:117–118. [Google Scholar]
- Lammerstma AA, Hume SP. Simplified reference tissue model for PET receptor studies. Neuroimage. 1996;4:153–158. doi: 10.1006/nimg.1996.0066. [DOI] [PubMed] [Google Scholar]
- Langston JW. The parkinson’s complex: Parkinsonism is just the tip of the iceberg. Ann Neurol. 2006;59:591–596. doi: 10.1002/ana.20834. [DOI] [PubMed] [Google Scholar]
- Lee CS, Samii A, Sossi V, et al. In vivo positron emission tomographic evidence for compensatory changes in presynaptic dopaminergic nerve terminals in parkinson’s disease. Ann Neurol. 2000;47:493–503. [PubMed] [Google Scholar]
- Lee HJ, Shin SY, Choi C, Lee YH, Lee SJ. Formation and removal of alpha-synuclein aggregates in cells exposed to mitochondrial inhibitors. J Biol Chem. 2002;277:5411–5417. doi: 10.1074/jbc.M105326200. [DOI] [PubMed] [Google Scholar]
- Liberatore GT, Jackson-Lewis V, Vukosavic S, et al. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- Logan J, Fowler JS, Volkow ND, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Graphical analysis of reversible radioligand binding from time-activity measurements applied to [N-11C-methyl]-(−)-cocaine PET studies in human subjects. J Cereb Blood Flow Metab. 1990;10:740–747. doi: 10.1038/jcbfm.1990.127. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Brundin P. Pathogenesis of parkinson’s disease: Dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci. 2002;3:932–942. doi: 10.1038/nrn983. [DOI] [PubMed] [Google Scholar]
- Maes F, Collignon A, Vandermeulen D, Marchal G, Suetens P. Multimodality image registration by maximization of mutual information. IEEE Trans Med Imaging. 1997;16:187–198. doi: 10.1109/42.563664. [DOI] [PubMed] [Google Scholar]
- Miller GW, Gainetdinov RR, Levey AI, Caron MG. Dopamine transporters and neuronal injury. Trends Pharmacol Sci. 1999;20:424–429. doi: 10.1016/s0165-6147(99)01379-6. [DOI] [PubMed] [Google Scholar]
- Mori F, Nishie M, Kakita A, Yoshimoto M, Takahashi H, Wakabayashi K. Relationship among alpha-synuclein accumulation, dopamine synthesis, and neurodegeneration in parkinson disease substantia nigra. J Neuropathol Exp Neurol. 2006;65:808–815. doi: 10.1097/01.jnen.0000230520.47768.1a. [DOI] [PubMed] [Google Scholar]
- Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T. Microglial activation and dopamine terminal loss in early parkinson’s disease. Ann Neurol. 2005;57:168–175. doi: 10.1002/ana.20338. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V, Baraldi M, Guilarte TR, et al. Translocator protein (18kDa): New nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006;27:402–409. doi: 10.1016/j.tips.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Pavese N, Gerhard A, Tai YF, et al. Microglial activation correlates with severity in Huntington disease: A clinical and PET study. Neurology. 2006;66:1638–1643. doi: 10.1212/01.wnl.0000222734.56412.17. [DOI] [PubMed] [Google Scholar]
- Pirker W, Djamshidian S, Asenbaum S, Gerschlager W, Tribl G, Hoffmann M, Brucke T. Progression of dopaminergic degeneration in parkinson’s disease and atypical parkinsonism: A longitudinal beta-CIT SPECT study. Mov Disord. 2002;17:45–53. doi: 10.1002/mds.1265. [DOI] [PubMed] [Google Scholar]
- Ravina B, Eidelberg D, Ahlskog JE, et al. The role of radiotracer imaging in Parkinson’s disease. Neurology. 2005;64:208–215. doi: 10.1212/01.WNL.0000149403.14458.7F. [DOI] [PubMed] [Google Scholar]
- Sidhu A, Wersinger C, Vernier P. Alpha-synuclein regulation of the dopaminergic transporter: A possible role in the pathogenesis of parkinson’s disease. FEBS Lett. 2004;565:1–5. doi: 10.1016/j.febslet.2004.03.063. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Miner LL, Sora I, Ujike H, Revay RS, Kostic V, Jackson-Lewis V, Przedborski S, Uhl GR. VMAT2 knockout mice: Heterozygotes display reduced amphetamine-conditioned reward, enhanced amphetamine locomotion, and enhanced MPTP toxicity. Proc Natl Acad Sci USA. 1997;94:9938–9943. doi: 10.1073/pnas.94.18.9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volles MJ, Lansbury PT., Jr Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry. 2002;41:4595–602. doi: 10.1021/bi0121353. [DOI] [PubMed] [Google Scholar]
- Volles MJ, Lee SJ, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, Lansbury PT., Jr Vesicle permeabilization by protofibrillar alpha-synuclein: Implications for the pathogenesis and treatment of parkinson’s disease. Biochemistry. 2001;40:7812–7819. doi: 10.1021/bi0102398. [DOI] [PubMed] [Google Scholar]
- Wong DF, Yung B, Dannals RF, et al. In vivo imaging of baboon and human dopamine transporters by positron emission tomography using [11C]-WIN 35, 428. Synapse. 1993;15:130–142. doi: 10.1002/syn.890150205. [DOI] [PubMed] [Google Scholar]
- Wu Y, Carson RE. Noise reduction in the simplified reference tissue model for neuroreceptor functional imaging. J Cereb Blood Flow Metab. 2002;22:1440–1452. doi: 10.1097/01.WCB.0000033967.83623.34. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Fukae J, Mori H, Mizuno Y, Hattori N. Positive immunoreactivity for vesicular monoamine transporter 2 in lewy bodies and lewy neurites in substantia nigra. Neurosci Lett. 2006;396:187–191. doi: 10.1016/j.neulet.2005.11.068. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Chen MK, Endres CJ, Ye W, Brasic JR, Alexander M, Crabb AH, Guilarte TR, Wong DF. An extended simplified reference tissue model for the quantification of dynamic PET with amphetamine challenge. Neuroimage. 2006;33:550–563. doi: 10.1016/j.neuroimage.2006.06.038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.