

Abstract
Purpose: The aim of the present investigation was preparation and characterization of sirolimus solid dispersions by solvent evaporation technique to improve its dissolution properties.
Methods: Polyvinylpyrrolidone (PVP), Poloxamer 188 and Cremophore RH40 were used to prepare the solid dispersions of sirolimus. In vitro dissolution study using USP type I apparatus, were performed in distilled water (containing SLS 0.4%) for pure sirolimus, physical mixtures, Rapamune and prepared solid dispersions. The characterization of solid dispersions was performed using Fourier Transform Infrared (FTIR) Spectroscopy and Differential Scanning Calorimetry (DSC).
Results: More than 75% of sirolimus was released within 30 minutes from all prepared solid dispersions. The dissolution rate of all prepared solid dispersion powders were more than physical mixtures. The absence of sirolimus peak in the DSC spectrum of solid dispersions indicated the conversion of crystalline form of sirolimus into amorphous form. The results from FT-IR spectroscopy showed that there was no significant change in the FT-IR spectrum of solid dispersions indicating absence of well-defined interaction between drug and carriers.
Conclusion: It was concluded that solid dispersion method, using PVP, Poloxamer 188 and Cremophore RH40 can improve dissolution rate of sirolimus.
Keywords: Rapamycin, Poor soluble, Immunosuppressive, Dissolution enhancement
Introduction
Oral route is the simplest and easiest way for drug administration because of the greater stability, smaller bulk and easy production process. Drugs with low aqueous solubility have low dissolution rates and hence suffer from poor oral bioavailability. The absorption rate of poorly water soluble drugs, formulated as an orally administered solid dosage form, is controlled by their dissolution rate in the fluid at the absorption site. Consequently poor solubility results in low bioavailability, increase in the dose, large inter- and intra-subject variation and large variations in blood drug concentrations under fed versus fasted conditions. Enhancement of solubility and dissolution rate is a challenging task in drug development.1-5 Absorption problem occur if the aqueous solubility is less than 1mg/mL. Sirolimus (rapamycin, SRL) is a lipophilic macrocyclic lactone with immunosuppressive properties. SRL shows poor water solubility (2.6 µg/mL) and slow dissolution rate so that dissolution of the drug is the rate limiting step for its absorption. Low solubility of SRL in biological fluids results in poor bioavailability (17%) after oral administration. This intrinsic physicochemical property of SRL and its low chemical stability, limits bioavailability and further drug development.6-10 Then it is important to improve the oral bioavailability of this drug by improving its solubility and dissolution rate. Various techniques were used to improve the dissolution rate of poorly water soluble drugs including micronization, solubilization, salt formation, complexation with polymers, physical form change, use of pro-drug and drug derivatization, addition of surfactants, formation of inclusion complexes with cyclodextrin, formation of amorphous drug, cosolvent approach and the formation of solid dispersions (SDs) with hydrophilic carriers.4,11-19 Solid dispersion is defined as dispersion of one or more active ingredients in an inert carrier or matrix at solid state prepared by melting, solvent, or melting-solvent method. Reduction or absence of aggregation and agglomeration, increasing wettability, obtaining a homogeneous distribution of a small quantity of drug in a solid state may also contribute to increase in dissolution rate. Molecular dispersion of the drug in polymeric carriers may lead to particle size reduction and surface area enhancement, which result in improvement in dissolution rate. Stabilization of unstable drugs is another advantage of solid dispersions.2,20-26 Polymers which are often used in SDs include polyethylene glycols, polyvinylpyrrolidone (PVP), Eudragits, Gelucires, Labrasol, Cremophore, Poloxamers, Gums, Mannitol, Urea and Cellulose derivatives. In the preparation of solid dispersions, solvent evaporation is the main process with large scale production possibilities. In many studies the solvent evaporation approach using different evaporation methods was reported. These methods include rotary evaporation, slow evaporation on a hot plate, evaporation under a stream of nitrogen, spin evaporation, spray drying, and the precipitation of solid dispersions using a super critical fluid.17,23,24,27-30 The solid dispersion technique has often proved to be the most successful in improving the dissolution and bioavailability of poorly soluble, active pharmaceutical ingredients because it is simple, economical and advantageous. The objective of the present investigation was to prepare SRL solid dispersions to improve the solubility of SRL, utilizing three polymers, Poloxamer 188, Polyvinylpyrrolidone and Cremophore RH40.
