

Abstract
The tumor microenvironment is a complex ecology of cells that evolves with and provides support to tumor cells during the transition to malignancy. Among the innate and adaptive immune cells recruited to the tumor site, macrophages are particularly abundant and are present at all stages of tumor progression. Clinical studies and experimental mouse models indicate these macrophages generally play a pro-tumoral role. In the primary tumor, macrophage can stimulate angiogenesis and enhance tumor cell invasion, motility and intravasation. During metastasis, macrophages prime the pre-metastatic site and promote tumor cell extravasation, survival and persistent growth. Macrophages are also immunosuppressive preventing tumor cell attack by natural killer and T cells during tumor progression and after recovery from chemo- or immuno-therapy. Therapeutic success in targeting these pro-tumoral roles in pre-clinical models and in early clinical trials suggests that macrophages are attractive targets as part of combination therapy in cancer treatment.
Introduction
Tumors engage the immune system from their inception. Initially this mainly involves cells of the innate system such as macrophages and mast cells with their prevalence dependent on tumor type. However, even early on there is also engagement of cells of the acquired system particularly T cells (Gajewski et al., 2013). Nevertheless, despite this adaptive response and data that suggests better prognosis with CD8+ T cell infiltration in some cancers there is little evidence of immune rejection in established tumors arguing that the local tumor microenvironment is immunosuppressive (Gajewski et al., 2013). Macrophages are among the most abundant normal cells in the tumor microenvironment. Substantial evidence indicates that macrophages rather than being tumoricidal as suggested after their activation in vitro (Fidler, 1988) adopt a pro-tumoral phenotype in vivo both in the primary and metastatic sites (Biswas et al., 2013). Indeed in lung cancer macrophages are polarised to a pro-tumoral phenotype at the time of tumor initiation (Redente et al., 2010). These activities include suppression of T cell responses (Coussens et al., 2013; Qian and Pollard, 2010). In addition, macrophages promote many important features of tumor progression including angiogenesis, tumor cell invasion, motility and intravasation as well as at the metastatic site, stimulation of tumor cell extravasation and persistent growth (Qian and Pollard, 2010). Each of these activities is delivered by an identifiable sub-population of macrophages (Qian and Pollard, 2010). These data together with experimental studies showing inhibition of tumor progression and metastasis by ablation of macrophages, argue that immune cell engagement by tumors is essential for their acquisition of a malignant phenotype. Consequently this cell type might represent an important therapeutic target for cancer treatment. Here we discuss the function of diverse macrophage sub-populations, their dynamic interplay with tumor cells that confer these pro-tumoral activities, and give particular emphasis to the immunoregulatory role of these cells. We suggest that ablation of or re-differentiation of macrophages within the tumor microenvironment will become an important prong of combination therapies designed to cure cancer.
Macrophages in the Primary Tumor
Cancer Initiation
Tumors acquire mutations in oncogenes or tumor suppressor genes that permit them to progress to malignancy. While most cancer research has focused upon these changes and most therapeutics are directed against these tumor cells it is now apparent that the non-malignant cells in the microenvironment evolve along with the tumor and provide essential support for their malignant phenotype (Joyce and Pollard, 2009). In fact both the systemic and local environment play a tumor-initiating role through the generation of a persistent inflammatory responses to a variety of stimuli (Balkwill and Mantovani, 2012). For example, obesity is associated with increased risk of many but not all cancers (Grivennikov et al., 2010) and is characterised by an enhanced systemic inflammatory response and locally, for example in the breast, to an increased number of inflammatory crown-like structures consisting of macrophage and adipocytes whose number strongly correlates with breast cancer risk (Howe et al., 2013). Similarly persistent inflammation referred to as “smouldering inflammation” caused by chronic infection with viruses such as Hepatitis B virus in liver, bacteria like Helicobacter pylori in the stomach, or due to continuous exposure to irritants such as asbestos in the lung is casually associated with cancer initiation (Balkwill et al., 2005; Brown et al., 2008). Furthermore, inflammatory conditions such as Crohn's disease dramatically increase the risk of colorectal cancer (Balkwill et al., 2005; Balkwill and Mantovani, 2012; Coussens and Werb, 2002; Grivennikov et al., 2010). Inflammation always has a substantial macrophage involvement through their production of molecules such as interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α)and interferon-γ (IFN-γ)(Brown et al., 2008; Grivennikov et al., 2010). To support this correlative data between macrophage-mediated inflammation and cancer induction, genetic ablation of the anti-inflammatory transcription factor Stat3 in macrophages results in a chronic inflammatory response in the colon that is sufficient to induce invasive adenocarcinoma (Deng et al., 2010). In addition, loss of the anti-inflammatory cytokine IL10 that acts through STAT3 enhances carcinogen-induced tumorigenesis in the intestine (Jobin, 2013). Mechanistically this inflammation can cause tumor initiation by creating a mutagenic microenvironment either directly through free radical generation or indirectly via alterations in the microbiome and barrier functions that allow access of genotoxic bacteria to the epithelial cells (Dedon and Tannenbaum, 2004; Jobin, 2013). Furthermore Langerhans cells a type of macrophage/DC can promote skin carcinogenesis by metabolic conversion of carcinogens to their activate mutagenic state (Modi et al., 2012). Macrophages also produce growth factors/cytokines that stimulate growth of epithelial cells that have spontaneously acquired cancer-associated mutations (Grivennikov et al., 2010). These mutations in turn may cause recruitment of inflammatory cells resulting in a vicious cycle that drives cancer progression (Balkwill and Mantovani, 2012; Qian and Pollard, 2010). Significant data therefore exists showing a causal role for macrophages in cancer initiation because of their central status as mediators of inflammation. However, it is unclear whether macrophages in some inflammatory situations can kill aberrant cells before they become tumorigenic and thus be anti-tumoral.
Macrophages involved in these cancer-initiating inflammatory responses are immune activated (Balkwill and Mantovani, 2012). However, once tumors are established the macrophages are educated to become pro-tumoral (Pollard, 2004; Qian and Pollard, 2010). During this transition from benign growth to an invasive cancer the microenvironment appears to be dominated by cytokines and growth factors that cause a bias away from this T helper 1(Th1)-like inflammatory response to create a Th2 type immune environment. This bias results in the polarisation of macrophages by a number of factors including IL4 synthesized by CD4+ T cells and/or tumor cells (Coussens et al., 2013; Gocheva et al., 2010) and growth factors produced by tumor cells such as CSF1 (Lin et al., 2002) and GM-CSF (Su et al., 2014). This Th2 environment is characterized by transforming growth factor-β1 (TGF-β1) and Arginase 1 as well as increased numbers of CD4+ T cells (DeNardo et al., 2009). It could be argued therefore that for tumors to prosper they need to acquire mutations and/or epigenetic changes that result in the synthesis of such factors that re-polarise resident macrophages or more likely recruit new monocytes (see below) so that they become differentiated into tumor-promoting cells and act as their handymen.
Origins of Tumor Associated Macrophages
It has recently been demonstrated that the historic description of adult resident tissue macrophages as being solely derived from bone marrow (BM) is not correct. In fact most tissue macrophages although with some exceptions such as the intestine, arise from yolk sac progenitors. In contrast macrophages involved in pathogen responses appear to come from circulating BM monocytes (Wynn et al., 2013). These different embryonic origins challenge the assumption that tumor-associated macrophages (TAMs) in the primary tumor originate from the BM. Evidence for different origins and responses has recently been shown in a mouse model of glioma with the presence of resident yolk-sac derived microglia and recruited BM-derived TAMs in the tumor microenvironment behaving differently to anti-macrophage therapies based on inhibition of the lineage regulating growth factor Colony Stimulating Factor-1 (CSF1) signalling. In this case the recruited TAMs appear to survive in response to another macrophage lineage regulating growth factor, granulocyte-macrophage colony stimulating factor (GM-CSF) (Pyonteck et al., 2013). Nevertheless, several recent studies suggest that most TAM sub-populations arise form the Ly6C+ population of circulating mouse monocytes in grafted tumors (Movahedi et al., 2010), primary mouse mammary tumors (Franklin et al., 2014) and in lung metastases (Qian et al., 2011). There has also been discussion about the origins of these monocytes with the suggestion that instead of coming directly from the BM they arise from extra-medullary haematopoiesis, particularly in the spleen. It is claimed that this gives a reservoir of monocytes that allows rapid mobilization to the tumor (Cortez-Retamozo et al., 2012). However, recent elegant experiments using photo-convertible fluorescent lineage tracing of spleen and BM monocytes suggest that the splenic contribution is minor and that BM is the primary source of monocytes that generate TAMs at least in the Lewis Lung carcinoma syngeneic transplant model (Shand et al., 2014).
