

Abstract
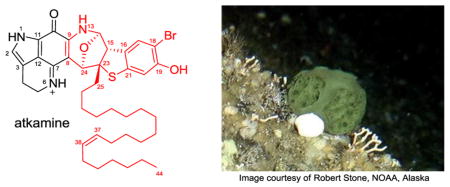
A new pyrroloiminoquinone alkaloid, named atkamine, with an unusual scaffold was discovered from a cold, deep water Alaskan sponge Latrunculia sp. collected from the Aleutian Islands. Olefin metathesis was utilized to determine the location of the double bond in the hydrocarbon chain. The absolute configuration was determined by using computational approaches combing with the ECD (electronic circular dichroism) spectroscopy.
The pyrroloiminoquinone alkaloids are known for their potent bioactivity toward various types of tumor cell lines1,2 and with the potential target of mammalian topoisomerase II in vivo.3 Other bioactivities of this alkaloid class include antimicrobial, antiviral, antimalarial, caspase inhibition, feeding deterrence, and immunomodulatory.4 These utilities together with the highly strained ring system have attracted a broad range of interests. Several total syntheses of this alkaloid class have been developed, including discorhabdin A,5 makaluvamine D,6 and other derivatives.7 Our discovery efforts searching for novel ring systems from the Alaskan marine region prompted us to focus on the cold water Alaska sponge Latrunculia sp. Although new discorhabdin alkaloids were discovered from our previous studies,8 an assessment of the extracts using LCMS revealed the generation of uniquely different formulas from those previously reported. As a result, from our recollection, a novel-type of pyrroloiminoquinone alkaloid was discovered. We report here a strained heterocyclic ring system named atkamine9 and elucidated with the assistance of chemical degradation, NMR, ECD spectra, and computational approaches.
Atkamine was isolated as a green-purple TFA salt soluble in methanol, DMSO, and dichloromethane. HRESIMS showed quasimolecular ions at m/z 734.3006 [M + H]+ and 736.3008 [M + H + 2]+ in a ratio of approximately 1:1.2 generating a reasonable molecular formula of C40H53BrN3O3S (±Δ 2.79 ppm). The UV spectrum showed an absorbance band range from 270 to 450 nm with the peak absorbance of 313 and 360 nm referring to a conjugated system. Acquiring from NMR experiments, the structure of the pyrroloiminoquinone motif was retained;8 the rest of this molecule was changed dramatically from other reported pyrroloiminoquinone alkaloids.1,4
Structural elucidation started from a tertiary carbon resonance (δ 81.1). The attached proton H (δ 5.42) showed a two-bond HMBC (Figure 1) correlation to C8; two 3-bond HMBC correlations to C7 and C9; and a weak 4-bond “W”-type HMBC correlation to C10, which together demonstrated that C24 was connected to the C8 on the pyrroloiminoquinone core. The chemical shift of C24 (δ 81.1) and H24 (δ 5.42) suggested an oxygen attachment. H24 was detected as a singlet by proton NMR with an additional HMBC correlation to a quaternary carbon (δ 78.3) thus establishing the covalent connection of C24– C23 (δ 78.3). On the basis of this connection, the other two HMBC correlations of H24 were three-bond correlations to two tertiary carbons, C (δ 90.2) and C (δ 69.0), through the ether linkage and through the quaternary C23, respectively. The characteristic chemical shift of C9 (δ 147.3) indicated an amino substitution on the α-position of the carbonyl group, which was commonly found in pyrroloiminoquinone derivatives.4–8 This assignment was verified by an HMBC correlation from H (δ 5.23) to C9; the carbon (δ 90.2) was thus arranged as C14 connecting to the pyrroloiminoquinone core through N13. This arrangement was also supported by the HMBC correlation from H14 to C24. Fused to the pyrroloiminoquinone core, the 1,3-oxazinane moiety constructed by C8, C9, N13, C14, O14, and C24 was established.
Figure 1.