Materials and Methods
Materials
Sirolimus was purchased from Poli Company (Lazio, Italy). Rapamune tablets were prepared from Wyeth Pharmaceuticals Company (Dublin, Ireland). Lactose, Avicel and Ac-di-sol were obtained from DMV (Veghel, Netherland). Magnesium stearate, Polyvinylpyrrolidone, Cremophore RH40, Poloxamer 188, Tocopheryl acetate, PEG 6000, Sodium lauryl sulphate, Aerosil, Mannitol, Ammonium acetate and Isopropyl alcohol were prepared from Merck Company (Darmstadt, Germany).
Preparation of physical mixtures (PMs)
Physical mixtures were prepared according to the carrier composition presented in Table 1. Accurately weighed quantities of drug (1 mg), excipients (Lactose: 175 mg, Aerosil: 0.4 mg, PEG 6000: 5 mg, Ac-di-sol: 10 mg, Stearate magnesium: 1 mg, Tocopheryl acetate: 1 mg) and carrier were taken in a glass bottle and mixed thoroughly for 5 min by triturating. The resulted product was stored in desiccator to carry out further analysis.
Table 1. Carrier composition of prepared formulations.
| Formulation code |
PVP (mg) |
Poloxamer 188 (mg) |
Cremophore RH40 (mg) |
| 1 | 0 | 14 | 4 |
| 2 | 18 | 0 | 0 |
| 3 | 4 | 13 | 1 |
| 4 | 13 | 4 | 1 |
| 5 | 14 | 0 | 4 |
| 6 | 8 | 8 | 2 |
| 7 | 11 | 4 | 3 |
| 8 | 0 | 18 | 0 |
| 9 | 4 | 11 | 3 |
Preparation of solid dispersion formulations
Solid dispersions of SRL were prepared by the solvent evaporation method. Accurately weighed quantities of SRL and the respective dispersion carrier(s) (Table 1) were transferred into a sufficient quantity of isopropyl alcohol to dissolve the ingredients. The mixture was then added to the blend of excipients according to the geometrical dilution method. The obtained granules were kept at room temperature for 24 hrs for drying followed by sieving (mesh no. 18). The prepared solid dispersions were stored in desiccator for further analysis.
Dissolution studies
The in vitro dissolution study was conducted for physical mixture powders, SDs and Rapamune (as an innovator’s product and standard formulation) using USP dissolution apparatus I. Samples equivalent to1 mg of SRL were added to 500 mL of distilled water (containing SLS 0.4%) which was maintained at 37.0 ± 0.5 °C and stirration rate of 100 rpm. An aliquot of 5 mL sample was withdrawn at 10, 20, 30, 45, 60 and 120 minutes.31 An equal volume of fresh dissolution medium was replaced immediately to keep the constant volume of dissolution medium. The filtered samples of SRL (through Whatmann filters) were assayed using our previously developed RP-HPLC method at 278 nm . Calibration curve was constructed in the range of 125-2000 ng/mL (r2=0.9992). Three replicates of each dissolution test were carried out.32
Fourier Transform Infrared (FTIR) spectroscopy
The IR spectra of pure drug, excipients, physical mixtures and solid dispersions were recorded using FTIR spectrophotometer (Bomem, Otava, Canada). Sample preparation was involved, mixing the sample thoroughly with potassium bromide in the ratio of 1:10 (Sample: KBr) and pressing under 160 KPa pressure. The scanning range was 4000 to 500 cm-1.
Differential scanning calorimetry (DSC)
The DSC thermograms of pure drug, carriers, physical mixtures and solid dispersions were recorded using differential scanning calorimeter (Shimadzu DSC-60, Japan). Five mg of each sample was heated in a sealed aluminum pan from 30 °C to 270 °C at a heating rate of 20 °C/min.