CSF1 is the major lineage regulator of most populations of macrophages whether they derive from the yolk sac or BM but in addition it is a chemotactic factor for macrophages (Chitu and Stanley, 2006). High CSF1 concentrations in tumors are associated with poor prognosis and expression is often found at the leading edge of tumors (Laoui et al., 2011; Qian and Pollard, 2010). In endometrial cancer its synthesis by tumor cells is an independent predictor of poor overall survival (Smith et al., 2013). Consistent with these clinical observations, deletion of CSF1 genetically from several models of cancer results in delayed initiation (cervical), progression (breast, pancreas) and metastasis (breast) associated with the loss of TAMs. Similarly the use of neutralizing antibodies, small molecule inhibitors or antisense RNA strategies to inhibit CSF1R signalling also affected tumor malignancy in both xenograft and GEM models of cancer (Abraham et al., 2010; Lin et al., 2001; Qian and Pollard, 2010; Quail and Joyce, 2013). Most of these strategies however, will have had systemic effects as well as local ones, making it difficult to determine whether the therapeutic effects are on the macrophage lineage and/or directly affecting the recruitment and survival of TAMs in the tumor. Direct evidence for CSF1 recruiting macrophages was provided in the mouse model of breast cancer caused by the mammary epithelial-restricted expression of the Polyoma Middle T oncoprotein (PyMT). In these studies organ-autonomous gain-of-function experiments whereby CSF1 was expressed in the mammary epithelium resulted in local macrophage recruitment and an acceleration of tumorigenesis in wild type mice and also the rescue of the loss-of Csf1 function mutation that had resulted in delayed tumor progression and reduced metastasis (Lin and Pollard, 2007; Wyckoff et al., 2007). Genetic gain-of-function of VEGFA over a loss-of function of CSF1 in the PyMT mouse model also resulted in a dramatic recruitment of macrophages and a rescue of angiogenesis that resulted in an acceleration of tumor progression to malignancy (Lin et al., 2007). VEGFA also recruits macrophage progenitors that then differentiate to TAMs under IL-4 influence in a xenograft model of skin cancer (Linde et al., 2012). Loss of these VEGF-recruited TAMs inhibited tumor growth, angiogenesis and invasion (Linde et al., 2012). These data indicate that CSF1 and VEGFA can be independent recruiters of macrophages to tumors in mouse models. This effect could be via recruitment of monocytes and/or through proliferation of recruited or resident cells. These growth factors probably act collaboratively with locally synthesized chemokines to reinforce recruitment or retention. For example CCL2 acting via its receptor CCR2 is a direct mediator of monocyte recruitment to the primary tumor and to metastases in the PYMT model (Cortez-Retamozo et al., 2012; Franklin et al., 2014; Qian et al., 2011) even though this recruitment requires CSF1 (Lin et al., 2001; Qian et al., 2011). Another example of chemokine-mediated TAM recruitment collaborating with GM-CSF is CCL18 acting via its receptor PITPNM3 in human breast cancer models (Su et al., 2014). Furthermore CCL9 acting through its receptor, CCR1, recruits immature myeloid cells in colon cancer models (Kitamura et al., 2010) (Kitamura et al., 2007). In each case ablation of these chemokines resulted in loss of monocytes and/or TAMs and a resultant inhibition of malignancy with effects particularly on tumor cell invasion and occasionally growth.
The origins of macrophages in many cancers particularly in early stages is still uncertain and further this recruitment and differentiation is likely to be different and more complex in those cancers exposed to microbial products such as in colon cancer than those in sterile sites. Nevertheless, while the understanding the origins of TAMs and their methods of recruitment, retention and differentiation is in its infancy understanding the mechanisms offers the tantalizing possibility of therapies targeted to recruited sub-populations of pro-tumoral macrophages that spares anti-tumoral ones and the resident macrophages associated with homeostasis.
Pro-tumoral Mechanisms of TAMs
Among the ways in which the micro-environmental support to tumors is the acquisition of a vasculature that provides oxygenation as well as the nutrition and waste disposal required for growth above a certain size in a process often referred to as the ‘angiogenic switch’ (Hanahan and Weinberg, 2011). CSF1 regulated macrophages regulate this switch in the PyMT model in part through production of VEGF (Lin and Pollard, 2007). In this model macrophage synthesized WNT7b targets vascular endothelial cells stimulating their production of VEGF resulting in the angiogenic switch (Yeo et al., 2014). Macrophages also promote neo-angiogenesis in glioblastoma models (Du et al., 2008) Characterisation of angiogenic TAMs show that they express TIE2. Genetic ablation of this population inhibits angiogenesis in a variety of models including glioblastoma and the PyMT model (De Palma et al., 2005). These Tie2+ macrophages are often aligned along the abluminal surface of blood vessels through endothelial cell expression of the TIE2 ligand ANG2. Targeting ANG2 or Tie2 releases this macrophage-vessel association and inhibits angiogenesis in the PyMT and RIP1-TAG models of breast and pancreatic cancer (Mazzieri et al., 2011). Interestingly, CSF1 up-regulates TIE2 on TAMs (Forget et al., 2014) indicating a link between CSF1, TIE2+ macrophages and the induction of the angiogenic switch. There are numerous additional reports of TAMs affecting angiogenesis in a wide range of models, mostly xenograft ones, and for further information the reader is referred to recent reviews on this topic (Coffelt et al., 2009; Nucera et al., 2011).
This population of TIE2+ macrophages aligned along the vessels also promotes another phenotype of malignancy, tumor cell intravasation into the circulation (Wyckoff et al., 2007). In fact macrophages promote directional tumor cell migration and invasion via a paracrine loop that consists of tumor cell synthesized CSF-1 and macrophage derived epidermal growth factor (EGF) or EGF family ligands. This causes tumor cells and macrophages to rapidly stream along collagen fibres in lock-step ending up in tumor cells clustering around blood vessels (Condeelis and Pollard, 2006; Wyckoff et al., 2007) (Figure 1). Upon arrival at the blood vessels, TIE2+ macrophages open up a gate for the tumor cells to escape. Macrophages also produce several other molecules that advance tumor cell invasion including Osteonectin (also known as SPARC) that increases tumor cell-ECM interaction and thus migration (Sangaletti et al., 2008), Cathepsin proteases that re-model the matrix and release sequestered growth factors (Laoui et al., 2011; Quail and Joyce, 2013) and TGF-β that promotes EMT of the invading tumor cells (Bonde et al., 2012). Thus these protumoral macrophages not only increase the invasive capacity of tumor cells but also increase the density of blood vessels giving a double whammy that increases the number of circulating tumor cells and thus metastasis (Figure 1). Consequently ablation of TAMs for example by genetic depletion of their major growth factor, CSF-I, diminishes the number of circulating tumor cells and reduces metastasis (Wyckoff et al., 2007). Importantly, an anatomical structure consisting of macrophages, endothelial, and tumor cells named the tumor microenvironment for metastasis (TMEM) is recognizable in histological sections and is predictive of metastatic potential in primary human breast cancers (Rohan et al., 2014).
Figure 1. Tumor-associated macrophages in the primary tumor promote malignancy.
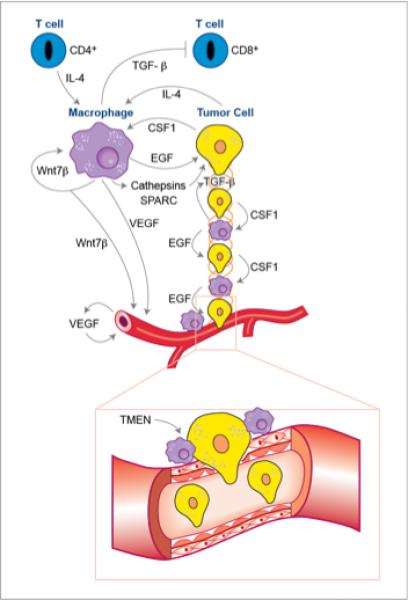
In the primary tumor microenvironment macrophages under the influence of IL-4 produced by CD4+ T cells and tumors and WNT7b promote tumor cell invasion. This invasion is mediated via a paracrine loop involving tumor-synthesized CSF1 and macrophage-produced EGF that drives migration of tumor cells in lock-step with macrophages along collagen fibres that act as highways towards blood vessels. This process also requires TGFβ that drives n epithelial-mesenchymal transition (EMT) in the tumor cells that promotes migration and matrix remodelling via Cathepsins and matrix adhesion of tumor cells via SPARC. This streaming of tumor cells results in their pileup on the vessels where macrophages promote their intravasation into the circulation through a structure named the “Tumor Microenvironment of Metastasis” (TMEN). In addition to effect on tumor cell migration and invasion TIE2+ macrophages produce VEGF and WNT7b that stimulates angiogenesis in the tumor. Thus there is an additive effect caused by macrophages of increased migration of tumor cells towards vessels and increased vascular targets that results in a large number of circulating tumor cells and thus increased malignancy. Macrophages also suppress cytotoxic T-cell responses through the mechanisms described in figure 2.