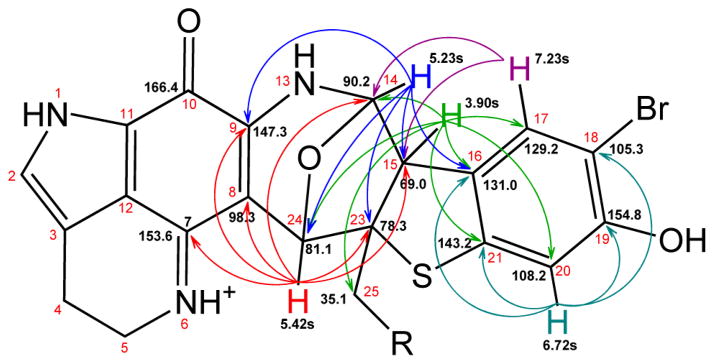
Key NMR chemical shifts and HMBC correlations of atkamine (R = FA side chain).
Based on this elucidation, the HMBC correlation from H24 to C (δ 69.0) must be a 3-bond correlation through the quaternary C23, and C (δ 69.0) was therefore attached to C23. The tertiary C (δ 69.0) was demonstrated to be also connected to C14 because H14 showed the HMBC correlations to C23 and C (δ 69.0). It was therefore elucidated as C15 (δ 69.0) located between C14 and C23. This arrangement can be verified by the HMBC correlations from H15 (δ 3.90) to C14 and C24. The bridged seven-membered ring (8-oxa-2-azabicyclo[3.2.1]oct-3-ene) constructed by C8, C9, N13, C14, C15, C23 and C24 was thus assigned.
H15 showed four strong HMBC correlations to a set of aromatic carbon resonances (δ 108.2, 129.2, 131.0, and 143.2). These data established the connection of C15 to a substituted benzene moiety. The arrangement of the aromatic carbons and substitution pattern of the benzene ring was established first by a key HMBC correlation from H15 to the aromatic C (δ 129.2). Based on the aromatic proton resonances and HMBC correlations, it was clear that the two sharp singlet resonances H (δ 7.23s) and H (δ 6.72s) belonged to the benzene ring and were para to each other. According to the chemical shifts, C (δ 105.3) was shielded and C (δ 154.8) was deshielded, which indicated the charge distribution pattern and corresponding bromine and the oxygenated substitution, respectively. The remaining substituted aromatic C (δ 143.2) was shown to be attached to a sulfur10 linked to the quaternary C23 (δ 78.3) based on the HMBC correlation from H15 to C21. The positions of the aromatic carbons were thus arranged as C16 (δ 131.0), C17 (δ 129.2), C18 (δ 105.3), 19 (δ 154.8), C20 (δ 108.2), and C21 (δ 143.2). To secure this arrangement, the HMBC correlations from the aromatic proton H17 to C15; from H20 to C16, C18, C19, and C21; from H14 to C16; and from H15 to C16, C17 and C20 were verified. Interestingly, the aromatic C18 and C20 signals were at unusually high field which referred to highly local negative charge distribution.
From the remaining proton NMR signals, three peak clusters including a deformed triplet of the terminal methyl resonance, CH3 (δ 0.81 t), a series of overlapped methylene signals, (CH2)n (δ 1.00–1.55), and two overlapped olefinic proton resonances, H (δ 5.28) and H (δ 5.30), could be clearly observed. This evidence supported a monounsatu-rated aliphatic side chain. Analyzing the 2D NMR spectra, the isolated spin system on the side chain can be readily deduced. This side chain was attached to the quaternary C23 based on the HMBC correlation from H15 to C25 (δ 35.1) and the correlations from H25a (δ 1.76) and H25b (δ 1.87) to C23. The chemical shifts of the allylic C36 (δ 26.9) and C39 (δ 27.0) along with the coupling constant of approximately 10 Hz between the two olefinic protons suggested a Z configuration of the double bond,11,12 which also matched well with the chemical shift prediction for the allylic carbons (Figure S15, Supporting Information). However, because of the overlapped signals on the aliphatic region, the position of this double bond was difficult to determine by using NMR. This issue was solved by fragmentation using an olefin metathesis (Figure 2).
Figure 2.