Results and Discussion
In Vitro dissolution studies
In vitro release studies revealed that there was marked increase in the dissolution rate of SRL from all solid dispersion formulations when compared to physical mixtures. From the in vitro drug release profiles (Figure 1), it is clear that all solid dispersions released more than 76% of containing drug during 30 min, whereas the released drug from Rapamune tablets and the physical mixtures was found to be 67% and 54% in 30 min respectively. However after 120 min, the released drug from Rapamune was higher than all solid dispersion formulations. In the case of unprocessed drug only approximately 7% was dissolved at 30 minutes. The in vitro drug release profiles, also demonstrated that formulation F3 and F7 containing all of three carriers, showed higher dissolution rate compared with other SD formulations. This may be attributed to the increase in drug wettability, reduction in particle size, conversion to amorphous form and solubilization of the drug due to the presence of hydrophilic carriers. From Figure 1 it is also clear that physical mixture showed an improvement in dissolution rate as compared to pure drug but not such an extent as by solvent evaporation technique as shown in the same figure. The dissolution rate enhancement for physical mixture (60±2.71% and 82.3±2.03% in 60 and 120 min respectively) was possibly due to close contact of the drug with hydrophilic polymer and surfactants brought about by the dry mixing process. This could result in increased dispensability and wettability of the drug leading to enhanced dissolution rate of SRL. In fact as the soluble carriers dissolve in the medium, the insoluble drug gets exposed to the medium as very fine particles resulting in rapid dissolution. To find out the most effective carrier and the best combination of carriers to enhance release rate of SRL from solid dispersion formulations, further research should be performed.
Figure 1 .
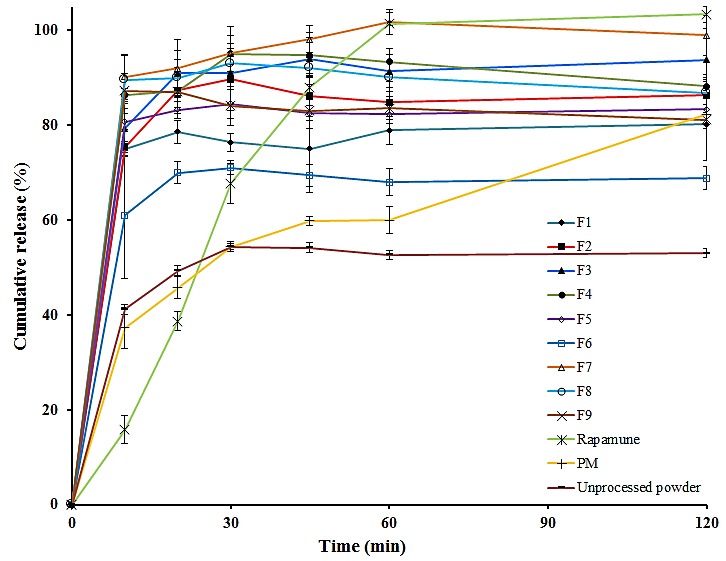
Comparative in vitro dissolution profiles of solid dispersions (F1-F9), physical mixture (PM) and Rapamune.
Fourier Transform Infrared spectroscopy
IR spectroscopic analysis provides information on chemical bonding, functional groups and presence or absence of changes in the crystalline structure of a compound. Spectra obtained using infrared spectroscopy allows to determine physical and chemical properties of the material by recording frequency of vibrations that correlate with common atomic bonds. Interactions observed between the solid dispersion components such as physical adsorption phenomenon, an effect of electrostatic forces, hydrogen bond and van der Waals interactions are in general reversible, while the chemical interactions, including ion exchange, protonation and complexation are irreversible changes.18,29,33-36 Changes in the IR spectrum confirm presence of these interactions and are recorded as new bands, bands disappearing, a shift or widening of the existing bands as well as a change in their intensity. Figure 2 shows the FTIR spectra of pure SRL, physical mixture of SRL with Poloxamer 188 and respective solid dispersions of SRL with Poloxamer 188 and PVP. The characteristic peaks of SRL (1636 cm-1, 1717 cm-1, 2950 cm-1), PVP (1669 cm-1, 3700 cm-1), Poloxamer 188 (3400 cm-1, 1110 cm-1, 3400 cm-1) and physical mixture revealed that there were no differences in the positions of the absorption bands in physical mixture indicating absence of any interaction between SRL and carriers in simple mixing process. Spectrum of solid dispersion prepared by PVP also didn’t represent any interaction. A positive shift in peak at 1717 cm-1 to 1720 cm-1 and elimination of peak at 3400 indicate formation of hydrogen bound between C=O group of SRL and O-H group of Poloxamer 188. However since no changes are seen in other drug bands, therefore the symmetry of drug molecule in solid dispersion prepared with Poloxamer 188 is the same as pure drug.