Once the barrier of the angiogenic switch has been surmounted tumors rapidly become invasive and thus characterised as malignant. This correlates with enhanced engagement of the acquired immune system indicating antigen recognition probably because the immune system has access to the products of mutated genes and/or recognizes tissue damage caused by invasion (Coussens and Pollard, 2011; Gajewski et al., 2013). However, despite data that suggest better prognosis with early T cell infiltration of some cancers, successful tumors that progress to kill the patients clearly are not rejected. This immunosuppression is at least in part mediated by macrophages or their progenitors (Figure 2), but also involves regulatory T cells, as well as tumor cell-mediated immune evasion (Coussens and Pollard, 2011; Gajewski et al., 2013; Movahedi et al., 2010). In this context and importantly, the combination of macrophages and a high ratio of CD4+ regulatory versus CD8+ T cells in human breast cancer is predictive of poorer survival (Ruffell et al., 2011).
Figure 2. Pro-tumor macrophage mechanisms of effector cells inhibition.

TAMs express an array of effector molecules that inhibit the anti-tumor immune responses; this includes cell surface receptors, cytokines, chemokines and enzymes. Inhibition of immune responses by direct cell-to cell-contact is based on the interaction of TAMs receptors ligands with their counterpart death/inhibitory receptors expressed by the target immune effector cells. TAMs express the ligand receptors for PD-1 and CTLA-4 that upon activation suppress cytotoxic functions of T- cell, NKT cells and NK cells. TAMs also express the ligand for the death receptors FAS and TRAIL that triggers in target cells caspase dependent cell death (apoptosis). TAMs also express the non-classical HLA-G that inhibits T cell function through interaction with the co-stimulatory signal of T cells ILT2, and HLA-E that inhibit NK cells through CD94 (also known as NKG2). TAMs secrete the cytokines IL-10 and TGFβ that inhibit T cells effector functions and induce regulatory functions, and chemokines CCL5, CCL20, CCL22 that recruit nTreg cells. TAMs secrete Arginase I that inhibit TCR ζ chain re-expression in activated T cells by the depletion of L-arginine.
Macrophages and DCs express classical and non-classical MHC-I molecules and this is normally associated with the presentation of antigens to T-cells. However macrophages can also express HLA molecules such as HLA-C (classical), HLA-E, and HLA-G (non-classical) membrane bound or soluble forms that can inhibit the activation of NK cells and a subsets of activated T cells upon their ligation to killer cell immunoglobulin like receptor CD94 (also known asNKG2) (Borrego et al., 1998) or the inhibitory leukocyte immunoglobulin-like receptors LIT-2 (HLA-E and HLA-G respectively). While some tumors express HLA-G (membrane bound or soluble) as part of their evasion mechanisms from NK and T cell lysis, others do not. These HLA-G negative tumors may rely on myeloid cell HLA-G expression as an effector of inhibitory mechanisms. An example of this is in glioblastoma and neuroblastoma where high concentrations of soluble HLA-G can be found in patient's serum. In this case microglia and circulating monocytes are the source of this secreted HLA-G (Kren et al., 2010; Morandi et al., 2007). The inhibition of effector CD8+ T cell activation in the lymph nodes by HLA-G expressing monocytes, macrophages or DCs may also be indirect. For example it has been shown that INF-γ secretion by activated NK cells that have migrated to the lymph node, is an important mediator of CD8+ T cell activation, and that HLA-G dependent inhibits this NK cells migration and INF-γ secretion (Kelly et al., 2002). In addition, HLA-G transfected APCs can inhibit CD4+ T cell activation and induce immunosuppressive differentiation in vitro (LeMaoult et al., 2004). Moreover, trogocytosis of HLA-G by activated CD4+ and CD8+ T cells during interaction with HLA-G+ APCs leads to rapid generation of T cell's suppressor functions (LeMaoult et al., 2007). Monocytes and macrophages can themselves express members of the LILRB inhibitory receptors family (LIT-2 and LIT-4) that upon binding HLA-G causes them to acquire immunosuppressive phenotype through the secretion of IL-10 and TGF-β1 (Brown et al., 2004; McIntire et al., 2004). However, the expression of inhibitory receptors and their HLA ligands by TAMs and their effect on TAMs immunosuppressive function are yet to determined.
In addition to these MHC molecules, macrophages express the ligands of the inhibitory receptors programmed cell death protein 1 (PD-1) and cytotoxic T-Lymphocyte Antigen 4 (CTLA-4). . These inhibitory ligands are normally up regulated in activated immune effector cells such as T cells, B cells, and NKT cells as part of a safety mechanism that controls the intensity of the immune response, and as part of inflammation resolution. Activation of PD-1 and CTLA-4 by their ligands (PD-L1, PD-L2 and B7-1 [D80], B7-1 [CD86] respectively) directly inhibits TCR and BCR signalling. This activation also inhibits T cell cytotoxic function, regulates their cell cycle, and inhibits their activation as CTLA4 competes with CD28 (co-stimulatory) binding. PD-L1 and PD-L2 are differentially expressed, with PD-L1 constitutively expressed by immune cells including T cells, B cells, macrophages, DCs, nonhematopoietic cells, and cancer cells. In contrast PD-L2 expression is limited to antigen-presenting cells (APCs). Its expression is induced in monocytes and macrophages by CSF1, IL-4, and INF-γ (Loke and Allison, 2003). Both PD-L1 and L2 are regulated in TAMs and myeloid-derived suppressor cells (See below –MDSC) (Belai et al., 2014; Duraiswamy et al., 2013). Recently Noman et al. showed that MDSCs and TAMs in hypoxic tumor regions up regulate the expression of PD-L1 as a consequence of HIF-1α signalling (Noman et al., 2014). Hypoxia acting via hypoxia inducible factor 1- α (HIF-1α) also induces T cell suppression by TAMS although the mechanism is unknown (Doedens et al., 2010). It has also been shown that monocytes from blood of glioblastoma patients express higher amounts of PD-L1 compared to healthy donors and that glioblastoma cell conditioned medium can up regulate PD-L1 expression in monocytes from healthy donors (Bloch et al., 2013). Similarly, monocytes from patients with hepatocellular carcinoma express PD-L1 that contributes to human tumor xenograft growth in vivo, while the blocking of PD-L1 reverses this effect (Kuang et al., 2009). The identification of B7-1 (CD80) as an additional inhibitory receptor for PD-L1 suggested the possibility of reverse signalling. Indeed, the culture of bone marrow derived DCs with anti PD-L1 antibody inhibits their activation, induces IL-10 expression, and suppresses co-cultured CD4+ T cell activation (Kuipers et al., 2006). However, it is challenging to determine the specific impact of TAM PD-1 ligand expression on effector cells inhibition in vivo since numerous cells in the tumor microenvironment express PD-L1 (Greaves and Gribben, 2013). Thus it is yet to be discovered if the signals from the PD-1 and PD-1 ligands contribute to TAMs immunosuppressive phenotype in vivo.
The CTLA-4 ligands B7-1 and B7-2 are differentially expressed by APCs. B7-2 is constitutively expressed in low amounts and it is up regulated during activation while B7-1 is expressed only upon APC activation. B7-1 and B7-2 are also the ligands of the T cell costimulatory CD28 however they bind with higher affinity to the inhibitory receptor CTLA-4. This differential affinity suggests direct competition for the ligand binding as a mechanism to induce suppression (Greenwald et al., 2002). TAM expression of B7-1 and B7-2 was shown to be dependent on their activation phenotype; both molecules are expressed by pro-inflammatory macrophages and are down-regulated by anti-inflammatory macrophages (Ding et al., 1993; Flores Villanueva et al., 1994; Kennedy et al., 2013). However, the specific inhibitory effect mediated by TAMs in vivo is still unknown and as with PD1 ligands, CTLA-4 ligands are expressed on some human tumors and other immune cells (Greaves and Gribben, 2013; Tamura et al., 2005; Tirapu et al., 2006). Finally, evidence from studies on the DC-T cell immunological synapse suggests that interaction of CTLA-4 with B7 ligands not only signals for the inhibition of T cells but also induces a DCs inhibitory phenotype (Butte et al., 2007; Mellor et al., 2004). Additional investigation is needed to determine whether such reverse signalling in TAMs is associated with a pro-inflammatory to anti-inflammatory switch.
B7-H4 is a relatively new member of the B7 superfamily that was implicated with suppression of T cells activation and is expressed on TAMs. The co-receptor for B7-H4 is currently unknown. In human ovarian cancer, TAMs expressing B7-H4 suppress the activation of antigen-specific T cells. Moreover, the inhibition of B7-H4 restores the stimulating function of TAMs and contributes to tumor regression (Kryczek et al., 2006). In addition, the expression of B7-H4 on TAMs was found to correlate with clinical stage of lung carcinoma and gastric cancer (Chen et al., 2012; Matsunaga et al., 2011).