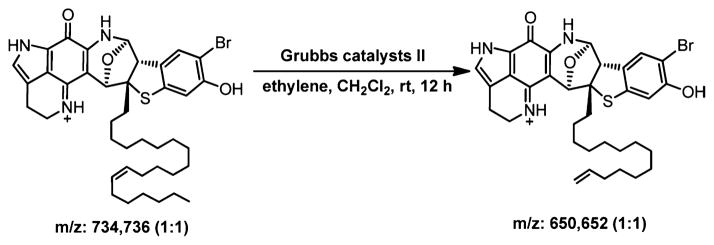
Olefin metathesis on atkamine.
The spatial relationship of the atoms was established based on the NOESY experiment, and the relative configuration was thus elucidated (Figure 3). From the observed NOESY correlations, it was clear that H14, H15, H24, and C25 were syn to each other, and the heterocycles were all cis-fused.
Figure 3.

Perspective structure and key NOESY correlations of atkamine with calculated distances (red, in angstroms).
The crystal for an XRD structure of atkamine was unachievable likely due to the limited amount of the sample and the flexible side chain. Alternatively, molecular modeling approaches were adopted. The rigid backbone with high electron density provided atkamine as an ideal candidate for molecular modeling using ab initio calculations. By using computational approaches and ECD experiments, the absolute configuration was determined. The MMFF-minimized structures were optimized in silico by using the hybrid DFT13 calculations with the B3LYP or BH&HLYP methods with the 6-31G(d,p) or TZVP basis set and the PCM14 solvation model (Figure 4), respectively, to enhance the reliability. Notably, resulting from the optimized structures, the dihedral angle of H14–C14–C15–H15 was nearly 90°, which explained the undetectable 3JH14–H15 on the proton NMR, and this also matched well with the Karplus equation. The optimized structures were then used for the excited-state TDDFT calculations, and the resulted excitation energies and rotational strengths were Boltzmann weighted and fit to a Gaussian function to generate a computed ECD spectrum and subsequently superimposed on the experiment data. Analyzing from the long wavelength region, the computed spectra showed two electronic transitions at 380 and 330 nm which were also recorded by the experiment. This absorption band was elucidated as a set of overlapped signals produced by the n → π* transitions from the hetero-atomic substations on the aromatic system. The π → π* transitions and the aromatic 1Lb transitions at the region of 250–300 nm were a more specific and characteristic presentation of the electronic structure of the conjugated system, and this was also reproduced by the calculations. From the overlaid ECD spectra (Figure 5), the absolute configuration of atkamine was thus assigned as 14R,15S,23S,24R.
Figure 4.
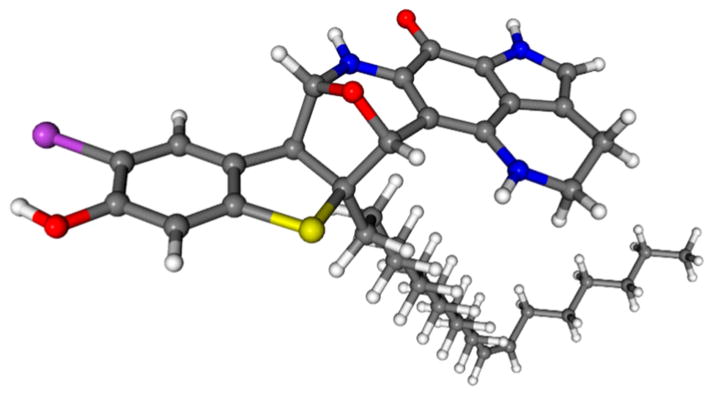
DFT-optimized structure of atkamine.
Figure 5.

Experimental ECD spectra and computed excited states and ECD curves by Boltzmann average.
Atkamine presented a new ring system. Intriguingly, the biogenesis of atkamine likely originates from just two standard amino acids and a monounsaturated fatty acid precursor without rearrangement (Figure 6). A proposed biogenesis is provided in the Supporting Information (Figure S11). Atkamine appears to utilize the common starting material and intermediate from the makaluvamine pathway.4 It may derive from makaluvamine M, and the aliphatic side chain has possibly originated from (Z)-15-docosenoic acid, a primary metabolite which was commonly found in sponge species with abundance.15 Several key steps included an acylation between the enamine moiety and the FA source,16 the formation of a quinone methide intermediate,17 consecutive conjugate additions,18 and a finally reductive cyclization. The incorporation of the sulfur in vivo is still under investigation, although it was achieved synthetically by nucleophilic addition,5 and two possible pathways were proposed here.