Figure 2 .
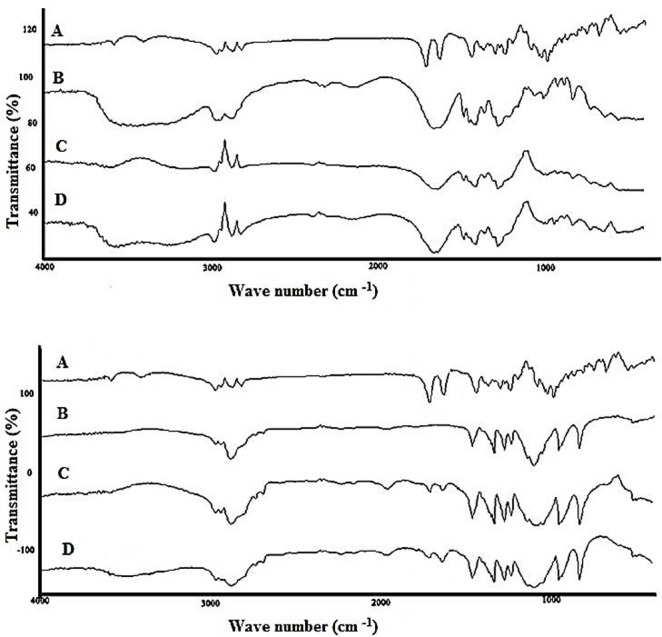
Top: FTIR spectra of pure SRL (A), PVP (B), physical mixture (C) and solid dispersion (D), Bottom: SRL (A), Poloxamer 188 (B), physical mixture (C) and solid dispersion (D).
Differential scanning calorimetry
Thermal analysis consists in measurement of changes in selected physical parameters of a substance when heated under conditions of a linear temperature increase. These changes are recorded as a function of time or temperature. DSC is considered to be a modern and accurate technique used in solid dispersion analysis. DSC enables the quantitative detection of all processes in which energy is required or produced (i.e., endothermic or exothermic phase transformations). It allows exploring the process of melting, crystallization, evaporation, phase equilibriums, sublimation, glassy and polymorphic transformations, dehydration, isomerization, adsorption and substance degradation.25,27,37,38 The thermal behavior of the prepared solid dispersions was studied by DSC. The DSC thermograms for pure SRL, PVP, their PM and SDs are shown in Figure 3. The pure SRL showed two endothermic peaks at 191.35 °C and 205.27 °C indicating crystallinity of SRL. Poloxamer 188 exhibited an endothermic melting peak at 55 °C. Exhibition of a wide peak in the thermogram of PVP at 90-140 °C, demonstrates dehydration of PVP in that range of temperature. Disappearance of endothermic peaks of SRL in the thermogram of solid dispersions prepared by Poloxamer 188 and PVP represents changing of crystalline form of SRL to amorphous form in the solid dispersions. Absence of these peaks in physical mixture may be due to the low concentration of SRL in PM or melting of SRL in Poloxamer 188 during the heating process.
Conclusion
The study revealed that optimum levels of hydrophilic carriers ensure a quick and complete dissolution of SRL from solid dispersions which are used in oral pharmaceutical formulations. The solid dispersions prepared with PVP, Poloxamer 188 and Cremophore RH40 as carrier using the solvent evaporation method improved the dissolution properties of SRL and the in vitro dissolution test showed a significant increase in the dissolution rate of solid dispersions as compared with pure SRL (P<0.05). The mechanism of improvement in the dissolution properties, on the basis of characterizations was predicted to be the changes in the crystal form to amorphous form of the drug molecules in the solid dispersions and wettability property of carriers. The DSC thermogram indicated that crystallinity of the drug was reduced in solid dispersion formulation with Poloxamer 188. Results from IR spectroscopy exhibited that there was interaction between SRL and Poloxamer 188.