TAMs also secrete an array of cytokines, chemokines and enzymes that can suppress CD4+ and CD8+ T cells effector function directly or indirectly by recruitment of natural regulatory T (nTreg) cells to the tumor microenvironment as well as by inducing the CD4+ regulatory fraction (iTreg) cells and sustaining their survival. Chemokine receptors CCR4, CCR5, CCR6 and CCR10 expressed by nTreg cells are involved in their migration into the tumor microenvironment (Adeegbe and Nishikawa, 2013). Curiel at al. demonstrated that CCL22 secreted by TAMs recruits CCR4+ nTreg cells to human ovarian carcinoma tumors and foster tumor growth (Curiel et al., 2004). In colorectal cancer, CCL20 secreted by TAMs recruit CCR6+ nTreg cells (Liu et al., 2011). In addition, CCL3, CCL4, CCL5 expressing myeloid-MDSC from a melanoma mouse model recruited nTreg cells through CCR5 signalling. TAMs in this mouse model expressed some of the CCR5 ligands (Schlecker et al., 2012). In addition, CCL5 is expressed by TAMs in other mouse tumor models (Biswas et al., 2006; Liou et al., 2013). The induction of iTreg cells in the tumor microenvironment is a complex process that is not completely understood. Nevertheless, TGF-β and IL-10 induce regulatory functions by the up regulation of the pivotal regulatory transcription factor, Foxp3, in CD4+ T cells (Adeegbe and Nishikawa, 2013). TAMs have been found to express IL-10 and TGF-β in different pathological scenarios including human and mouse cancers (Pollard, 2004). Macrophages in the intestinal immune system were shown to induce iTreg cells by the secretion of IL-10 and TGF-β (Denning et al., 2007). Savage and co-workers investigated the ability of human macrophages to induce regulatory T cells and showed that IL-10 expressing anti-inflammatory macrophages but not pro-inflammatory macrophages are responsible for induction of iTreg cells (Savage et al., 2008). In addition, TAMs isolated from human renal cell carcinoma induce the expression of CTLA4 and Foxp3 in CD4+ T cells (Daurkin et al., 2011). TGF-β and IL-10 are also involved in direct modulation of T cells functions. TGF-β inhibits cytotoxic T lymphocyte, Th1 and Th2 CD4+ T cells (Oh and Li, 2013) while IL-10 inhibits Th1 and Th2 CD4+ T cell helper functions (Ng et al., 2013).
TAMs can also suppress T cell activity by the depletion of L-arginine in the tumor microenvironment. Nitric-oxide synthase (NOS) and arginase I (ARGI) are L-arginine processing enzymes that were shown to be differentially secreted by macrophages as a function of their activation state (pro-inflammatory and anti-inflammatory respectively) (Biswas and Mantovani, 2010). TAMs secrete ARGI into the microenvironment in different human cancers and mouse cancer models (Doedens et al., 2010; Sharda et al., 2011). ARGI metabolizes L-arginine to urea and L-ornithine hence depleting it from the tumor microenvironment. L-arginine is necessary for T cells function and its depletion inhibits the re-expression of the CD3 ζ chain after internalization caused by antigen stimulation and TCR signalling (Rodriguez et al., 2004; Rodriguez et al., 2003). In fact the expression of ARGI is considered to be the hallmark of anti-inflammatory macrophages, so-called M2 macrophages (see below), in mice and a marker of many TAM populations (Sica and Mantovani, 2012).
In addition to bona fide macrophages there is an extensive literature on a group of cells collectively called myeloid-derived suppressor cells (MDSC) that accumulate in the spleen and tumors during malignant progression. These cells in ex-vivo CTL assays can suppress T cell responses (Gabrilovich et al., 2012). Furthermore, in vivo MDSCs block DC maturation at the invasive edge of tumors (Gabrilovich et al., 2012). In mice MDSCs are defined as being CD11b+ and Gr1+ . These markers define both monocytic and granulocytic cells (both Ly6c and Ly6G antigens are recognised by the anti-GR1 antibody). The consensus view is that MDSCs consist of a mixed population (Gabrilovich et al., 2012). The majority of MDSCs being Ly6G+ immature granulocytes that will not be further discussed in this review and they have been well reviewed recently (Gabrilovich et al., 2012). The minority population is Ly6C+ Ly6G- suggesting they are monocytic in origin and thus have been termed monocytic MDSCs (M-MDSC). These M-MDSCs have greater immunosuppressive potency than the granulocytic ones and are further defined as F4/80+ a marker also found on inflammatory monocytes (Gabrilovich and Nagaraj, 2009; Gabrilovich et al., 2012). It has long been recognised that monocytes can be immunosuppressive but it is unclear in cancer whether such cells accumulate in excessive numbers as a transient to mature macrophages or whether M-MDSCs represent a novel monocyte-derived terminal cell type. These cells are MHClow and co-stimulatory molecule low or negative suggesting they do not directly induce anti-T cell activity. Instead they highly express TGF-β and ARG1 that contributes to non-specific immune suppression (Gabrilovich and Nagaraj, 2009; Gabrilovich et al., 2012; Yang et al., 2008). Despite the obvious distinction between monocytic and granulocytic sub-types the usual lack of discrimination between these groups in experiments and the lack of unique markers on M-MDSCs precluding specific ablation of these cells makes the specific in vivo function M-MDSCs in immunosuppression hard to define. Consequently they will not be further discussed here and there are excellent reviews defining their functions and classification elsewhere (Gabrilovich and Nagaraj, 2009; Gabrilovich et al., 2012; Montero et al., 2012). These studies with M- MDSCs also calls into question the cell type that can present antigens to the incoming T cells in tumors and thus cause recognition of tumors at early stages. Krummel and colleagues developed a system to detect OVA antigen presentation in the PyMT mouse model and using this model defined an APC that was Cd11c+ F4/80+ in the tumor margin that could be either a TAM or DC but not an M-MDSC. However, this antigen presentation by this TAM/DC population to T cells while present was abortive (Engelhardt et al., 2012). In fact these DC-like cells and CD8+ T cells appear to be ‘trapped’ in the tumor margin even in xenograft models in the face of chemotherapy, suggesting a novel immunosuppressive mechanism (Boissonnas et al., 2013; Engelhardt et al., 2012).
Altogether, TAM expression of cell surface receptors, secreted cytokines, chemokines and enzymes suggest they have an important role in recruitment and activation of Treg cells and the suppression of effector cells in the tumor microenvironment (Figure 2). Nevertheless the dominant mechanisms in vivo even in simple xenograft mouse models are unknown. This failure is not surprizing given the exact myeloid cell type(s) that engages the acquired immune system is ill-defined and because most experiments use homogeneous, transplanted tumor models that are inherently immunogenic due to up-regulation of latent retroviruses and other epigenetic changes caused by cell culture. Better definition of these immunosuppressive mechanisms needs complex evolving autochthonous and thus “self” models in which immune response can be tracked as the tumor evolves. These models will allow specific definition of antigen presentation and the means whereby cells of the monocyte/macrophage lineage suppresses this response.
Macrophages at the Metastatic Site
Once tumor cells escape from the primary site they passage through the lymphatic and/or circulatory system and ultimately a few establish at distant sites to give metastases. These sites vary according to cancer for example in breast they primarily go to bone then lung and brain. It is this process that is essential to understand as 95% of deaths from solid tumors in the developed world are due to metastasis. Monocytes/macrophages are essential metastasis promoters acting both to prepare sites and also to promote the extravasation, survival and persistent growth of metastatic cells (Joyce and Pollard, 2009; Qian et al., 2009). Even before tumor cells arrive, the frequency and site specificity of metastatic growth can be influenced by primary tumors through the formation of sites that enhance homing of circulating tumor cells known as pre-metastatic niches (Psaila and Lyden, 2009). These niches are populated by Cd11b+ VEGFR1+ myeloid cells whose recruitment is promoted by Lysyl Oxidase and S110A and whose ablation inhibits the formation of these sites (Psaila and Lyden, 2009). Several other factors have been shown to be important for pre-metastatic niche formation, most recently, tumor derived exosomes that program the myeloid cells to be pro-tumoral and pro-angiogenic through activation of the receptor tyrosine kinase MET (Peinado et al., 2012). Exosomes derived from different melanoma strains can also re-direct metastatic cell target tropisms from one tissue to another (Peinado et al., 2011). The formation of the niche is also dependent on platelets that presumably deposit fibrin in the target tissues that attracts myeloid cells. Consequently pre-metastatic niche formation is blocked by anti-coagulants (Gil-Bernabe et al., 2012).