Figure 6.
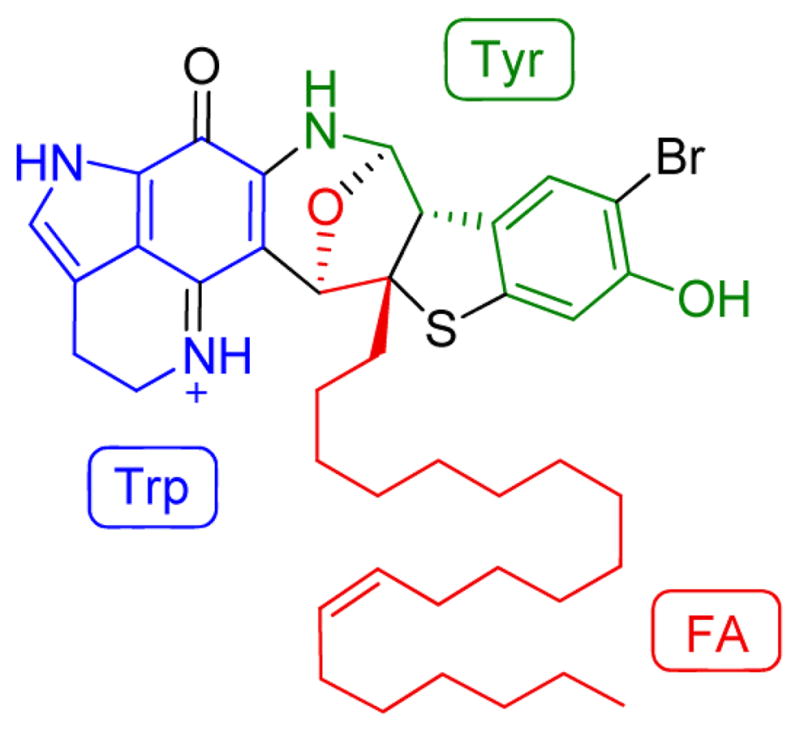
Core components of atkamine.
Atkamine represents a unique pyrroloiminoquinone alkaloid core. The highly fused rings joined with several heteroatoms will provide a synthetically challenging target. The bioactivity of atkamine has not been presented due to the remote collection site and the challenge of sample recovery. A total synthesis will be essential to further investigate the chemistry and effectively characterize the biological potential for this new ring system.
Supplementary Material
Acknowledgments
We thank Dr. James W. Sims (Department of Pharmacognosy, School of Pharmacy, University of Mississippi) and Dr. Robert Stone (NOAA Alaskan Field Surveys) for the sample collection and the Mississippi Center for Supercomputing Research (MCSR) for supercomputing time.
Footnotes
The authors declare no competing financial interest.