Figure 3 .
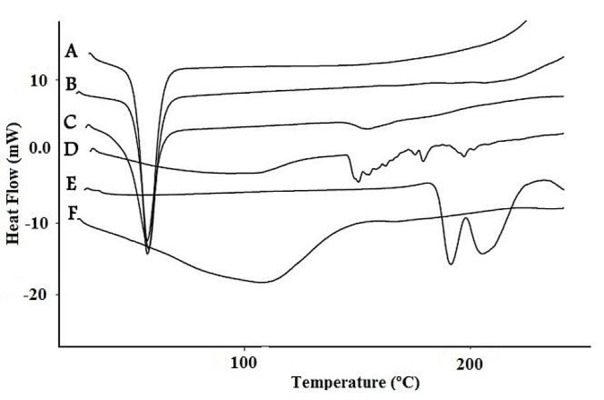
DSC thermograms of Poloxamer 188 (A), physical mixture of Poloxamer 188 and SRL (B), solid dispersion with Poloxamer 188 (C), solid dispersion with PVP (D) , SRL (E) and PVP (F).
Acknowledgments
This article is based on a thesis submitted for Pharm D degree (No.3580) in Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran.
Conflict of Interest
The authors report no conflict of interests.
References
- 1.Thorat AA, Dalvi SV. Liquid antisolvent precipitation and stabilization of nanoparticles of poorly water soluble drugs in aqueous suspensions: Recent developments and future perspective. Chem Eng J . 2012;181–182:1–34. [Google Scholar]
- 2.Kawakami K. Modification of physicochemical characteristics of active pharmaceutical ingredients and application of supersaturatable dosage forms for improving bioavailability of poorly absorbed drugs. Adv Drug Deliv Rev . 2012;64(6):480–95. doi: 10.1016/j.addr.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 3.Elder DP, Holm R, Diego HL. Use of pharmaceutical salts and cocrystals to address the issue of poor solubility. Int J Pharm . 2013;453(1):88–100. doi: 10.1016/j.ijpharm.2012.11.028. [DOI] [PubMed] [Google Scholar]
- 4.Desai PP, Date AA, Patravale VB. Overcoming poor oral bioavailability using nanoparticle formulations - opportunities and limitations. Drug Discov Today Technol . 2012;9(2):e87–95. doi: 10.1016/j.ddtec.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Merisko-Liversidge E, Liversidge GG. Nanosizing for oral and parenteral drug delivery: A perspective on formulating poorly-water soluble compounds using wet media milling technology. Adv Drug Deliv Rev . 2011;63(6):427–40. doi: 10.1016/j.addr.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Simamora P, Alvarez JM, Yalkowsky SH. Solubilization of rapamycin. Int J Pharm . 2001;213(1–2):25–9. doi: 10.1016/s0378-5173(00)00617-7. [DOI] [PubMed] [Google Scholar]
- 7.Hu X, Lin C, Chen D, Zhang J, Liu Z, Wu W. et al. Sirolimus solid self-microemulsifying pellets: Formulation development, characterization and bioavailability evaluation. Int J Pharm . 2012;438(1–2):123–33. doi: 10.1016/j.ijpharm.2012.07.055. [DOI] [PubMed] [Google Scholar]
- 8.Sen HN, Larson TA, Meleth AD, Smith WM, Nussenblatt RB. Subconjunctival sirolimus for the treatment of chronic active anterior uveitis: Results of a pilot trial. Am J Ophthalmol . 2012;153(6):1038–42. doi: 10.1016/j.ajo.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghanbarzadeh S, Valizadeh H, Zakeri-Milani P. Application of factorial designs and response surface methodology in formulation development of sirolimus liposome prepared by thin film hydration technique. BioImpacts . 2013;3(2):75–81. doi: 10.5681/bi.2013.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghanbarzadeh S, Valizadeh H, Zakeri-Milani P. The effects of lyophilization on the physico-chemical stability of sirolimus liposomes. Adv Pharm Bull . 2013;3(1):25–9. doi: 10.5681/apb.2013.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahuja N, Katare OP, Singh B. Studies on dissolution enhancement and mathematical modeling of drug release of a poorly water-soluble drug using water-soluble carriers. Eur J Pharm Biopharm . 