Studies of lung metastasis show that upon their arrival at the target site tumor cells together with associated platelets recruited via their expression of tissue factor form micro-clots and arrest in the target tissue vessels (Gil-Bernabe et al., 2012). This arrest enables CCL2 synthesized by the tumor cells to generate a chemoattractive gradient that recruits Ly6C monocytes through their expression of the CCL2 receptor, CCR2 (Cortez-Retamozo et al., 2012; Qian et al., 2011). In addition clotting up-regulates VECM1 on endothelial cells that promotes myeloid cell attachment and thus their recruitment (Ferjancic et al., 2013). These recruited monocytes enhance extravasation of tumor cells in part by expression of VEGF that cause vascular permeability. Consistent with this is that inhibition of CCR2 signalling blocks tumor cell extravasation and inhibits metastasis (Qian et al., 2011). These recruited monocytes differentiate into CCR2+, VEGFR1+ Ly6C- F4/80+ metastasis-associated macrophages (MAMs) (Figure 3). Ablation of this MAM population using genetic and chemical means inhibits metastatic seeding and persistent growth, the latter effect being evident even after the metastases have been established (Qian et al., 2009; Qian et al., 2011). Mechanistically this is via the maintenance of CSF1 signalling in macrophages and through the enhancement of tumor cell survival (Qian et al., 2009) via engagement of VCAM1 expressed upon the tumor cells that generates an AKT mediated anti-apoptotic signal (Chen et al., 2011). Myeloid cells also promotes mesenchymal to epithelial transition (MET) and tumor growth by inhibiting TGF-β signalling in these epithelial metastatic cells (Gao et al., 2012).
Figure 3. Macrophages promote metastasis.

Arrest of tumor cells in the vasculature of target organs through the formation of microclots (1) results in CCL2-mediated recruitment of CCR2-expressing circulating inflammatory monocytes (2). These monocytes differentiate into metastasis-associated macrophages (MAMs) that mediate tumor cell extravasation via VEGF that increases vascular permeability (3). MAMS under the influence of CSF-1 further promote tumor cell survival (4) and persistent growth associated by angiogenesis and may also prevent T cell cytotoxicity (5).
Many cancers also metastasise to the bone such as breast and prostate. In this process another cell from the mononuclear phagocytic lineage, the osteoclast, plays an important role. This cell is lineage regulated by CSF1 followed by differentiation and proliferation in response to RANK Ligand that lead to the multi-nuclear functional osteoclast. These cells are often activated by metastatic cells to degrade bone and release growth factors resulting in a vicious cycle. As this process is dependent on a different cell type to classical macrophages it will not be reviewed further here but readers are referred to recent reviews that discuss process and therapeutic opportunities (Camacho and Pienta, 2014; Esposito and Kang, 2014; Mundy, 2002).
Macrophages as Therapeutic Targets
Macrophages are exceptionally diverse in their functions reflecting the different origins, local environment and responses to challenges (Wynn et al., 2013). Consideration of macrophage function in immunity let to the proposal of two classes of macrophages: 1) the activated macrophages responding to IFN-γ, TNF-α and toll-like receptor 4 (TLR4) activation capable of killing pathogens through mechanisms such as iNOS; 2) Alternatively activated macrophages responding to IL-4 and IL-13 involved in anti-parasitic immunity and in asthma (Gordon, 2003). The original in vitro characterizations were extended to in vivo models by Mills and co-workers who called these states M1 (activated) and M2 (alternatively activated) (Mills, 2012). These descriptions were captured to suggest that TAMs could be either tumor killing (M1) or tumor promoting (M2) (Sica et al., 2008). However, while these extreme forms of polarization are seductive, the already described multiple phenotypes of TAMs activity engaged in different biological functions in the tumor suggested such definitions are limiting and probably do not exist in the complex tumor microenvironment (Qian and Pollard, 2010). In fact different macrophages associated with diverse phenotypes and particular to different tumor types argues for a plethora of different populations. Furthermore, in most large scale transcriptome analysis macrophages have a mixed phenotype expressing both M1 and M2 markers (Qian and Pollard, 2010). In addition there have been no definitive experiments where unique ablation of macrophages designated as M1 or M2 has been achieved and thus their role in tumor promotion is unknown. In contrast, ablation of specific sub-populations such as TIE2+ or MAMs can be demonstrated to affect specific activities such as angiogenesis or metastatic seeding. We have always proposed that sub-populations should be defined by biology rather than enforcing pre-existing nomenclature upon function (Qian and Pollard, 2010). Thus despite ongoing discussion on nomenclature, the clinical challenge remains to block macrophage trophic phenotypes together with their immunosuppressive behaviours and enhance their activation anti-tumoral activities.
Several recent studies suggest that such an approach is feasible and therapeutic (Coussens et al., 2013; De Palma and Lewis, 2013). The major strategy so far is based upon genetic experiments whereby inhibition of CSF1 signalling in PYMT models inhibits tumor progression and metastasis (Lin et al., 2001) and uses anti-CSF1 receptor neutralizing antibodies or small molecule inhibitors to interfere with this pathway (Coussens et al., 2013). Strikingly inhibition of CSF1R in glioblastoma mouse models results in a dramatic reduction in tumor volume and long-term survival of the mice. This CSF1R inhibition did not kill the TAMs but caused them to re-polarize to a state regulated by GM-CSF that has been suggested to be anti-tumoral (Quail and Joyce, 2013). Similar results can be seen in cervical and breast cancer models (Strachan et al., 2013). Small molecule inhibitors to CSF1R also have been shown to deplete some populations of TAMs and in established tumors to dramatically enhance responses to chemotherapy. This effect is at least in part due to the removal of macrophage-mediated immunosuppression during the tumor recovery period (DeNardo et al., 2011; Mitchem et al., 2013). These effects seem not to be restricted to chemotherapy since Tie+ positive TAMs limit the efficacy of anti-vascular reagents and their ablation strongly increases the therapeutic efficacy of these agents (Priceman et al., 2010; Welford et al., 2011). In other models, M-MDSCs modulate the efficacy of anti-vascular therapies (Shojaei et al., 2007). Furthermore low-dose irradiation of tumors programs macrophages to an activated state that orchestrate T-cell immunotherapy (Klug et al., 2013). Macrophages also enhance the therapeutic efficacy of monoclonal antibodies (De Palma and Lewis, 2013). In addition, the chemotherapeutic agent Trabectedin directly kills monocytes/macrophages and has therapeutic efficacy against tumors in mouse models (Germano et al., 2013). Similarly amphotericin B enhances macrophage-mediated inhibition of glioma tumor-initiating cells (Sarkar et al., 2014). Most importantly a recent clinical trial reports objective clinical responses in diffuse-type giant cell tumors that over-express CSF1 by using a neutralizing antibody to the CSF1R in a single molecule approach and this response is characterized by an increase in the CD8+/CD4+ T cell ratio (Ries et al., 2014). This dramatic result together with the other examples given above strongly support targeting the destruction or re-differentiation of macrophages as an important part of combinatorial therapies in human cancer patients.
Perspectives
We have argued previously that TAMs recapitulate the roles of macrophages in tissue development and repair that is coupled with suppression of immune responses to the tissue damage caused by invading epithelial structures (Pollard, 2004). Gene profiling of TAMs supports this hypothesis while at the same time define many subpopulations with different pro-tumoral functions (Qian and Pollard, 2010). The pre-clinical experimental data described above suggest that targeting TAMs either by ablation or re-polarization can be beneficial in cancer therapy. This is an attractive approach since these diploid normal cells do not have the enhanced mutation rates of tumor cells that inevitably lead to drug resistance. Indeed several clinical trials are underway targeting CSF1R signalling as a means of removing macrophage pro-tumoral support and the most recent of these studies reports clinical efficacy (Ries et al., 2014). However, these pan-macrophage therapeutic approaches will have systemic toxicities as they target all macrophages. As we move forward the realization of diverse origins of macrophages with recruited ones being different from resident ones (Wynn et al., 2013) suggest that more sophisticated therapies that only target TAMs or MAMs may be possible (Modi et al., 2012). Importantly a definition of macrophage sub-populations in different human cancers and in different sub-types of cancer in a particular tissue is needed to advance these options. Another exciting therapeutic approach is to enhance chemotherapy or immunotherapy by removing the immunosuppressive activities of macrophages. In this arena pre-clinical data (Figure 4) indicates several strategies that can be combined to improve the already encouraging anti-tumoral clinical results obtained by inhibiting regulatory T cell mechanisms through the use of neutralizing anti-PD1, -PD-L1 or -CTLA4 antibodies (Page et al., 2014). Further definition of the regulation of immunoregulatory mechanisms in macrophages should allow the development of a whole new range of therapeutics.
Figure 4. Re-programming Macrophages to be Anti-Tumoral.

Macrophages in the tumor in general are immunoregulatory and suppress immune responses to tumor-derived antigens. However, in some circumstances particularly with appropriate therapeutic interventions, macrophages can be anti-tumoral by direct tumor cell killing, the removal of vital support such as inhibition of angiogenesis or by the activation T cells. This differential polarization is under the control of many stimuli as shown that alters the differentiated state of the macrophages. Some factors such as GM-CSF act in pro- or anti-tumoral fashion dependent on context (Pyonteck et al., 2013; Su et al., 2014). Therapeutic interventions can re-polarize these cells to become immunostimulatory macrophages that on their own can cause tumor regression or that enhance the activity of chemo-, anti-vascular or immuno-therapies. References to these polarizing agents can be found in (De Palma and Lewis, 2013; Sica and Mantovani, 2012).