Supporting Information Available. Experimental procedure, NMR spectropic data, and computational data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Antunes EM, Copp BR, Davies-Coleman MT, Samaai T. Nat Prod Rep. 2005;22:62. doi: 10.1039/b407299p. [DOI] [PubMed] [Google Scholar]
- 2.Crews P, Gerwick W, Schmitz F, France D, Bair K, Wright A, Hallock Y. Pharm Biol (Lisse, Neth) 2003;41:39. [Google Scholar]
- 3.Radisky DC, Radisky ES, Barrows LR, Copp BR, Kramer RA, Ireland CM. J Am Chem Soc. 1993;115:1632. [Google Scholar]
- 4.Hu JF, Fan H, Xiong J, Wu SB. Chem Rev (Washington, DC, US) 2011;111:5465. doi: 10.1021/cr100435g. [DOI] [PubMed] [Google Scholar]
- 5.Tohma H, Harayama Y, Hashizume M, Iwata M, Kiyono Y, Egi M, Kita Y. J Am Chem Soc. 2003;125:11235. doi: 10.1021/ja0365330. [DOI] [PubMed] [Google Scholar]
- 6.White JD, Yager KM, Yakura T. J Am Chem Soc. 1994;116:1831. [Google Scholar]
- 7.(a) Rives A, Delaine T, Legentil L, Delfourne E. Tetrahedron Lett. 2009;50:1128. [Google Scholar]; (b) Iwao M, Motoi O, Fukuda T, Ishibashi F. Tetrahedron. 1998;54:8999. [Google Scholar]; (c) Morooka Y, Fukuda T, Iwao M. Tetrahedron Lett. 1999;40:1713. [Google Scholar]; (d) Izawa T, Nishiyama S, Yamamura S. Tetrahedron. 1994;50:13593. [Google Scholar]; (e) Wada Y, Otani K, Endo N, Harayama Y, Kamimura D, Yoshida M, Fujioka H, Kita Y. Tetrahedron. 2009;65:1059. doi: 10.1021/ol901471r. [DOI] [PubMed] [Google Scholar]; (f) Oshiyama T, Satoh T, Okano K, Tokuyama H. Tetrahedron. 2012;68:9376. [Google Scholar]
- 8.Na M, Ding Y, Wang B, Tekwani BL, Schinazi RF, Franzblau S, Kelly M, Stone R, Li XC, Ferreira D, Hamann MT. J Nat Prod. 2010;73:383. doi: 10.1021/np900281r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Named after a major Aleutian Island known as Atka Island.
- 10.This sulfur substitution is commonly found in pyrroloiminoquinone alkaloids.1,4
- 11.Gunstone FD, Pollard MR, Scrimgeour CM, Vedanayagam HS. Chem Phys Lipids. 1977;18:115. doi: 10.1016/0009-3084(77)90031-7. [DOI] [PubMed] [Google Scholar]
- 12.Bus J, Sies I, Lie Ken Jie MSF. Chem Phys Lipids. 1977;18:130. doi: 10.1016/0009-3084(77)90032-9. [DOI] [PubMed] [Google Scholar]
- 13.Density functional theory: Kohn W, Sham LJ. Phys Rev. 1965;140:A1133.Hohenberg P, Kohn W. Phys Rev. 1964;136:B864.Calais JL. Int J Quantum Chem. 1993;47:101.Comstock MJ, editor. In The Challenge of d and f Electrons. Vol. 394. American Chemical Society; Washington, DC: 1989. p. i.
- 14.Polarizable continuum model: Pascualahuir JL, Silla E, Tuñon I. J Comput Chem. 1994;15:1127.Cossi M, Barone V, Cammi R, Tomasi J. Chem Phys Lett. 1996;255:327.
- 15.Denis C, Wielgosz-Collin G, Bretéché A, Ruiz N, Rabesaotra V, Boury-Esnault N, Kornprobst JM, Barnathan G. Lipids. 2009;44:655. doi: 10.1007/s11745-009-3291-9. [DOI] [PubMed] [Google Scholar]
- 16.(a) Denic V, Weissman JS. Cell. 2007;130:663. doi: 10.1016/j.cell.2007.06.031. [DOI] [PubMed] [Google Scholar]; (b) Jakobsson A, Westerberg R, Jacobsson A. Prog Lipid Res. 2006;45:237. doi: 10.1016/j.plipres.2006.01.004. [DOI] [PubMed] [Google Scholar]; (c) Su HM, Moser AB, Moser HW, Watkins PA. J Biol Chem. 2001;276:38115. doi: 10.1074/jbc.M106326200. [DOI] [PubMed] [Google Scholar]
- 17.Bolton JL, Turnipseed SB, Thompson JA. Chem-Biol Interact. 1997;107:185. doi: 10.1016/s0009-2797(97)00079-3. [DOI] [PubMed] [Google Scholar]
- 18.Begley TP, Xi J, Kinsland C, Taylor S, McLafferty F. Curr Opin Chem Biol. 1999;3:623. doi: 10.1016/s1367-5931(99)00018-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.