2007;65(1):26–38. doi: 10.1016/j.ejpb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 12.Chakraborty S, Shukla D, Mishra B, Singh S. Lipid--an emerging platform for oral delivery of drugs with poor bioavailability. Eur J Pharm Biopharm . 2009;73(1):1–15. doi: 10.1016/j.ejpb.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 13.Chen H, Khemtong C, Yang X, Chang X, Gao J. Nanonization strategies for poorly water-soluble drugs. Drug Discov Today . 2011;16(7–8):354–60. doi: 10.1016/j.drudis.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 14.Gao L, Liu G, Ma J, Wang X, Zhou L, Li X. Drug nanocrystals: In vivo performances. J Control Release . 2012;160(3):418–30. doi: 10.1016/j.jconrel.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 15.Rojtanatanya S, Pongjanyakul T. Propranolol–magnesium aluminum silicate complex dispersions and particles: Characterization and factors influencing drug release. Int J Pharm . 2010;383(1–2):106–15. doi: 10.1016/j.ijpharm.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 16.Shegokar R, Müller RH. Nanocrystals: Industrially feasible multifunctional formulation technology for poorly soluble actives. Int J Pharm . 2010;399(1–2):129–39. doi: 10.1016/j.ijpharm.2010.07.044. [DOI] [PubMed] [Google Scholar]
- 17.Wu C, Wang Z, Zhi Z, Jiang T, Zhang J, Wang S. Development of biodegradable porous starch foam for improving oral delivery of poorly water soluble drugs. Int J Pharm . 2011;403(1–2):162–9. doi: 10.1016/j.ijpharm.2010.09.040. [DOI] [PubMed] [Google Scholar]
- 18.Zidan AS, Rahman Z, Sayeed V, Raw A, Yu L, Khan MA. Crystallinity evaluation of tacrolimus solid dispersions by chemometric analysis. Int J Pharm . 2012;423(2):341–50. doi: 10.1016/j.ijpharm.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 19.Valizadeh H, Zakeri-Milani P, Barzegar-Jalali M, Mohammadi G, Danesh-Bahreini MA, Adibkia K. et al. Preparation and characterization of solid dispersions of piroxicam with hydrophilic carriers. Drug Dev Ind Pharm . 2007;33(1):45–56. doi: 10.1080/03639040600814965. [DOI] [PubMed] [Google Scholar]
- 20.Baird JA, Taylor LS. Evaluation of amorphous solid dispersion properties using thermal analysis techniques. Adv Drug Deliv Rev . 2012;64(5):396–421. doi: 10.1016/j.addr.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 21.Chauhan B, Shimpi S, Paradkar A. Preparation and evaluation of glibenclamide-polyglycolized glycerides solid dispersions with silicon dioxide by spray drying technique. Eur J Pharm Sci . 2005;26(2):219–30. doi: 10.1016/j.ejps.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 22.Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm . 2000;50(1):47–60. doi: 10.1016/s0939-6411(00)00076-x. [DOI] [PubMed] [Google Scholar]
- 23.Kim EJ, Chun MK, Jang JS, Lee IH, Lee KR, Choi HK. Preparation of a solid dispersion of felodipine using a solvent wetting method. Eur J Pharm Biopharm . 2006;64(2):200–5. doi: 10.1016/j.ejpb.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 24.Kolasinac N, Kachrimanis K, Homsek I, Grujic B, Duric Z, Ibric S. Solubility enhancement of desloratadine by solid dispersion in poloxamers. Int J Pharm . 2012;436(1–2):161–70. doi: 10.1016/j.ijpharm.2012.06.060. [DOI] [PubMed] [Google Scholar]