Acknowledgements
The author's work has been supported by NIH grants RO1 CA172451 and PO1 CA100324, The Rowley breast cancer scientific research project #1003251030, NY and the Wellcome Trust, UK.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: JWP has patents pending on some aspects of macrophage inhibition in tumor therapy.
References
- Abraham D, Zins K, Sioud M, Lucas T, Schafer R, Stanley ER, Aharinejad S. Stromal cell-derived CSF-1 blockade prolongs xenograft survival of CSF-1-negative neuroblastoma. Int. J. Cancer. 2010;126:1339–1352. doi: 10.1002/ijc.24859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adeegbe DO, Nishikawa H. Natural and induced T regulatory cells in cancer. Frontiers in immunology. 2013;4:190. doi: 10.3389/fimmu.2013.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin. Cancer Biol. 2012;22:33–40. doi: 10.1016/j.semcancer.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Belai EB, de Oliveira CE, Gasparoto TH, Ramos RN, Torres SA, Garlet GP, Cavassani KA, Silva JS, Campanelli AP. PD-1 blockage delays murine squamous cell carcinoma development. Carcinogenesis. 2014;35:424–431. doi: 10.1093/carcin/bgt305. [DOI] [PubMed] [Google Scholar]
- Biswas SK, Allavena P, Mantovani A. Tumor-associated macrophages: functional diversity, clinical significance, and open questions. Seminars in immunopathology. 2013;35:585–600. doi: 10.1007/s00281-013-0367-7. [DOI] [PubMed] [Google Scholar]
- Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, Bottazzi B, Doni A, Vincenzo B, Pasqualini F, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation). Blood. 2006;107:2112–2122. doi: 10.1182/blood-2005-01-0428. [DOI] [PubMed] [Google Scholar]
- Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nature immunology. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:3165–3175. doi: 10.1158/1078-0432.CCR-12-3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissonnas A, Licata F, Poupel L, Jacquelin S, Fetler L, Krumeich S, Thery C, Amigorena S, Combadiere C. CD8+ tumor-infiltrating T cells are trapped in the tumor-dendritic cell network. Neoplasia. 2013;15:85–94. doi: 10.1593/neo.121572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonde AK, Tischler V, Kumar S, Soltermann A, Schwendener RA. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC cancer. 2012;12:35. doi: 10.1186/1471-2407-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrego F, Ulbrecht M, Weiss EH, Coligan JE, Brooks AG. Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. The Journal of experimental medicine. 1998;187:813–818. doi: 10.1084/jem.187.5.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D, Trowsdale J, Allen R. The LILR family: modulators of innate and adaptive immune pathways in health and disease. Tissue antigens. 2004;64:215–225. doi: 10.1111/j.0001-2815.2004.00290.x. [DOI] [PubMed] [Google Scholar]
- Brown ER, Charles KA, Hoare SA, Rye RL, Jodrell DI, Aird RE, Vora R, Prabhakar U, Nakada M, Corringham RE, et al. A clinical study assessing the tolerability and biological effects of infliximab, a TNF-alpha inhibitor, in patients with advanced cancer. Ann. Oncol. 2008;19:1340–1346. doi: 10.1093/annonc/mdn054. [DOI] [PubMed] [Google Scholar]
- Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho DF, Pienta KJ. A multi-targeted approach to treating bone metastases. Cancer Metastasis Rev. 2014 doi: 10.1007/s10555-013-9476-y. [DOI] [PubMed] [Google Scholar]
- Chen C, Zhu YB, Shen Y, Zhu YH, Zhang XG, Huang JA. Increase of circulating B7-H4-expressing CD68+ macrophage correlated with clinical stage of lung carcinomas. Journal of immunotherapy (Hagerstown, Md. : 1997) 2012;35:354–358. doi: 10.1097/CJI.0b013e31824212c4. [DOI] [PubMed] [Google Scholar]
- Chen Q, Zhang XH, Massague J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer cell. 2011;20:538–549. doi: 10.1016/j.ccr.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitu V, Stanley ER. Colony-stimulating factor-1 in immunity and inflammation. Curr. Opin. Immunol. 2006;18:39–48. doi: 10.1016/j.coi.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Coffelt SB, Hughes R, Lewis CE. Tumor-associated macrophages: effectors of angiogenesis and tumor progression. Biochim. Biophys. Acta. 2009;1796:11–18. doi: 10.1016/j.bbcan.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, Ryan RJ, Iwamoto Y, Marinelli B, Gorbatov R, et al. Origins of tumor-associated macrophages and neutrophils. Proc. Natl. Acad. Sci. U. S. A. 2012;109:2491–2496. doi: 10.1073/pnas.1113744109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Pollard JW. Leukocytes in mammary development and cancer. Cold Spring Harbor perspectives in biology. 2011;3 doi: 10.1101/cshperspect.a003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339:286–291. doi: 10.1126/science.1232227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- Daurkin I, Eruslanov E, Stoffs T, Perrin GQ, Algood C, Gilbert SM, Rosser CJ, Su LM, Vieweg J, Kusmartsev S. Tumor-associated macrophages mediate immunosuppression in the renal cancer microenvironment by activating the 15-lipoxygenase-2 pathway. Cancer Res. 2011;71:6400–6409. doi: 10.1158/0008-5472.CAN-11-1261. [DOI] [PubMed] [Google Scholar]
- De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer cell. 2013;23:277–286. doi: 10.1016/j.ccr.2013.02.013. [DOI] [PubMed] [Google Scholar]
- De Palma M, Venneri MA, Galli R, Sergi LS, Politi LS, Sampaolesi M, Naldini L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer cell. 2005;8:211–226. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Dedon PC, Tannenbaum SR. Reactive nitrogen species in the chemical biology of inflammation. Arch. Biochem. Biophys. 2004;423:12–22. doi: 10.1016/j.abb.2003.12.017. [DOI] [PubMed] [Google Scholar]
- DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, Coussens LM. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer cell. 2009;16:91–102. doi: 10.1016/j.ccr.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer discovery. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Zhou JF, Sellers RS, Li JF, Nguyen AV, Wang Y, Orlofsky A, Liu Q, Hume DA, Pollard JW, et al. A novel mouse model of inflammatory bowel disease links mammalian target of rapamycin-dependent hyperproliferation of colonic epithelium to inflammation-associated tumorigenesis. Am. J. Pathol. 2010;176:952–967. doi: 10.2353/ajpath.2010.090622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8:1086–1094. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- Ding L, Linsley PS, Huang LY, Germain RN, Shevach EM. IL-10 inhibits macrophage costimulatory activity by selectively inhibiting the up-regulation of B7 expression. J Immunol. 1993;151:1224–1234. [PubMed] [Google Scholar]
- Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, Coussens LM, Karin M, Goldrath AW, Johnson RS. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010;70:7465–7475. doi: 10.1158/0008-5472.CAN-10-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z, Bergers G. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res. 2013;73:6900–6912. doi: 10.1158/0008-5472.CAN-13-1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, Egeblad M, Krummel MF. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer cell. 2012;21:402–417. doi: 10.1016/j.ccr.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito M, Kang Y. Targeting tumor-stromal interactions in bone metastasis. Pharmacol. Ther. 2014;141:222–233. doi: 10.1016/j.pharmthera.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferjancic S, Gil-Bernabe AM, Hill SA, Allen PD, Richardson P, Sparey T, Savory E, McGuffog J, Muschel RJ. VCAM-1 and VAP-1 recruit myeloid cells that promote pulmonary metastasis in mice. Blood. 2013;121:3289–3297. doi: 10.1182/blood-2012-08-449819. [DOI] [PubMed] [Google Scholar]
- Fidler IJ. Macrophage therapy of cancer metastasis. Ciba Found. Symp. 1988;141:211–222. doi: 10.1002/9780470513736.ch12. [DOI] [PubMed] [Google Scholar]
- Fidler IJ. The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis revisited. Nature reviews. Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- Flores Villanueva PO, Reiser H, Stadecker MJ. Regulation of T helper cell responses in experimental murine schistosomiasis by IL-10. Effect on expression of B7 and B7-2 costimulatory molecules by macrophages. J Immunol. 1994;153:5190–5199. [PubMed] [Google Scholar]