- 25.Sethia S, Squillante E. Solid dispersion of carbamazepine in PVP K30 by conventional solvent evaporation and supercritical methods. Int J Pharm . 2004;272(1–2):1–10. doi: 10.1016/j.ijpharm.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 26.Mohammadi G, Barzegar-Jalali M, Valizadeh H, Nazemiyeh H, Barzegar-Jalali A, Siahi Shadbad MR. et al. Reciprocal powered time model for release kinetic analysis of ibuprofen solid dispersions in oleaster powder, microcrystalline cellulose and crospovidone. J Pharm Pharm Sci . 2010;13(2):152–61. doi: 10.18433/j3jg61. [DOI] [PubMed] [Google Scholar]
- 27.Barmpalexis P, Kachrimanis K, Georgarakis E. Solid dispersions in the development of a nimodipine floating tablet formulation and optimization by artificial neural networks and genetic programming. Eur J Pharm Biopharm . 2011;77(1):122–31. doi: 10.1016/j.ejpb.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 28.Karavas E, Georgarakis E, Bikiaris D. Application of PVP/HPMC miscible blends with enhanced mucoadhesive properties for adjusting drug release in predictable pulsatile chronotherapeutics. Eur J Pharm Biopharm . 2006;64(1):115–26. doi: 10.1016/j.ejpb.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 29.Newa M, Bhandari KH, Li DX, Kwon TH, Kim JA, Yoo BK. et al. Preparation, characterization and in vivo evaluation of ibuprofen binary solid dispersions with poloxamer 188. Int J Pharm . 2007;343(1–2):228–37. doi: 10.1016/j.ijpharm.2007.05.031. [DOI] [PubMed] [Google Scholar]
- 30.Sarmento B, Ferreira D, Veiga F, Ribeiro A. Characterization of insulin-loaded alginate nanoparticles produced by ionotropic pre-gelation through DSC and FTIR studies. Carbohydr Polym . 2006;66(1):1–7. [Google Scholar]
- 31.U.S. Food and Drug Administration. Dissolution Methods. 2013; Available from: http://www.accessdata.fda.gov/scripts/cder/dissolution/index.cfm.
- 32.Islambulchilar Z, Ghanbarzadeh S, Emami S, Valizadeh H, Zakeri-Milani P. Development and validation of an HPLC method for the analysis of sirolimus in drug products. Adv Pharm Bull . 2012;2(2):135–9. doi: 10.5681/apb.2012.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jamrogiewicz M. Application of the near-infrared spectroscopy in the pharmaceutical technology. J Pharm Biomed Anal . 2012;66:1–10. doi: 10.1016/j.jpba.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 34.Rahman Z, Zidan AS, Khan MA. Risperidone solid dispersion for orally disintegrating tablet: Its formulation design and non-destructive methods of evaluation. Int J Pharm . 2010;400(1–2):49–58. doi: 10.1016/j.ijpharm.2010.08.025. [DOI] [PubMed] [Google Scholar]
- 35.Zahedi P, Lee PI. Solid molecular dispersions of poorly water-soluble drugs in poly(2-hydroxyethyl methacrylate) hydrogels. Eur J Pharm Biopharm . 2007;65(3):320–8. doi: 10.1016/j.ejpb.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 36.Krishnan M, Flanagan DR. FTIR-ATR spectroscopy for monitoring polyanhydride/anhydride-amine reactions. J Control Release . 2000;69(2):273–81. doi: 10.1016/s0168-3659(00)00312-6. [DOI] [PubMed] [Google Scholar]
- 37.Wu JX, Yang M, Berg F, Pajander J, Rades T, Rantanen J. Influence of solvent evaporation rate and formulation factors on solid dispersion physical stability. Eur J Pharm Sci . 2011;44(5):610–20. doi: 10.1016/j.ejps.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 38.Vippagunta SR, Maul KA, Tallavajhala S, Grant DJ. Solid-state characterization of nifedipine solid dispersions. Int J Pharm . 2002;236(1–2):111–23. doi: 10.1016/s0378-5173(02)00019-4. [DOI] [PubMed] [Google Scholar]