- Forget MA, Voorhees JL, Cole SL, Dakhlallah D, Patterson IL, Gross AC, Moldovan L, Mo X, Evans R, Marsh CB, Eubank TD. Macrophage colony-stimulating factor augments tie2-expressing monocyte differentiation, angiogenic function, and recruitment in a mouse model of breast cancer. PloS one. 2014;9:e98623. doi: 10.1371/journal.pone.0098623. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO. The Cellular and Molecular Origin of Tumor-Associated Macrophages. Science. 2014 doi: 10.1126/science.1252510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nature reviews. Immunology. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumors. Nature reviews. Immunology. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nature immunology. 2013;14:1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Joshi N, Choi H, Ryu S, Hahn M, Catena R, Sadik H, Argani P, Wagner P, Vahdat LT, et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res. 2012;72:1384–1394. doi: 10.1158/0008-5472.CAN-11-2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, Erba E, Uboldi S, Zucchetti M, Pasqualini F, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer cell. 2013;23:249–262. doi: 10.1016/j.ccr.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Gil-Bernabe AM, Ferjancic S, Tlalka M, Zhao L, Allen PD, Im JH, Watson K, Hill SA, Amirkhosravi A, Francis JL, et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood. 2012;119:3164–3175. doi: 10.1182/blood-2011-08-376426. [DOI] [PubMed] [Google Scholar]
- Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL, Berman T, Joyce JA. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010;24:241–255. doi: 10.1101/gad.1874010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nature reviews. Immunology. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Greaves P, Gribben JG. The role of B7 family molecules in hematologic malignancy. Blood. 2013;121:734–744. doi: 10.1182/blood-2012-10-385591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald RJ, Latchman YE, Sharpe AH. Negative co-receptors on lymphocytes. Current opinion in immunology. 2002;14:391–396. doi: 10.1016/s0952-7915(02)00341-2. [DOI] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Howe LR, Subbaramaiah K, Hudis CA, Dannenberg AJ. Molecular pathways: adipose inflammation as a mediator of obesity-associated cancer. Clin. Cancer Res. 2013;19:6074–6083. doi: 10.1158/1078-0432.CCR-12-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobin C. Colorectal cancer: looking for answers in the microbiota. Cancer discovery. 2013;3:384–387. doi: 10.1158/2159-8290.CD-13-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nature reviews. Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JM, Darcy PK, Markby JL, Godfrey DI, Takeda K, Yagita H, Smyth MJ. Induction of tumor-specific T cell memory by NK cell-mediated tumor rejection. Nature immunology. 2002;3:83–90. doi: 10.1038/ni746. [DOI] [PubMed] [Google Scholar]
- Kennedy BC, Showers CR, Anderson DE, Anderson L, Canoll P, Bruce JN, Anderson RC. Tumor-associated macrophages in glioma: friend or foe? Journal of oncology. 2013;2013:486912. doi: 10.1155/2013/486912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Fujishita T, Loetscher P, Revesz L, Hashida H, Kizaka-Kondoh S, Aoki M, Taketo MM. Inactivation of chemokine (C-C motif) receptor 1 (CCR1) suppresses colon cancer liver metastasis by blocking accumulation of immature myeloid cells in a mouse model. Proc. Natl. Acad. Sci. U. S. A. 2010;107:13063–13068. doi: 10.1073/pnas.1002372107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Kometani K, Hashida H, Matsunaga A, Miyoshi H, Hosogi H, Aoki M, Oshima M, Hattori M, Takabayashi A, et al. SMAD4-deficient intestinal tumors recruit CCR1+ myeloid cells that promote invasion. Nat. Genet. 2007;39:467–475. doi: 10.1038/ng1997. [DOI] [PubMed] [Google Scholar]
- Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, Pfirschke C, Voss RH, Timke C, Umansky L, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer cell. 2013;24:589–602. doi: 10.1016/j.ccr.2013.09.014. [DOI] [PubMed] [Google Scholar]
- Kren L, Muckova K, Lzicarova E, Sova M, Vybihal V, Svoboda T, Fadrus P, Smrcka M, Slaby O, Lakomy R, et al. Production of immune-modulatory nonclassical molecules HLA-G and HLA-E by tumor infiltrating ameboid microglia/macrophages in glioblastomas: a role in innate immunity? Journal of neuroimmunology. 2010;220:131–135. doi: 10.1016/j.jneuroim.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Kryczek I, Zou L, Rodriguez P, Zhu G, Wei S, Mottram P, Brumlik M, Cheng P, Curiel T, Myers L, et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. 2006;203:871–881. doi: 10.1084/jem.20050930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 2009;206:1327–1337. doi: 10.1084/jem.20082173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuipers H, Muskens F, Willart M, Hijdra D, van Assema FB, Coyle AJ, Hoogsteden HC, Lambrecht BN. Contribution of the PD-1 ligands/PD-1 signaling pathway to dendritic cell-mediated CD4+ T cell activation. European journal of immunology. 2006;36:2472–2482. doi: 10.1002/eji.200635978. [DOI] [PubMed] [Google Scholar]
- Laoui D, Movahedi K, Van Overmeire E, Van den Bossche J, Schouppe E, Mommer C, Nikolaou A, Morias Y, De Baetselier P, Van Ginderachter JA. Tumor-associated macrophages in breast cancer: distinct subsets, distinct functions. Int. J. Dev. Biol. 2011;55:861–867. doi: 10.1387/ijdb.113371dl. [DOI] [PubMed] [Google Scholar]
- LeMaoult J, Caumartin J, Daouya M, Favier B, Le Rond S, Gonzalez A, Carosella ED. Immune regulation by pretenders: cell-to-cell transfers of HLA-G make effector T cells act as regulatory cells. Blood. 2007;109:2040–2048. doi: 10.1182/blood-2006-05-024547. [DOI] [PubMed] [Google Scholar]
- LeMaoult J, Krawice-Radanne I, Dausset J, Carosella ED. HLA-G1-expressing antigen-presenting cells induce immunosuppressive CD4+ T cells. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:7064–7069. doi: 10.1073/pnas.0401922101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin EY, Gouon-Evans V, Nguyen AV, Pollard JW. The macrophage growth factor, CSF-1, in mammary gland development and tumor progression. J. Mammary Gland Biol. Neoplasia. 2002;7:147–162. doi: 10.1023/a:1020399802795. [DOI] [PubMed] [Google Scholar]
- Lin EY, Li J, Bricard G, Wang W, Deng Y, Sellers R, Porcelli SA, Pollard JW. Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages. Molecular Oncology. 2007;1:288–302. doi: 10.1016/j.molonc.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001;193:727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin EY, Pollard JW. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 2007;67:5064–5066. doi: 10.1158/0008-5472.CAN-07-0912. [DOI] [PubMed] [Google Scholar]
- Linde N, Lederle W, Depner S, van Rooijen N, Gutschalk CM, Mueller MM. Vascular endothelial growth factor-induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. J. Pathol. 2012;227:17–28. doi: 10.1002/path.3989. [DOI] [PubMed] [Google Scholar]
- Liou GY, Doppler H, Necela B, Krishna M, Crawford HC, Raimondo M, Storz P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappaB and MMPs. The Journal of cell biology. 2013;202:563–577. doi: 10.1083/jcb.201301001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zhang N, Li Q, Zhang W, Ke F, Leng Q, Wang H, Chen J, Wang H. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PloS one. 2011;6:e19495. doi: 10.1371/journal.pone.0019495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A. 2003;100:5336–5341. doi: 10.1073/pnas.0931259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga T, Saito H, Ikeguchi M. Increased B7-H1 and B7-H4 Expressions on Circulating Monocytes and Tumor-Associated Macrophages are Involved in Immune Evasion in Patients with Gastric Cancer. Yonago acta medica. 2011;54:1–10. [PMC free article] [PubMed] [Google Scholar]
- Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, Politi LS, Gentner B, Brown JL, Naldini L, De Palma M. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer cell. 2011;19:512–526. doi: 10.1016/j.ccr.2011.02.005. [DOI] [PubMed] [Google Scholar]
- McIntire RH, Morales PJ, Petroff MG, Colonna M, Hunt JS. Recombinant HLA-G5 and -G6 drive U937 myelomonocytic cell production of TGF-beta1. Journal of leukocyte biology. 2004;76:1220–1228. doi: 10.1189/jlb.0604337. [DOI] [PubMed] [Google Scholar]
- Mellor AL, Chandler P, Baban B, Hansen AM, Marshall B, Pihkala J, Waldmann H, Cobbold S, Adams E, Munn DH. Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. International immunology. 2004;16:1391–1401. doi: 10.1093/intimm/dxh140. [DOI] [PubMed] [Google Scholar]
- Mills CD. M1 and M2 Macrophages: Oracles of Health and Disease. Crit. Rev. Immunol. 2012;32:463–488. doi: 10.1615/critrevimmunol.v32.i6.10. [DOI] [PubMed] [Google Scholar]
- Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73:1128–1141. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi BG, Neustadter J, Binda E, Lewis J, Filler RB, Roberts SJ, Kwong BY, Reddy S, Overton JD, Galan A, et al. Langerhans cells facilitate epithelial DNA damage and squamous cell carcinoma. Science. 2012;335:104–108. doi: 10.1126/science.1211600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero AJ, Diaz-Montero CM, Kyriakopoulos CE, Bronte V, Mandruzzato S. Myeloid-derived suppressor cells in cancer patients: a clinical perspective. J. Immunother. 2012;35:107–115. doi: 10.1097/CJI.0b013e318242169f. [DOI] [PubMed] [Google Scholar]
- Morandi F, Levreri I, Bocca P, Galleni B, Raffaghello L, Ferrone S, Prigione I, Pistoia V. Human neuroblastoma cells trigger an immunosuppressive program in monocytes by stimulating soluble HLA-G release. Cancer research. 2007;67:6433–6441. doi: 10.1158/0008-5472.CAN-06-4588. [DOI] [PubMed] [Google Scholar]
- Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, Mack M, Pipeleers D, In't Veld P, De Baetselier P, Van Ginderachter JA. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010;70:5728–5739. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nature reviews. Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- Ng TH, Britton GJ, Hill EV, Verhagen J, Burton BR, Wraith DC. Regulation of adaptive immunity; the role of interleukin-10. Frontiers in immunology. 2013;4:129. doi: 10.3389/fimmu.2013.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014;211:781–790. doi: 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucera S, Biziato D, De Palma M. The interplay between macrophages and angiogenesis in development, tissue injury and regeneration. Int. J. Dev. Biol. 2011;55:495–503. doi: 10.1387/ijdb.103227sn. [DOI] [PubMed] [Google Scholar]
- Oh SA, Li MO. TGF-beta: guardian of T cell function. J Immunol. 2013;191:3973–3979. doi: 10.4049/jimmunol.1301843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annu. Rev. Med. 2014;65:185–202. doi: 10.1146/annurev-med-092012-112807. [DOI] [PubMed] [Google Scholar]
- Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, Garcia-Santos G, Ghajar C, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012;18:883–891. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin. Cancer Biol. 2011;21:139–146. doi: 10.1016/j.semcancer.2011.01.002. [DOI] [PubMed] [Google Scholar]
- Pollard JW. Tumor educated macrophages promote tumor progression and metastasis. Nature Reviews Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- Priceman SJ, Sung JL, Shaposhnik Z, Burton JB, Torres-Collado AX, Moughon DL, Johnson M, Lusis AJ, Cohen DA, Iruela-Arispe ML, Wu L. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood. 2010;115:1461–1471. doi: 10.1182/blood-2009-08-237412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nature reviews. Cancer. 2009;9:285–293. doi: 10.1038/nrc2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, Lang RA, Pollard JW. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PloS one. 2009;4:e6562. doi: 10.1371/journal.pone.0006562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumor metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redente EF, Dwyer-Nield LD, Merrick DT, Raina K, Agarwal R, Pao W, Rice PL, Shroyer KR, Malkinson AM. Tumor progression stage and anatomical site regulate tumor-associated macrophage and bone marrow-derived monocyte polarization. Am. J. Pathol. 2010;176:2972–2985. doi: 10.2353/ajpath.2010.090879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I, et al. Targeting Tumor-Associated Macrophages with Anti-CSF-1R Antibody Reveals a Strategy for Cancer Therapy. Cancer cell. 2014 doi: 10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, Delgado A, Correa P, Brayer J, Sotomayor EM, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–5849. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, Ochoa JB, Ochoa AC. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol. 2003;171:1232–1239. doi: 10.4049/jimmunol.171.3.1232. [DOI] [PubMed] [Google Scholar]
- Rohan TE, Xue X, Lin HM, D'Alfonso TM, Ginter PS, Oktay MH, Robinson BD, Ginsberg M, Gertler FB, Glass AG, et al. Tumor microenvironment of metastasis and risk of distant metastasis of breast cancer. J. Natl. Cancer Inst. 2014;106 doi: 10.1093/jnci/dju136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell B, Au A, Rugo HS, Esserman LJ, Hwang ES, Coussens LM. Leukocyte composition of human breast cancer. Proc. Natl. Acad. Sci. U. S. A. 2011 doi: 10.1073/pnas.1104303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangaletti S, Di Carlo E, Gariboldi S, Miotti S, Cappetti B, Parenza M, Rumio C, Brekken RA, Chiodoni C, Colombo MP. Macrophage-derived SPARC bridges tumor cell-extracellular matrix interactions toward metastasis. Cancer Res. 2008;68:9050–9059. doi: 10.1158/0008-5472.CAN-08-1327. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Doring A, Zemp FJ, Silva C, Lun X, Wang X, Kelly J, Hader W, Hamilton M, Mercier P, et al. Therapeutic activation of macrophages and microglia to suppress brain tumor-initiating cells. Nat. Neurosci. 2014;17:46–55. doi: 10.1038/nn.3597. [DOI] [PubMed] [Google Scholar]
- Savage ND, de Boer T, Walburg KV, Joosten SA, van Meijgaarden K, Geluk A, Ottenhoff TH. Human anti-inflammatory macrophages induce Foxp3+ GITR+ CD25+ regulatory T cells, which suppress via membrane-bound TGFbeta-1. J Immunol. 2008;181:2220–2226. doi: 10.4049/jimmunol.181.3.2220. [DOI] [PubMed] [Google Scholar]
- Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, Cerwenka A. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol. 2012;189:5602–5611. doi: 10.4049/jimmunol.1201018. [DOI] [PubMed] [Google Scholar]
- Shand FH, Ueha S, Otsuji M, Koid SS, Shichino S, Tsukui T, Kosugi-Kanaya M, Abe J, Tomura M, Ziogas J, Matsushima K. Tracking of intertissue migration reveals the origins of tumor-infiltrating monocytes. Proc. Natl. Acad. Sci. U. S. A. 2014;111:7771–7776. doi: 10.1073/pnas.1402914111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharda DR, Yu S, Ray M, Squadrito ML, De Palma M, Wynn TA, Morris SM, Jr., Hankey PA. Regulation of macrophage arginase expression and tumor growth by the Ron receptor tyrosine kinase. J Immunol. 2011;187:2181–2192. doi: 10.4049/jimmunol.1003460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, Blanchard D, Bais C, Peale FV, van Bruggen N, et al. Bv8 regulates myeloid-cell-dependent tumor angiogenesis. Nature. 2007;450:825–831. doi: 10.1038/nature06348. [DOI] [PubMed] [Google Scholar]
- Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P, Mantovani A. Macrophage polarization in tumor progression. Semin. Cancer Biol. 2008;18:349–355. doi: 10.1016/j.semcancer.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HO, Stephens ND, Qualls CR, Fligelman T, Wang T, Lin CY, Burton E, Griffith JK, Pollard JW. The clinical significance of inflammatory cytokines in primary cell culture in endometrial carcinoma. Mol Oncol. 2013;7:41–54. doi: 10.1016/j.molonc.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, Daniel D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8 T cells. Oncoimmunology. 2013;2:e26968. doi: 10.4161/onci.26968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S, Liu Q, Chen J, Chen J, Chen F, He C, Huang D, Wu W, Lin L, Huang W, et al. A Positive Feedback Loop between Mesenchymal-like Cancer Cells and Macrophages Is Essential to Breast Cancer Metastasis. Cancer cell. 2014;25:605–620. doi: 10.1016/j.ccr.2014.03.021. [DOI] [PubMed] [Google Scholar]
- Tamura H, Dan K, Tamada K, Nakamura K, Shioi Y, Hyodo H, Wang SD, Dong H, Chen L, Ogata K. Expression of functional B7-H2 and B7.2 costimulatory molecules and their prognostic implications in de novo acute myeloid leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11:5708–5717. doi: 10.1158/1078-0432.CCR-04-2672. [DOI] [PubMed] [Google Scholar]
- Tirapu I, Huarte E, Guiducci C, Arina A, Zaratiegui M, Murillo O, Gonzalez A, Berasain C, Berraondo P, Fortes P, et al. Low surface expression of B7-1 (CD80) is an immunoescape mechanism of colon carcinoma. Cancer Res. 2006;66:2442–2450. doi: 10.1158/0008-5472.CAN-05-1681. [DOI] [PubMed] [Google Scholar]
- Welford AF, Biziato D, Coffelt SB, Nucera S, Fisher M, Pucci F, Di Serio C, Naldini L, De Palma M, Tozer GM, Lewis CE. TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. The Journal of clinical investigation. 2011;121:1969–1973. doi: 10.1172/JCI44562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, Segall JE, Pollard JW, Condeelis J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67:2649–2656. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, Carbone DP, Matrisian LM, Richmond A, Lin PC, Moses HL. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo EJ, Cassetta L, Qian BZ, Lewkowich I, Li JF, Stefater JA, 3rd, Smith AN, Wiechmann LS, Wang Y, Pollard JW, Lang RA. Myeloid WNT7b Mediates the Angiogenic Switch and Metastasis in Breast Cancer. Cancer Res. 2014;74:2962–2973. doi: 10.1158/0008-5472.CAN-13-2421. [DOI] [PMC free article] [PubMed] [Google Scholar]